

Abstract
CFTR (ABCC7), unique among ABC exporters as an ion channel, regulates ion and fluid transport in epithelial tissues. Loss of function due to mutations in the cftr gene causes cystic fibrosis (CF). The most common CF-causing mutation, the deletion of F508 (ΔF508) from the first nucleotide binding domain (NBD1) of CFTR, results in misfolding of the protein and clearance by cellular quality control systems. The ΔF508 mutation has two major impacts on CFTR: reduced thermal stability of NBD1 and disruption of its interface with membrane-spanning domains (MSDs). It is unknown if these two defects are independent and need to be targeted separately. To address this question we varied the extent of stabilization of NBD1 using different second site mutations and NBD1 binding small molecules with or without NBD1/MSD interface mutation. Combinations of different NBD1 changes had additive corrective effects on ΔF508 maturation that correlated with their ability to increase NBD1 thermostability. These effects were much larger than those caused by interface modification alone and accounted for most of the correction achieved by modifying both the domain and the interface. Thus, NBD1 stabilization plays a dominant role in overcoming the ΔF508 defect. Furthermore, the dual target approach resulted in a locked-open ion channel that was constitutively active in the absence of the normally obligatory dependence on phosphorylation by protein kinase A. Thus, simultaneous targeting of both the domain and the interface, as well as being non-essential for correction of biogenesis, may disrupt normal regulation of channel function.
Keywords: Cystic fibrosis, CFTR, thermal stability, ion channel, protein folding
Introduction
Cystic fibrosis, the most common genetic disease in the Caucasian population results from mutations in the gene coding for the CFTR anion channel protein, important in epithelial ion and fluid homeostasis. Most patients have a Phe508 deletion mutation (ΔF508) causing a loss of function and a folding defect in the channel protein 1; 2; 3. A great deal of mechanistic insight has been gained into how this single residue deletion affects the biosynthesis and assembly of the large multi-domain membrane protein and its handling by cellular quality control and proteolytic systems 4; 5; 6; 7; 8. The impact of the mutation is mitigated at sub-physiological temperature 9 and studies with the isolated first nucleotide binding domain (NBD1) in which F508 resides revealed a large reduction in thermodynamic stability 10; 11. This shift is reflected at the level of the channel function of the full-length protein 12; 13; 14; 15. Restoration of stability is achieved when the protein is expressed in cells kept at reduced temperatures 9, exposed to osmolytes 16; 17 and by a number of second site mutations 6.
The effectiveness of these manipulations in experimental cell culture systems has motivated extensive screening efforts to identify small molecules that can mimic the effects of low temperature and osmolytes and serve as lead compounds for the development of pharmaceutical treatments of the disease 18; 19; 20. These cell based screens for the appearance of the protein or its function on the cell surface have yielded several encouraging compounds 21, the most effective thus far being VX-809, discovered by Vertex Pharmaceuticals 22. Although there is some evidence that these compounds may bind directly to the nascent CFTR polypeptide 23; 24; 25, their precise binding sites have not yet been defined and some may act indirectly as so-called proteostasis regulators 26.
The existing corrector compounds are much more effective in supporting the maturation of ΔF508 CFTR in cells at temperatures well below 37°C than at the temperature where correction is required in patients’ tissues 23. These results strongly imply that the compounds do not act primarily by restoring the diminished thermodynamic stability, although this does not exclude the possibility that increasing temperature might reduce the binding affinity of corrector compounds and thus diminish their efficiency.
The 3D structure of NBD1 determined by X-ray crystallography 27; 28 showed that F508 is located on the surface of NBD1 and structures of the full-length protein determined by homology modeling 29; 30; 31; 32 and partially confirmed by Cys-pair cross-linking 29 indicated that the residue participates in a hydrophobic contact with the fourth cytoplasmic loop (CL4) of the C-terminal membrane-spanning domain. Studies in which the ΔF508 protein was modified by second-site changes in NBD1 and the NBD1/CL4 interface separately or in combination have shown that the two types of modifications together are more effective than either alone 33; 34 and led to the idea that both should be targeted for effective correction of the defect. However, it has not been determined whether more effective means of stabilizing either the domain or the interface may be sufficient, nor has the influence of these protein maturation-promoting changes on channel function been analyzed in detail. Here, we present data showing that the combination of several additive second site changes in NBD1 without any direct alteration of the NBD1/CL4 interface can effectively restore ΔF508 CFTR maturation and stability and that small molecules binding to a specific NBD1 site have weaker but similar effects. Furthermore, the combination of maximal NBD1 stabilization and interface modification by the paradoxical R1070W interface mutation disrupts the normal control of channel activity by phosphorylation. Thus, while the targeting of both the domain and the interface is a rational therapeutic approach, it may not be essential and could have unintended consequences.
Results
Multiple NBD1 stabilizing mutations have additive corrective effects on ΔF508 CFTR maturation, cell surface appearance and channel function
Teem and colleagues originally used genetic methods to identify second site suppressor mutations of the ΔF508 CFTR defect in NBD1 35. Since then, a range of additional NBD1 modifications having similar effects have been described. These include several single residue substitutions that improve the solubility of isolated NBD1 27, deletion of the regulatory insertion (RI) peptide 13; 36, and the introduction of proline residues at several mobile sites 14. Rabeh et al 33 found that while several of the solubilizing and suppressor mutations caused only a modest promotion of ΔF508 CFTR maturation, their effects were greater when they were combined with the NBD1/CL4 interface substitutions R1070W or V510D, the latter also having been shown to have a direct effect on NBD1 thermostability 10. These and related studies of Mendoza et al 34 have led to the proposal that two separate steps are required to correct the ΔF508 mutation i.e., stabilization of NBD1 and modification of its interface with CL4. Here we addressed the relative contributions of each of these steps and how they are inter-related. For NBD1 stabilization within the full-length mutant protein, proline introduction, RI deletion, and the original suppressor mutations were used in different combinations. In general, combination of any two of these stabilizing changes increased ΔF508 maturation (greater band C/B ratio) from about 30% to 50% of the wild-type level (Fig.1a, top panel, lanes 4–6). When all three modifications were combined (ΔF508/combo, lane 7), the amounts of both B (Fig. 1a, second panel) and C (Fig. 1a, third panel) bands were further increased resulting in a somewhat lesser increment in C/B ratio (Fig. 1a, bottom panel). Nevertheless, the amount of C band reached ~ 75% of wild-type level. In contrast, each of the interface substitutions, V510D (lane 8) and R1070W (lane 9) had much smaller effects. Their addition to the maximally stabilized NBD1, i.e., lanes 10 and 11, respectively compared to lane 7 caused a further increase in band C to levels approaching that of the wild-type but this increment amounted to only a further increase in C/B ratio of about 25% and an even lesser increase in C band amount. These data are consistent with both NBD1 and NBD1/CL4 correction ameliorating impaired ΔF508 CFTR maturation but they also indicate that NBD1 stabilization makes the larger contribution and appears to play the dominant role.
Figure 1. Combinations of NBD1 stabilizing mutants are additive in promoting ΔF508 CFTR maturation and channel function.

(a). Western blot analysis of BHK cells stably expressing WT and ΔF508 CFTR with various rescuing mutants grown at 37°C. The intensity of complex glycosylated mature CFTR (C band) and core glycosylated immature CFTR (B band) was quantified and C/B ratio was calculated. Data plotted are mean and standard deviation (N=3). 2PT: S492P/A534P/I539T; 4PT: 2PT+S422P/S434P; 3SS: G550E/R553M/R555K; 4SS: 3SS + I539T; ΔRI: deletion of regulatory insertion amino acids 404–435; combo: ΔRI + 2PT + 3SS. (b). Pulse chase labeling of BHK cells stably expressing CFTR variants. Autoradiograms shown were quantified electronically, and rates of disappearance of immature precursors (B band) and appearance of mature products (C band) were graphed. N=3. ΔF/combo: ΔF508 with 2PT/ΔRI/3SS mutations. (c). Cell surface expression of CFTR on BHK cells expressing WT and Δ F508 CFTR variants with an epitope tag were measured by on cell Western. (d). Channel activity measurement by iodide efflux assay on BHK cells expressing CFTR variants. The values represent the mean ± SD of the amount of iodide released from the cells during the 1 min interval (N=4).
To supplement the evaluation of maturation from the steady-state amounts of the immature and mature forms of the protein, their inter-conversion was followed in pulse-chase experiments (Fig. 1b). The rates of disappearance of the immature forms of the differently stabilized ΔF508 variants were not appreciably different from each other or from the wild-type. However, at the 1 h and 4 h chase times the amount of mature product formed by the NBD1 stabilized variant (ΔF/combo: ΔF/2PT/ΔRI/3S) was as high as that of the wild-type although somewhat less at the 2 h chase time. The rates of appearance of the mature products and the levels reached were increased further when the R1070W mutation was added to the combined NBD1 changes but not when V510D was added. Thus, the combined NBD1 changes without interface changes result in a maturation level approaching that of wild-type with added interface modification causing only a small further increment.
The amounts of CFTR reaching the cell surface also was measured employing external epitope labeled forms and functionally in an iodide efflux assay. The surface level of ΔF508 CFTR with the combined NBD1 stabilizing mutations (ΔF/combo) reached ~90% that of WT CFTR (Fig. 1c), and the level was somewhat further increased when V510D or R1070W was added to the combination (ΔF/combo/V510D and ΔF/combo/R1070W). Forskolin-stimulated iodide efflux from cells expressing ΔF508/2PT CFTR was delayed compared to WT CFTR and the peak efflux rate was ~40% of the WT level (Fig. 1d, top panel). Additional NBD1 stabilizing mutations (ΔF508/combo) increased the peak to ~ 60% of WT, and the lag of peak appearance typical for rescued ΔF508 CFTR was no longer present. Functional activity measured using this iodide efflux assay was similar for the ΔF508/combo and the ΔF508/4PT/R1070W, indicating the effectiveness of the combination of multiple NBD1 stabilizing mutations without interface modification in promoting ΔF508 CFTR channel activity. The addition of V510D to the ΔF508/combo did not further increase its peak of iodide efflux. It was initially surprising to observe that, the addition of R1070W to the ΔF508/combo apparently greatly diminished iodide efflux (Fig. 1d, bottom panel). However, evaluation of the intracellular iodide level of these cells after six rinses, prior to the stimulation of efflux, revealed only about 1/3 the amount of iodide as the WT and other mutants. This apparent inability of BHK cells expressing ΔF508/combo/R1070W CFTR to be effectively stimulated by forskolin cocktail suggested that the ΔF508/combo/R1070W CFTR channel might be constitutively active, a possibility that was subsequently confirmed at the single channel level (see Fig. 5 below).
Figure 5. Combined NBD1 stabilizing mutations promote maturation and function of R1070W CFTR in both ΔF508 and wild-type backgrounds.

(a). Single channel current tracings of the wild type, ΔF508/Combo and ΔF508/Combo/R1070W CFTRs at 25°C. All points histograms, single channel conductances, and open probailities are shown as in Fig. 4. (b) Single channel currents at 37°C showing the influence of combined NBD1 stabilizing changes on ΔF508 and wild-type CFTR without and with the R1070W mutation. X-scale bar below traces is 10s.
Enhancement of full-length ΔF508 CFTR maturation by NBD1 stabilizing mutations correlate with their increase in NBD1 Tm
The additive effects of different NBD1 stabilizing mutations in promoting ΔF508 CFTR maturation and function prompted us to investigate whether their relative impacts correlated with their ability to thermally stabilize NBD1. In addition to the S492P substitution found in three non-mammalian species that are relatively insensitive to the destabilizing influence of the ΔF508 mutation13 we also included a second Q-loop proline substitution, S495P present in shark CFTR. As evident from the heat capacity profiles (Fig. 2a, upper panel) and Tm values determined by differential scanning calorimetry (DSC) (Fig. 2a, lower panel), additive effects of the various NBD1 stabilizing mutations were found to increase the Tm of isolated NBD1. S492P or I539T alone slightly increased the Tm of NBD1 (ΔTm 2–3°C) similar to the affect of the solubilization mutations F494N and Q637R combined. The S492P and I539T substitutions had additive affects such that ΔTm increased to 4.4 °C, and ΔTm was further increased to 8.4 °C when the additional mutations A534P/G550E/R553M/R555K were introduced. The S495P substitution alone in the isolated NBD1 of human CFTR caused the largest increase in Tm (5.99 ± 0.33°C) of any single change tested (Fig.2a, lower panel). Therefore, we compared the effect of S495P with that of other NBD1 stabilizing mutations on the maturation of ΔF508 CFTR. As shown in Fig. 2b, the S495P substitution in ΔF508 CFTR resulted in substantial maturation. The effect of this single mutation was in contrast to that of S492P which only increased maturation substantially when present together with I539T. Combination of I539T with S495P, however, did cause a further increase in maturation and the effects of the two proline substitutions also were additive. Importantly, the extent of improvement in maturation by the different changes paralleled the magnitudes of the Tm increases in NBD1 that they caused (Figs. 2a and b). This tight correlation between the effects of a full spectrum of known NBD1 second-site mutations on the thermal stability of purified NBD1 determined by DSC and the biosynthetic maturation of full-length ΔF508 CFTR (Fig. 2c) strongly supports the idea that thermal stabilization of NBD1 is able to bring about virtually complete rescue of ΔF508 CFTR.
Figure 2. Correlation between NBD1 thermal stability and ΔF508 CFTR maturation.
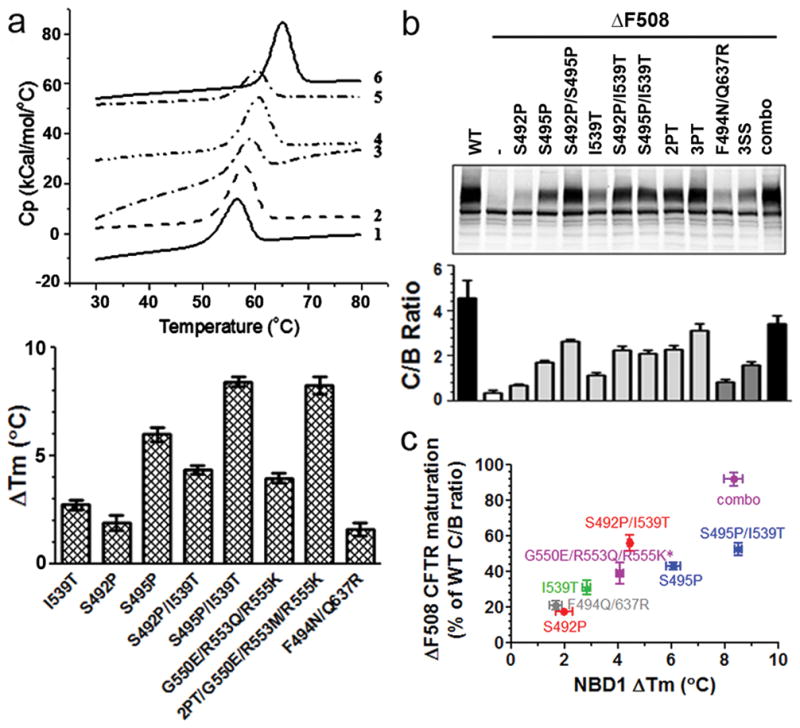
(a). Upper panel: DSC heat capacity profiles for WT-(ΔRI, ΔRE)-NBD1 and sequence-matched NBD1 stabilizing mutants in buffer containing 2 mM ATP and 3 mM MgCl2 at a scan rate of 2 °C/min. 1-WT; 2-S492P; 3-I539T; 4. S492P/I539T; 5-G550E/R553Q/R555K; 6-combo. Lower panel: The Tm difference between ΔRI/ΔRE-NBD1 with those carrying various NBD1 stabilizing mutations. The values represent the mean ± SD (N=2–3) (b). HEK cells transiently transfected with WT and ΔF508 CFTR with various NBD1 stabilizing mutants were grown at 37°C for 48 hr. The intensity of mature C band and immature B band was quantified and the C/B ratio was calculated. The values represent the mean ± SD (N=3–6). 2PT: S492P/A534P/I539T; 3PT: 2PT+S495P. (c). The rescue of ΔF508 CFTR by NBD1 stabilizing mutants were expressed as the percent of WT C/B ratio and plotted against the Tm increase caused by corresponding mutants in ΔRI/ΔRE-NBD1. * In full length CFTR R553M was introduced instead of R553Q in isolated NBD1. Based on our single mutation analysis, the Tm difference between G550E/R553Q/R555K and G550E/R553M/R555K is less than 1°C.
NBD1 binding small molecules also increase ΔF508 CFTR maturation
Since amino acid substitutions at several sites in NBD1 restore the thermal stability and maturation of ΔF508 CFTR, we asked whether small molecule binders could have a similar effect. Interestingly, none of the small molecule correctors of ΔF508 turned up in cell based screens so far appear to act on NBD1, perhaps partially explaining why they have little effect on the thermal stability of the full-length mutant protein 23; 25. Therefore we employed two related compounds (5-bromoindole-3-acetic acid, BIA and its analog 5′-bromo-3-ethoxyindole- acetic acid, BEIA) found earlier to bind to a specific site on NBD1 during co-crystallization experiments (Fig. 3a), performed by SGX Pharmaceuticals with the support of Cystic Fibrosis Foundation Therapeutics Inc. These compounds caused small but significant increases in the Tm of purified NBD1 (Fig. 3b). BIA increased the Tm of WT and ΔF508 NBD1 by 0.9 ± 0.2 and 1.4 ± 0.2°C, respectively, while the influence of BEIA was slightly larger, causing Tm increases of 1.7 ± 0.1 and 2.4 ± 0.2°C, respectively. Exposure of cells expressing ΔF508 CFTR to BIA caused a dose-dependent promotion of maturation (Fig. 3c). However this increase occurred over the high concentration range of 0.1 to 1.5 mM, reflecting low affinity binding. BEIA had a similar but more pronounced affect, causing appreciable maturation at 0.5 mM, consistent with its slightly higher binding affinity to NBD1 and increase in Tm of NBD1. Notably, this compound at a concentration of 1 mM, not yet toxic to cells, caused a large elevation in the amounts of both the immature (band B) and mature (band C) forms of the mutant protein (Fig. 3c).
Figure 3. Known binders of NBD1 promote the maturation of ΔF508 CFTR and the effect is additive with corrector VX-809 and stabilizing mutations.

(a). Structures of NBD1 binders BIA and BEIA and the site of BIA binding to NBD1. b). BIA slightly increased Tm of both WT- and ΔF-(ΔRI, ΔRE) NBD1 Dotted lines: ΔF; solid lines: WT; black lines: without compound; red lines: with 4 mM BIA and blue lines: with 4 mM BEIA in a buffer with 1 mM ATP, 1.5 mM MgCl2 and 5% v/v DMSO, at a scan rate of 2 °C/min. (c and d). BHK cells expressing ΔF508 CFTR were treated with various doses of these compounds in the absence (c) and presence of 3 μM VX-809 (d) for 24 hrs at 37°C. The intensity of mature C band and immature B band was quantified and the C/B ratio was calculated. N=3. (e&f) BHK cells expressing ΔF508 CFTR with NBD1 stabilizing mutants (e) and interface patch mutant (f) were treated with BIA and BEIA at various doses for 24 hrs at 37°C. The intensity of mature C band and immature B band was quantified and the C/B ratio was calculated. N=3.
To test the action of these NBD1 binding compounds together with a corrector compound not known to act on NBD1, we exposed cells to these indole compounds and an optimal concentration of the VX-809 corrector. The results shown in Fig. 3d clearly revealed an elevated response to both of the NBD1 binders in the presence of VX-809, consistent with the corrector and the binders having different sites and modes of action.
To further probe the action of these indole-based compounds, we assessed their affects on ΔF508 CFTR already partially corrected by second site mutations in NBD1 or at the NBD1/CL4 interface. Fig. 3e show that both BIA and BEIA further strongly increased maturation of the NBD1 stabilized variant ΔF508/2PT (S492P/A534P/I539T). Thus, it appears that binding to a site on NBD1 separate from the sites changed by mutation is additive with their summed impact. Turning to the interface modified variants (Fig. 3f), BEIA also caused a large further increase in the maturation of the ΔF508/V510D protein. The BEIA influence on the other interface substituted form of the protein, R1070W was less pronounced, particularly at the lower concentrations of the compound. Overall, however the NBD1 binding indole compounds enhanced maturation of ΔF508 CFTR with or without genetic stabilization of NBD1 or the NBD1/CL4 interface, providing the first direct evidence that NBD1 binding small molecules other than nucleotides can counteract the folding defect. In addition, iodide efflux assays showed that BIA increased the functional activity of ΔF508 CFTR to a similar level as that rescued by 2PT mutations (Fig. S1), which also was further increased by VX-809.
The disease-associated CL4 mutation R1070W differentially influences wild-type and ΔF508 CFTR
R1070W is a relatively rare disease-associated mutation in the CL4 region of CFTR. Although the mutation does not cause complete loss of function, it does diminish biosynthetic maturation and channel activity 37, resulting in a moderate CF phenotype in patients 38. Homology models of CFTR 3D structure indicated that the R1070-containing segment of CL4 was proximal to F508 on the NBD1 surface 29; 30; 31; 32 and Thomas and colleagues reasoned that the introduction of the aromatic tryptophan side chain by R1070W substitution might compensate for the loss of the phenylalanine side chain due to the absence of F508 at the interface between domains 8. They showed that the R1070W mutation substantially improved the maturation of ΔF508 CFTR. This effect has been widely confirmed and is integral to the proposed two-step strategy for correction of the ΔF508 defect 34. In this model, step 1 is NBD1 stabilization and step 2, NBD1/CL4 modulation.
Contrasting effects of R1070W on wild-type and ΔF508 CFTR is very evident at the level of biosynthetic processing as shown in Fig. 4a, where maturation of the wild-type is reduced while that of ΔF508 is promoted. In both cases, however, addition of the combined NBD1 stabilizing changes restored maturation to levels similar to wild-type. The compensatory effect of a stabilized NBD1 on the CL4 mutation as well as on the ΔF508 mutation within the domain is indicative of allosteric propagation of a conformational signal across the NBD1/CL4 interface. While the opposite effects of R1070W on WT and ΔF508 CFTR maturation have been well documented, the corresponding functional consequences have not. We have analyzed the single channel activities of these species. The R1070W mutation alone resulted in very low channel open probabilities at both 25°C and 35°C (Fig. 4b). Remarkably, there was no significant increase in the Po values with increasing temperature suggesting that this variant may be thermally unstable. This appeared to be the case, as activity was completely lost after holding at 35°C for approximately 15 min. In contrast, robust channel gating was observed with the combined ΔF508/R1070W variant with the expected elevation of Po as temperature was increased from 25°C to 35°C (Fig. 4c). This increased frequency of gating was also observed in experiments where temperature was continuously ramped upward from below 25°C to above 35°C (Fig. 4d). However, full conductance gating abruptly ceased shortly after reaching the higher temperature (note arrow in lower tracing of Fig. 4d). Therefore, as with the much less active R1070W/WT form, this more active double mutant (ΔF508/R1070W) has a similar short functional life-time at 35°C confirming that it remains thermally unstable 15.
Figure 4. The R1070W mutation in CL4 has different effects on the gating of wild-type and ΔF508 CFTR channels.

(a). Western blot indicating enhancement of maturation of mutant R1070W and ΔF508 CFTRs by combined NBD1 stabilizing mutants (combo). (b and c). Single channel recordings of wild type (b) and ΔF508 CFTR (c) containing the R1070W mutant at 25 °C and 35°C. All point histograms used to calculate single channel parameters are shown to the left of each tracing. Time scale bar of 10 s is shown below the last tracing. Single channel currents (gamma, γ) and open probabilities (Po) are shown above each tracing. (d). Wild-type and ΔF508/R1070W CFTR single-channel recordings during continuous temperature ramp of 1 °C/min from 30 to 35 °C, maintained at 30 °C for 3 min before initiating the ramp and held at 35 °C for 2 min. The moment of ΔF508/R1070W functional inactivation is shown by an arrow above the tracing and appeared after about one minute at 35 °C. X-scale bar below trace is 60 s and Y-scale bar is 1 pA.
Increased NBD1 thermal stability promotes the CFTR channel open state
In the course of analyzing the channel activity of various constructs that improve mutant CFTR maturation we observed a tendency towards progressively increased open probabilities. This behavior was especially evident when the single channel activities of ΔF508 CFTR modified by combined NBD1 stabilizing changes with and without the R1070W interface mutation were compared with the unmodified wild-type at both 25°C (Fig. 5a) and 37°C (Fig. 5b). At the lower temperature (Fig. 5a), NBD1 stabilization alone caused ΔF508 to have a Po of 0.58, exceeding that of the wild-type (0.25) but with obviously reduced gating kinetics as we had observed previously due to the introduction of only the stabilizing proline residues into NBD1 14. Strikingly, addition of the R1070W mutation to the NBD1 stabilized combination resulted in a very high open probability (0.91) even at this lower temperature. At mammalian physiological temperature (Fig. 5b), the kinetics of the NBD1 stabilized ΔF508 variant was accelerated as expected and the open probability of 0.78 (Fig. 5b, second tracing) was quite similar to that of the wild-type (0.81) (Fig. 5b; top tracing). With the addition of R1070W a nearly completely locked open state was achieved with a Po of 0.98 (Fig. 5b, third tracing). Thus, the NBD1 and NBD1/CL4 interface modification, together very effective in improving ΔF508 CFTR maturation, also has a profound influence on channel function. The lower two tracings in Fig. 5b indicate that the combined stabilizing modifications of NBD1 also elevate the open probability of the wild-type channel with and without the R1070W mutation.
Combined NBD1 stabilization and NBD1/CL4 interface modification result in constitutive ion channel activity of ΔF508 CFTR
In iodide efflux experiments from whole cells expressing ΔF508/combo/R1070W CFTR we had observed that the cells could not be efficiently loaded with the anion prior to the stimulation of cAMP production to activate PKA (Fig. 1d), suggesting the possibility that the apparent locked-open state might exist in the absence of the normally obligatory phosphorylation by PKA. To test this possibility directly, membrane vesicles were treated with a phosphatase to remove any possible activating phosphorylation of R domain sites prior to bilayer fusion. Unmodified wild-type CFTR is entirely dependent on phosphorylation by PKA 39 as illustrated in Fig. S2. Not surprisingly, as seen in Fig. 6, the alkaline phosphatase treated channels of both ΔF508/combo/ CFTR (Fig. 6a, upper trace) and the wild type based combo CFTR channels (Fig. 6b, upper trace) had very low open probability. In contrast, the NBD1 combo stabilized versions of the ΔF508 CFTR channels (Fig. 6a, middle tracing) and wild-type (Fig. 6b, middle tracing) responded very differently to addition of the R1070W mutation, the former channel becoming essentially locked open while the open probability of the latter was increased only modestly.
Figure 6. Combined NBD1 stabilizing mutations and R1070W result in phosphorylation-independent opening of ΔF508 CFTR channels.

(a). Single channel recordings at 37°C of alkaline phosphatase treated ΔF508 CFTR containing combined NBD1 stabilizing mutations without (upper tracing) and with R1070W (middle tracing) as indicated. Lower tracing: single channel recording at 37°C of ΔF508/combo/R1070W with all 15 PKA phosphorylation sites mutated (15SA). X-scale bar below traces is 10s. (b). Tracings for same variants of wild-type rather than ΔF508 CFTR.
To further verify that the high open probability state of ΔF508/combo/R1070W CFTR was reached independently of phosphorylation by PKA we employed a construct in which serines or threonines at all 15 PKA phosphorylation sites were mutated to Ala (15SA) 39. While the activity of the combo/R1070W/WT CFTR was little changed by the 15SA modifications (Fig. 6b, lower tracing), the 15SA version of the ΔF508/combo/R1070W CFTR channel remained fully active even in the absence of PKA phosphorylation sites (Fig. 6a, lower tracing), confirming that the maximally NBD1 stabilized and NBD1/CL4 interface modified form of ΔF508 CFTR has lost normal regulatory control by phosphorylation.
Discussion
In this study we attempted to determine whether correction of the ΔF508 defect in CFTR required modification of both the NBD1 domain in which F508 resides and the NBD1/CL4 interface, to which the residue also contributes or if targeting only one of these locations is sufficient. Our thinking was influenced by the fact that CFTRs of several non-mammalian species, including avia and elasmobranchs, that are little influenced by the ΔF508 mutation, have stabilizing sequence differences in NBD1 but not at the interface 14. We had previously made second site changes mimicking some of these NBD1 differences in human ΔF508 CFTR and found them to be strongly stabilizing, although not quantitatively restoring a wild-type level of maturation 14. Furthermore, we also had shown that pairs of cysteine residues introduced on either side of the interface could be cross-linked after but not before these stabilizing NBD1 second site changes were made in ΔF508 CFTR 23. Thus, it appeared that a remodeled interface formed on stabilization of ΔF508 NBD1 was adequate for assembly and function of the full-length mutant protein.
To further pursue this hypothesis here we combined several of the most effective second site changes in NBD1 with or without the interface stabilizing mutant R1070W and compared their effects on maturation, traffic to the cell surface and channel function. It was found that the outcome depended on which and how many stabilizing NBD1 changes were made. While most individual changes and even the combination of two different ones caused relatively modest increases in percent maturation, the combination of three different sets of changes caused much larger increases to levels reaching at least 2/3 of wild-type (Fig. 1). Strikingly, the introduction of a proline residue at position 495 (S495P) in the Q-loop was found to cause the largest increase in the domain Tm and have the strongest restorative effect on maturation of the full-length protein of any single NBD1 second site change and its influence was additive with others (Fig. 2). This tight correlation between the capability of NBD1 mutations to increase the thermal stability of NBD1 and enhance ΔF508 CFTR maturation might explain the lack of effective rescue of ΔF508 CFTR by the combination of NBD1 mutations which increase NBD1 thermal stability to a lesser extent 33.
The practical objective of efforts to understand how to overcome or compensate for the ΔF508 defect is the development of efficacious therapeutic methods. The effectiveness of the stabilizing mutagenic changes in NBD1 provide encouragement that small molecules binding specifically to the domain might have similar effects. However, other than nucleotides that are the domain’s natural ligands, stabilizing NBD1 binders had not been identified. Odolczyk et al 40 reported the docking to NBD1 of several compounds identified by virtual screening but their binding has not been confirmed experimentally. We took advantage of two related compounds found to co-crystallize with NBD1 by SGX Pharmaceuticals. Although requiring high non-pharmaceutical concentrations, BIA and BEIA caused a small increase in the Tm of NBD1 and promoted maturation of full-length ΔF508 CFTR (Fig. 3). These data provide proof of principle that NBD1 binding small molecules can be stabilizing and corrective. Importantly, their effects are additive with those of the VX-809 corrector that interacts at sites other than NBD1 23; 24; 25. These findings emphasize the need to discover other NBD1 binders with higher affinity than these indole compounds. A strategy of targeting flexible regions of NBD1 may be suggested by the effects of proline introduction into three of these regions: the RI, the SDR and the Q-loop 14, which enhance maturation and thermal stability. As demonstrated in the current work the S495P mutation, two residues C-terminal of the Q-loop glutamine has the greatest impact. This glutamine is known to contribute to the organization of the catalytic ATP binding site in ABC proteins and although the exact function and mechanism of action of the loop that follows it is not fully understood, it joins the ATP binding core and alpha-helical domains and may undergo conformational changes during the catalytic cycle, possibly switching between more and less extended states 41. In addition to changing the side chain, a proline substitution might be expected to influence the conformational mobility of the loop. Side chains of the loop residues interact with the membrane-spanning domains in other ABC proteins and we have observed cross-linking between cysteines replacing the Q-loop residue W496 and T1064 in CL4 of CFTR as well as between M498 and L1065 42. Thus, Q-loop stabilization by prolines (at positions 495 or 492) could possibly act by improving the connection between NBD1 and CL4 that is compromised by the absence of F508.
These considerations focus attention on the influence of the “corrective” modifications of NBD1 or the NBD1/CL4 interface on the function of the “corrected” mutant protein. Our attention was drawn to this issue by the observations that progressive improvement in human ΔF508 CFTR maturation with the introduction of increasing numbers of proline substitutions resulted in slowed gating kinetics 14. Therefore, while restoration of the thermal stability deficit caused by ΔF508 clearly is necessary, it appeared that hyper-stabilization could be detrimental to function. This suggestion is extended by the current results in which further stabilization of NBD1 by combining multiple different kinds of modifications (”combo”) shifts the rescued ΔF508 CFTR channel gating to a higher than wild-type open probability state (Fig. 5).
Turning to the impact of modifying the NBD1/CL4 interface, which may be a site of action of the VX-809 corrector 23; 25, the R1070W mutation, shown by others to improve ΔF508 CFTR maturation 8, was the main tool we employed. We observed that the mutation alone promoted only a low level of maturation as did the other known interface change, V510D 43. Not only were their effects small compared to those of NBD1 stabilizing changes but R1070W also caused only a small increment above the correction caused by combined NBD1 stabilization (Fig. 1). From a functional perspective, most significant was the finding that, when modified by combined NBD1 mutations and R1070W, the efficiently assembled ΔF508 channel was constitutively active, not requiring phosphorylation by PKA (Fig. 6). Although the mechanism underlying this effect remains to be elucidated, it occurs in the stabilized mutant protein only when the NBD1/CL4 interface is altered by both the absence of F508 and the presence of R1070W (Fig. 5 and 6). The disease-associated R1070W mutation alone is destabilizing of wild-type CFTR but does not cause it to be constitutively active without phosphorylation even when it contains all the NBD1 stabilizing changes (Fig. 6). Clearly the different interface, that is formed when the aromatic F508 residue is removed and an aromatic tryptophan replaces R1070, alters channel regulation from that occurring when only one or neither of these changes is made.
These findings do not detract from the idea of therapeutically targeting the interface in different ways since it clearly plays a role in the propagation of stabilizing effects between domains as evidenced by the fact that impaired maturation caused by R1070W can be overcome by NBD1 stabilizing changes (Fig. 4a). Furthermore, from a therapeutic perspective it may be extremely challenging to achieve the high degree of NBD1 stabilization by small molecules to obviate the need to also target the interface. However, because the interface mediates a domain-swapping interaction fundamental to the structure and function of all members of the ABC exporter class of proteins, its modification may have to be done delicately and precisely with monitoring of both assembly/stability and function. Less delicate maneuvers such as covalent cross-linking across the interface arrest channel gating 29. It remains uncertain whether a therapeutic strategy targeting NBD1 and its interface with membrane-spanning domains may be necessary or optimal as NBD1 stabilization alone can have a strong corrective effect and combining both modifications can disrupt regulation of the rescued mutant channel.
Materials and Methods
Construction and expression of mutants
Various site-specific mutations were introduced into the CFTR construct in pcDNA3 vector by the Stratagene Quick Exchange protocol as described44. Point mutations were confirmed by automated DNA sequencing (Eurofins MWG Operon, Huntsville, AL). Human embryonic kidney 293 (HEK) cells were transiently transfected using Jet PEI transfection reagent (Polyplus Transfection Inc., NY) according to the manufacturer’s instructions. Cells were harvested 48 hrs post transfection. For stable expression, constructs were cotransfected with pNUT plasmid into baby hamster kidney (BHK-21) cells, which were selected and maintained in methotrexate (500 μM) containing DMEM-F12 media. For all experiments, cells were grown at 37°C. In some experiments, cells were treated with BIA (5-bromo-3-indole acetic acid, Sigma-Aldrich, St. Louis, MO) and BEIA (5-bromo-4-ethoxyindole acetic acid, PharmaBlock, Nanjing, China) with or without VX-809 for 24 hrs. Cell lysates were prepared in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 1% deoxycholate, pH 7.4) plus protease inhibitor cocktail (1 μg/ml leupeptin, 2 μg/ml aprotinin, 3.57 μg/ml E64, 156.6 μg/ml benzamidine and 2 mM Pefablock). Protein concentrations were measured using bicinchoninic acid assay (BCA reagent from Pierce Thermo Scientific). Twenty micrograms of total proteins were loaded on 7.5% SDS-PAGE and subjected to Western blot analysis to determine CFTR protein expression and maturation. The amount of mature and immature CFTR was measured with a Odyssey Imaging system (Li-Cor Bioscience, Lincoln, Nebraska).
Cell surface detection
BHK cells expressing WT and ΔF508 CFTR variants with an external epitope tag on extracellular loop 4 were plated at 30,000 cells per well in 96 well plates and grown overnight. Cells were washed once with PBS and fixed for 10 minutes at room temperature with 2% paraformaldehyde in PBS. Cells were then washed and blocked with 5% normal goat serum + 1% BSA in PBS for 1 hour. Primary antibody against the external epitope tag was diluted in blocking solution and incubated for 2 hours. After washing, cells were incubated with Alexa Fluor 790 goat- anti-mouse antibody (Molecular Probes) and Syto 60 nucleic acid stain (Li-Cor Biosciences) for 1 hour. Plates were read using an Odyssey infrared scanner and the ratio of fluorescence reading at 800/700 was calculated to measure the surface expression of CFTR.
Metabolic pulse chase
Short term pulse chase experiment was performed as described44 to compare the maturation efficiency of WT and mutant CFTR. Briefly, BHK cells stably expressing WT CFTR or ΔF508 CFTR with various stabilizing mutations grown in 6 well plates were pulse labeled for 20 minutes using 500 ul of 100 mCi/ml 35S-methionine (MP Biomedicals, Solon, OH) in methionine free DMEM. Cells were then washed 2 times with PBS and chased with growth medium supplemented with 2 mM cold methionine for the indicated times. The chase was stopped by washing 2 times with cold PBS and solubilizing the cells with RIPA buffer containing the protease inhibitor cocktail. CFTR was immunoprecipitated using antibody 596. The immunoprecipitated proteins were then run on a 6% polyacrylamide gel, fixed with acetic acid-methanol and soaked in 1 M sodium salycilate for radiography. Band intensity was quantified by directly measuring the radioactivity using a Packard Instant Imager.
Iodide efflux assay
BHK cells stably expressing WT and mutant CFTR grown to ~ 100% confluence in 6 well plates were incubated in an iodide loading buffer (136 mM NaI, 3 mM KNO3, 2 mM Ca(NO3)2, 11 mM glucose and 20 mM HEPES, pH 7.4) for 1 hour at room temperature. Extracellular iodide was removed by rinsing the cells 6 times with iodide-free efflux buffer (same as the loading buffer except NaNO3 replaced NaI). Samples were collected by completely replacing the efflux buffer (1 ml volume) with fresh solution at 1 min intervals. The first four samples were used to establish the baseline. Iodide efflux upon stimulation with PKA agonists (10 μM forskolin, 100 μM dibutyryl cyclic AMP and 1 mM 3-isobuty-1-methylxanthine) was measured using an iodide selective electrode (Thermo Fisher.). At the end of each assay, efflux buffer containing 0.1% Triton X-100 was added to release leftover iodide, as shown with the * in the figure legend. In case of compound treatment, cells were incubated with compound at doses indicated in figure legends for 24 hr at 37°C, and compounds were present during loading and throughout the efflux assay.
Human NBD1 Expression and Purification
Human NBD1 constructs, residues 387–646 minus the RI residues 405–436 (ΔRI/ΔRE-NBD1), containing the selected mutations were expressed as SUMO fusions in BL21(DE3) Codon Plus cells and purified as described previously10; 28. Briefly, fusion protein was first purified by prepacked Ni Sepharose column, followed by removal of the SUMO tag using Ulp1 protease and dialysis. The sample was then passed through a second Ni column to remove residual SUMO tag, followed by a Superdex 200 gel filtration column to collect the monomeric protein. Purified proteins were stored at −80 °C in buffer containing 20mM HEPES, pH 7.5, 150 mM NaCl, 10%glycerol, 10%ethylene glycol, 2mM Tris(2-carboxymethyl)-phosphine, 2 mM ATP, and 3 mM MgCl2. Protein concentration was determined with the Pierce 660 nm assay in microtiter plate format, calibrated using B. subtilis NAD synthetase. ATP concentration in the sample was determined by HPLC on a Shimadzu HPLC system (Shimadzu Corp. USA) equipped with a 100 x 4.6 mm Synergi 4μ Polar-RP column (Phenomenex Inc. USA).
Differential Scanning Calorimetry
Calorimetry was carried out on the VP-Capillary DSC System (MicroCal Inc., GE HealthCare, U.S.A.), in 0.130 mL cells, at a heating rate of 2 °C/min. Unless otherwise indicated, the buffer for NBD1 DSC experiments was the same as its storage buffer (see above). Prior to each DSC experiment, ATP concentration of the sample was checked and adjusted if necessary to 2 ± 0.1 mM. A buffer-only heat capacity curve was subtracted from the protein curve, and data were analyzed using the MicroCal Origin 7.0 software, from which the unfolding temperature (Tm), and the calorimetric (ΔHc) unfolding enthalpy were obtained. At least two measurements were performed on each construct. The general reproducibility of NBD1 Tm values is within ± 0.3 °C.
Planar bilayer based single channel measurement
Planar lipid bilayers were prepared by painting a 0.2 mm hole drilled in a Teflon cup with a phospholipids solution in n-decane containing a 3:1 mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (Avanti Polar Lipids). The lipid bilayer separated 1.0 ml of solution in the Teflon cup (cis side) from 5.0 ml of a solution in an outer glass chamber (trans side). Both chambers were magnetically stirred and thermally insulated. Heating and temperature control were established by a temperature control system (TC2BIP, Cell Micro Controls). CFTR ion channels were transferred into the preformed lipid bilayer by spontaneous fusion of membrane vesicles containing CFTR variants. To maintain uniform orientation and functional activity of CFTR channels transferred into the bilayer, 5 mM ATP, 50 nM PKA and membrane vesicles were added in the cis compartment only. Calf intestinal alkaline phosphatase (200 units/ml, New England Biolabs, Ipswich, MA) was substituted for PKA and ATP in the membrane vesicle stock solution to generate control vesicles with nonphosphorylated CFTR protein. After 30 min treatment at room temperature alkaline phosphatase was washed out by sedimentation and resuspension and then used for further single channel recording without PKA in the cis compartment. All measurements were done in symmetrical salt solution (300mM Tris–HCl, pH 7.2, 5mM MgCl2 and 1mM EGTA) under voltage-clamp conditions by using an Axopatch 200B amplifier. The membrane voltage potential of −75 mV is the difference between cis and trans (ground) compartments. The output signal was filtered with an eight-pole Bessel low-pass filter, LPBF-48DG (NPI Electronic, Tamm, Germany), with a cutoff frequency of 50 Hz, digitized with a sampling rate of 500 Hz and recorded with pClamp 9.2 software. Origin 75 software (Origin Lab Corp., Northampton, MA) was used to fit all point histograms (pClamp 9.2, Axon Instruments) by multi-peak Gaussians. Single-channel current was defined as the distance between peaks on the fitting curve and used for the calculation of the single-channel conductance. The probability of the single channel being open (Po) was calculated as a ratio of the area under the peak for the open state to the total area under both peaks on the fitting curve.
Supplementary Material
Highlights.
Is combined targeting of NBD1 and NBD1/MSD an ideal strategy to rescue ΔF508 CFTR?
Multiple NBD1 second site changes alone additively restored ΔF508 CFTR maturation.
Maturation of ΔF508 CFTR correlated quantitatively with NBD1 Tm increase.
Combined NBD1 and NBD1/MSD mutations resulted in constitutive channel opening.
Therefore targeting of NBD1 should be a priority for ΔF508 CFTR correction.
Acknowledgments
We thank SGX Pharmaceuticals/Eli Lilly and Co. and Cystic Fibrosis Foundation Therapeutics Inc. for generously providing information on NBD1 binders and giving us permission to test these compounds. We also thank Qingxian Zhou for technical assistance in purification of NBD1. Access to the Cap-DSC was provided by the Biocalorimetry Lab supported by the Shared Facility Program of the UAB Comprehensive Cancer Center, Grant # 316851.This work was supported by grants from the National Institutes of Health to J.R.R. (R01 DK051619, R01 DK051870, P01 HL110873) and to both C.G.B. and J.R.R. from the Cystic Fibrosis Foundation.
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- ABC
ATP-binding cassette protein
- ΔF508
deletion of phenylalanine 508
- NBD1
first nucleotide binding domain
- MSD
membrane spanning domain
- CL4
cytoplasmic loop 4
- ΔRI
deletion of regulatory insertion
- DSC
differential scanning calorimetry
- Tm
melting temperature
- BIA
5-bromoindole-3-acetic acid
- BEIA
5′-bromo-3-ethoxyindole- acetic acid
- PKA
protein kinase A
- BHK
baby hamster kidney cells
- HEK
human embryonic kidney 293 cells
References
- 1.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 2.Du K, Sharma M, Lukacs GL. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 3.Riordan JR. CFTR Function and Prospects for Therapy. Annu Rev Biochem. 2008;77:701–26. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 4.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79:S167–73. doi: 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- 5.Kim SJ, Skach WR. Mechanisms of CFTR Folding at the Endoplasmic Reticulum. Front Pharmacol. 2012;3:201. doi: 10.3389/fphar.2012.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, Thomas PJ, Sheppard DN, Amaral MD. Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem Biol. 2008;15:62–9. doi: 10.1016/j.chembiol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR, 3rd, Balch WE. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–15. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 8.Thibodeau PH, Richardson JM, 3rd, Wang W, Millen L, Watson J, Mendoza JL, Du K, Fischman S, Senderowitz H, Lukacs GL, Kirk K, Thomas PJ. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem. 2010;285:35825–35. doi: 10.1074/jbc.M110.131623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 10.Protasevich I, Yang Z, Wang C, Atwell S, Zhao X, Emtage S, Wetmore D, Hunt JF, Brouillette CG. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010;19:1917–31. doi: 10.1002/pro.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X, Atwell S, Spencer Emtage J, Wetmore DR, Brouillette CG, Hunt JF. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 2010;19:1932–47. doi: 10.1002/pro.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hegedus T, Aleksandrov A, Cui L, Gentzsch M, Chang XB, Riordan JR. F508del CFTR with two altered RXR motifs escapes from ER quality control but its channel activity is thermally sensitive. Biochim Biophys Acta. 2006;1758:565–72. doi: 10.1016/j.bbamem.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV, Riordan JR. Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J Mol Biol. 2010;401:194–210. doi: 10.1016/j.jmb.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aleksandrov AA, Kota P, Cui L, Jensen T, Alekseev AE, Reyes S, He L, Gentzsch M, Aleksandrov LA, Dokholyan NV, Riordan JR. Allosteric modulation balances thermodynamic stability and restores function of DeltaF508 CFTR. J Mol Biol. 2012;419:41–60. doi: 10.1016/j.jmb.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, O’Donnell N, Landstrom A, Skach WR, Dawson DC. Thermal instability of DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) channel function: protection by single suppressor mutations and inhibiting channel activity. Biochemistry. 2012;51:5113–24. doi: 10.1021/bi300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- 17.Howard M, Fischer H, Roux J, Santos BC, Gullans SR, Yancey PH, Welch WJ. Mammalian osmolytes and S-nitrosoglutathione promote Delta F508 cystic fibrosis transmembrane conductance regulator (CFTR) protein maturation and function. J Biol Chem. 2003;278:35159–67. doi: 10.1074/jbc.M301924200. [DOI] [PubMed] [Google Scholar]
- 18.Verkman AS, Lukacs GL, Galietta LJ. CFTR chloride channel drug discovery--inhibitors as antidiarrheals and activators for therapy of cystic fibrosis. Curr Pharm Des. 2006;12:2235–47. doi: 10.2174/138161206777585148. [DOI] [PubMed] [Google Scholar]
- 19.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PD, Negulescu P. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–30. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 20.Hanrahan JW, Sampson HM, Thomas DY. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol Sci. 2013;34:119–25. doi: 10.1016/j.tips.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb Perspect Med. 2013:3. doi: 10.1101/cshperspect.a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–8. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He L, Kota P, Aleksandrov AA, Cui L, Jensen T, Dokholyan NV, Riordan JR. Correctors of DeltaF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013;27:536–45. doi: 10.1096/fj.12-216119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren HY, Grove DE, De La Rosa O, Houck SA, Sopha P, Van Goor F, Hoffman BJ, Cyr DM. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell. 2013;24:3016–24. doi: 10.1091/mbc.E13-05-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, Hegedus T, Beekman JM, Lukacs GL. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol. 2013;9:444–54. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb Perspect Biol. 2011:3. doi: 10.1101/cshperspect.a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23:282–93. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF, Emtage S. Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem. 2005;280:1346–53. doi: 10.1074/jbc.M410968200. [DOI] [PubMed] [Google Scholar]
- 29.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008;105:3256–61. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mornon JP, Lehn P, Callebaut I. Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci. 2009;66:3469–86. doi: 10.1007/s00018-009-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendoza JL, Thomas PJ. Building an understanding of cystic fibrosis on the foundation of ABC transporter structures. J Bioenerg Biomembr. 2007;39:499–505. doi: 10.1007/s10863-007-9117-7. [DOI] [PubMed] [Google Scholar]
- 32.Dalton J, Kalid O, Schushan M, Ben-Tal N, Villa-Freixa J. New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J Chem Inf Model. 2012;52:1842–53. doi: 10.1021/ci2005884. [DOI] [PubMed] [Google Scholar]
- 33.Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A, Lukacs GL. Correction of Both NBD1 Energetics and Domain Interface Is Required to Restore DeltaF508 CFTR Folding and Function. Cell. 2012;148:150–63. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA, Thomas PJ. Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–74. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teem JL, Berger HA, Ostedgaard LS, Rich DP, Tsui LC, Welsh MJ. Identification of revertants for the cystic fibrosis ĐF508 mutation using STE6-CFTR chimeras in yeast. Cell. 1993;73:335–346. doi: 10.1016/0092-8674(93)90233-g. [DOI] [PubMed] [Google Scholar]
- 36.Atwell S, Brouillette CG, Conners K, Emtage S, Gheyi T, Guggino WB, Hendle J, Hunt JF, Lewis HA, Lu F, Protasevich, Rodgers LA, Romero R, Wasserman SR, Weber PC, Wetmore D, Zhang FF, Zhao X. Structures of a minimal human CFTR first nucleotide-binding domain as a monomer, head-to-tail homodimer, and pathogenic mutant. Protein Eng Des Sel. 2010 doi: 10.1093/protein/gzq004. [DOI] [PubMed] [Google Scholar]
- 37.Seibert FS, Linsdell P, Loo TW, Hanrahan JW, Clarke DM, Riordan JR. Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J Biol Chem. 1996;271:15139–45. doi: 10.1074/jbc.271.25.15139. [DOI] [PubMed] [Google Scholar]
- 38.Krasnov KV, Tzetis M, Cheng J, Guggino WB, Cutting GR. Localization studies of rare missense mutations in cystic fibrosis transmembrane conductance regulator (CFTR) facilitate interpretation of genotype-phenotype relationships. Hum Mutat. 2008;29:1364–72. doi: 10.1002/humu.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hegedus T, Serohijos AW, Dokholyan NV, He L, Riordan JR. Computational Studies Reveal Phosphorylation-dependent Changes in the Unstructured R Domain of CFTR. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Odolczyk N, Fritsch J, Norez C, Servel N, da Cunha MF, Bitam S, Kupniewska A, Wiszniewski L, Colas J, Tarnowski K, Tondelier D, Roldan A, Saussereau EL, Melin-Heschel P, Wieczorek G, Lukacs GL, Dadlez M, Faure G, Herrmann H, Ollero M, Becq F, Zielenkiewicz P, Edelman A. Discovery of novel potent DeltaF508-CFTR correctors that target the nucleotide binding domain. EMBO Mol Med. 2013;5:1484–501. doi: 10.1002/emmm.201302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones PM, George AM. Mechanism of the ABC transporter ATPase domains: catalytic models and the biochemical and biophysical record. Crit Rev Biochem Mol Biol. 2013;48:39–50. doi: 10.3109/10409238.2012.735644. [DOI] [PubMed] [Google Scholar]
- 42.He L, Aleksandrov AA, Serohijos AW, Hegedus T, Aleksandrov LA, Cui L, Dokholyan NV, Riordan JR. Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J Biol Chem. 2008;283:26383–90. doi: 10.1074/jbc.M803894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loo TW, Bartlett MC, Clarke DM. The V510D suppressor mutation stabilizes DeltaF508-CFTR at the cell surface. Biochemistry. 2010;49:6352–7. doi: 10.1021/bi100807h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang XB, Tabcharani JA, Hou YX, Jensen TJ, Kartner N, Alon N, Hanrahan JW, Riordan JR. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all ten PKA consensus phosphorylation sites. J Biol Chem. 1993;268:11304–11311. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.