

Abstract
Background
Src homology 2 domain–containing inositol 5-phosphatase 1 (SHIP-1) controls the intracellular level of the phosphoinositide 3–kinase product phosphotidylinositol-3,4,5-trisphosphate and functions as a negative regulator of cytokine and immune receptor signaling. Emerging evidence suggests that the phosphoinositide 3-kinase pathway might be involved in allergic inflammation in the lung. However, the functional relevance of SHIP-1 in the TH2 activation pathway has not been established. SHIP-1−/− mice have spontaneous myeloproliferative inflammation in the lung, the nature of which has not been elucidated. We hypothesized that SHIP-1 plays an important role as a regulator in pulmonary allergic inflammation and in maintaining lung homeostasis.
Objective
To test our hypothesis, we characterized the pulmonary phenotype of SHIP-1−/− mice.
Results
Analyses of lung histopathology and bronchoalveolar lavage cellularity revealed that the majority of SHIP-1−/− mice had progressive and severe pulmonary inflammation of macrophages, lymphocytes, neutrophils, and eosinophils; mucous hyperplasia; airway epithelial hypertrophy; and subepithelial fibrosis. These pathologic changes were accompanied by exaggerated production of TH2 cytokines and chemokines, including IL-4, IL-13, eotaxin, and monocyte chemoattractant protein 1, in the lung. Furthermore, the number of mast cells significantly increased, and many of these cells were undergoing degranulation, which was correlated with increased content and spontaneous release of histamine in the lung tissue of SHIP-1−/− mice.
Conclusion
These findings provide strong evidence that mice lacking SHIP-1 have an allergic inflammation in the lung, suggesting that SHIP-1 plays an important role in regulating the TH2 signaling pathway and in maintaining lung homeostasis.
Clinical implications
SHIP-1 as a regulator might be a potential therapeutic target for controlling allergic inflammation in diseases such as asthma.
Keywords: Phosphatase, phosphoinositide 3-kinase, signaling, TH2 inflammation, asthma, allergy
Src homology 2 domain–containing inositol 5-phosphatase 1 (SHIP-1) is a negative regulator in a variety of cytokine, immunoreceptor, and growth factor signaling pathways. SHIP-1 has important functions in T cells, B cells, mast cells, basophils, and neutrophils.1-7 The phosphoinositide 3-kinase (PI3K) pathway is critical in many biologic processes, including cell proliferation, apoptosis, and migration. Activated PI3K catalyzes the formation of the lipid product phosphotidylinositol-3,4,5-trisphosphate (PIP3), which mediates downstream responses. As one of the negative regulators in this pathway, SHIP-1 controls the intracellular level of PIP3. The major mechanism of SHIP-1 has been elucidated in the interaction between SHIP-1 and the inhibitory receptor FcγRIIB of B lympho-cytes.8,9 On B-cell antigen receptor–FcγRIIB coligation, SHIP-1 is recruited to the immunoreceptor tyrosine–based inhibitory motif domain of the receptor. After localizing to the cell membrane, SHIP-1 dephosphorylates PIP3 to PI-3,4-bisphosphate, effectively reducing or terminating the downstream signaling of the PI3K pathway.
Asthma is a chronic inflammatory disorder of the lung. TH2 inflammation of the airway is a major component in the pathogenesis of asthma. In animal models of allergic asthma, proper allergens can elicit TH2 inflammation in the airways and can cause a series of pathophysiologic changes in the lung. The TH2 cytokines IL-4 and IL-13 are essential in this response. Both cytokines signal through IL-4 receptor complexes and activate signal transducer and activator of transcription 6 pathway and downstream target genes. However, several recent studies have suggested that the PI3K pathway might also be involved in the TH2 inflammatory response in mouse models of allergic asthma.10,11 Either PI3K inhibitors10 or expression of a dominant-negative PI3K subunit reduced the inflammatory response of the airway to antigen challenge.11 The intracellular level of PIP3 is a critical point for regulation. SHIP-1 and phosphatase and tensin homolog deleted on chromosome 10 are known to be able to hydrolyze PIP3. Recently, phosphatase and tensin homolog deleted on chromosome 10 has been suggested to play a role in a mouse model of allergic asthma.12 An in vitro study also showed that IL-4 induced tyrosine phosphorylation of SHIP-1 in 32D cells,13 suggesting an interaction between SHIP-1 and the IL-4 signaling pathway. However, a functional relationship between SHIP-1 and the TH2 signaling pathway remains undefined.
In previous studies it was noticed that some SHIP-1−/− mice had inflammatory infiltration of macrophages and neutrophils in the lung,3,4 but the nature of the inflammation has not been characterized. Whether eosinophils and other allergic responses were present in the lungs of these mice was not examined, and the role of SHIP-1 in lung biology is not clear.
We hypothesized that SHIP-1 is a negative regulator of lung inflammation and maintenance of lung homeostasis. We investigated the nature of the inflammatory response in the lungs of SHIP-1−/− mice, and our results demonstrate that mice lacking SHIP-1 spontaneously have a remarkable allergic inflammatory response in the lung, accompanied by TH2 cytokine and chemokine upregulation, suggesting that under physiologic conditions, SHIP-1 is an essential negative regulator in maintaining lung homeostasis.
Methods
Animals
SHIP-1−/− mice on a genetic background of C57BL/6 × 129sv were generated as previously described.2 Heterozygous mice (SHIP-1+/−) were bred to generate SHIP-1−/− and SHIP-1+/+ (wild-type [WT]) mice, which were used as control animals. Mice were used at 9 to 11 weeks of age. All mice were housed in cages with microfilters in the specific pathogen-free environment. All procedures performed on mice were in accordance with the National Institutes of Health guidelines for humane treatment of animals and were approved by the Johns Hopkins University Institutional Animal Use and Care Committee.
Lung and bronchoalveolar lavage fluid samples
Lung tissue and bronchoalveolar lavage (BAL) fluid samples were obtained as previously described.14,15 Briefly, mice were anesthetized, the trachea was isolated by means of blunt dissection, and small-caliber tubing was inserted and secured in the airway. Three successive volumes of 0.75 mL of PBS with 0.1% BSA were instilled and gently aspirated and pooled. BAL fluid samples were centrifuged, and supernatants were stored at −70°C until use. The number and types of cells in the pellet were determined. The lung was perfused with cold PBS through the right ventricle with cut vena cava until the pulmonary vasculature was clean. The whole lung was excised either for RNA and protein analyses or inflated with fixatives for histology.
Histologic evaluation
Hematoxylin and eosin, periodic acid–Schiff (PAS), Masson's trichrome, and toluidine blue stains were performed on lung sections after pressure fixation with Streck solution (Streck Laboratories, La Vista, Neb), as previously described.15 The same microscopic magnification was used for the sample slides from WT and SHIP-1−/− mice under comparison. Mast cells in the lung tissues were identified by metachromatic granules stained with toluidine blue and were quantified in 20 distinct microscopic fields per lung section per mouse.
mRNA analysis
Total cellular RNA from lungs was obtained with Trizol Reagent (Invitrogen, Carlsbad, Calif). The mRNA of specific genes was evaluated by means of RT-PCR with specific primers, as previously described.15 β-Actin was used as an internal control. Amplified PCR products were analyzed by means of electrophoresis, and the intensity of the bands and the ratio of specific mRNA to β-actin were analyzed with the Bio-Rad Gel Doc system and the Quantity One 4.4.1 software (Bio-Rad Laboratories, Hercules, Calif).
Cytokines, chemokines, and total IgE levels
The levels of cytokines, chemokines, and total IgE in the BAL fluid were determined by using commercial ELISA kits per the manufacturer's instructions (R&D Systems, Inc, Minneapolis, Minn).
Chitinase and histamine assays
Chitinase activity in the BAL fluid was determined by using a fluorogenic substrate, as described previously.16 Total histamine and spontaneously released histamine levels were determined by using single-cell suspensions from spleens and lungs, as previously described17 and in as described detail in the Methods section in the Online repository at www.jacionline.org.
Statistics
Data were assessed by using the Student t test and expressed as means ± SEM. Differences between samples with P values of less than .05 were considered statistically significant.
Results
Lung inflammation
To define the nature of the inflammation seen in SHIP-1−/− mice, we compared the lung histopathology and BAL fluid cellularity of WT and SHIP-1−/− mice. Lung sections from WT mice had no inflammation. In contrast, cellular infiltration of the airway and lung parenchyma was readily seen in lung sections from SHIP-1−/− mice. Some infiltration involved focal areas and others more diffuse areas, yet some involved nearly an entire lobe (Fig 1). Further analysis of a set of 12 SHIP-1−/− mice demonstrated that there was considerable variation in the degree of severity of inflammation among these mice (see Table E1 in the Online Repository at www.jacionline.org). In some mice (16.6%) no obvious inflammation was detected by means of either lung histology or BAL fluid cell counts. However, in some mice (16.6%) inflammation was only detected on the basis of increased BAL fluid cell counts. Nonetheless, the majority of the SHIP-1−/− mice (66%) by the age of 9 to 11 weeks had obvious pulmonary inflammation, varying in degree from mild to severe (see Table E1). Under high-power viewing, clusters of eosinophils and activated macrophages were seen in the lung (Fig 2, A). The presence of large numbers of eosinophils in the lung is a clear sign of allergic inflammation. Consistent with these findings, BAL fluid cellularity analysis showed significant increases in all cell types, including macrophages, eosinophils, lymphocytes, and neutrophils, in SHIP-1−/− mice when compared with that seen in WT mice (Fig 2, B). These results indicate that the pulmonary inflammation seen in SHIP-1−/− mice is a characteristic TH2 response.
Fig 1.
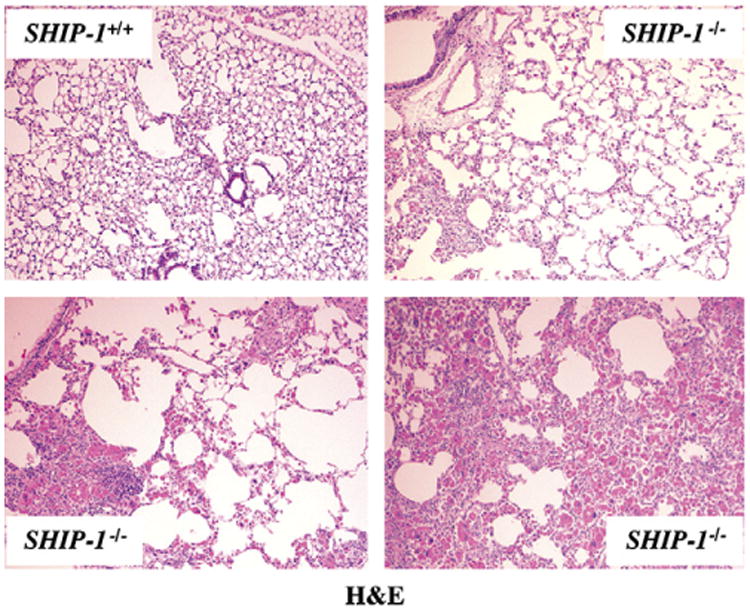
Pulmonary inflammation of SHIP-1−/− mice. Hematoxylin and eosin (H&E) staining of lung sections from WT and SHIP-1−/− mice is shown. Lung sections from WT mice showed normal structure of airways and alveoli. In contrast, mild, medium, and severe cellular infiltration was seen in the airways and lung parenchyma of SHIP-1−/− mice.
Fig 2.
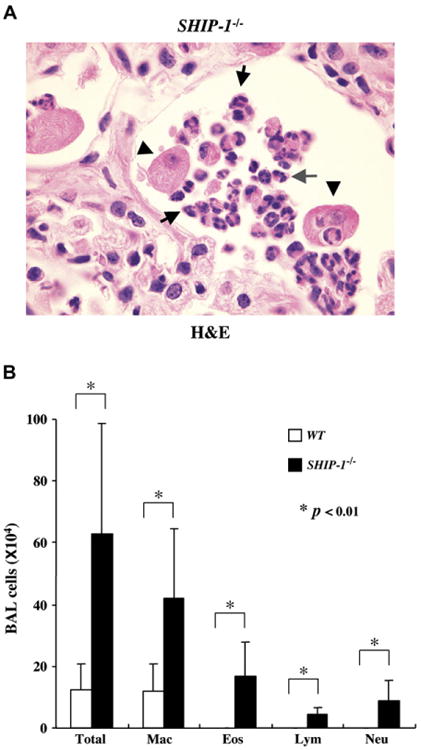
Pulmonary eosinophilia in SHIP-1−/−mice. A, High-power view of a hematoxylin and eosin (H&E)–stained lung section from a SHIP-1−/− mouse. Enlarged and activated macrophages (arrowheads) and eosinophils forming clusters (arrows) are indicated. B, BAL cellularity analysis. Total cell counts and differentials were obtained from WT mice (open column) and SHIP-1−/− mice (solid column). Each group had 6 mice. *P < .01.
Mucus hyperplasia/metaplasia
To determine whether mucus hyperplasia was present, we analyzed lung sections from WT and SHIP-1−/− mice using PAS staining for mucin. As expected, no PAS-positive staining was found in the lung sections of WT mice (Fig 3, A). In contrast, markedly increased PAS-positive cells were readily seen in the airways of SHIP-1−/− mice (Fig 3, A). In some areas airway epithelia were almost completely covered by goblet cells (Fig 3, A). To further understand the mechanisms, we assessed the expression of the mucin gene MUC5AC at mRNA levels in the lung. RT-PCR results showed that low levels of MUC5AC mRNA was seen in the lungs of WT mice, whereas increased MUC5AC mRNA was found in SHIP-1−/− mice (Fig 3, B). Densitometric analysis confirmed the RT-PCR results (see Fig E1 in the Online Repository at www.jacionline.org). These results indicate that SHIP-1 deficiency resulted in goblet cell hyperplasia, mucus hyperproduction, and upregulation of MUC5AC gene expression in the airways.
Fig 3.
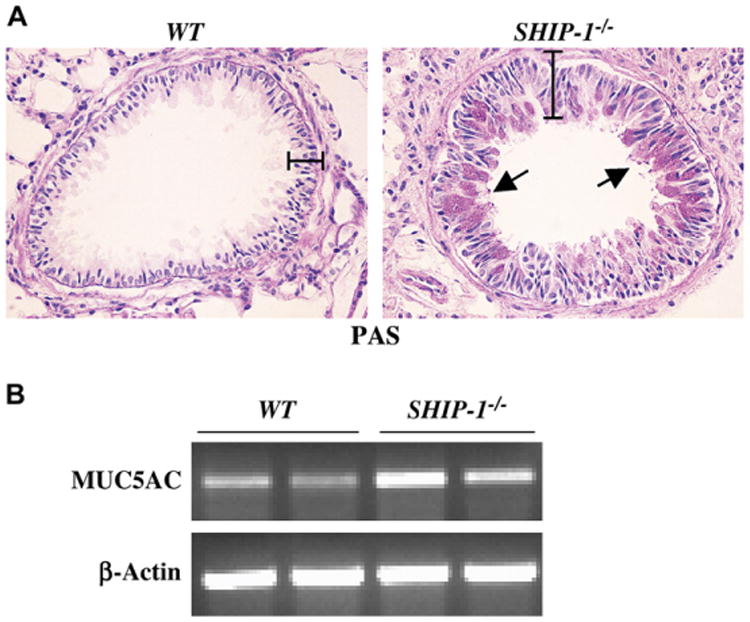
Mucus metaplasia and airway epithelial hypertrophy. A, PAS staining of lung sections from a WT mouse and a SHIP-1−/− mouse is shown. PAS-positive cells in the airways and thickened airway epithelium were readily seen in SHIP-1−/− mice. The bars show the thickness of the airway epithelium. B, RT-PCR analysis of MUC5AC gene expression in WT and SHIP-1−/− mice is shown. Specific primers were used to amplify MUC5AC mRNA. Shown are representative results of 3 experiments.
Epithelial hypertrophy
Airway epithelial hypertrophy can be found in mouse models of allergic asthma and in IL-13–transgenic mice.14 When airways of comparablesize from WT and SHIP-1−/− mice were examined, significantly thickened epithelia were readily seen in the lungs of SHIP-1−/− mice (Figs 3, A, and 4, A). Under the same magnification, the average thickness of the airway epithelia of SHIP-1−/− mice was more than double that of WT mice (Figs 3, A, and 4, A). The size of the airway lumen was severely reduced (Fig 3, A).
Fig 4.
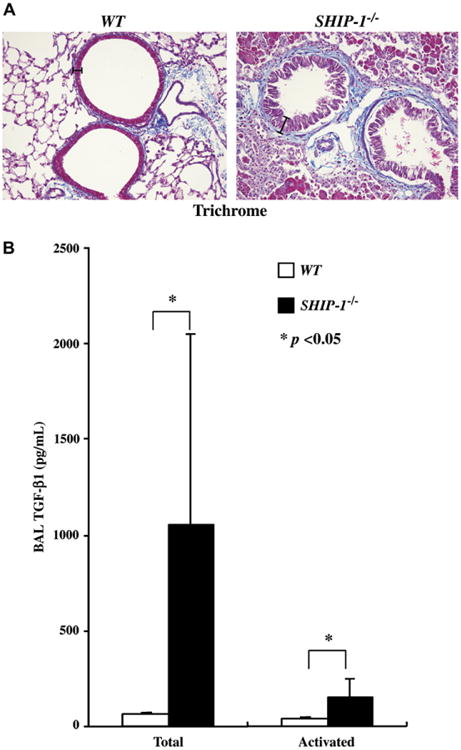
Subepithelial fibrosis and TGF-β1 expression in SHIP-1−/− mice. A, Masson's trichrome staining of lung sections from a WT mouse and a SHIP-1−/− mouse is shown. The bars show the thickness of the airway epithelium. B, TGF-β1 protein levels in the BAL fluid of WT (open column) and SHIP-1−/− (solid column) mice are shown. ELISA was used to determine the total and spontaneously activated TGF-β1 levels in the BAL fluid and expressed as means ± SEM (n = 6 for each group, *P < .05).
Subepithelial fibrosis in the lung
To assess fibrotic changes in the lung, Masson's trichrome staining was used to detect collagen deposition. As shown in Fig 4, A, trichrome positively stained thin layers of collagen, as part of the normal structure, can be seen around the airways of WT mice (Fig 4, A). However, significantly increased collagen deposition was easily seen in the subepithelial areas of the airways of SHIP-1−/− mice (Fig 4, A). To understand the molecular mechanisms of the fibrotic change, we determined the expression of TGF-β1 in the lungs of WT and SHIP-1−/− mice. As shown in Fig 4, B, either total or activated TGF-β1 was barely detectable in the BAL fluid from WT mice. In contrast, TGF-β1 production was highly increased in the lungs of SHIP-1−/− mice, including spontaneously activated TGF-β1 (Fig 4, B). These data indicate that pulmonary fibrosis, as part of the inflammation, resulted from SHIP-1 deficiency.
Chitinase upregulation
Chitinase and chitinase-like genes were significantly upregulated in SHIP-1−/− mice (see the Results section in the Online Repository at www.jacionline.org).
Increased IgE levels in the lung
The concentration of total IgE was significantly increased in the BAL fluid of SHIP-1−/− mice compared with that of WT mice (see Fig E2 in the Online Repository at www.jacionline.org).
Increased and activated mast cells in the lung
We examined numbers of mast cells in the lung tissues of SHIP-1−/− mice and compared these with levels seen in WT mice by using toluidine blue staining. Few mast cells were found in the lungs of WT mice. In contrast, mast cells were readily seen in the lungs of SHIP-1−/− mice (Fig 5, A). These cells were forming clusters and distributed in the lung parenchyma. More importantly, mast cells in the lungs of WT mice were intact, whereas nearly all mast cells in the lungs of SHIP-1−/− mice appeared in a degranulating state (Fig 5, A). Quantification showed that the number of mast cells was significantly higher in the lungs of SHIP-1−/− mice than in those of WT mice (Fig 5, B).
Fig 5.
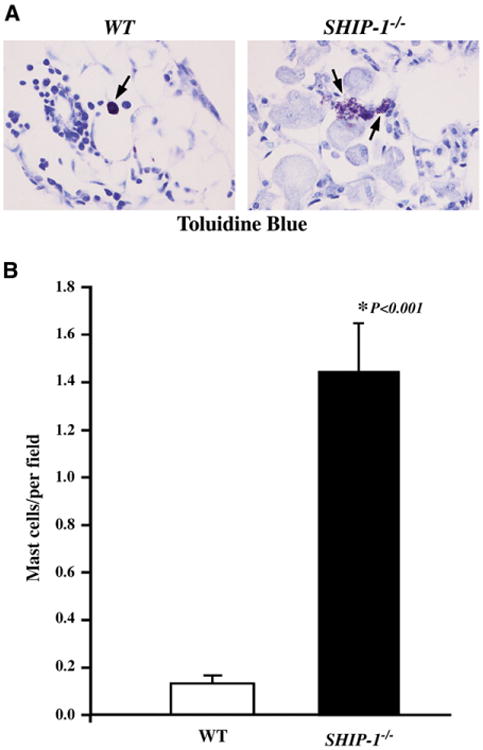
Mast cells and histamine content in the lung. A, Toluidine blue staining of lung sections from WT and SHIP-1−/− mice is shown. Degranulating mast cells were readily seen in the lungs of SHIP-1−/− mice. B, Quantification of mast cells is shown. Mast cells were counted and expressed as mean ± SEM numbers of mast cells per view field (n = 4 for each group).
Histamine content and release
Measurement of histamine in tissue cells isolated from spleens and lungs of SHIP-1−/− mice and compared with that of WT mice showed that total histamine content in these cells was 3-fold and 1.5-fold higher in the spleens and lungs, respectively (see Fig E3 in the Online Repository at www.jacionline.org). Furthermore, cells from SHIP-1−/− mice released more histamine after incubation in culture for 24 hours (Fig E3). These findings are consistent with the histologic observations above.
Cytokines and chemokines in the lung
To understand the underlying molecular mechanisms of the pulmonary inflammation in SHIP-1−/− mice, we investigated the cytokine expression profiles in the lung. As determined by means of RT-PCR, the expression of IL-4, IL-5, and IL-13 was increased at the mRNA level, whereas the expression of IFN-γ was not, in the lungs of SHIP-1−/− mice (Fig 6, A). Densitometric analysis confirmed the RT-PCR results (see Fig E4 in the Online Repository at www.jacionline.org). ELISA results showed that both IL-4 and IL-13 proteins were significantly increased in the BAL fluid of SHIP-1−/− mice (Fig 6, B). IL-5 levels did not change at the protein level. IFN-γ levels did not show any differences between WT and SHIP-1 mice (Fig 6, B). Next we examined the chemokine expression in the lung. As shown in Figs 7 and E5 (see Fig E5 in the Online Repository at www.jacionline.org), TH2-related chemokines, such as eotaxin and monocyte chemoattractant protein (MCP) 1, MCP-2, MCP-3, and MCP-5,18 were upregulated in the lungs of SHIP-1−/− mice at the mRNA level, and some were significantly increased at the protein level in the BAL fluid of SHIP-1−/− mice. However, the expression of the IFN-γ–inducible 10 kD protein (IP-10) gene did not change.
Fig 6.
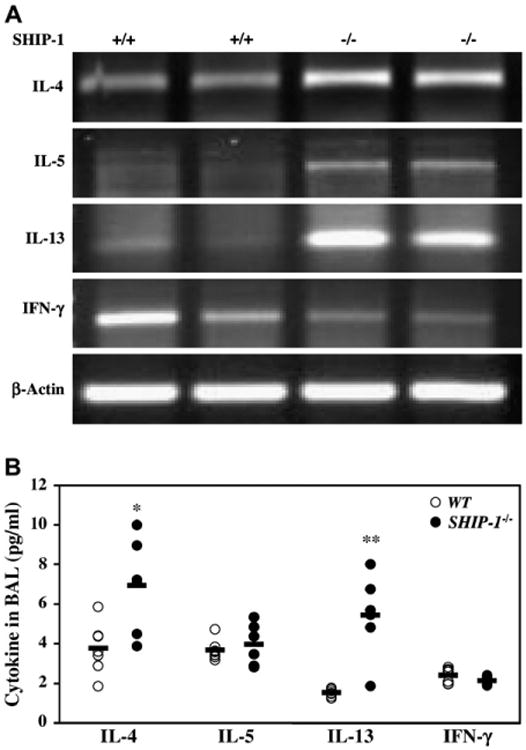
Cytokine expression in the lung. A, RT-PCR analysis of total lung RNA from WT and SHIP-1−/− mice using cytokine gene-specific primers is shown. Amplified products were analyzed by means of electrophoresis. Shown is a representative of 3 experiments. B, ELISA analysis of cytokines in the BAL fluid of WT and SHIP-1−/− mice is shown (n = 6-7 mice for each group, *P = .01 and **P < .01 vs WT group).
Fig 7.
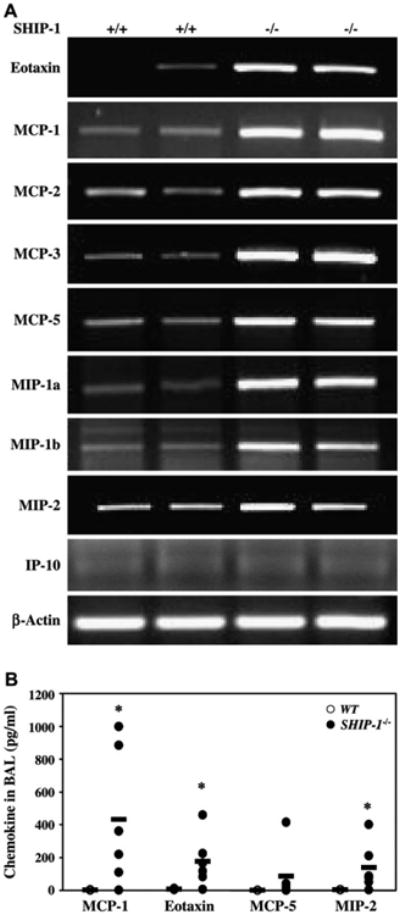
Chemokine expression in the lung. A, RT-PCR analysis of total lung RNA from WT and SHIP-1−/− mice using chemokine gene-specific primers is shown. Amplified products were analyzed by means of electrophoresis. Shown is a representative of 3 experiments. MIP, macrophage inflammatory protein. B, Chemokine concentrations in the BAL fluid of WT and SHIP-1−/− mice were determined by means of ELISA (n = 6-7 mice for each group, *P < .05 vs WT group).
Discussion
Several recent studies have suggested that the PI3K pathway is involved in pulmonary TH2 inflammation in animal models.10-12 Several lines of evidence have suggested that SHIP-1, as a regulator of the PI3K pathway, might have a role in allergic response. In vitro studies have shown that SHIP-1 functioned as a negative regulator in mast cell degranulation in mouse cells19 and human cells.20 IL-4 stimulation of 32D cells resulted in phosphorylation of SHIP-1,13 suggesting its involvement in the TH2 signaling. Dendritic cells from Lyn-deficient mice had decreased phosphorylation of SHIP-1, suggesting that SHIP-1 might participate in regulating dendritic cell (DC) activation in response to allergen.21 SHIP-1 knockout mice exhibited a general myeloproliferative response that affects several tissues and organs. Some SHIP-1– null mice had a spontaneous inflammatory response in the lung, indicating that SHIP-1 is an important regulator in the lung.2,3 Recently, it was reported that macrophages isolated from SHIP-1−/− mice display an alternatively activated (M2) phenotype, suggesting that SHIP-1 is a negative regulator of M2 generation.22 However, the nature of the inflammation, the cell types infiltrating the lung, and the pathologic changes in the lung are not clear. Furthermore, whether SHIP-1 plays a role in regulating the TH2 signaling pathway, particularly IL-4 receptor–mediated signaling, is not clear. Whether SHIP-1 has any function in maintaining lung homeostasis also remains unknown.
To define the nature of the inflammation seen in SHIP-1−/− mice, we analyzed the lung pathology in detail. Our results showed that several features characteristic of allergic inflammation were present in the SHIP-1−/− mice. Cellular infiltration of the airway and lung parenchyma with varying degrees of severity was seen in SHIP-1−/− mice (Fig 1 and see Table E1 in the Online Repository at www.jacionline.org). Detailed analyses showed that although some mice did not show obvious inflammation, either by histology or BAL fluid cell counts, the majority of SHIP-1−/− mice had mild, moderate, or severe pulmonary inflammation by 9 to 11 weeks of age (see Table E1). This observation is in accordance with the results of survival analysis in a previous study, in which 40% of SHIP-1−/− mice died from lung inflammation by the age of 12 weeks.3 Pulmonary eosinophilia is a major feature of TH2 inflammation commonly seen in patients with allergic asthma and in animal models of asthma. Further histologic analysis at high power revealed large numbers of inflammatory cells, particularly eosinophils, in the airways and lung parenchyma of SHIP-1−/− mice (Fig 2, A). BAL analysis confirmed these findings (Fig 2, B). In addition, genetic background might not have a major role in the development of pulmonary inflammation because SHIP-1−/− mice on a BALB/c background also showed similar lung pathology (data not shown).
Mucus hyperproduction is an important component of TH2 response and a part of airway remodeling in patients with allergic asthma and in mouse models of asthma.14 PAS staining of lung sections revealed increased numbers of mucin-secreting goblet cells in the airways of SHIP-1−/− mice (Fig 3, A), accompanied by upregulated MUC5AC mRNA (Fig 3, B, and see Fig E1 in the Online Repository at www.jacionline.org). In addition, epithelial hypertrophy was noticed in the lungs of SHIP-1−/− mice (Figs 3, A, and 4, A). One of the consequences of mucus hyperproduction and airway epithelial hypertrophy is severely decreased size of the airway lumen (Figs 3, A, and 4, A), which could lead to restricted airflow.
Pulmonary fibrosis is a process of airway remodeling usually seen in the chronic phase of asthma. Lung sections from SHIP-1−/− mice had increased collagen deposition (Fig 4, A). This is accompanied by significantly increased production of TGF-β1, particularly spontaneously activated TGF-β1 in SHIP-1−/− mice (Fig 4, B).
The expression of several chitinases is closely associated with the TH2 phenotype of the lung.23-25 Chitinase-like proteins YM-1 and YM-2 had been found in crystal forms in the lungs in mouse models of asthma.23,24 Crystal particles were readily appreciated in the BAL fluid and in the lungs of SHIP-1−/− mice (see Fig E6, A, in the Online Repository at www.jacionline.org). RT-PCR confirmed that acidic mammalian chitinase (AMCase), YM-1, and YM-2, but not chitotriosidase, were upregulated in the lungs of SHIP-1−/− mice (Fig E6, B). Increased chitinase activity was also seen in the BAL fluid of SHIP-1−/− mice (Fig E6, C).
SHIP-1−/− mice showed increased B-cell function and increased serum Ig levels, including IgG2a and IgG2b levels.26 However, IgE levels were not determined in that study. We found that SHIP-1−/− mice had increased total IgE levels in the BAL fluid. It is likely that the increased IgE levels in the lung tissue of SHIP-1−/− mice are a result of increased B-cell function and increased Ig production in general. It is also possible, however, that they are part of the TH2 inflammatory response. Mast cells have increased sensitivity in the absence of SHIP-1,19 and increased IgE levels in the lung might further increase the mast cell response to various stimulations.
Taken together, these results demonstrated that the inflammation seen in SHIP-1−/− mice is a typical TH2 inflammation in the lung. To understand the underlying mechanisms of the lung phenotype in SHIP-1−/−mice, we investigated the cytokines and chemokines that might be involved. In the lungs of SHIP-1−/− mice, levels of the TH2 cytokines IL-4 and IL-13 were increased at both the mRNA and protein levels, whereas levels of the TH1 cytokine IFN-γ were not (Fig 6). Consistent with the TH2 cytokine profile, chemokines also showed a TH2-dominant pattern (Fig 7).
The current view of the initiation of TH2 inflammation is that after allergen stimulation, antigen-presenting cells interact with naive CD4+ T cells. In the presence of IL-4, these cells differentiate into TH2 cells. When encountering the allergen again, TH2 cells respond by proliferating and producing TH2 cytokines, which activate downstream genes, leading to tissue inflammation and other effects. However, it is possible that the differentiation phase might be bypassed. Under experimental conditions, direct administration or transgenic overexpression of the effector cytokines IL-4 and IL-13 can elicit TH2 inflammation in the lung.14,27,28 The inflammation seen in the lungs of SHIP-1−/− mice has many features of TH2 immune responses and occurs in the absence of overt immunologic sensitization and challenge. It is unclear whether the pulmonary phenotype requires TH2 cell differentiation and activation or is only part of the TH2 effector response. The role of T and B cells in the process is not clear. Mast cells and basophils are part of the innate immune system. Mast cells can release mediators and have an important role in mediating allergic responses. Recently, mast cells and basophils have been recognized to be able to produce TH2 cytokines that might be important for TH2 cell differentiation and TH2 effector functions.29-31 Given the fact that SHIP-1 has a regulatory role in mast cells, it is reasonable to speculate that SHIP-1 acts as a negative regulator to keep mast cells or basophils in check. When lacking SHIP-1, mast cells and basophils might have increased sensitivity or a decreased threshold for releasing mediators and cytokines, such as IL-4 and IL-13, which might be responsible for the TH2 phenotype in SHIP-1−/− mice. Our histologic analysis and mediator experiments showed that the number of mast cells was significantly increased in the lungs of SHIP-1−/− mice, the majority of these cells were activated and in a degranulation state, and total and spontaneously released histamine by the lung cells and splenocytes was significantly increased (Fig 5 and see Fig E3 in the Online Repository at www.jacionline.org). Whether mast cells are the source of the increased TH2 cytokines seen in the lungs of SHIP-1−/− mice is under investigation. These observations, however, do not exclude the potential contributions by other cell types, such as T cells, lung epithelial cells, or dendritic cells. In addition, the low magnitude of increase in TH2 cytokines and the relatively higher increase in chemokines in SHIP-1−/− mice suggests that dysregulation of the signaling downstream of IL-4Rα is potentially more important in generating the pulmonary phenotype.
In conclusion, our studies demonstrated that the inflammatory response seen in the lungs of SHIP-1−/− mice has many features of allergic inflammation. These pathologic changes are accompanied by increased and activated mast cells in the lung and upregulation of TH2 cytokines and chemokines. These findings strongly support a role for SHIP-1 in regulating TH2 immune response and in maintaining lung homeostasis. A potential role of SHIP-1 in allergen-induced response is under investigation. Further studies of the functions of SHIP-1 will provide valuable insights into the regulation of the TH2 signaling pathway and potential therapeutic targets for the development of effective treatment of TH2 inflammatory diseases in the lung.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health (NIH) grant RO1HL074095 and an American Lung Association Research Grant to Z.Z. and NIH Grant KO8 to T.Z. D.L.B. is a National Cancer Institute of Canada Research Scientist supported by the National Cancer Institute of Canada and the Canadian Institutes of Health Research.
Abbreviations
- BAL
Bronchoalveolar lavage
- MCP
Monocyte chemoattractant protein
- PAS
Periodic acid–Schiff
- PI3K
Phosphoinositide 3-kinase
- PIP3
Phosphotidylinositol-3,4,5-trisphosphate
- SHIP-1
Src homology 2 domain–containing inositol 5-phosphatase 1
- WT
Wild type
Footnotes
Disclosure of potential conflict of interest: D. L. Barber has received grant support from Hoffman-La Roche. The rest of the authors have declared that they have no conflict of interest.
References
- 1.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383:263–6. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 2.Liu Q, Oliveira-Dos-Santos AJ, Mariathasan S, Bouchard D, Jones J, Sarao R, et al. The inositol polyphosphate 5-phosphatase ship is a crucial negative regulator of B cell antigen receptor signaling. J Exp Med. 1998;188:1333–42. doi: 10.1084/jem.188.7.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–20. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Sasaki T, Kozieradzki I, Wakeham A, Itie A, Dumont DJ, et al. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13:786–91. doi: 10.1101/gad.13.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacDonald S, Vonakis B. Association of the Src homology 2 domain-containing inositol 5′ phosphatase (SHIP) to releasability in human basophils. Mol Immunol. 2002;38:1323–7. doi: 10.1016/s0161-5890(02)00082-2. [DOI] [PubMed] [Google Scholar]
- 6.Daigle I, Yousefi S, Colonna M, Green DR, Simon HU. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat Med. 2002;8:61–7. doi: 10.1038/nm0102-61. [DOI] [PubMed] [Google Scholar]
- 7.Freeburn RW, Wright KL, Burgess SJ, Astoul E, Cantrell DA, Ward SG. Evidence that SHIP-1 contributes to phosphatidylinositol 3,4,5-trisphosphate metabolism in T lymphocytes and can regulate novel phosphoinositide 3-kinase effectors. J Immunol. 2002;169:5441–50. doi: 10.4049/jimmunol.169.10.5441. [DOI] [PubMed] [Google Scholar]
- 8.Chacko GW, Tridandapani S, Damen JE, Liu L, Krystal G, Coggeshall KM. Negative signaling in B lymphocytes induces tyrosine phosphorylation of the 145-kDa inositol polyphosphate 5-phosphatase, SHIP. J Immunol. 1996;157:2234–8. [PubMed] [Google Scholar]
- 9.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 10.Myou S, Leff AR, Myo S, Boetticher E, Tong J, Meliton AY, et al. Blockade of inflammation and airway hyperresponsiveness in immune-sensitized mice by dominant-negative phosphoinositide 3-kinase-TAT. J Exp Med. 2003;198:1573–82. doi: 10.1084/jem.20030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan W, Aguinaldo Datiles AM, Leung BP, Vlahos CJ, Wong WS. An anti-inflammatory role for a phosphoinositide 3-kinase inhibitor LY294002 in a mouse asthma model. Int Immunopharmacol. 2005;5:495–502. doi: 10.1016/j.intimp.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Kwak YG, Song CH, Yi HK, Hwang PH, Kim JS, Lee KS, et al. Involvement of PTEN in airway hyperresponsiveness and inflammation in bronchial asthma. J Clin Invest. 2003;111:1083–92. doi: 10.1172/JCI16440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamorano J, Keegan AD. Regulation of apoptosis by tyrosine-containing domains of IL-4R alpha: Y497 and Y713, but not the STAT6-docking tyrosines, signal protection from apoptosis. J Immunol. 1998;161:859–67. [PubMed] [Google Scholar]
- 14.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–93. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo L, Johnson RS, Schuh JC. Biochemical characterization of endogenously formed eosinophilic crystals in the lungs of mice. J Biol Chem. 2000;275:8032–7. doi: 10.1074/jbc.275.11.8032. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder JT, Saini S. Assay methods for measurement of mediators and markers of allergic inflammation. In: Detrick B, Hamilton RG, Folds JD, editors. Manual of molecular and clinical laboratory immunology. 7th. Washington (DC): ASM; 2006. pp. 964–74. [Google Scholar]
- 18.Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol Today. 1998;19:568–74. doi: 10.1016/s0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 19.Huber M, Kalesnikoff J, Reth M, Krystal G. The role of SHIP in mast cell degranulation and IgE-induced mast cell survival. Immunol Lett. 2002;82:17–21. doi: 10.1016/s0165-2478(02)00012-3. [DOI] [PubMed] [Google Scholar]
- 20.Vonakis BM, Gibbons S, Jr, Sora R, Langdon JM, MacDonald SM. Src homology 2 domain-containing inositol 5′ phosphatase is negatively associated with histamine release to human recombinant histamine-releasing factor in human basophils. J Allergy Clin Immunol. 2001;108:822–31. doi: 10.1067/mai.2001.119159. [DOI] [PubMed] [Google Scholar]
- 21.Beavitt SJ, Harder KW, Kemp JM, Jones J, Quilici C, Casagranda F, et al. Lyn-deficient mice develop severe, persistent asthma: Lyn is a critical negative regulator of Th2 immunity. J Immunol. 2005;175:1867–75. doi: 10.4049/jimmunol.175.3.1867. [DOI] [PubMed] [Google Scholar]
- 22.Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–74. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, et al. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–82. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 24.Webb DC, McKenzie AN, Foster PS. Expression of the Ym2 lectin-binding protein is dependent on interleukin (IL)-4 and IL-13 signal transduction: identification of a novel allergy-associated protein. J Biol Chem. 2001;276:41969–76. doi: 10.1074/jbc.M106223200. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, et al. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–74. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helgason CD, Kalberer CP, Damen JE, Chappel SM, Pineault N, Krystal G, et al. A dual role for Src homology 2 domain-containing inositol-5-phosphatase (SHIP) in immunity: aberrant development and enhanced function of b lymphocytes in ship -/- mice. J Exp Med. 2000;191:781–94. doi: 10.1084/jem.191.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–3. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 29.Min B, Prout M, Hu-Li J, Zhu J, Jankovic D, Morgan ES, et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med. 2004;200:507–17. doi: 10.1084/jem.20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gessner A, Mohrs K, Mohrs M. Mast cells, basophils, and eosinophils acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are sufficient for rapid cytokine production. J Immunol. 2005;174:1063–72. doi: 10.4049/jimmunol.174.2.1063. [DOI] [PubMed] [Google Scholar]
- 31.Weiss DL, Brown MA. Regulation of IL-4 production in mast cells: a paradigm for cell-type-specific gene expression. Immunol Rev. 2001;179:35–47. doi: 10.1034/j.1600-065x.2001.790104.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.