

Abstract
Metabotropic glutamate receptor 1 (mGlu1), which belongs to class C G protein-coupled receptors (GPCRs), can be coupled with G protein to transfer extracellular signal by dimerization and allosteric regulation. Unraveling the dimer packing and allosteric mechanism can be of great help for understanding specific regulatory mechanism and designing more potential negative allosteric modulator (NAM). Here, we report molecular dynamics simulation studies of the modulation mechanism of FITM on the wild type, T815M and Y805A mutants of mGlu1 through weak interaction analysis and free energy calculation. The weak interaction analysis demonstrates that van der Waals (vdW) and hydrogen bonding play an important role on the dimer packing between six cholesterol molecules and mGlu1 as well as the interaction between allosteric sites T815, Y805 and FITM in wild type, T815M and Y805A mutants of mGlu1. Besides, the results of free energy calculations indicate that secondary binding pocket is mainly formed by the residues Thr748, Cys746, Lys811 and Ser735 except for FITM-bound pocket in crystal structure. Our results can not only reveal the dimer packing and allosteric regulation mechanism, but also can supply useful information for the design of potential NAM of mGlu1.
The metabotropic glutamate receptor 1 (mGlu1) is one member of class C G protein-coupled receptors (GPCRs) and is considered to be a promising target to treat Alzheimer’s disease, anxiety, cancer, schizophrenia autism and chronic pain1,2,3. It can form constitutive homodimer to activate the downstream signal modulator at the cell surface4. The mGlu1 receptor contains orthosteric binding site in Venus flytrap domain (VFD) and allosteric binding site in the transmembrane (7TM) domain3. The orthosteric ligands can activate or inactivate the mGlu1 through binding to VFD. The allosteric modulators can alter the binding affinity of orthosteric ligands in negative, neutral and positive ways by binding to allosteric sites of mGlu14,5. Some recent studies prove that only a single 7TM is promoted to couple with G protein in the agonist-bound homodimeric mGlu16,7,8.
The recent reported crystal structure of mGlu1 has provided useful information to understand the structural features and activation mechanism of class C GPCRs3,9. However, it is still difficult to know about the dynamical structural characteristics of mGlu1 at atomic level based on the crystal structure. As a complementary method to experimental study, molecular dynamics (MD) simulations can provide a useful and reliable way to study the dynamical behavior of mGlu1 at atomic level. Computational methods have also been successfully used to predict the thermostable mutants of mGlu1 to form a single stable crystal conformational state bound to ligands10. MD and metadynamics simulations are also proved to be very useful to understand the regulation mechanism of smoothened (SMO) receptor, which has the similar long and narrow pocket with mGlu1 in the 7TM domain11. Based on the known crystal structures and MD simulation, MD simulation methods have been successfully used to study the dynamical behavior and elucidate the allosteric regulation mechanism12,13,14,15,16,17,18.
The mGlu1 has been considered to be an important target of the known structure of class C GPCRs for structure-based drug design19. Due to the specific allosteric sites in the 7TM domain of mGlu1, negative allosteric modulators (NAM) can be designed based on the crystal structure or known NAM3,20,21. The cholesterols are considered as the lipid rafts to pack the lipids and membrane proteins tightly22. The dynamical dimer mediated by cholesterols and allosteric mechanism of mGlu1 are still elusive. It is difficult to distinguish the main interactive way such as hydrogen bonding, steric repulsion or van der Waals (vdW) between mGlu1 and NAM. It is worthwhile to study the detailed dynamical dimer interaction and allosteric mechanism between the homodimers of mGlu1 and allosteric ligands.
Here, we perform molecular dynamics simulations on wild type, T815M and Y805A mutants of mGlu1. The weak interaction and free energy calculation are employed to analyze the dimerization and allosteric mechanism. The results of weak interaction analysis indicate that T815 and Y805 in wild type mGlu1 have stronger vdW and hydrogen bonding interaction than that in T815M and Y805A mutants of mGlu1. The free energy profiles energy barrier in the first and secondary pockets for binding of FITM. Our results supply a detail structural basis for the allosteric and homodimer mechanism of mGlu1.
Methods
Model preparation
To study the dimer structure of metabotropic glutamate receptor 1 (mGlu1), the crystal structure of mGlu1 is obtained from PDB database (ID: 4OR2)3. The redundant residues are deleted except mGlu1 and its ligand. The dimer of mGlu1 is surrounded by the explicit 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids with length of 68 Å and width of 101 Å. The complex of lipid and protein is immersed into TIP3P water23 box with the size of 68 Å × 101 Å × 90 Å. The ionic concentration of system is set to be 0.15mol/L according to the physiological ion concentration and neutralized by the sodium and chloride ions. The final system contains a total of ~46,000 atoms per periodic cell. The charges and Lennard-Jones parameters are assigned according to the atomic types which are defined in CHARMM force field. The CHARMM force field parameterizations of (4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazol-2-yl)-N-methylbenzamide) FITM are generated by using CGenFF24 and SwissParam25. Once the parameters of models are completely built, the T815 and Y805 are mutated into M815 and A805 to construct another two simulated systems by using VMD software26.
Molecular dynamics simulations
To equilibrate the wild type and two mutants of mGlu1 in complex with FITM, firstly, for each system, all atoms are fixed except for lipid tail to minimize 10 ps and equilibrate 500 ps. Secondly, the mGlu1 and FITM are constrained, the system is minimized for 10 ps and equilibrated for 500 ps. Thirdly, these three entire systems are all released to carried out 5 ns equilibrated MD simulations. At last, the 50 ns MD simulations are performed on these three systems under constant temperature (310 K) and constant pressure (1 bar).
Our MD simulations are run with the time step of 2 fs by using NAMD27 (version 2.9b3) software with CHARMM 27 force field28 in periodically infinite explicit water and lipid. The conjugate gradient method is employed for the minimization. The electrostatic interaction is computed with a 12 Å nonbonded cutoff based on the particle-mesh Ewald (PME)29 method. All MD simulations are based on the langevin thermostat and langevin barostat30 methods for the constant temperature of 310 K and pressure of 1 bar, respectively. The simulated trajectory frames are saved every 5 ps for analysis.
Weak interaction analysis
The weak interaction, which involves noncovalent interaction31,32, can be used to find the main interactive way between receptor and ligand from hydrogen bonding, electrostatic, steric repulsion and van der Waals (vdW) forces. The hydrogen bonds occupancy are popularly used to study the interaction between the ligands and receptors33. However, the hydrogen bonds occupancy can not distinguish other noncovalent interaction between protein and ligands such as steric repulsion and vdW forces and vdW may not contribute a favorable force on the binding between receptor and ligands. The weak interaction can detail the favorable and unfavorable contributions of protein-protein, protein-ligand or protein and nucleic acids clearly. The weak interaction analysis can be shown by plotting reduced density gradient (RDG or s(r)) with respect to the function of electron density ρ(r) (see eq. 1):
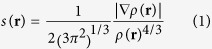 |
However, the weak interaction on one frame of trajectory reflects the limited information for protein complex. The average weak interaction, which bases on the full trajectory of molecular dynamics simulations, can provide the more accuracy and smooth isosurfaces between protein and protein or protein and ligand. The average weak interaction analysis34 is calculated based on averaged density gradient 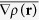 and averaged density
and averaged density 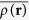 (see eq. 2):
(see eq. 2):
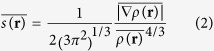 |
The Multiwfn35 software is used to analyze the weak interaction for wild type and mutated mGlu1 in complex with FITM. The number of grids is set to 200 × 200 × 200 in three dimensional spaces. The 5000 frames of trajectories are extracted to average the density. The representation and color of averaged weak interaction are shown in VMD software26. To analyze traditional hydrogen bonds occupancy, the angle and distance between the donors and acceptors are set no more than 35° and 3.5 Å, respectively36.
Free energy calculation
Adaptive biasing force (ABF)37,38 are popularly used to calculate the free energy during the dissociation process of ligand in the pocket of protein. The equal probability and reversible process sampling are used to improve accuracy of free energy calculation along reaction coordinate. The free energy ΔG is obtained from the reaction coordinate a to b as shown in equation 3:
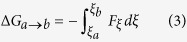 |
where Fξ is the biasing force. Because the model of mGlu1 is constructed along Z axis, the free energy of FITM is calculated along Z axis in this study. To study the dissociation process of FITM in the pocket of mGlu1, the reference residues are chosen Cys671, Thr793, Thr824 and Leu763. The reaction coordinate of ABF is divided into three non-overlapping windows. Each window is set to 8 Å with 0.2 Å/bin. The wall force constant is assigned to 50 kcal/mol/Å2 for the boundary potential. The 500 samples are applied before a biasing force. The 20 ns MD simulations are carried out on each window. A total of 360 ns ABF simulations are performed on the systems of wild type and mutated mGlu1 in complex with FITM.
Results and Discussion
Structural features and fluctuation of mGlu1 dimers
The function structure of mGlu1 is formed by asymmetric parallel monomer which contacts with each other by six cholesterol molecules packed against the helices I and II of mGlu1 (Fig. 1A,B). In the extracellular part of mGlu1, residue C657 forms disulfide bond with C746 to connect the β-hairpin and helix III. The negative allosteric modulator FITM locates at the pocket which surrounds by seven helixes of mGlu1 (Fig. 1A).
Figure 1. The dimer structure of mGlu1.

(A) The cartoon representation of mGlu1. (B) The model of mGlu1 in complex with ions, FITM, cholesterols, lipids and water. The red and blue spheres are the sodium and chloride ions.
To study the structural fluctuations of the entire system, the wild type, T815M and Y805A mutants of mGlu1 in complex with ions, FITM, cholesterols, lipids and water are built as the initial structure for molecular dynamics simulations (Fig. 1B). As shown in Fig. 2A, the root mean square deviation (RMSD) of backbone atoms of dimer A, B or the entire mGlu1 (dimer A and B) indicates that the whole system reaches equilibrium phase in the 50 ns simulation time. The residues K678 and E783 are considered as “ionic lock” between helix III and VI of mGlu1. The RMSD and distance of K678-E783 indicate the mGlu1 still keep stable inactive conformation during the molecular dynamics simulations (see Fig. 2A,B). Besides, the residues S625 and N780 have interaction in the intercellular part of mGlu1. Figure 2B shows S625 keeps stable distance with N780 in 50 ns molecular dynamics simulations. These results indicate that the mGlu1 systems have reached the equilibrium phase during the 50 ns molecular dynamics simulations.
Figure 2. The structural fluctuation of mGlu1.

(A) The RMSD of backbone atoms of dimer A, B, residues K678-E783 and two dimers of wild type mGlu1 versus simulation time. (B) The distances of S625-N780 and K678-E783. (C) The cartoon representation of occupied surface area of FITM and six cholesterols on mGlu1. (D) The occupied surface area of FITM and six cholesterols with respect to simulation time in dimer A and B, respectively.
The weak interaction analysis for cholesterols in wild type mGlu1
The mGlu1 is in form of dimer A and B which interact with each other by six cholesterol molecules. The ligand FITM is a negative allosteric modulator (NAM) which can alter the activity of mGlu1 through interaction with the allosteric residues. The cholesterols and FITM studies can profile the dimerization and allosteric regulation mechanism of mGlu1. As shown in Fig. 2C,D, the occupied surface areas of ligand FITM and six cholesterol molecules on mGlu1 are calculated by VolArea39. The cholesterol molecules occupy an area about 450 Å2 in dimer A or B during the molecular dynamics simulations. It indicates that the cholesterol molecules can interact with dimer A and B. The allosteric modulator FITM also possesses relatively stable area in the pocket of mGlu1. It indicates that cholesterol molecules and FITM can form the dynamical stable interaction with mGlu1.
The weak interaction analysis can detail favorable and unfavorable interactions for protein and ligand. It can also complements with hydrogen bonding analysis, steric repulsion and van der Waals (vdW) interaction. Here, we employ the weak interaction analysis to profile the dimers packing and mGlu1-cholesterols interaction mechanism. Due to the plane symmetry on six cholesterol fragments (Fs), the fragments 1-4 (F1-4) are chosen to analyze the weak interaction. The average reduced density gradient (aRDG) of F1-4, Fs and W588 is calculated versus averaging effective density based on molecular dynamics simulations (Fig. 3A,B and Supplementary Figure S1). The left and right spikes indicate the attracting and steric repulsive effects between two molecules, respectively. As shown in Fig. 3C, the green and light blue are major colors on isosurfaces between the major parts of cholesterol fragments. It indicates that the vdW and weak hydrogen bonds play an important role on the packing of main part of cholesterol fragments. The part of head and tail of cholesterol fragments shows the red and orange. It indicates the head and tail parts have the dynamical friction between cholesterol fragments during the molecular dynamics simulations. W588 is considered as an important residue to interact with six cholesterol molecules3. Figure 3C shows W588 in dimer A have strong hydrogen bonding and vdW interaction with F1-F3. The isosurfaces show the steric repulsive effect between F4 and W588. However, according to the symmetry, the W588 in dimer B can interact with F4-F6 through hydrogen bond and vdW interaction. Two residues W588 of dimer A and B are like the buttons to connect with each other tightly by cholesterol molecules. The results indicate that the vdW and hydrogen bonding can pack the main parts of six cholesterol molecules and mGlu1 stably.
Figure 3. The weak interaction analysis for cholesterol fragments.

(A) The average reduced density gradient (aRDG) versus average effective density between cholesterol fragment 1 (F1) and 2 (F2). (B) The average reduced density gradient (aRDG) versus average effective density between fragments 1, 2, 3, 4, 5, 6 (Fs) and residue W588. (C) Gradient isosurfaces for F1, F2, F3, F4 and residue W588. The color bar shows the blue-green-red scale ranging from −0.04 to 0.02 au. Blue indicates the strong attractive interaction, while red indicates repulsive interaction. The letters A, R and V in color bar represents attraction, repulsion and van der Waals interaction.
The weak interaction analysis for FITM in wild type and mutated mGlu1
The residues Thr815 and Tyr805 are the notable and key sites for the allosteric modulation of FITM in mGlu13. To the study the allosteric modulation mechanism of mGlu1, the weak interaction analysis is employed. As shown in Fig. 4A,B, FITM has the high hydrogen bonds occupancy with Thr815 and Tyr805 in dimer A and B of mGlu1 during molecular dynamics simulations. The nitrogen and hydrogen atoms of FITM form the dynamical hydrogen bonds with the hydrogen atom of Tyr805 and oxygen atom of Thr815, respectively (see Fig. 4B). It indicates that there is a strong attraction interaction between FITM and allosteric sites. To explore other possible interaction mode between FITM and residues Thr815, Tyr805, the weak interaction analysis is used to generate the gradient isosurfaces between residues Thr815, Tyr805 and allosteric modulator FITM in dimer A and B. The scatter diagram is drawn to show the spikes which represent the weak interaction between FITM and residues of dimer A and B (Supplementary Figure S2). It is obviously that the vdW effect is also the main interactive way between FITM and allosteric residues Thr815 and Tyr805 (see Fig. 4C,D). Some parts of gradient isosurfaces show orange and red between FITM and allosteric residues. It indicates that there is the steric repulsion between FITM and allosteric residues during the molecular dynamics simulations. However, the attracted force is greater than the repulsive force totally. These results show that vdW and hydrogen bonds effects can keep the stable allosteric binding between FITM and residues Thr815, Tyr805 together even though some repulsive interaction exists between FITM and allosteric sites during molecular dynamics simulations.
Figure 4. The weak interaction analysis in wild type mGlu1.

(A) The hydrogen bonds occupancy between key residues Thr815, Tyr805 of dimer A, B and NAM FITM in 50 ns simulation time. (B) The representation of hydrogen bonds between residues Thr815, Tyr805 and NAM FITM. (C,D) Gradient isosurfaces between residues Thr815, Tyr805 and NAM FITM in dimer A and B of mGlu1.
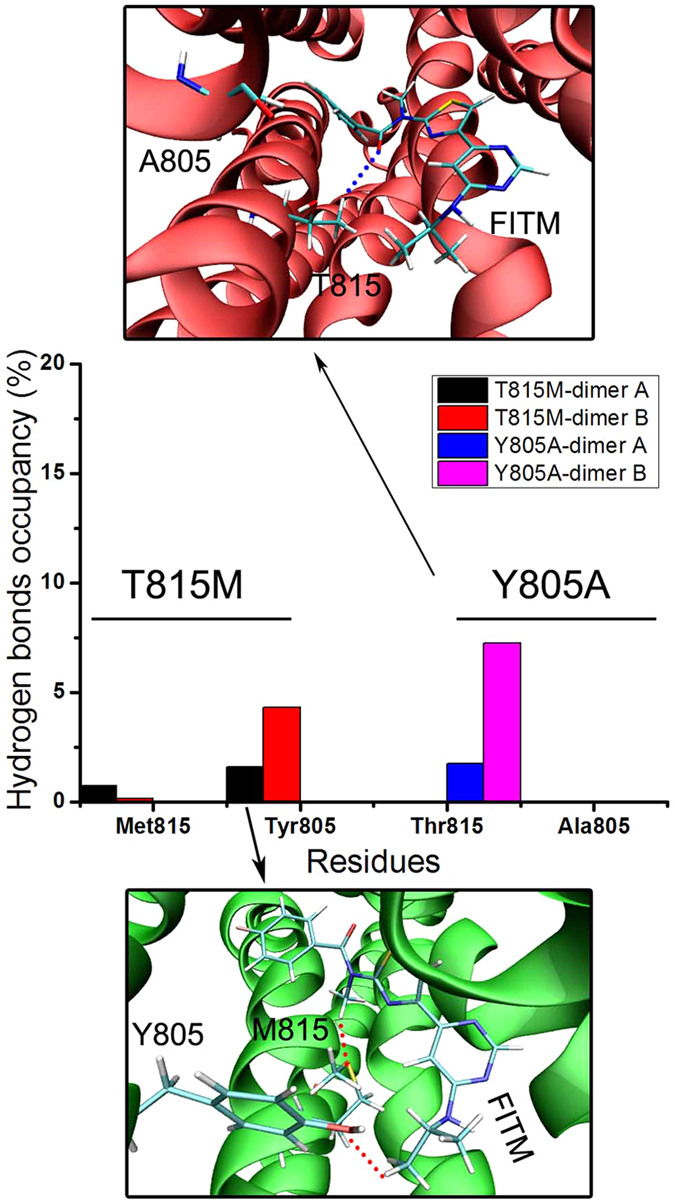
The T815M and Y805A mutants are reported to notably reduce the binding affinity of FITM3. To further detail allosteric mechanism of FITM in mGlu1, 100 ns MD simulations are further performed on the mutated systems. The RMSDs of T815M and Y805A mutants show the mutated systems get into the equilibrium phase during 50 ns MD simulations (Supplementary Figure S3). The weak interaction analysis is calculated based on the simulation trajectories (Fig. 5A–D and Supplementary Figure S4). In the T815M mutant, the residue M815 of mGlu1 forms two different conformations to interact with FITM in dimer A and B. As shown in Fig. 5A, because the methyl group of M815 turns round to FITM in dimer A, M815 has the weak vdW and hydrogen bonds with FITM except the strong repulsive interaction. In dimer B, the methyl group of M815 stays away from FITM and it does not show the vdW effect except attracting and strong repulsive interaction between M815 and FTIM (Fig. 5B). The small blue and green gradient isosurfaces indicate that the vdW and hydrogen bonds forces are also reduced between allosteric residue Y805 and FITM in the T815M mutant during molecular dynamics simulations. It infers that the M815 has longer side chain than T815 to enhance the steric repulsion between FITM and allosteric sites.
Figure 5. The weak interaction analysis in T815M and Y805A mutants of mGlu1.

(A,B) Gradient isosurfaces between residues Met815, Tyr805 and FITM of dimer A, B in T815M mutant of mGlu1. (C,D) Gradient isosurfaces between residues Thr815, Ala805 and FITM of dimer A, B in Y805A mutant of mGlu1.
In the Y805A mutant, the residue A805 has no hydrogen bond and vdW interaction with FITM in mGlu1 during molecular dynamics simulations (see Fig. 5C,D). The green and blue, which only occupancy few area between residue T815 and FITM, indicate that T815 in the Y805A mutant has very weak vdW and hydrogen bonds interaction with FITM compared with T815 in wild type mGlu1 (Figs 4C,D and 5C,D). The hydrogen bonds occupancy is used to analyze the interaction between residues M815, A805 and FITM. As shown in Fig. 6, in T815M mutant, the residues M815 occupy less than 1% hydrogen bonds occupancy in dimer A and B during molecular dynamics simulations. In Y805A mutant, the residue A805 does not form the hydrogen bonds with FITM with molecular dynamics simulations. The residue T815 forms less than 9% hydrogen bonds occupancy during molecular dynamics simulations. The results of weak interaction analysis and hydrogen bonds occupancy analysis show that the allosteric sites T815 and Y805 are important residues for negative allosteric modulator design of mGlu1.
Figure 6. The hydrogen bonds occupancy between FITM and key residues T815, M815, Y805, A805 during molecular dynamics simulations.
Free energy calculations
The weak interaction analysis can provide information about the interaction between allosteric ligand and key residues in the binding pocket of mGlu1. Free energy calculation can enhance the understanding of the NAM behavior during the dissociation process of FITM in the binding pocket of mGlu1. Adaptive biasing force (ABF) is employed here to calculate the free energy when FITM dissociates from dimer A and B of mGlu1. Figure 7A,B shows the free energy of FITM during the dissociation process from the pocket of wild type, T815M and Y805A mutants of mGlu1, respectively. It need conquer about 35 kcal/mol energy when FITM gets out of the pocket in dimer A and B of wild type mGlu1. It is obviously that FITM in dimer A and B of wild type mGlu1 need overcome higher barrier energy than in dimer A and B of T815M and Y805A mutants of mGlu1. It indicates that FITM tends to bind the pocket of wild type mGlu1 than in T815M and Y805A mutants of mGlu1. The average hydrogen bonding occupancies of wild type, T815M and Y805A (aWildtype, aT815M, aY805A) mutants of mGlu1 are calculated between FITM and mGlu1 along the dissociation route of FITM (Fig. 7C,D). The dynamical dissociation process of FITM from the pocket of mGlu1 is shown in Supplementary Movie S1. The main residues, which interact with FITM along the dissociation path, are Tyr805, Thr815, Thr748, Cys746, Lys811 and Ser735. It is obviously that the residue Tyr805 in wild type of mGlu1 has higher hydrogen bond occupancy than that in T815M and Y805A mutants of mGlu1 (Fig. 7C). Due to the small volume of Ala805, FITM forms the lowest number of hydrogen bonds with Ala805 in Y805A mutant. Residue Thr748 in the extracellular part has the higher hydrogen bonds occupancy in wild type, T815M and Y805A mutants of mGlu1 because the residue Thr748 forms a secondary pocket with Lys811, Cys746 and Ser735 in β-hairpin above the pocket of mGlu1 (Fig. 7C,D).
Figure 7. Free energy calculations.

(A,B) The free energy calculations are profiled when FITM separates from dimer A, B of wild type, T815M and Y805A mGlu1 system, respectively. (C) The average hydrogen bonds occupancy on dimer A and B of mGlu1 along the separated route of FITM. (D) The cartoon diagram of the separated path of FITM and key interactive residues of overlapped structures of wild type, T815M and Y805A mutants of mGlu1. The T(M)815 represents residue T815 or M815. And Y(A)805 represents residue Y805 or A805.
Figure 7A,B and D show the two main energy deep wells in dimer A and B of wild type mGlu1. The lowest energy deep well corresponds to the FITM-bound pocket in the crystal structure of mGlu1 in the first state. FITM binds to allosteric site residues T815 and Y805 stably. The state 2 shows that FITM places into another higher energy deep well after conquering the energy barrier of ~20 kcal/mol. FITM binds to a secondary pocket which mainly forms by the residues Thr748, Cys746, Lys811 and Ser735. In the state 3, the FITM has been pulled out of mGlu1 completely through crossing over ~20 kcal/mol energy barrier. It can be seen that the residues in secondary pocket play an important role on the dynamical binding between mGlu1 and negative allosteric modulator.
Conclusions
In this work, we studied the dimeric packing and allosteric regulation mechanism of mGlu1 through weak interaction analysis and free energy calculation based on the molecular dynamics simulations. The results from MD simulation and weak interaction analysis indicate that vdW and hydrogen bond interaction play an important role on the packing between six cholesterol molecules and the dimer of mGlu1. Besides, the weak interaction analysis also indicates that residues T815 and Y805 in wild type mGlu1 can form stronger vdW and hydrogen bonding interaction with FITM than that in T815M and Y805A mutants of mGlu1. The free energy calculation shows that residues Thr748, Cys746, Lys811 and Ser735 are important residues before the ligand escapes from the pocket of mGlu1. Our results reveal the dimer packing and allosteric mechanism of mGlu1, which will be useful for the allosteric modulator design.
Additional Information
How to cite this article: Bai, Q. and Yao, X. Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. Sci. Rep. 6, 21763; doi: 10.1038/srep21763 (2016).
Supplementary Material
Acknowledgments
The work is supported by the Fundamental Research Funds for the Central Universities (Grant No. lzujbky-2015-166). The computational parts of this work are supported by the computing facilities of Extremadura Research Centre for Advanced Technologies (CETA-CIEMAT), funded by the European Regional Development Fund (ERDF). CETA-CIEMAT belongs to CIEMAT and the Government of Spain.
Footnotes
Author Contributions X.Y. and Q.B. conceived the project. Q.B. and X.Y. designed the experiments. Q.B. carried out the research and analysis of data. Q.B. and X.Y. contributed to the analytic tools. The article was written by Q.B. and X.Y.
References
- Niswender C. M. & Conn P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50, 295–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G., Carpenter R. L., Ocain T. D. & Bear M. F. Mechanism-based approaches to treating fragile X. Pharmacol Ther 127, 78–93 (2010). [DOI] [PubMed] [Google Scholar]
- Wu H. et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J., Prezeau L., Rondard P., Pin J. P. & Goudet C. Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol Ther 130, 9–25 (2011). [DOI] [PubMed] [Google Scholar]
- Doumazane E. et al. Illuminating the activation mechanisms and allosteric properties of metabotropic glutamate receptors. Proc Natl Acad Sci USA 110, E1416–1425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock C. et al. Activation of a dimeric metabotropic glutamate receptor by intersubunit rearrangement. J. Biol. Chem. 282, 33000–33008 (2007). [DOI] [PubMed] [Google Scholar]
- Huang S. et al. Interdomain movements in metabotropic glutamate receptor activation. Proc Natl Acad Sci USA 108, 15480–15485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L. et al. Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nat Chem Biol (2014). [DOI] [PubMed] [Google Scholar]
- Dore A. S. et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 (2014). [DOI] [PubMed] [Google Scholar]
- Bhattacharya S., Lee S., Grisshammer R., Tate C. G. & Vaidehi N. Rapid Computational Prediction of Thermostabilizing Mutations for G Protein-Coupled Receptors. J Chem Theory Comput 10, 5149–5160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q., Shen Y., Jin N., Liu H. & Yao X. Molecular modeling study on the dynamical structural features of human smoothened receptor and binding mechanism of antagonist LY2940680 by metadynamics simulation and free energy calculation. Biochim Biophys Acta 1840, 2128–2138 (2014). [DOI] [PubMed] [Google Scholar]
- Kean J., Bortolato A., Hollenstein K., Marshall F. H. & Jazayeri A. Conformational thermostabilisation of corticotropin releasing factor receptor 1. Sci Rep 5, 11954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmas R. E., Yurtsever M. & Durdagi S. Investigation of Inhibition Mechanism of Chemokine Receptor CCR5 by Micro-second Molecular Dynamics Simulations. Sci Rep 5, 13180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M. & Okada T. Structural conservation among the rhodopsin-like and other G protein-coupled receptors. Sci Rep 5, 9176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. et al. Residues remote from the binding pocket control the antagonist selectivity towards the corticotropin-releasing factor receptor-1. Sci Rep 5, 8066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q. et al. Ligand induced change of beta2 adrenergic receptor from active to inactive conformation and its implication for the closed/open state of the water channel: insight from molecular dynamics simulation, free energy calculation and Markov state model analysis. Phys. Chem. Chem. Phys. 16, 15874–15885 (2014). [DOI] [PubMed] [Google Scholar]
- Dror R. O. et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 (2013). [DOI] [PubMed] [Google Scholar]
- Arkhipov A., Shan Y., Kim E. T., Dror R. O. & Shaw D. E. Her2 activation mechanism reflects evolutionary preservation of asymmetric ectodomain dimers in the human EGFR family. Elife 2, e00708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A. & Zhang K. Y. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 71C, 26–37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. P. et al. A novel class of succinimide-derived negative allosteric modulators of metabotropic glutamate receptor subtype 1 provides insight into a disconnect in activity between the rat and human receptors. ACS Chem Neurosci 5, 597–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurevich M., Talhami A., Shalev D. E. & Gilon C. Allosteric inhibition of g-protein coupled receptor oligomerization: strategies and challenges for drug development. Curr Top Med Chem 14, 1842–1863 (2014). [DOI] [PubMed] [Google Scholar]
- Simons K. & Ehehalt R. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 110, 597–603 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W. & Klein M. L. Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79, 926–935 (1983). [Google Scholar]
- Vanommeslaeghe K., Raman E. P. & MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 52, 3155–3168 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Cuendet M. A., Grosdidier A. & Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 (2011). [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A. & Schulten K. VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38 (1996). [DOI] [PubMed] [Google Scholar]
- Phillips J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D. et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins†. The Journal of Physical Chemistry B 102, 3586–3616 (1998). [DOI] [PubMed] [Google Scholar]
- Darden T., York D. & Pedersen L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. The Journal of Chemical Physics 98, 10089–10092 (1993). [Google Scholar]
- Feller S. E., Zhang Y., Pastor R. W. & Brooks B. R. Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics 103, 4613–4621 (1995). [Google Scholar]
- Johnson E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Garcia J. et al. NCIPLOT: a program for plotting non-covalent interaction regions. J Chem Theory Comput 7, 625–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q. et al. Computational study on the interaction between CCR5 and HIV-1 entry inhibitor maraviroc: insight from accelerated molecular dynamics simulation and free energy calculation. Phys. Chem. Chem. Phys. 16, 24332–24338 (2014). [DOI] [PubMed] [Google Scholar]
- Wu P., Chaudret R., Hu X. & Yang W. Noncovalent Interaction Analysis in Fluctuating Environments. J Chem Theory Comput 9, 2226–2234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. & Chen F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012). [DOI] [PubMed] [Google Scholar]
- Espinosa E., Molins E. & Lecomte C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 285, 170–173 (1998). [Google Scholar]
- Darve E., Rodriguez-Gomez D. & Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 128, 144120 (2008). [DOI] [PubMed] [Google Scholar]
- Héni J. r., Fiori G., Chipo C & Klei M. L. Exploring Multidimensional Free Energy Landscapes Using Time-Dependent Biases on Collective Variables. Journal of Chemical Theory and Computation 6, 35–47 (2009). [DOI] [PubMed] [Google Scholar]
- Ribeiro J. V., Tamames J. A., Cerqueira N. M., Fernandes P. A. & Ramos M. J. Volarea - a bioinformatics tool to calculate the surface area and the volume of molecular systems. Chem Biol Drug Des 82, 743–755 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.