

Abstract
Cancer is a heterogeneous disease caused by diverse genomic alterations in oncogenes and tumor suppressor genes. Despite recent advances in high-throughput sequencing technologies and development of targeted therapies, novel cancer drug development is limited due to the high attrition rate from clinical studies. Patient-derived xenografts (PDX), which are established by the transfer of patient tumors into immunodeficient mice, serve as a platform for co-clinical trials by enabling the integration of clinical data, genomic profiles, and drug responsiveness data to determine precisely targeted therapies. PDX models retain many of the key characteristics of patients’ tumors including histology, genomic signature, cellular heterogeneity, and drug responsiveness. These models can also be applied to the development of biomarkers for drug responsiveness and personalized drug selection. This review summarizes our current knowledge of this field, including methodologic aspects, applications in drug development, challenges and limitations, and utilization for precision cancer medicine.
Keywords: cancer drug development, cancer genomics, patient-derived xenografts, precision medicine
INTRODUCTION
Emerging evidence has indicated that cancer is a genomic disease caused by diverse alterations in oncogenes and tumor suppressors associated with oncogenic signaling pathways (Garraway et al., 2013). Recent advances in high-throughput sequencing technologies have elucidated the mutational landscape of several cancers and provided a comprehensive catalogue of cancer genes (Bell et al., 2011; Koboldt et al., 2012; Muzny et al., 2012; Verhaak et al., 2010; Weinstein et al., 2013). These and other genomic analyses of cancers have demonstrated that cancer is often a highly heterogeneous disease, even with respect to genomic mutation levels and tumor histology in the same cancer type. However, traditional cancer classification based on the cellular origin of tumors and on tumor histology cannot explain these heterogeneous characteristics. This widespread heterogeneity can explain, to some degree, why some patients with a given cancer type are more or less resistant to a particular therapy than other patients. The concept of precision medicine, whereby patients are grouped into subpopulations based on sophisticated genomic profiling, aims to address this problem by enabling development of therapies designed to specifically target the unique cancer of an individual or subgroup of individuals.
For some cancers, extensive genomic profiling has yielded identification of many cancer-related genes, which in turn, has led to identification of genomic alterations that can be targeted for therapy. Examples of drugs that target specific genomic alternations and have been translated to the clinic include imatinib, which targets the BCR-ABL1 fusion protein in chronic myeloid leukemia (Quintas-Cardama and Cortes, 2009), trastuzumab, which targets HER2 amplification in breast and gastric cancer (Slamon et al., 2011), gefitinib and erlotinib targeting the EGFR mutation in lung cancer (Pao et al., 2004), and crizotinib targeting the EML4-ALK fusion protein in lung cancer (Shaw et al., 2011). In addition to enabling the translation of such drugs to the clinic, the accelerated development of targeted therapies has promoted the concept that treatment decisions might eventually be guided by the molecular profiling of individual tumors (Macconaill and Garraway, 2010). The integration of genomic profiling and targeted therapy are now broadly investigated in almost all kinds of cancer types.
However, drug development in oncology is usually hampered by high clinical attrition rates (DiMasi et al., 2013). The clinical approval success rates of cancer drugs are lower than 15%, especially lower in solid tumors (DiMasi et al., 2013). Despite the great promise of genomic alteration targeted therapy, it has proven difficult to identify targetable genomic alterations in quite a lot of cancers. Genomic instability of cancer increases the complexity of the cancer phenotype, as genomic instability in a given cancer can result in simultaneous alterations in several oncogenes and tumor suppressors involved in various cancer-associated signaling pathways. In addition, a tumor is a mixture of heterogeneous cells including cancer cells, fibroblasts, vessel cells, and immune cells. Even cancer cells from a single patient exhibit diverse characteristics in the genomic and transcriptomic signature, contributing to the phenomenon of the intra-tumoral heterogeneity of cancer. Therefore, in many cases, drug responses cannot be fully anticipated via simple genetic profiling.
To overcome these limitations, more precise and comprehensive profiling of genomic alterations in tumors, and of drug responsiveness, is needed. Patient-derived xenografts (PDX), a research platform whereby patient tumors are transplanted into immunodeficient mice, are designed to meet this need by providing clinically predictive models of human cancers. This review delineates recent advances of PDX models and their significance for precision cancer medicine.
PATIENT-DERIVED XENOGRAFT MODELS: OVERVIEW
PDX models are generated by implanting sectioned patient tumor fragments into immunodeficient mice, subcutaneously or orthotopically (into the organ of the cancer origin). Although previously used methods for drug testing such as in vitro cell culture and organoid cultures are advantageous in easy genetic manipulation and high-throughput screening, they showed strong selective proliferation, adaptation to culture condition and loss of tumor heterogeneity (Gao and Chen, 2015). An important advantage of PDX models is that they retain key characteristics of patients’ tumors. The histologic characteristics and genomic signature of the patient tumor, and the heterogeneity of cancer cells, are highly preserved in PDX tumors (Kopetz et al., 2012). PDX tumors also contain stromal and immune cells originating from the patient (Kopetz et al., 2012). Thus, PDX models are the most clinically relevant cancer models developed to date, and represent a highly predictive drug response platform that recapitulates the therapeutic outcome in human patients.
The procedures for the generation of PDX are very straight-forward, comprising the collection of fresh surgical tumor-containing tissue, division of the tumor into pieces, and then implantation, either subcutaneously or orthotopically, into an immunodeficient mouse (Fig. 1; Tentler et al., 2012). In some cases, investigators put tumors into the renal capsule for effective blood flow. The tumors, which are established in immunodeficient mice, are cut into 2–4 mm fragments and reimplanted into new hosts for next passage. Several different mouse strains are used for generation of PDX models, and they differ with respect to the degree of immunosuppression, research benefits, and experimental considerations (Table 1). Nude, B6 Rag 1 and BALB scid mice have intact innate immunity and NK cells, and can be used for engraftment of human and mouse cancer cell lines. On the contrary, NOD scid and NOD/scid/IL2γ-receptor null (NSG) mice have defects in both humoral and innate immunity, and support implantation of human hematopoietic cells and solid tumors. Human acute myeloid leukemia and acute lymphoblastic leukemia primary cells showed faster leukemic symptom and more leukemic cells in NSG mice compared to other immunodeficient mice (Agliano et al., 2008). The time needed for engraftment of PDX mice differs according to tumor type, site of implantation, and mouse strain, but in general it takes between two and four months (Morton and Houghton, 2007). For drug treatment testing, tumor-bearing mice can be subsequently expanded from early passages.
Fig. 1.
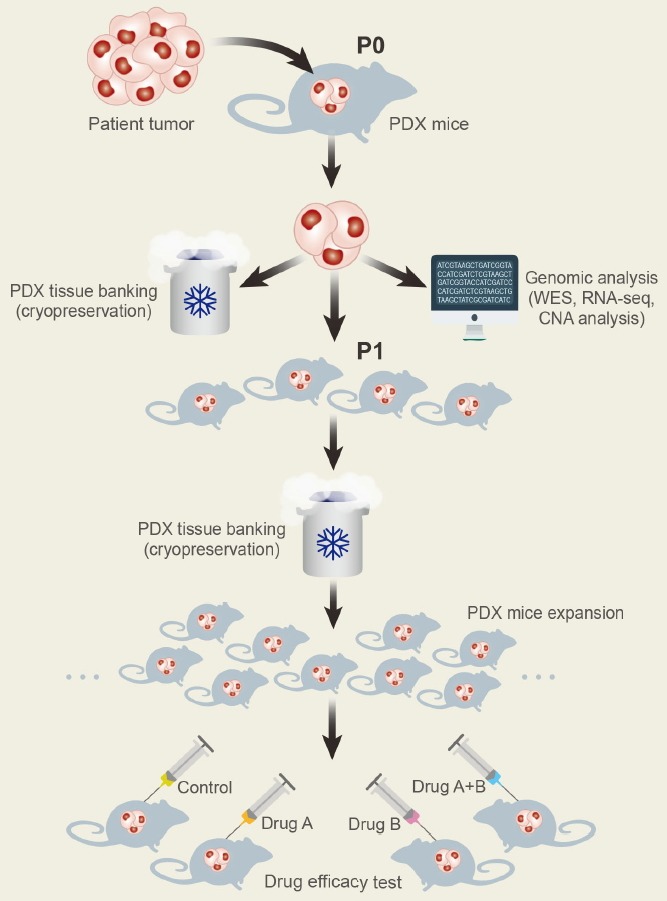
Generation of PDX models. Surgical specimens from cancer patients are divided into small pieces and transplanted into immunodeficient mice (P0). When tumors are grown in P0 mice, xenografts are used for genomic analysis including whole exome sequencing (WES), RNA sequencing (RNA-seq), and copy number alteration (CNA) analysis, and then maintained in cryo-banks for preservation. After expanding tumor xenografts in immunodeficient mice (P1 and more), in vivo drug responsiveness is screened in these models.
Table 1.
Comparison of immunodeficient mouse strains [Modified from The Jackson Laboratory’s JAX® Mice and Services website (https://www.jax.org/jax-mice-and-services/find-and-order-jax-mice/most-popular-jax-mice-strains/immunodeficient-mouse-and-xenograft-host-comparisons)].
| NOD scid γ (NSG) | NOD scid | BALB scid | B6 Rag1 | Outbred nude | Inbred nude | |
|---|---|---|---|---|---|---|
| Mature B cells | Absent | Absent | Absent | Absent | Present | Present |
| Mature T cells | Absent | Absent | Absent | Absent | Absent | Absent |
| Dendritic cells | Defective | Defective | Present | Present | Present | Present |
| Macrophages | Defective | Defective | Present | Present | Present | Present |
| Natural killer cells | Absent | Defective | Present | Present | Present | Present |
|
| ||||||
| Complement | Absent | Absent | Present | Present | Present | Present |
| Leakiness | Very low | Low | Low | Absent | N/A | N/A |
| Irradiation tolerance | Low | Low | Low | High | High | High |
|
| ||||||
| Lymphoma incidence | Low | High (thymic lymphoma) | High (thymic lymphoma) | Low | Low | Low |
| Median survival | > 89 weeks | 36 weeks | Not determined | Not determined | Not determined | Not determined |
| Benefits |
|
|
|
|
|
|
|
| ||||||
| Considerations |
|
|
|
|
|
|
Reported success rates of PDX models estimated by obtaining successful PDX tumors for next passages have ranged between 23% and 75%, depending on the tumor type (Table 2; Siolas and Hannon, 2013). Generally, colorectal (64–89%) and pancreatic (62%) tumors have had high engraftment rates, but breast cancers (13–27%), especially estrogen receptor-positive breast cancers, have shown low success rates, even in the laboratories most successful with PDX engraftment in general (Hidalgo et al., 2014; Williams et al., 2013). Clinically aggressive and metastatic cancers have exhibited high PDX model engraftment rates compared to less aggressive and nonmetastatic cancers (Zhao et al., 2012), and patients with tumors that had a high engraftment rate in PDX mice showed lower overall survival and increased metastatic characteristics compared to patients whose tumors showed a low engraftment rate (Garrido-Laguna et al., 2011), suggesting that the engraftment rate can sometimes be considered a predictive marker of disease prognosis (DeRose et al., 2011). Engraftment success can also be affected by experimental protocols. In breast cancer, supplementation with estradiol pellets enhanced stable xenograft take rate from 2.6% to 21.4% (Zhang et al., 2013), and in prostate cancer, male NOD scid mice supplemented with testosterone showed successful engraftment in 38.9% (7/18) of samples (Lin et al., 2014). Among immunodeficient mouse strains, scid-based strains, such as BALB scid, NOD scid, and NOD/scid/IL2γ-receptor null (NSG), demonstrated more successful engraftment compared to nude (nu) mutation-based strains due to further immune suppression (Landis et al., 2013). Therefore, tumor type, cancer aggressiveness, hormonal supplements and mouse host strains are important experimental factors to determine the engraftment rates.
Table 2.
Summary of engraftment rates of PDX tumors
| Tumor type | Mice strain | Implantation site | Engraftment rate | References |
|---|---|---|---|---|
| Breast cancer | Nude | Subcutaneous | 13% | Marangoni et al., 2007 |
| NOD scid | Mammary fat pad | 27% | DeRose et al., 2011 | |
| scid/Beige, NSG | Mammary fat pad | 19–21% | Zhang et al., 2013 | |
| NOD scid | Humanized mammary fat pad | 13% | Li et al., 2013 | |
|
| ||||
| Colorectal cancer | NOD scid | Subcutaneous | 87% | Bertotti et al., 2011 |
| Nude | Subcutaneous | 64% | Julien et al., 2012 | |
| Nude | Orthotopic | 89% | Aytes et al., 2012 | |
|
| ||||
| Head and neck cancer (squamous cell carcinoma) | NSG | Subcutaneous | 85% | Kimple et al., 2013 |
| Nude | Subcutaneous | 54% | Keysar et al., 2013 | |
|
| ||||
| Medulloblastoma | Rag2 scid | Orthotopic | 52% | Zhao et al., 2012 |
|
| ||||
| Non-small cell lung cancer | NOD scid | Subcutaneous | 25% | Fichtner et al., 2008 |
| NOD scid | Renal capsule | 90% | Dong et al., 2010 | |
|
| ||||
| Pancreatic cancer (ductal adenocarcinoma) | Nude | Subcutaneous | 61% | Garrido-Laguna et al., 2011 |
| Nude | Orthotopic | 62% | Reyes et al., 1996 | |
| ICR scid | Subcutaneous | 67% | Mattie et al., 2013 | |
|
| ||||
| Uveal melanoma | NOD scid | Subcutaneous | 28% | Némati et al., 2010 |
The tumor implantation site is one of considering factors for the generation of PDX models. Subcutaneously transplanted PDX models have been widely developed due to their simple procedure and easy measurement of tumor size, and suitable for massive expansion of PDX models. Orthotopic models are anticipated to show more similar behavior of patient tumors, especially in metastasis (Hoffman, 2015), but well-trained surgical skill is required for appropriate PDX models generation. The subrenal capsule site is considered as tumor plantation site due to its hypervascularity (Lin et al., 2014), and exhibited successful PDX generation in several cancers including prostate and ovarian cancer (Lin et al., 2014; Scott et al., 2013). Considerable evidence indicates that PDX tumors retain the histological and genomic characteristics of the original human tumor (Rosfjord et al., 2014). At low passages, the morphological features and chromosomal stability of PDX tumors are comparable with the corresponding patient tumors (Fig. 2; DeRose et al., 2011; Rosfjord et al., 2014; Zhang et al., 2013). In addition, comprehensive gene-expression data analyses have indicated that PDX tumors cluster with their original tumors in clustering studies, and that the majority of the key genes and signaling pathway activity in original tumors are preserved in PDX models (Reyal et al., 2012; Tentler et al., 2012). In breast cancer PDX models, even spontaneous metastatic patterns appear to be recapitulated to clinically relevant sites including thymus, lung, bone, and peritoneum (DeRose et al., 2011). In serial passage experiments in which the repeated transplantation of the same tumor has been performed, a relatively high degree of genomic aberration has been preserved in colorectal, breast, and pancreatic cancers (Julien et al., 2012; Mattie et al., 2013; Reyal et al., 2012), but the degree of preservation is dependent on the particular cancer indication studied (Rosfjord et al., 2014). In addition, some studies have reported PDX-unique single nucleotide mutations, which are caused either by adaptation of tumor cells to a new microenvironment or by overgrowth of subclones below detectable limits in the original tumor (Ding et al., 2010; Li et al., 2013).
Fig. 2.
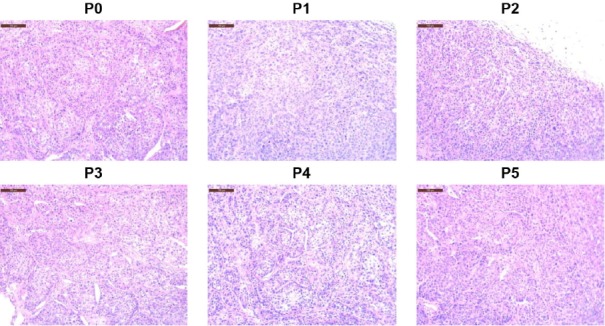
Histologic comparison among PDX tumors from passage 0 (P0) to passage 5 (P5). Breast cancer PDX model #45356922 was generated in NSG mice and PDX tumors were serially implanted from P0 to P5. Tissues were formalin-fixed and stained by H&E staining. Scales bars: 100 μm.
from these studies shows a notable similarity of drug activity between PDX mice and patients, indicating remarkable predictive value of PDX models for clinics (Hidalgo et al., 2014; Rosfjord et al., 2014). For example, PDX models and human patients exhibited comparable responsiveness rates in anti-EGFR antibody cetuximab treatment for colorectal cancers and gemcitabine treatment for pancreatic cancers (Bertotti et al., 2011; Garrido-Laguna et al., 2011; Julien et al., 2012). In addition, selected drugs that were effective in PDX models produced similar responsiveness for patients in terms of resistance and sensitivity (Hidalgo et al., 2011).
In predicting the therapeutic effects in clinic, PDX models are valuable research tools which can overcome well-recognized limitations of conventional cell line-derived xenograft (CDX; Johnson et al., 2001; Kung, 2007). Although the anti-angiogenic agent, bevacizumab prolonged survival in glioblastoma (GBM) CDX study (de Groot et al., 2010), bevacizumab failed to extend overall survival of patient in clinic (Lai et al., 2011). Like these outcomes, the survival length of GBM PDX was not altered by bevacizumab treatment and tumor became more aggressive and invasive (Joo et al., 2013).
In sum, these evidences indicate that PDX models can be a stable preclinical research platform that retains the characteristics of original patients’ tumors with regard to histology, genomic and transcriptomic signatures, and drug responsiveness.
APPLICATIONS OF PDX MODELS
PDX models have proven to be valuable platforms for preclinical drug testing in many types of cancer. The effectiveness of the combination of nab-paclitaxel and gemcitabine in pancreatic cancer was demonstrated in a PDX experiment (Von Hoff et al., 2011), and later this regimen was reported to provide a survival benefit for patients with advanced pancreatic cancer in a randomized phase III study (Von Hoff et al., 2013). PDX models are especially advantageous in preclinical studies of targeted agents, because, after evaluating the genomic characteristics of a PDX model, a subset of PDX models with the same genomic characteristics can be tested for targeted therapeutics. For example, the effect of the pan-RAF and SRC-family kinase dual inhibitor was evaluated by previously known RAF inhibitor-sensitive and resistant melanoma PDX models (Girotti et al., 2015). In addition, combination treatments of targeted drugs and cytotoxic drugs can also be tested in previously characterized PDX models. For example, our group recently validated the efficacy of combination therapies using a BCL2L1 inhibitor and irinotecan in genomically defined gastric cancer PDX models (Fig. 3; Park et al., 2015).
Fig. 3.
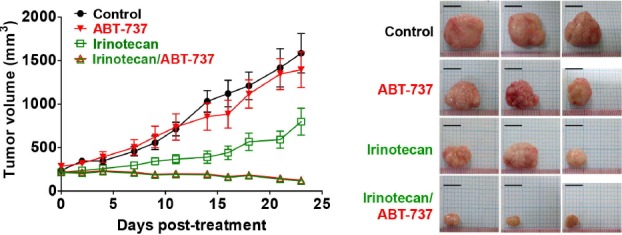
In vivo drug efficacy testing of combination therapy using PDX models. Combination effects of BCL2L1 inhibitor (ABT-737) and cytotoxic drug (irinotecan) were tested in BCL2L1-amplified (11 copies) gastric cancer PDX models. Average tumor sizes of treated groups are plotted (left panel) and representative tumors after treatment are shown (right panel). Scale bar: 10 mm. (reproduced from Park et al., 2015).
For more systematic application of PDX models to drug development process, biorepositories, or biobank of PDX tumors are required. Especially, genomically well-characterized PDX repositories are valuable resources for developing improved diagnotics and therapeutics of cancers (Dowst et al., 2015). Recently, several groups, both in academia and industry, developed PDX repositories, and have now tried to develop collaborative networks for PDX biobanking (Hidalgo et al., 2014). For example, EurOPDX is a consortium of centers working on PDX models in Europe, and aims at the development of network of clinically relevant and annotated PDX models (Hidalgo et al., 2014).
With genomically well-defined PDX collections, biomarkers associated with drug sensitivity and resistance can be identified, which facilitates precision cancer medicine (Fig. 4). PDX tumors recapitulated previously known sensitive biomarkers including KRAS wild-type status for cetuximab sensitivity in colorectal cancers (Bertotti et al., 2011; Julien et al., 2012) and KRAS wild-type status for erlotinib sensitivity in lung cancers (Fichtner et al., 2008). By comparing drug-sensitive and -resistant PDX groups in colorectal cancers, amplification of HER2 was identified as a resistant biomarker for cetuximab treatment (Bertotti et al., 2011). PDX models are also valuable tools for generating drug-resistant tumor models, which is achieved by repeated administration of a given drug. These resistant tumors are more clinically relevant compared to previous in vitro cancer cell culture systems, and are used for investigation of drug resistance mechanisms and identification of drug resistance biomarkers. For example, continuous administration of the BRAF inhibitor vemurafenib resulted in vemurafenib-resistant melanoma models with BRAF mutations, and elevated mutant BRAF protein resulting from this continuous drug treatment was reported as a critical factor in vemurafenib resistance (Das Thakur et al., 2013).
Fig. 4.
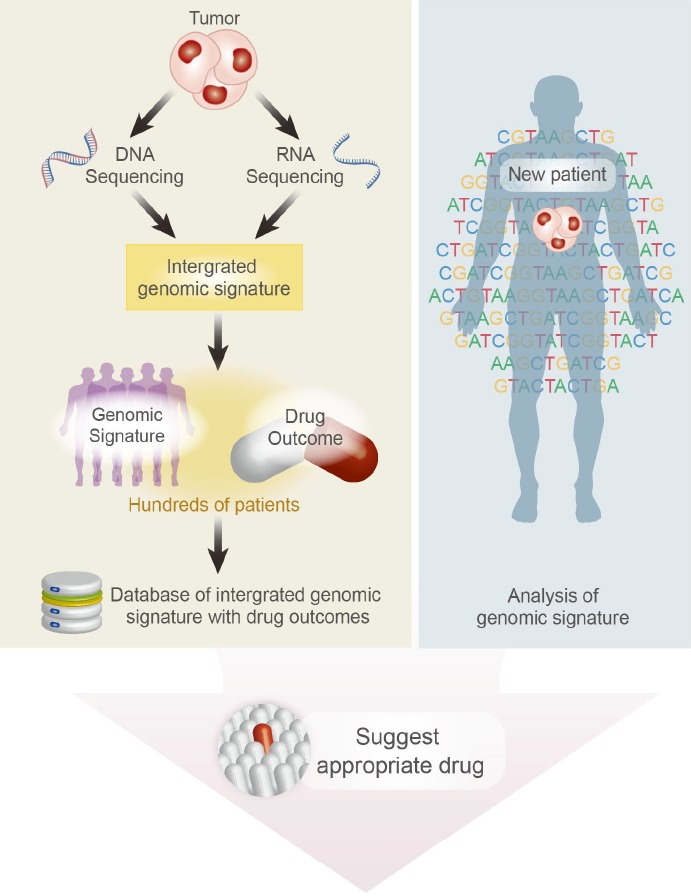
An integrated strategy for precision medicine using PDX models. Database of integrated genomic signature with drug outcome can be generated from hundreds of PDX models. By integrating clinical data from patients, genomic profiling data, and drug screening data, PDX models are valuable platforms for precision cancer medicine and personalized drug selection.
The predictive power of PDX models is now being applied to co-clinical trials for novel cancer therapeutics. Phase I/II clinical trials take more than 5 years to complete, and during these trials, real-time analysis and integration of data for stratification of responders and resistant populations are limited. To overcome this hurdle, co-clinical trials are carried out, whereby drug treatment experiments for mouse tumor models are performed in parallel with clinical trials, and preclinical and clinical data are integrated (Nardella et al., 2011). The concept of co-clinical trials was first developed for genetically engineered mouse models, but this strategy can be applied to genetically well-defined PDX models (Hidalgo et al., 2014). The co-clinical trial approach using PDX models will facilitate rapid stratification of responders, prioritization of attractive drug combinations, identification of resistance mechanisms, and determination of biomarkers (Nardella et al., 2011).
Although genomic profiling of tumors provides significant improvement in patient stratification and targeted drug selection, additional decision-making processes may be required when multiple potential targets are identified or no targetable mutation is found. To facilitate drug selection in such cases, PDX models are used as “Avatar” models for personalized cancer treatment. The avatar models are mouse substitutes of specific human patients for drug testing. In one prospective study, avatar models were generated from 14 patients, and effective treatment options were explored with 63 drugs in 232 treatment regimens (Hidalgo et al., 2011). Following identification of the most effective treatment regimens in the xenograft model, the 17 regimens were tried in 11 patients, and durable partial remission was detected in 15 treatments.
And in another study, PDX clinical trial (PCT) was demonstrated to be assessed by comparing the targeted drug response in PDX to responses in patients. The 67% melanoma PDX with BRAF mutation showed response to encorafenib, a selective BRAF inhibitor and this was concordant with the reported phase II clinical trial response rates in patients. And also, insensitive BRAF-mutated melanoma PDX to BRAF inhibitor responded to combined therapy with MEK inhibitor. This is consistent with combination phase I/II trial in BRAF-mutant melanoma patients (Gao et al., 2015). Thus, personalized PDX models showed high positive predictive value for drug testing, and can be beneficial for therapeutic decision-making and guiding treatment options.
PDX tumors can recapitulate the intratumoral heterogeneity of original patients’ tumors, and a growing body of evidence shows that tumors contain a distinct subpopulation, usually termed cancer stem cells (CSCs). CSCs have the capacity for self-renewal and differentiation, and have been reported to be more resistant than other cancer cells to chemotherapy and radiation (Rosen and Jordan, 2009). A large number of CSCs can be obtained through expansion of PDX tumors in mice without compromising the heterogeneity of the original tumor (Rosfjord et al., 2014). Using specific surface markers, CSCs have been purified and characterized in several types of PDX tumors (Bertolini et al., 2009; Schatton et al., 2008). In addition, the heterogenetic profiles of patients’ tumors evolve in response to environmental changes, and the microenvironment of tumors in a mouse background provides selection pressure for tumors, recapitulating tumor evolution and clonal selection in patients (Whittle et al., 2015). PDX tumor from primary basal-like breast cancer retained the primary tumor mutations and showed additional de novo mutations similar with the brain metastatic lesion, suggesting that the PDX models exhibit genomic adjustments compatible with cancer evolution in patients (Ding et al., 2010). The clonal dynamics in PDX propagation was evaluated through deep-genome and single-cell sequencing, and the results showed that the degree of clonal selection varied from extremely selective engraftment of minor clones to moderate, polyclonal engraftment, and similar clonal expansion patterns were reproduced in independent grafts of the same tumors (Eirew et al., 2015). In sum, the above studies demonstrate that PDX models can be applied to the study of tumor heterogeneity, CSCs, and cancer evolution.
PDX models in early passages retain stromal components and acidic conditions of microenvironment in patient tumors, and can be used to study the effects of microenvironment on tumor biology (Choi et al., 2014). The microenvironment-targeting agent, ibrutinib was effective at inhibiting tumor growth and increasing survival in PDX models of pancreatic ductal adenocarcinoma (Masso-Valles et al., 2015). Tumor architecture can be conserved through multiple passages by recruitment of murine stromal cells instead of human stromal elements (Delitto et al., 2015).
PDX models can also be applied to study the effect of cancer immunotherapy. For example, mice engrafted with patient-derived gastric cancer tissue and peripheral mononuclear cells from same patient exhibited slow tumor growth by combination therapy of anti-hCD137 and anti-hPD1 antibodies (Fernandez de Sanmamamed et al., 2015). However, humanized PDX models, which have patient-derived tumor and full repertoire of human immune cells by engrafting human hematopoietic stem cells, are required for proper preclinical models of cancer immunotherapy.
CHALLENGES IN PDX MODELS
Although PDX models maintain the histology, genomic architecture, and drug responsiveness of the original tumors, during engraftment into immunodeficient mice the clonal profiles of PDX tumors can change, in comparison to the patients’ tumors. Analysis of genome-wide variant allele frequencies in serial passages of PDX tumors showed that clonal selection occurs more frequently in initial engraftment steps than in propagation steps, but the detailed clonal dynamics differs depending on the different tumor samples of the same tumor type (Eirew et al., 2015). This clonal dynamics in PDX tumors is probably generated by selection acting on preexisting clones, rather than the generation of new clones (Aparicio et al., 2015). As a result, it is probable that the more aggressive clones become dominant in PDX tumors, and in some cases, PDX models indeed showed the genomic and transcriptomic signature of metastatic and relapsed cancers (Rosfjord et al., 2014). These aggressive clones could be particularly important targets in cancer therapeutics.
For use of PDX models in decision-making processes of cancer therapeutics, engraftment failure and the long periods of time required for PDX establishment are limiting factors. Because success rates of PDX models vary depending on the cancer type and tumor aggressiveness as detailed above, there is a probability that PDX collections are skewed toward more aggressive type of cancers (Hidalgo et al., 2014). The time for establishment and expansion of PDX models for drug efficacy testing—four to eight months, as noted above—is usually not feasible for immediate application in patient decision-making for therapeutics. An alternate strategy would be to perform PDX-based drug testing during the period of initial standard therapy and pursue the second-line treatments based on PDX drug screening.
The depletion of human stromal and immune cells in engrafted tumors of late passages is a major limitation of PDX models for studies of tumor microenvironments and metastasis. The interaction of tumor-associated cells, such as vascular cells, fibroblasts, and inflammatory cells, with malignant cells plays an important role in tumor biology, and recent studies have shown that tumor microenvironments are also associated with drug responsiveness (Ostman, 2012). In addition, the application of immune-modulating agents such as anti-PD1 antibody into PDX models is limited, as immunocompromised mice used for PDX generation lack the essential components of an immune system (Hidalgo et al., 2014). To overcome this hurdle, immunocompromised mice that are transplanted with human hematopoietic stem and progenitor cells, i.e., humanized mice, are promising options. Some studies have demonstrated that diverse lineages of functional human immune cells have developed via the infusion of human hematopoietic stem cells, and of human immune and stromal cells, in tumor xenografts (Morton et al., 2015; Rongvaux et al., 2014).
Another pitfall of PDX models is lymphomagenesis of human tumors in mice. This phenomenon has been reported in PDX models of several cancer types including non-small cell lung cancers, hepatocellular carcinomas, prostate cancers, and gastric cancers (Chen et al., 2012; John et al., 2012; Wetterauer et al., 2015; Zhang et al., 2015). Most of these lymphomas were human-derived lymphomas expressing human CD45 markers, consistent with B-cell subtype lymphomas (Chen et al., 2012; Zhang et al., 2015). Further, Epstein-Barr virus (EBV) infections are highly associated with lymphomagenesis (Chen et al., 2012; Zhang et al., 2015). Tissue inflammation in parent tumors increases the risk of having a human-derived lymphoma in the PDX models, and gastric cancers exhibit a much higher rate of lymphoma proliferation than colorectal cancers partly due to higher rate of baseline inflammation (Zhang et al., 2015). A suggested mechanism of lymphoma formation is that, without immunosurveillance in immunodeficient mice, EBV can infect and transform B lymphocytes into a proliferative state (Zhang et al., 2015). Therefore, exclusion of human lymphomagenesis in PDX models requires validation of established PDX tumors in immunodeficient mice by histology and human CD34 markers (Zhang et al., 2015).
CONCLUSION
Precision medicine for cancer treatment requires the integration of massive volumes of diverse data from genomic profiling and drug responsiveness. Recent advances in high-throughput sequencing techniques have enabled genomic analysis of individual patients’ cancer. PDX models retain the histology, genomic characteristics, and even drug responsiveness of the original human patient tumors, and can be used as ‘Avatars’ for drug responsiveness studies. These models are valuable for diverse drug development processes including biomarker development, preclinical drug testing, co-clinical trials, and personalized drug selection. Finally, development of mutation-specified PDX collections by integrating sequencing data and avatar experiments will provide novel opportunities for therapeutic optimization, improved clinical outcomes, and precision cancer medicine.
Acknowledgments
Thanks to Stephen Sampson of JAX Scientific Program Development (SPD) for editing and review of manuscript and Zoe Reifsnyder for assistance with Figs. 1 and 4. This work was supported by grants from a Ewha Womans University Research Grant; the National Cancer Institute under award number P30CA034196; the Korean Healthcare Technology R&D project through the Korean Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant No. HI13C2148); and the International Research & Development Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (grant No. 2015K1A4A3047851).
REFERENCES
- Agliano A., Martin-Padura I., Mancuso P., Marighetti P., Rabascio C., Pruneri G., Shultz L.D., Bertolini F. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rγ null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int. J. Cancer. 2008;123:2222–2227. doi: 10.1002/ijc.23772. [DOI] [PubMed] [Google Scholar]
- Aparicio S., Hidalgo M., Kung A.L. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer. 2015;15:311–316. doi: 10.1038/nrc3944. [DOI] [PubMed] [Google Scholar]
- Aytes A., Molleví D.G., Martinez-Iniesta M., Nadal M., Vidal A., Morales A., Salazar R., Capellà G., Villanueva A. Stromal interaction molecule 2 (STIM2) is frequently overexpressed in colorectal tumors and confers a tumor cell growth suppressor phenotype. Mol. Carcinog. 2012;51:746–753. doi: 10.1002/mc.20843. [DOI] [PubMed] [Google Scholar]
- Bell D., Berchuck A., Birrer M., Chien J., Cramer D.W., Dao F., Dhir R., DiSaia P., Gabra H., Glenn P., et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini G., Roz L., Perego P., Tortoreto M., Fontanella E., Gatti L., Pratesi G., Fabbri A., Andriani F., Tinelli S., et al. Highly tumorigenic lung cancer CD133(+) cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A., Migliardi G., Galimi F., Sassi F., Torti D., Isella C., Cora D., Di Nicolantonio F., Buscarino M., Petti C., et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- Chen K., Ahmed S., Adeyi O., Dick J.E., Ghanekar A. Human solid tumor xenografts in immunodeficient mice are vulnerable to lymphomagenesis associated with Epstein-Barr virus. PLoS One. 2012;7:e39294. doi: 10.1371/journal.pone.0039294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.Y.C., Lin D., Gout P.W., Collins C.C., Xu Y., Wang Y.Z. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Adv. Drug Deliver. Rev. 2014;79–80:222–237. doi: 10.1016/j.addr.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Das Thakur M., Salangsang F., Landman A.S., Sellers W.R., Pryer N.K., Levesque M.P., Dummer R., McMahon M., Stuart D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot J.F., Fuller G., Kumar A.J., Piao Y., Eterovic K., Ji Y.J., Conrad C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro. Oncol. 2010;12:233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delitto D., Pham K., Vlada A.C., Sarosi G.A., Thomas R.M., Behrns K.E., Liu C., Hughes S.J., Wallet S.M., Trevino J.G. Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. Am. J. Pathol. 2015;185:1297–1303. doi: 10.1016/j.ajpath.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose Y.S., Wang G.Y., Lin Y.C., Bernard P.S., Buys S.S., Ebbert M.T.W., Factor R., Matsen C., Milash B.A., Nelson E., et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011;17:1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi J.A., Reichert J.M., Feldman L., Malins A. Clinical approval success rates for investigational cancer drugs. Clin. Pharmacol. Ther. 2013;94:329–335. doi: 10.1038/clpt.2013.117. [DOI] [PubMed] [Google Scholar]
- Ding L., Ellis M.J., Li S.Q., Larson D.E., Chen K., Wallis J., Harris C.C., McLellan M.D., Fulton R.S., Fulton L.L., et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Guan J., English J.C., Flint J., Yee J., Evans K., Murray N., Macaulay C., Ng R.T., Gout P.W., et al. Patient-derived first generation xenografts of non-small cell lung cancers: promising tools for predicting drug responses for personalized chemotherapy. Clin. Cancer Res. 2010;16:1442–1451. doi: 10.1158/1078-0432.CCR-09-2878. [DOI] [PubMed] [Google Scholar]
- Dowst H., Pew B., Watkins C., McOwiti A., Barney J., Qu S., Becnel L. B. Acquire: an open-source comprehensive cancer biobanking system. Bioinformatics. 2015;31:1655–1662. doi: 10.1093/bioinformatics/btv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirew P., Steif A., Khattra J., Ha G., Yap D., Farahani H., Gelmon K., Chia S., Mar C., Wan A., et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518:422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de Sanmamamed M., Lopez Rodriguez I., Schalper K.A., Onate C., Azpilikueta A., Rodriguez-Ruiz M.E., Morales-Kastresana A., Labiano S., Perez-Gracia J.L., Martin-Algarra S., et al. Nivolumab and urelumab enhance antitumor activity of human T lymphocytes engrafted in Rag2-/-IL2Rγnull immunodeficient mice. Cancer Res. 2015;75:3466–3478. doi: 10.1158/0008-5472.CAN-14-3510. [DOI] [PubMed] [Google Scholar]
- Fichtner I., Rolff J., Soong R., Hoffmann J., Hammer S., Sommer A., Becker M., Merk J. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin. Cancer Res. 2008;14:6456–6468. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- Gao D., Chen Y. Organoid development in cancer genome discovery. Curr. Opin. Genet. Dev. 2015;30:42–48. doi: 10.1016/j.gde.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Korn J.M., Ferretti S., Monahan J.E., Wang Y., Singh M., Zhang C., Schnell C., Yang G., Zhang Y., et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Garraway L.A., Verweij J., Ballman K.V. Precision oncology: an overview. J. Clin. Oncol. 2013;31:1803–1805. doi: 10.1200/JCO.2013.49.4799. [DOI] [PubMed] [Google Scholar]
- Garrido-Laguna I., Uson M., Rajeshkumar N.V., Tan A.C., de Oliveira E., Karikari C., Villaroel M.C., Salomon A., Taylor G., Sharma R., et al. Tumor engraftment in nude mice and enrichment in stroma-related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res. 2011;17:5793–5800. doi: 10.1158/1078-0432.CCR-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti M.R., Lopes F., Preece N., Niculescu-Duvaz D., Zambon A., Davies L., Whittaker S., Saturno G., Viros A., Pedersen M., et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27:85–96. doi: 10.1016/j.ccell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M., Bruckheimer E., Rajeshkumar N.V., Garrido-Laguna I., De Oliveira E., Rubio-Viqueira B., Strawn S., Wick M.J., Martell J., Sidransky D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol. Cancer Ther. 2011;10:1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M., Amant F., Biankin A.V., Budinska E., Byrne A.T., Caldas C., Clarke R.B., de Jong S., Jonkers J., Maelandsmo G.M., et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R.M. Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer. 2015;15:451–452. doi: 10.1038/nrc3972. [DOI] [PubMed] [Google Scholar]
- John T., Yanagawa N., Kohler D., Craddock K.J., Bandarchi-Chamkhaleh B., Pintilie M., Sykes J., To C., Li M., Panchal D., et al. Characterization of lymphomas developing in immunodeficient mice implanted with primary human non-small cell lung cancer. J. Thorac. Oncol. 2012;7:1101–1108. doi: 10.1097/JTO.0b013e3182519d4d. [DOI] [PubMed] [Google Scholar]
- Johnson J.I., Decker S., Zaharevitz D., Rubinstein L.V., Venditti J., Schepartz S., Kalyandrug S., Christian M., Arbuck S., Hollingshead M., et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo K.M., Kim J., Jin J., Kim M., Seol H.J., Muradov J., Yang H., Choi Y.L., Park W.Y., Kong D.S., et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013;3:260–273. doi: 10.1016/j.celrep.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Julien S., Merino-Trigo A., Lacroix L., Pocard M., Goere D., Mariani P., Landron S., Bigot L., Nemati F., Dartigues P., et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin. Cancer Res. 2012;18:5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- Kimple R.J., Harari P.M., Torres A.D., Yang R.Z., Soriano B.J., Yu M., Armstrong E.A., Blitzer G.C., Smith M.A., Lorenz L.D., et al. Development and characterization of HPV-positive and HPV-negative head and neck squamous cell carcinoma tumorgrafts. Clin. Cancer Res. 2013;19:855–864. doi: 10.1158/1078-0432.CCR-12-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keysar S.B., Astling D.P., Anderson R.T., Vogler B.W., Bowles D.W., Morton J.J., Paylor J.J., Glogowska M.J., Le P.N., Eagles-Soukup J.R., et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol. Oncol. 2013;7:776–790. doi: 10.1016/j.molonc.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Fulton R.S., McLellan M.D., Schmidt H., Kalicki-Veizer J., McMichael J.F., Fulton L.L., Dooling D.J., Ding L., Mardis E.R., et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopetz S., Lemos R., Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin. Cancer Res. 2012;18:5160–5162. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung A.L. Practices and pitfalls of mouse cancer models in drug discovery. Adv. Cancer Res. 2007;96:191–212. doi: 10.1016/S0065-230X(06)96007-2. [DOI] [PubMed] [Google Scholar]
- Lai A., Tran A., Nghiemphu P.L., Pope W.B., Solis O.E., Selch M., Filka E., Yong W.H., Mischel P.S., Liau L.M., et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J. Clin. Oncol. 2011;29:142–148. doi: 10.1200/JCO.2010.30.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis M.D., Lehmann B.D., Pietenpol J.A., Chang J.C. Patient-derived breast tumor xenografts facilitating personalized cancer therapy. Breast Cancer Res. 2013;15:201. doi: 10.1186/bcr3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.Q., Shen D., Shao J.Y., Crowder R., Liu W.B., Prat A., He X.P., Liu S.Y., Hoog J., Lu C., et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D., Wyatt A. W., Xue H., Wang Y., Dong X., Haegert A., Wu R., Brahmbhatt S., Mo F., Jong L., et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74:1271–1283. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
- Macconaill L.E., Garraway L.A. Clinical implications of the cancer genome. J. Clin. Oncol. 2010;28:5219–5228. doi: 10.1200/JCO.2009.27.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni E., Vincent-Salomon A., Auger N., Degeorges A., Assayag F., de Cremoux P., de Plater L., Guyader C., De Pinieux G., Judde J.G., et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin. Cancer Res. 2007;13:3989–3998. doi: 10.1158/1078-0432.CCR-07-0078. [DOI] [PubMed] [Google Scholar]
- Masso-Valles D., Jauset T., Serrano E., Sodir N.M., Pedersen K., Affara N.I., Whitfield J.R., Beaulieu M.E., Evan G.I., Elias L., et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res. 2015;75:1675–1681. doi: 10.1158/0008-5472.CAN-14-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattie M., Christensen A., Chang M.S., Yeh W., Said S., Shostak Y., Capo L., Verlinsky A., An Z.L., Joseph I., et al. Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia. 2013;15:1124–1136. doi: 10.1593/neo.13922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton C.L., Houghton P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007;2:247–250. doi: 10.1038/nprot.2007.25. [DOI] [PubMed] [Google Scholar]
- Morton J.J., Bird G., Keysar S.B., Astling D.P., Lyons T.R., Anderson R.T., Glogowska M.J., Estes P., Eagles J.R., Le P.N., et al. XactMice: humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene. 2015. (Epub ahead of print) [DOI] [PMC free article] [PubMed]
- Muzny D.M., Bainbridge M.N., Chang K., Dinh H.H., Drummond J.A., Fowler G., Kovar C.L., Lewis L.R., Morgan M.B., Newsham I.F., et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella C., Lunardi A., Patnaik A., Cantley L.C., Pandolfi P.P. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 2011;1:108–116. doi: 10.1158/2159-8290.CD-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Némati F., Sastre-Garau X., Laurent C., Couturier J., Mariani P., Desjardins L., Piperno-Neumann S., Lantz O., Asselain B., Plancher C., et al. Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clin. Cancer Res. 2010;16:2352–2362. doi: 10.1158/1078-0432.CCR-09-3066. [DOI] [PubMed] [Google Scholar]
- Ostman A. The tumor microenvironment controls drug sensitivity. Nat. Med. 2012;18:1332–1334. doi: 10.1038/nm.2938. [DOI] [PubMed] [Google Scholar]
- Pao W., Miller V., Zakowski M., Doherty J., Politi K., Sarkaria I., Singh B., Heelan R., Rusch V., Fulton L., et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H., Cho S.Y., Kim H., Na D., Han J.Y., Chae J., Park C., Park O.K., Min S, Kang J., et al. Genomic alterations in BCL2L1 and DLC1 contribute to drug sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA. 2015;112:12492–12497. doi: 10.1073/pnas.1507491112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A., Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619–1630. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyal F., Guyader C., Decraene C., Lucchesi C., Auger N., Assayag F., De Plater L., Gentien D., Poupon M.F., Cottu P., et al. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Res. 2012;14:R11. doi: 10.1186/bcr3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes G., Villanueva A., García C., Sancho F.J., Piulats J., Lluís F., Capellá G. Orthotopic xenografts of human pancreatic carcinomas acquire genetic aberrations during dissemination in nude mice. Cancer Res. 1996;56:5713–5719. [PubMed] [Google Scholar]
- Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S.V., Teichmann L.L., Saito Y., Marches F., Halene S., Palucka A.K., et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014;32:364–U230. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen J.M., Jordan C.T. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosfjord E., Lucas J., Li G., Gerber H.P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem. Pharmacol. 2014;91:135–143. doi: 10.1016/j.bcp.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Schatton T., Murphy G.F., Frank N.Y., Yamaura K., Waaga-Gasser A.M., Gasser M., Zhan Q., Jordan S., Duncan L.M., Weishaupt C., et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C.L., Becker M.A., Haluska P., Samimi G. Patient-derived xenograft models to improve targeted therapy in epithelial ovarian cancer treatment. Front. Oncol. 2013;3:295. doi: 10.3389/fonc.2013.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A.T., Yeap B.Y., Solomon B.J., Riely G.J., Gainor J., Engelman J.A., Shapiro G.I., Costa D.B., Ou S.H.I., Butaney M., et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siolas D., Hannon G.J. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon D., Eiermann W., Robert N., Pienkowski T., Martin M., Press M., Mackey J., Glaspy J., Chan A., Pawlicki M., et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 2011;365:1273–1283. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler J.J., Tan A.C., Weekes C.D., Jimeno A., Leong S., Pitts T.M., Arcaroli J.J., Messersmith W.A., Eckhardt S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak R.G.W., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D.D., Ramanathan R.K., Borad M.J., Laheru D.A., Smith L.S., Wood T.E., Korn R.L., Desai N., Trieu V., Iglesias J.L., et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J. Clin. Oncol. 2011;29:4548–4554. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N., et al. Increased survival in pancreatic cancer with nabpaclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetterauer C., Vlajnic T., Schuler J., Gsponer J.R., Thalmann G.N., Cecchini M., Schneider J., Zellweger T., Pueschel H., Bachmann A., et al. Early development of human lymphomas in a prostate cancer xenograft program using triple knock-out immunocompromised mice. Prostate. 2015;75:585–592. doi: 10.1002/pros.22939. [DOI] [PubMed] [Google Scholar]
- Whittle J.R., Lewis M.T., Lindeman G.J., Visvader J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S.A., Anderson W.C., Santaguida M.T., Dylla S.J. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab. Invest. 2013;93:970–982. doi: 10.1038/labinvest.2013.92. [DOI] [PubMed] [Google Scholar]
- Zhang X.M., Claerhout S., Prat A., Dobrolecki L.E., Petrovic I., Lai Q., Landis M.D., Wiechmann L., Schiff R., Giuliano M., et al. A Renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.H., Liu Y.Q., Wang X.H., Tang Z.Y., Li S.X., Hu Y., Zong X.L., Wu X.J., Bu Z.D., Wu A.W., et al. The extent of inflammatory infiltration in primary cancer tissues is associated with lymphomagenesis in immunodeficient mice. Sci. Rep. 2015;5:9447. doi: 10.1038/srep09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.M., Liu Z.G., Yu L.T., Zhang Y.J., Baxter P., Voicu H., Gurusiddappa S., Luan J., Su J.M., Leung H.C.E., et al. Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro. Oncol. 2012;14:574–583. doi: 10.1093/neuonc/nos061. [DOI] [PMC free article] [PubMed] [Google Scholar]