

Abstract
Human mesenchymal stem cells (hMSCs) are intrinsically heterogeneous and comprise subpopulations that differ in their proliferation, multi-potency, and functional properties, which are commonly demonstrated by culturing hMSCs at different plating densities. The objective of this study was to investigate the metabolic profiles of different subpopulations of hMSC by testing the hypothesis that the clonogenic hMSC subpopulation, which is selectively enriched in clonal density (CD) and low density (LD) culture (10 and 100 cells per square centimeter, respectively), possesses a metabolic phenotype that differs from that of hMSC in medium- or high-density (MD: 1,000 and HD: 3,000 cells per square centimeter, respectively). Cells at CD and LD conditions exhibited elevated expression of CD146 and colony forming unit-fibroblast compared with cells at MD- or HD. Global metabolic profiles revealed by gas chromatography-mass spectrometry of cell extracts showed clear distinction between LD and HD cultures, and density-dependent differences in coupling of glycolysis to the TCA cycle. Metabolic inhibitors revealed density-dependent differences in glycolysis versus oxidative phosphorylation (OXPHOS) for ATP generation, in glutamine metabolism, in the dependence on the pentose phosphate pathway for maintaining cellular redox state, and sensitivity to exogenous reactive oxygen species. We also show that active OXPHOS is not required for proliferation in LD culture but that OXPHOS activity increases senescence in HD culture. Together, the results revealed heterogeneity in hMSC culture exists at the level of primary metabolism. The unique metabolic characteristics of the clonogenic subpopulation suggest a novel approach for optimizing in vitro expansion of hMSCs.
Keywords: Mesenchymal stem cells, Cellular proliferation, Hypoxia, Cellular therapy
Introduction
The potential for clinical applications of human mesenchymal stem cells (MSCs) or multipotential stromal cells (hMSCs) for treating many devastating diseases, from ischemic cardiovascular diseases and stroke to traumatic bone and cartilage injuries, requires efficient cell expansion while preserving their therapeutic potential [1–3]. However, culture-induced changes in hMSC phenotype have been well-documented and the suitability of culture-expanded hMSCs for eventual therapeutic application is a significant barrier that must be addressed [4]. hMSCs are intrinsically heterogeneous and composed of slow and rapidly dividing subpopulations that exist even at the clonal level with varying differentiation capacity among cells derived from a single colony [5, 6]. Immediately after isolation and upon culture expansion, hMSCs acquire and accumulate genetic and phenotypic changes in culture and that many proliferating cells progressively enlarge, exit the cell cycle, and become senescent [7, 8]. The content of the clonogenic and multipotent subset in heterogeneous hMSC culture is not only correlated with cell proliferation but also with therapeutic outcome in various disease models. For example, a clonally purified hMSC population was more potent to repair infracted myocardium compared with the parent population due to their enhanced cardiomyocyte and endothelial cell differentiation [9]. Rapidly proliferating hMSCs cytometrically sorted from low density (LD) culture (100 cells per square centimeter) exhibit enhanced migration and engraftment into the hippocampal region in a mice model [10]. Early progenitor MSCs sorted from LD culture (100 cells per square centimeter) showed increased clonogenicity and had better retention in heart in mice with myocardial infarcts [11]. In contrast, non-clonal/confluent expanded hMSCs are prone to abrogate the therapeutic potential due to their increased senescent population that senescent hMSCs exhibited impaired migratory capacity in response to proinflammatory signals and failed to produce a therapeutic effect in a LPS-induced lethal endotoxemia model [12]. Thus, preserving hMSC clonal population during culture expansion is an important goal in hMSC application.
Stem cells have unique metabolic properties that actively shape cell fate and their adaptation to their immediate microenvironment [13, 14]. The key features of the stem cell metabolism include a glycolytic phenotype and metabolic plasticity that ensure progenitor pool renewal, resistance to oxidative stress, and capacity of metabolic reconfiguration upon differentiation. hMSCs have been shown to use both glycolysis and oxidative phosphorylation (OXPHOS) for ATP generation, in which metabolic substrates such as oxygen tension and glucose are key regulators [15–17]. MSCs derive a significant portion of ATP (approximately 75%) from OXPHOS under normoxia condition but are able to proliferate without compromising viability and colony forming capacity under significantly reduced ATP conditions by switching to glycolysis under low oxygen condition [18]. The enhanced hMSC proliferation and maintenance of colony forming and multipotency by hypoxia is mediated through hypoxia-inducible factors (HIFs), a known promoter of active glycolysis [16, 19, 20]. Glycolysis also has prosurvival function beyond energy production and maintains hMSC viability in an ischemic environment [21]. Moreover, MSC have the capacity to reconfigure their metabolic profile to sustain the increased energy demand in differentiation and functions [22, 23].
To date, investigations of hMSC metabolism have been performed under standard culture conditions that are known to comprise cells of varying degrees of multipotentiality, stage of lineage commitment, and senescence [16, 20, 23]. As a result, the hMSC metabolic profiles only reflect the average of a heterogeneous population with limited insight into the metabolism of the clonogeneic subset that contributes most to hMSC expansion [5]. Extensive studies have shown that plating density not only influences hMSC morphology but also their phenotypic and genotypic properties. The early MSC progenitors enriched in low passage and low density (LD) culture (50–150 cells per square centimeter) expressed a unique set of surface proteins including podocalyxin-like protein (PODXL), CD49f, CD49d, c-Met, CXCR4, and CX3CR1, which are all previously shown to be involved in cell trafficking, homing, and mobilization [11]. Gene expression of three pluripotency markers (Sox2, Nanog, and Oct4), Osterix and Msx2 of LD culture were also reverted to levels of early-passage, whereas the expressions of osteogenic-related genes were reduced [24]. Therefore, the analysis of density-dependent hMSC metabolism can provide contrasting profiles of early hMSC progenitors representing the most proliferative subset versus expanded hMSC at stationary phase. The objectives of this study are to investigate the metabolic profiles of hMSC expanded under low-plating density and to test the hypothesis that the clonogenic hMSC subset selectively enriched in clonal density (CD) and LD culture (10–100 cells per square centimeter) possesses a unique metabolic phenotype compared with hMSC in standard culture (1,000–3,000 cells per square centimeter).
Materials and Methods
Culture of hMSCs
Frozen hMSCs at passage 1 in freezing media (1 × 106 cell per milliliter per vial in minimum essential medium α (α-MEM), 2 mM l-glutamine, 30% fetal bovine serum (FBS), and 5% dimethyl sulfoxide) were obtained from the Tulane Center for Gene Therapy and cultured following the method outlined in our prior publications [25, 26]. The MSCs were isolated from the bone marrow of healthy donors ranging in age from 19 to 49 years based on plastic adherence, negative for CD34, CD45, CD117 (all less than 2%) and positive for CD29, CD44, CD49c, CD90, CD105, and CD147 markers (all greater than 95%), and possess tri-lineage differentiation potential upon induction in vitro [27]. Briefly, hMSCs at passage 1 were expanded and maintained in α-MEM supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA, https://www.atlantabio.com) and 1% Penicillin/Streptomycin (Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) in a standard CO2 incubator (37°C and 5% CO2). Cells were grown to 70%–80% confluence and then harvested by incubation with 0.25% trypsin/EDTA (Invitrogen). Harvested cells were replated at density of 1,500 cells per square centimeter and subcultured up to P5 then plated at CD (10 cells per square centimeter), LD (100 cells per square centimeter), medium density (MD, 1,000 cells per square centimeter), and high density (HD, 3,000 cells per square centimeter), and maintained for 5 days till assays. hMSCs from three different donors were used in the study and the results were reported in the main text and the Supporting Information Materials. All reagents were purchased from Sigma Aldrich (St. Louis, MO, http://www.sigmaaldrich.com) unless otherwise noted.
Cell Number, Growth Inhibition Effect, CFU-F Assay, and Glucose and Lactate Measurement
Cell number and colony forming unit-fibroblast (CFU-F) were determined following the methods reported previously [27, 28]. Briefly, harvested cells were lysed using proteinase K overnight and Picogreen (Molecular Probes, Eugene, OR, http://www.lifetechnologies.com) was added to the samples and read using a Fluror Count (PerkinElmer, Boston, MA, http://www.perkinelmer.com). Growth inhibition effect was determined by calculating the percentage difference in total cell numbers between control and treated groups normalized to control groups at the end of each treatment,
For CFU-F assay, MSCs from LD culture and HD culture were plated in 60 cm2 culture dishes at the density of 10 cells per square centimeter in α-MEM supplemented with 10% FBS with medium change every 3 days. At the end of 15 days culture, cells were stained with 20% crystal violet solution in methanol for 15 minutes at room temperature (RT). After phosphate-buffered saline (PBS) wash, the numbers of individual colonies were counted. Glucose and lactate concentration of medium samples were measured using an YSI 2700 Biochemistry Select Analyzer (YSI, Yellow Spring, OH, https://www.ysi.com). The consumption rates were then normalized by total cell number and divided by the time of incubation.
13C-Glucose Labeling and Metabolite Extraction and Derivatization
13C-glucose labeling and metabolite extraction and derivatization followed the established methods reported in our prior publication [27]. Briefly, glucose-free Dulbecco's modified Eagle's medium (DMEM) medium (Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) supplemented with a 2:2:1 mixture of unlabeled, U-13C- and 13C1-labeled glucose (Cambridge Isotopes Laboratories, Andover, MA, https://www.isotope.com) at the same concentration as used for hMSC expansion (1.0 g/l glucose). Cells were seeded and cultured for 2 days in DMEM with unlabeled medium, and then medium was replaced with isotope-enriched medium and cultured for additional 3 days. Cell collection started by removing medium from culture plates, washing with PBS, and flash-frozen by addition of liquid nitrogen to the plate to quench metabolic activities. Plates were stored in −80°C overnight and intracellular metabolites were extracted with an acenotrile:water (1:1) solution followed by addition of the internal standard (norleucine 0.085 mg/ml solution). The extract was then centrifuged at 4,000 rpm for 10 minutes at 48°C, and the supernatant was transferred to a silanized glass Reacti-Vial (Wheaton, Millville, NJ, http://wheaton.com) and stored at −80°C. Before derivatization, frozen extracts were dried under vacuum overnight and dissolved in 20 μl pyridine and 20 μL N-methyl-N-(tert butyldimethylsilyl) trifluoro-acetamide containing 1% tert-butyldimethylchlorosilane (Thermo Scientific, Rockford, IL, http://www.thermoscientific.com). The reaction was performed under a stream of argon, Reacti-vials were closed and heated to 75°C for 60 minutes and then cooled to RT. Injection of derivatized extracts in the gas chromatography-mass spectrometry (GC-MS) was completed within 24 hours.
GC-MS Analysis of Derivatized Extracts
Derivatized samples (1 μl) were injected in splitless mode in an HP Agilent 6890 series gas chromatograph coupled with an HP Agilent 5973 mass selective detector and separated on a 30-m DB5 column (J&W Scientific, Folsom, CA, http://www.agilent.com). Mass spectra were collected over a range of m/z 50–650 at a rate of 2 Hz. Metabolites were identified by comparison with standards and unknowns were identified with their retention time and by searching of the spectra in the NIST02 mass spectral library, using tools available in the software Wsearch32 (www.wsearch.com.au). Peak areas were calculated from the [M-57]+·and [M-159]+· ions for aminoacids and [M-57]+· and [M-189]+· in the carboxylic acids by fitting the elution profile to a Gaussian, eliminating the baseline and summing over all isotope peaks for a specific ion. The area was then normalized to the peak area of the internal standard norleucine which was calculated in the same way and divided by the cell number.
Detailed methods and examples of calculating isotope incorporation are provided in the Supporting Information.
Intracellular ATP, ROS
hMSCs were centrifuged, resuspended in de-ionized water, and heated immediately in a boiling water bath for 10 minutes. After cooling on ice for 30 seconds, the mixture was centrifuged and supernatant collected. Upon measurement, 10 μl of ATP solution was mixed with 100 μl of the luciferin-luciferase reagent, and the ATP bioluminescence was measured using an Orion Microplate Luminometer (Titertek-Berthold, Pforzheim, Germany, http://www.titertek-berthold.com) after 15 minutes incubation. ATP content was normalized to protein content per cell. For reactive oxygen species (ROS), aliquots of cell suspension were incubated with 25 μM carboxy-H2DCFDA at 37°C for 30 minutes. The intracellular ROS of MSCs was assessed by flow cytometry (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com).
Immunocytochemistry and MMP by Flow Cytometry
Trypsinized MSCs were washed in PBS, and fixed at 4% paraformaldehyde at RT. Nonspecific antigens were blocked by incubating the cells in PBS containing 1% bovine serum albumin at RT. Aliquots of cell suspension were incubated with fluorochrome-conjugated, anti-mouse monoclonal antibodies. For HIF-1α analysis, cells were scraped from the dish on ice, and cell suspension was washed once in ice-cold PBS. Cells were then fixed and permeabilized in 0.2% triton X-100 PBS for 10 minutes at RT. Non-specific binding sites were blocked in PBS with 1% bovine serum albumin, 10% goat serum, 4% nonfat dry milk for 15 minutes at RT. After washing with PBS, cells were incubated with anti-HIF-1α antibody at RT for 2 hours, following with 1 hour incubation with fluorescein isothiocyanate (FITC)-conjugated secondary antibody at RT. Labeled samples were washed in PBS followed by flow cytometry analysis with the isotype controls run in parallel at the same concentration used for each antibody. For mitochondrial membrane potential (MMP) measurement, trypsinized MSCs were washed by centrifugation in warm Hanks' balanced saline solution (HBSS). Cell suspension was incubated with tetramethylrhodamine, methyl ester (TMRM) (Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) at 37°C, washed with HBSS, and analyzed by flow cytometry (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com).
SA-βGal activity
Cells were grown on 10 cm2 dishes for 5 days. SA-βGal activity was then determined as described in the manufacturer's instruction (Sigma, St. Louis, MO, http://www.sigmaaldrich.com).
LDH Activity, NADPH, NADP+, G6PD Activity, and Glutathione
MSCs were lysed and incubated at RT on a shaker in lysing buffer. Supernatant was used to determine the lactate dehydrogenase (LDH) activity normalized by total cell number according to the manufacturer's instructions (Roche Applied Science, Indianapolis, IN, https://lifescience.roche.com). NADPH and NADP+ measurement were carried out as described in the manufacturer's instruction (Sigma, St. Louis, MO, http://www.sigmaaldrich.com). Glucose-6-phosphate dehydrogenase (G6PD) activity and glutathione measurements were carried out as described in the manufacturer's instruction (Cayman, Ann Arbor, MI, https://www.caymanchem.com; Cell Biolab, San Diego, CA, http://www.cellbiolabs.com).
Real-Time Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was isolated using the Rneasy Plus kit (Qiagen, Valencia, CA, https://www.qiagen.com) following instructions. Reverse transcription was carried out using 2 μg of total RNA, anchored oligo-dT primers (Operon, Huntsville, AL, http://www.operon.com), and Superscript III (Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com). Primers specific for target genes were designed using the software Oligo Explorer 1.2 (Genelink, Hawthorne, NY, http://www.genelink.com), and Betaactin was used as an endogenous control for normalization. Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) reactions were performed on an ABI7500 instrument (Applied Biosystems), using SYBR Green PCR Master Mix. The amplification reactions were performed and the quality and primer specificity were verified. Fold variation in gene expressions were quantified using the comparative Ct method: 2-(CtTreatment -CtControl), which is based on the comparison of the expression of the target gene (normalized to beta-actin).
Statistics/Data Analysis
Unless otherwise noted, all experiments were performed at least in triplicate (n = 3), and representative data are reported. Experimental results are expressed as means ± SD of the samples. Statistical comparisons were performed by one-way analysis of variance and Tukey's post hoc test for multiple comparisons, and significance was accepted at p < 0.05.
Results
Enrichment of Clonogenic Subset of hMSCs in LD Culture
Cell morphology, size distribution, proliferation rate, and CFU-F formation were measured from passage 5–7 hMSCs plated at 10, 100, 1,000, or 3,000 cells per square centimeter (referred to as CD), LD, MD, and HD, respectively). As expected, hMSCs plated at LD were more uniformly small and spindle-shaped, whereas hMSCs plated at MD or HD were more flattened and enlarged and exhibited a broader size distribution (Fig. 1A). LD cells expanded more rapidly (with doubling time around 1.5 days) compared with cells at MD or HD, which exhibited relatively constant cell numbers during the same period (Fig. 1B). CD and LD cells displayed enhanced colony forming (CFU-F) ability compared with that of MD and HD cultures (Fig. 1C). LD cells had higher expression of CD146, a surrogate cell-surface marker for hMSC clonogenity [29], compared with HD culture (58% vs. 10%, respectively), although no significant difference was observed for CD271 among the samples (Fig. 1D). In addition, LD cells exhibited significant higher Oct4 gene expression compared with HD cells, although no significant difference was observed for Nanog and Sox2 (Fig. 1E). LD cells exhibited comparable osteogenesis and adipogenesis ability compared with HD cells (Supporting Information Figs. S1–S4). Analysis of surface marker expressions by flow cytometry showed both LD and HD cells were positive for hMSCs phenotypic markers (CD105, CD90, CD166, HLA, CD147, CD49c, and CD29), but negative for hematopoietic stem cell marker CD117 (Supporting Information Table S1). Both LD and HD cells showed certain expression of PODXL and CD49f, which have been suggested to be associated with hMSCs migration and homing abilities, though no significant difference was observed between LD and HD cells (Supporting Information Table S1) [11]. hMSCs in both LD and HD were negative for CD184 and CD3 and had comparable expression of CD49b (approximately 8%) (Supporting Information Table S1). Together, the density-dependent proliferation, colony forming potential, and CD146 expression demonstrate the enrichment of the clonogenic subpopulation with improved multipotentiality in CD and LD culture compared with MD and HD.
Figure 1.
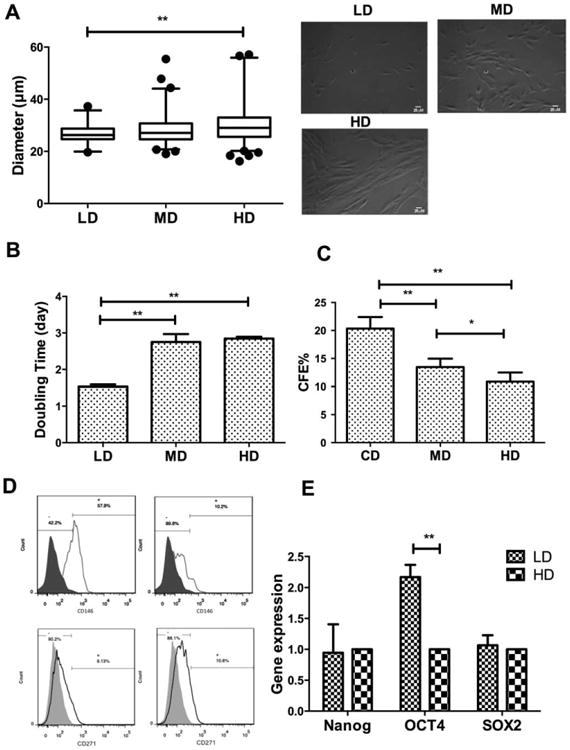
Density-dependent changes in stem cell growth, morphology, and surface markers. (A): Cell diameter distributions and representative images of cells at different densities taken at 10×.; (B): Cell doubling time over 3 days; (C): CFU-F efficiency after 15 days of culture at different densities; (D): Flow-cytometric measurement of cells positive for CD146 (top row) or CD271 (bottom row) at low density (left column) or high density (right column); (E): Quantitative reverse transcriptase-polymerase chain reaction measurement of stem cell gene Nanog, OCT-4, SOX-2 mRNA level. Data represent the mean of at least three independent determinations; errors represent the standard error in the mean. *, p < 0.05; **, p < 0.01. Abbreviations: CD, clonal density; CFE, CFU-F efficiency; HD, high density; LD, low density; MD, medium density.
Density-Dependent Energy Metabolism
Metabolic profiles for hMSC grown at LD and HD were generated by extracting the polar metabolites and analyzing the mixture using GC-MS. Peak areas were determined for known metabolites and compared using principal component analysis (PCA). The profiles for cells grown in HD cluster differently in the scores plot from those grown in LD (Fig. 2A) when plotting the first two principal components. The first principal component, which accounted for approximately 63% of the total sample variance, clearly distinguishes the profiles based on culture density.
Figure 2.
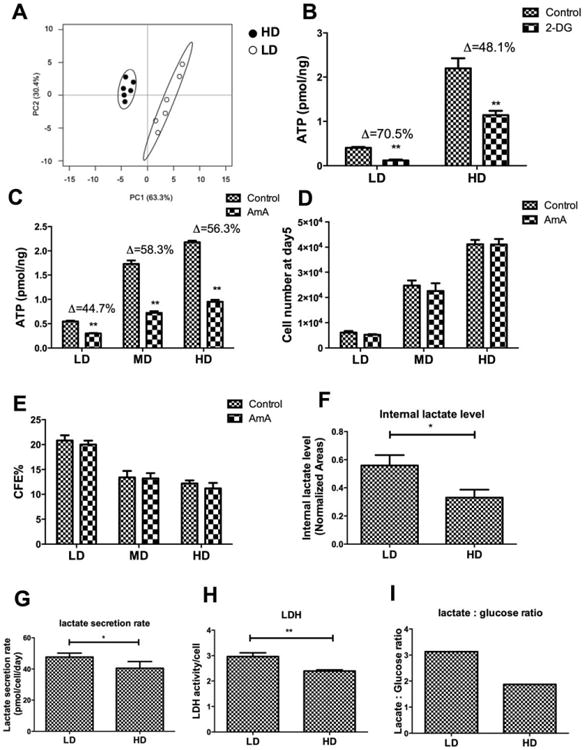
Density-dependent differences in energy metabolism. (A): Principal component analysis scores plot for peak areas from 35 metabolites extracted from cells grown in low density (LD) or high density (HD) culture; Ellipses represent the two-dimensional 95% confidence intervals obtained from a bivariate Gaussian fit to the peak clusters; (B): ATP content normalized to protein content/cell for different cell densities grown in standard media or in the presence of 2 mM 2-deoxyglucose for 48 hours; (C): ATP content normalized to protein content/cell for different cell densities grown in standard media or in the presence of 3 μM AmA for 48 hours; (D): Cell number as a function of plating density in the absence and presence of 3 μM AmA for 48 hours; (E): CFU-F efficiency as a function of plating density in the absence and presence of 3 μM AmA for 48 hours; (F): Peak area for the [M-15]+· of the t-BuDMS derivative of lactate extracted from LD or HD cells, normalized to the area of internal standard, norleucine; (G): Lactate amount increase in spent medium normalized to cell count and incubation time as a function of plating density; (H): Lactate dehydrogenase activity normalized to cell count as a function of plating density; (I): Ratio of lactate to glucose in spent medium as a function of plating density. Data represent the mean of at least three independent determinations; errors represent the standard error in the mean. *, p < 0.05; **, p < 0.01. Abbreviations: CFE, CFU-F efficiency; 2-DG, 2-deoxyglucose; HD, high density; LD, low density; LDH, lactate dehydrogenase; MD, medium density.
It is well established that stem cell proliferation and multilineage potency are enhanced under conditions that favor a glycolytic rather than OXPHOS origin for ATP synthesis [13, 30, 31]. To determine whether differences in ATP generation accounts for the different metabolic profiles, LD or HD culture was treated with 2-deoxyglucose (2-DG) or antimycin A (AmA), known inhibitors of glycolysis and OXPHOS, respectively. ATP content was more significantly reduced in LD culture exposed to 2-DG than to AmA (reduced by 70% and 48%, respectively), whereas this situation was reversed in HD culture (reduced by 45% and 56%, respectively) (Fig. 2B, 2C). Interestingly, AmA treatment had relatively minor impact on cell growth or CFU-F at either density (Fig. 2D, 2E). Internal lactate level, lactate secretion rate, LDH activity, and the lactate:glucose ratio all were higher in LD culture than in HD (Fig. 2F–2I). Collectively, these data indicate that hMSCs in CD and LD have higher proportion of glycolysis for ATP production while cells in MD and HD have higher proportion of OXPHOS for ATP production.
LD cells exhibit a lactate:glucose ratio (3.2) that exceeds the “theoretical” maximum of 2 observed in the HD cells. To determine the contribution of glutaminolysis to this elevated lactate:-glucose ratio, LD and HD cells were glutamine-deprived for 48 hours. Glutamine deprivation completely abolished hMSC proliferation and CFU-F in LD and CD but had minimal effect in HD culture (Fig. 3A, 3B). Glutamine-deprivation reduced ATP production to similar extents in both LD and HD culture (Fig. 3C). Treatment with aminooxyacetate, an inhibitor of glutamine-dependent transaminases, also inhibited proliferation in LD but had limited effect in HD (Fig. 3D). Thus, it appears that hMSC can use both glycolysis and OXPHOS for ATP production, starting from glucose and glutamine as carbon source, respectively.
Figure 3.
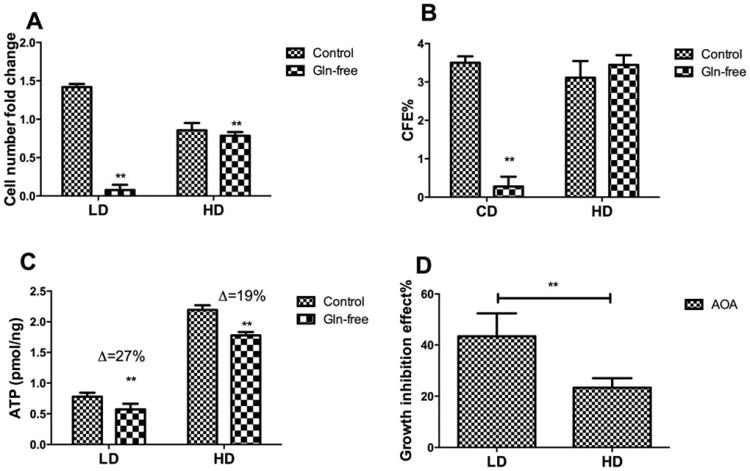
Density-dependent glutamine metabolism. (A): Fold-change in cell number as a function of plating density in the presence or absence of glutamine in the medium for 48 hours; (B): CFU-F efficiency as a function of plating density in the presence or absence of glutamine in the medium for 48 hours; (C): ATP content as a function of plating density in the presence or absence of glutamine in the medium for 48 hours; (D): Growth inhibition as a function of plating density in the presence of 100 μM aminooxyacetate for 48 hours. Data represent the mean of at least three independent determinations; errors represent the standard error in the mean. Δ: percentage difference of ATP between control and additional treatment divided by control. *, p < 0.05; **, p < 0.01. Abbreviations: AOA, aminooxyacetate; CFE, CFU-F efficiency; HD, high density; LD, low density.
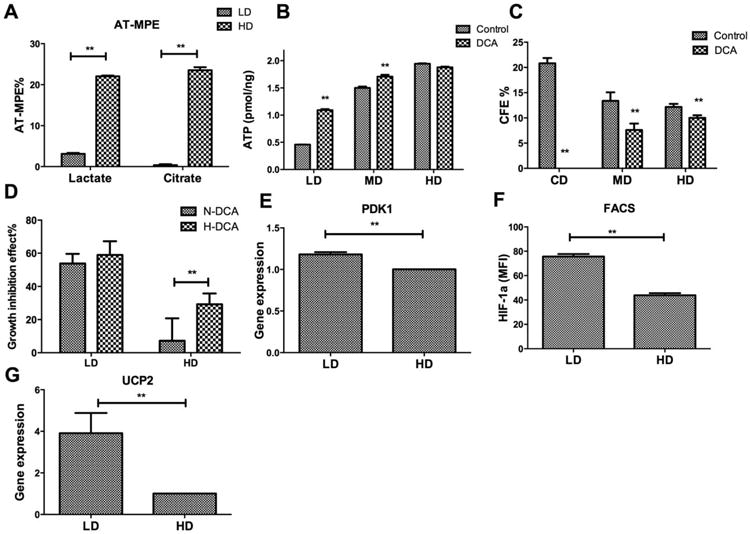
To investigate whether coupling of glycolysis to the TCA cycle was impaired in CD or LD cultures compared with HD, hMSC were cultured at LD or HD in the presence of 13C-labelled glucose and the isotope enrichment of lactate and citrate determined by GC-MS. As shown in Figure 4A, the mole fraction of isotope enrichment in lactate and citrate was approximately equal in HD culture. On the other hand, the citrate pool was significantly less labeled than lactate in LD culture, indicating poor transmission of labeled pyruvate from glycolysis to the TCA cycle under these conditions. To investigate whether this differential enrichment correlated with reduced activity of pyruvate dehydrogenase (PDH), cells in CD, LD, and HD culture were exposed to 2 mM dichloroacetate (DCA). DCA is known to enhance the activity of PDH, thereby increasing the coupling of glycolysis to the TCA cycle. As expected, DCA treatment significantly increased cellular ATP levels in LD cultures but this effect was less pronounced at MD and HD (Fig. 4B). Surprisingly, DCA treatment reduced CFU-F in MD and HD culture (by approximately 43% and 18%, respectively) but abolished CFU-F in CD (no colonies were observed) (Fig. 4C). The pronounced growth-inhibitory effects of DCA were independent of oxygen level in LD but were more pronounced under hypoxic conditions in HD (Fig. 4D). In agreement with reduced OXPHOS in LD cells, expression of pyruvate dehydrogenase kinase 1 (PDK1) was also found to be upregulated in LD cells compared with HD cells (Fig. 4E). To investigate the regulatory mechanism of the coupling of glycolysis and tricarboxylic acid (TCA) cycle between LD and HD cells, both HIF-1α and uncoupling protein 2 (UCP2) were examined. While no significant difference was found for mRNA level of HIF-1α between LD and HD cells (Supporting Information Fig. S5), a significant higher HIF-1α protein level was found in LD cells than HD cells (Fig. 4F; Supporting Information Fig. S6). In addition, an approximate approximately threefold increase was observed for UCP2 mRNA level in LD compared with HD cells (Fig. 4G).
Figure 4.
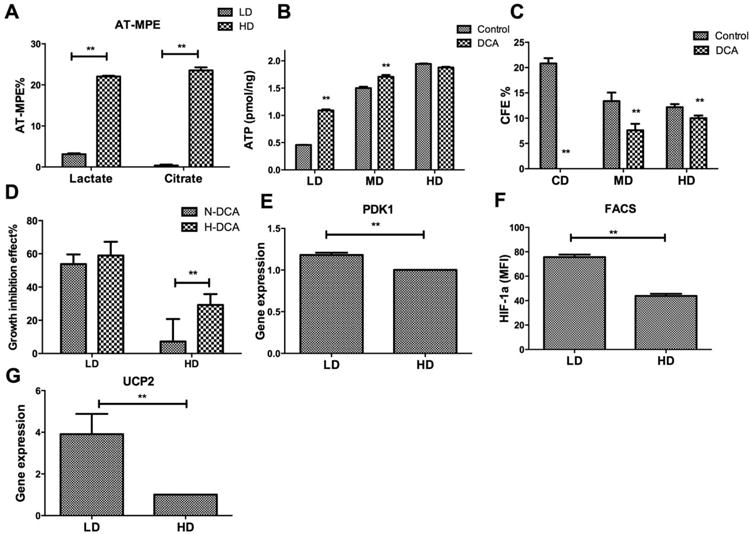
Density-dependent coupling of glycolysis to the tricarboxylic acid (TCA) cycle. (A): Absolute-total molar percent enrichment of 13C-glucose atoms in lactate and citrate for hMSC in low density and high density; (B): ATP content as a function of plating density in the absence or presence of dichloroacetate (DCA); (C): CFU-F efficiency as a function of plating density in the absence or presence of DCA; (D): Growth inhibition as a function of plating density in nomoxia or hypoxia in the presence of DCA. (E): Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) measurement of pyruvate dehydrogenase kinase 1 (PDK1) mRNA level; (F): Flow cytometry measurement of HIF-1α protein level; (G): qRT-PCR measurement of uncoupling protein 2 (UCP2) mRNA level. Data represent the mean of at least three independent determinations; errors represent the standard error in the mean. *, p < 0.05; **, p < 0.01. Abbreviations: AT-MPE, absolute total molar percent enrichment; DCA, dichloroacetate; FACS, fluorescence-activated cell sorting; HD, high density; LD, low density; MD, medium density; PDK1, pyruvate dehydrogenase kinase 1; UCP2, uncoupling protein 2.
Density-Dependent ROS Metabolism
We hypothesized that the growth-inhibitory effects of DCA in LD culture resulted from increased electron transport that increased the level of ROS and, consequently, apoptosis. ROS level was significantly higher in HD than LD. DCA treatment significantly increased ROS level in LD but not in HD culture (Fig. 5A). Background caspase 3/7 expression was significantly higher in HD than LD. DCA treatment significantly increased caspase 3/7 expression in LD (Fig. 5B) but not in MD or HD culture. DCA had relatively little effect on the inner MMP in HD cells, measured by TMRM intensity using flow cytometry, compared with the significant decreased MMP in LD culture (Fig. 5C).
Figure 5.
Density-dependent response to reactive oxygen species (ROS). (A): Flow-cytometric measurement of ROS as a function of plating density in the presence or absence of dichloroacetate (DCA) in the medium for 48 hours; (B): Caspase 3/7 activity normalized to cell count as a function of plating density in the absence and presence of DCA for 48 hours. (C): Flow cytometry measurement of mitochondrial membrane potential of low density (left column) or high density (right column) in absence or presence of DCA for 48 hours; (D): SA-βGal activity normalized to cell count as a function of plating density; (E): SA-βGal activity normalized to cell count as a function of plating density in the absence or presence of DCA; (F): SA-βGal activity normalized to cell count as a function of plating density in the absence or presence of hydrogen peroxide. Data represent the mean of at least three independent determinations; errors represent the standard error in the mean. *, p < 0.05; **, p < 0.01. Abbreviations: DCA, dichloroacetate; HD, high density; H2O2, hydrogen peroxide; LD, low density; MD, medium density; MMP, mitochondrial membrane potential; ROS, reactive oxygen species.
High levels of ROS are known to induce senescence in hMSC culture [32, 33]. HD culture resulted in a significantly higher level of SA-βGal activity (Fig. 5D), indicating a higher percentage of senescent cells in HD than in LD culture. Treatment with DCA (2 mM) or hydrogen peroxide (H2O2) (100 μM) both increased senescent expression in LD and HD culture (Fig. 5E, 5F), suggesting OXPHOS and ROS play significant roles in cellular senescence during expansion. This also suggested that LD cells, compared with HD cells, are better able to counteract the effects of ROS during expansion. Indeed, LD cells contained a significantly higher pool of NADPH plus NADP+, a higher NADPH/NADP+ ratio (Fig. 6A), and exhibited higher level of glutathione (Fig. 6B). mRNA level of glutathione synthetase (GSS) was also modestly higher in LD cells (Fig. 6G). Treatment with dehydroepiandosterone (DHEA), a known inhibitor of glucose-6-phosphate dehydrogenase, which catalyzes the first and committed step in the pentose phosphate pathway (PPP) [34], reduced cell proliferation and increased caspase 3/7 expression more significantly in LD than HD culture (Fig. 6C, 6D). Cells were cultured in the presence of a 2:2:1 mixture of unlabeled, 1-13C- and U-13C-glucose and the isotopomer distribution in lactate under LD and HD conditions was analyzed using GC-MS. The M3:M2:M1 isotopomer ratio should equal 4:0:1 if all of the glucose is metabolized through glycolysis; changing these ratios is consistent with an increase in glucose metabolized through the PPP. The M3:M1 ratio in HD culture was nearly 4:1 with M2 accounting for <10% of the labeled isotopomers (Fig. 6E). On the other hand, LD culture exhibited a significant increase in the M2 isotopomer (increasing to approximately 40% of the labeled isotopomers) with a corresponding decrease in M3 and M1, suggesting increased PPP activity in LD cells. These results are corroborated by the higher levels of G6PD enzymatic activity (Fig. 6F) and the expression of G6PD, 6-phosphogluconate dehydrogenase (6PGD) genes (Fig. 6G) in LD cells compare with HD cells. Collectively, these data indicate increased activity of the PPP in LD culture, with a corresponding increased ability to combat the deleterious effects of ROS.
Figure 6.
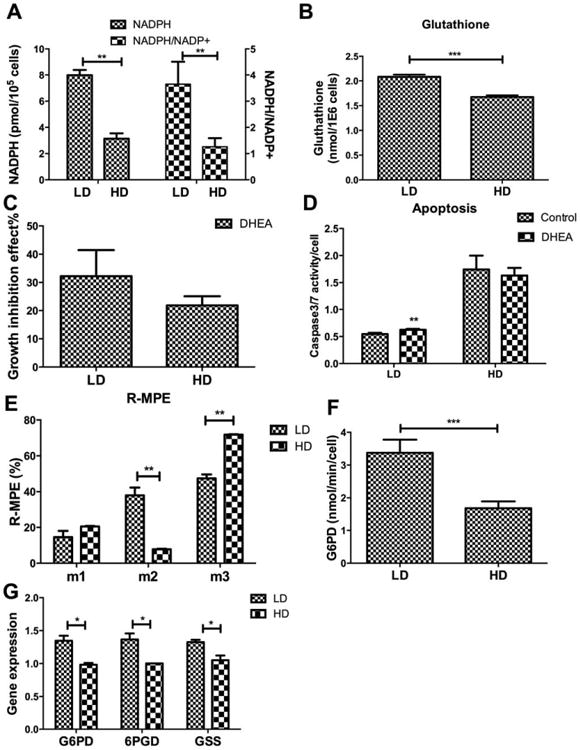
Density-dependent activity of pentose phosphate pathway. (A): NADPH or NADPH to NADP+ ratio as a function of plating density; (B): Level of glutathione as a function of plating density; (C): Growth inhibition as a function of plating density in the presence of dehydroepiandosterone (DHEA) (200 μM) for 48 hours; (D): Caspase 3/7 activity normalized to cell count as a function of plating density in the absence or presence of DHEA; (E): Relative-molar percent enrichment of 13C-glucose atoms in lactate for hMSC as a function of labeled isotopomers in low density or high density (HD); (F): Glucose-6-phosphate dehydrogenase (G6PD) activity as a function of plating density; (G): Quantitative reverse transcriptase-polymerase chain reaction measurement of mRNA level for G6PD, 6-phosphogluconate dehydrogenase (6PGD), and glutathione synthetase (GSS). Data represent the mean of at least three independent determinations; errors represent the standard error in the mean. *, p< 0.05; **, p < 0.01. Abbreviations: DHEA, dehydroepiandosterone; G6PD, glucose-6-phosphate dehydrogenase; GSS, glutathione synthetase; HD, high density; LD, low density; 6PGD, 6-phosphogluconate dehydrogenase; R-MPE, relative molar percent enrichment.
Discussion
Density-Dependent Enrichment of the Clonogenic Subset in hMSC Culture
hMSC culture in vitro comprises a heterogeneous population with significant phenotypic differences even within a single colony [5]. A small subset of clonogneic cell was found to contribute most significantly toward population expansion but also progressively increase in cell size and expression of senescent markers [7]. As heterogeneity of culture-expanded hMSCs significantly influences hMSC properties and therapeutic potency [4, 35], identification of the metabolic demand of the proliferative subset and manipulating the culture condition to amplify the clonogenic subset are the approachable strategies to enhance hMSC expansion while best preserving their clonogenic properties.
Enrichment of the clonogenic subset in LD hMSC culture is supported by the inverse correlation between plating density and proliferation, CFU-F forming efficiency, expression of CD146, expression of pluripotent transcription factor Oct4, and is in agreement with prior studies [36–38]. In the absence of a definitive set of surface markers that can unequivocally identify the multipotent subset of hMSC culture [39], hMSCs' ability to undergo clonal growth, for example, the ability to form single-cell-derived colonies when plated at very LD, has been used as a measure of clonogenicity of hMSC and is found to closely correlate with the concentration of multipotent progenitors in hMSC culture [5, 40]. The impact of plating density on hMSC phenotypic properties has been explained by the unique properties of the early progenitors to undergo density-independent proliferation at clonal or LD, a process that occurs most readily at clonal density but can be interrupted by neighboring cells as plating density increases [39]. The density-dependent enrichment of clonogneic subset was further confirmed by higher CD146+ expression in LD culture. CD146, also a pericyte marker known as melanoma cell adherent molecule, is thought to reflect the perivascular origin of the postnatal MSCs and distinguishes CFU-Fs cells from other osteogenic strains and stromal cells in bone marrow [41, 42]. However, we did not observe density-dependent variation in CD271+ population, which is another surface marker that identifies the highly clonogenic cells in unfractioned bone marrow, presumably owing to its rapid down-regulation upon ex vivo expansion of hMSCs as reported previously [43]. Oct4 is a transcript factor that was found in embryonic stem cell but not in their differentiated daughter cells and is suggested to be one of the specific markers for pluripotency [44]. The higher expression of Oct4 in LD cells compared with HD cells also confirmed density-dependent enrichment of clonogenic subset. Thus, the range plating density used in this study enables density-dependent proliferation and enrichment of the clonogenic subset.
Density-Dependent Energy Metabolism in hMSC
The results of this study extend the concept of heterogeneity of cultured hMSC to primary metabolism and point to several specific metabolic differences between cells cultured at clonogenic or LD and those cultured at MD and HD.
First, hMSC at low plating density exhibit a pronounced reliance on aerobic glycolysis for ATP production, but still are capable of utilizing electron transfer and OXPHOS for ATP production. Low plating density also correlates with reduced coupling of glycolysis to the TCA cycle as shown by decreased incorporation of glucose-derived 13C atoms into citrate compared with that seen at HD. This metabolic phenotype is similar to that observed for hMSC in hypoxic culture [15, 16, 23, 27] and provides a rationale for the proliferative effects of hypoxic culture. However, the metabolic profile observed at low culture exists at normoxic (e.g., 20%) oxygen levels. Furthermore, the low glucose uptake, residual ATP production even in the presence of 2-DG, the high lactate:glucose ratio, and the low enrichment of lactate from glucose carbons all suggest an alternative source of energy, which we argue to be glutamine based on the glutamine-deprivation results. Again, this reliance on glutaminolysis resembles the effects of hypoxia on hMSC at higher density. The increased metabolism of glutamine would effectively overcome reduced carbon flux into the TCA cycle due to the (partial) inhibition of PDH noted above, provide carbons for oxidative metabolism in the TCA cycle that is coupled to ATP production through OXPHOS, and augment ATP produced in aerobic glycolysis. Interestingly, this additional source of ATP does not appear to be required for proliferation, as hMSC showed similar proliferation rates whether glutamine was present in the growth medium or not. Together, the results of this study show that the metabolic phenotype in LD culture resembles that of aerobic glycolysis and further strengthens the concept that low level of ATP produced from glycolysis alone is sufficient to maintain the proliferative, undifferentiated state of multipotent hMSCs.
Second, LD cells exhibit increased activity of the PPP compared with HD culture. The increased activity in the PPP was shown most directly by the increased expression of the PPP genes G6PD and 6GPD and G6PD enzymatic activity and by the increased sensitivity to inhibition of G6PD by DHEA. The lactate isotopomer pattern in LD cells also suggests increased PPP. In the oxidative phase of the PPP, glucose C1 is lost as CO2, ultimately producing ribulose-5-phosphate, which can be isomerized to ribose-5-phosphate for incorporation into nucleic acids, or converted into glycolytic intermediates for further metabolism in glycolysis. On the other hand, glucose metabolized through glycolysis will not lose the C1 atom and the resulting isotopomer pattern in lactate, which is in equilibrium with the pyruvate pool, will reflect the relative activity of glycolysis versus the PPP [45–47]. Together, the increased PPP activity and glutamine metabolism contribute to a significantly higher level of NADPH in the clonogenic subset, including an increased total pool size, for example, NADPH plus NADP+, and a higher NADPH / NADP+ ratio.
Finally, the LD cells show a significantly higher capacity for reducing ROS and redox balancing compared with HD culture. Metabolism in LD cells appears optimized to reduce the deleterious effects of ROS production by limiting coupling of glycolysis to the TCA cycle and by increasing the reducing potential with elevated NADPH production and glutathione, a ROS scavenger, through PPP and glutamine metabolism. Indeed, enhanced OXPHOS by DCA treatment leads to significantly increased ROS and apoptosis in LD-hMSCs. In contrast, both H2O2 and DCA treatment increased cellular senescence in HD-hMSC, which can be attenuated by hypoxia treatment (data not shown), suggesting ROS accumulation from active OXPHOS in HD-hMSCs is the primary factor for inducing replicative senescence. These results are in agreement with previous reports that oxidative stress is a primary factor that induces hMSC differentiation and senescence [19, 48]. The divergent responses of LD- and HD-hMSCs to DCA treatment may be due to the differences of mitochondria structure, physiology, and cellular distribution among the subsets in the heterogeneous hMSC culture. For embryonic stem cells, low mitochondria activity found during the early embryonic development has been used as a biochemical index of stemness for the selection of more primitive stem cells [49, 50]. Similar to MSCs, human HSCs reside in the same hypoxic in vivo microenvironment and preferentially use glycolysis to obtain energy [51]. HSCs are also a heterogeneous set of cells with a hierarchically structured population including long-term HSCs with long-lasting self-renewal capacity with low mitochondrial mass to more specialized progenitors containing high levels of mitochondrial mass [52, 53]. In monkey MSCs, culture expansion induced significant changes in the intracellular distribution and function of mitochondria with increasing tendency to depart from the perinuclear region to an evenly distributed pattern accompanied by increase in cellular ATP content [54]. Thus, detailed analysis of hMSC mitochondria with regards to their distribution in the heterogeneous hMSC population is warranted to delineate their role in hMSC metabolic responses.
We observed a reduced mitochondrial potential (or increased leakiness), increased ROS level, and increased caspase 3/7 expression after DCA treatment under LD conditions, suggesting that high levels of electron-transport and OXPHOS would result in significant ROS production in the clonogenic subset with increased apoptosis. We did not observe this leakiness under HD conditions and others have observed a functional maturation and increased number of mitochondria upon expansion in culture [54, 55]. The enhanced glycolysis to TCA flux coupled with a more pronounced dependence on OXPHOS for ATP synthesis in HD cells would provide more opportunity for ROS generation. However, the increased energy production via OXPHOS is not beneficial to cells grown in HD culture because the hMSC proliferation potential decreases with increasing passage number. Indeed, cells appear to be unable to keep up with the increased ROS, as evidenced by higher expression of caspase 3/7, a reduced total NADPH pool, and reduced glutathione level. The accumulated cellular damage under these conditions may then lead to senescence and reduced multipotency. In an attempt to identify the mechanisms underlying the distinct metabolism in LD and HD cultures, the expressions of two glycolysis or TCA cycle associated regulators, HIF-1α and UCP2, were examined. HIF-1α mediated reconfiguration towards glycolytic metabolism enhances stem cell proliferation and maintains their multi-potency [56]. It is noted that protein level of HIF-1α was significantly higher in LD cells under normoxia and that this aerobic stabilization of HIF-1α has been suggested as an indicator of clonogenicity and a feature of hMSC to maintain their innate properties in vitro [57]. UCP2 was also found to be up-regulated in LD culture compared with HD culture. UCP2 is evidenced to mediate proton conductance in mammalian cells, of which the biological role includes modulation of metabolic signals and pathways via control of ATP production, as well as redox regulation via control of ROS production [58, 59]. The up-regulated expression of UCP2 in LD culture is in agreement with the reduced ATP content, and reduced ROS production. However, we did not observe any difference in mitochondrial inner membrane potential between LD and HD cultures. It is possible that LD culture has other mechanisms such as glycolysis ATP hydrolysis instead of electron transport to maintain mitochondrial membrane potential, as seen in human pluripotent stem cell [60].
Metabolic and Phenotypic Heterogeneity in hMSC Expansion
It has long been observed that the clonogenic subset or early passage hMSCs have higher potential in proliferation, secretion of trophic factors, engraftment, and differentiation but preserving this subset in culture has been a challenge [61, 62]. Conditions that stimulate glycolysis and suppress OXPHOS such as hypoxia or other activators of HIF, are known to prolong hMSC proliferation and better preserve the clonogenic properties. The results of this study show that the clonogenic subset enriched in clonal and LD culture has a glycolytic phenotype even under normoxic culture, but undergoes a metabolic shift towards OXPHOS as cell density increases, contributing to reduced clonogenic subset at high plating density.
The mechanisms underlying the metabolic shift as plating density increases may be due to reduced proliferation of the clonogenic subset whose growth is progressively inhibited by the presence of neighboring cells as culture density increases. On the other hand, chronic exposure to high concentration of mitogenic factors in culture may lead to metabolic switch from a predominant glycolytic phenotype to increased OXPHOS during passaging. Indeed, long-term and individual cell tracking of hMSC in culture provides direct evidence that a small percentage of small (approximately 9%) and yet highly proliferative hMSC attached at day 1 account for the majority of progenies produced over the course of hMSC culture [7]. As culture progresses, however, the small cells progressively enlarge and exhibit senescent biomarkers, concomitant with increase in population doubling time and increase in senescent subset. Although direct experimental evidence is required, the declining glycolytic subset in hMSC culture observed in current study coincides with the disappearing small “super-replicator” observed in hMSC tracking experiment, suggesting a potential link between metabolic and morphological phenotype [7].
Identification of the metabolic phenotype of the clonogenic subset in the heterogeneous MSC population provides the foundation for an implementable strategy to preserve and amplify the clonogenic subset in culture expansion. A recent study showed that hMSCs isolated from pediatric bone marrow and umbilical cord blood have higher levels of HIF-1α mRNA that are stabilized under normoxic conditions and a correspondingly larger number of glycolytic HIF target genes and increased glycolysis [57]. Although expressed at low level and not sufficient to stabilize HIF-1α, HIF-1α mRNA was also induced in normoxic adult bone marrow MSC with comparable levels of individual glycolytic HIF target genes as in the pediatric samples. These results support the notion that the glycolytic phenotype reflects hMSC's developmental origin but that hMSCs undergo a culture-induced metabolic transition towards increased OXPHOS activity, contributing to replicative senescence. In addition to oxygen tension and plating density, other factors that contribute to the metabolic transition remain to be elucidated. It is well known that hMSCs are mechano-sensitive and actively respond to their immediate biomechanical cues and topographic features by modulating their metabolic properties [63]. Thus, the clonogenic subset that undergoes density-independent proliferation or the “super-replicators” identified in hMSC culture by cell tracking may possess not only unique metabolic but also mechano-sensing properties that influence their fate in vitro. Elucidating these contributing factors will significantly improve hMSC culture strategy that maximizes their regenerative potential.
Conclusion
Our data indicate that heterogeneity in hMSCs culture exists at the level of primary metabolism. Clonogenic hMSC subpopulation enriched in low density culture possesses a distinct metabolic pattern compared to hMSC in medium- or high-density in the coupling of glycolysis versus oxidative phosphorylation for ATP generation, in glutamine metabolism, and in the dependence on the pentose phosphate pathway for maintaining cellular redox state. The study of hMSCs metabolism has important implications for developing an implementable metabolic strategy to maximize hMSCs therapeutic potential.
Supplementary Material
Significance Statement.
Application of human mesenchymal stem cells (hMSCs) in cell therapy requires a therapeutically competent population but hMSCs are intrinsically heterogeneous and comprise of subpopulations of varying properties and therapeutic potency. The metabolic characteristics of hMSC have been shown to influence their trophic properties and lineage-specific differentiation at population level. This study, for the first time, demonstrated that heterogeneity in hMSC culture exists at the level of primary metabolism and that the metabolic phenotype of hMSC subpopulation underpins the population heterogeneity during expansion. The results establish the foundation for an implementable metabolic strategy to identify, preserve, and amplify the clonogenic subset during hMSC expansion to maximize therapeutic outcome.
Acknowledgments
This work was supported by James King Biomedical Research Program (4KB09 and 3KF05 [to T.M.]).
Footnotes
Author Contributions: Y.L.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; N.M.: collection and/or assembly of data, data analysis and interpretation; B.A.B.: data collection, data analysis; T.M.L.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript; T.M.: conception and design, financial support, administrative support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest: The authors indicate no potential conflicts of interest.
See www.StemCells.com for supporting information available online.
References
- 1.Prockop DJ, Olson SD. Clinical trials with adult stem/progenitor cells for tissue repair: Let's not overlook some essential precautions. Blood. 2007;109:3147–3151. doi: 10.1182/blood-2006-03-013433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prockop DJ, Kota DJ, Bazhanov N, et al. Evolving paradigms for repair of tissues by adult stem/progenitor cells (MSCs) J Cell Mol Med. 2010;14:2190–2199. doi: 10.1111/j.1582-4934.2010.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng. 2010;12:87–117. doi: 10.1146/annurev-bioeng-070909-105309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bara JJ, Richards RG, Alini M, et al. Bone marrow-derived mesenchymal stem cells change phenotype following in vitro culture: Implications for basic research and the clinic. Stem Cells. 2014;32:1713–1723. doi: 10.1002/stem.1649. [DOI] [PubMed] [Google Scholar]
- 5.Russell KC, Phinney DG, Lacey MR, et al. In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells. 2010;28:788–798. doi: 10.1002/stem.312. [DOI] [PubMed] [Google Scholar]
- 6.Muraglia A, Cancedda R, Quarto R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J Cell Sci. 2000;113:1161–1166. doi: 10.1242/jcs.113.7.1161. [DOI] [PubMed] [Google Scholar]
- 7.Whitfield MJ, Lee WCJ, Van Vliet KJ. Onset of heterogeneity in culture-expanded bone marrow stromal cells. Stem Cell Res. 2013;11:1365–1377. doi: 10.1016/j.scr.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Wagner W, Horn P, Castoldi M, et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PloS One. 2008;3:e2213. doi: 10.1371/journal.pone.0002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang SH, Ge JB, Sun AJ, et al. Comparison of various kinds of bone marrow stem cells for the repair of infarcted myocardium: Single clonally purified non-hematopoietic mesenchymal stem cells serve as a superior source. J Cell Biochem. 2006;99:1132–1147. doi: 10.1002/jcb.20949. [DOI] [PubMed] [Google Scholar]
- 10.Lee RH, Hsu SC, Munoz J, et al. A subset of human rapidly self-renewing marrow stromal cells preferentially engraft in mice. Blood. 2006;107:2153–2161. doi: 10.1182/blood-2005-07-2701. [DOI] [PubMed] [Google Scholar]
- 11.Lee RH, Seo MJ, Pulin AA, et al. The CD34-like protein podxl and alpha6-integrin (CD49f) identify early progenitor MSCs with increased clonogenicity and migration to infarcted heart in mice. Blood. 2009;113:816–826. doi: 10.1182/blood-2007-12-128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sepulveda JC, Tome M, Fernandez ME, et al. Cell senescence abrogates the therapeutic potential of human mesenchymal stem cells in the lethal endotoxemia model. Stem Cells. 2014;32:1865–1877. doi: 10.1002/stem.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folmes CD, Dzeja PP, Nelson TJ, et al. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grayson WL, Zhao F, Izadpanah R, et al. Effects of hypoxia on human mesenchymal stem cell expansion and plasticity in 3d constructs. J Cell Physiol. 2006;207:331–339. doi: 10.1002/jcp.20571. [DOI] [PubMed] [Google Scholar]
- 16.Dos Santos F, Andrade PZ, Boura JS, et al. Ex vivo expansion of human mesenchymal stem cells: a more effective cell proliferation kinetics and metabolism under hypoxia. J Cell Physiol. 2010;223:27–35. doi: 10.1002/jcp.21987. [DOI] [PubMed] [Google Scholar]
- 17.Choudhery MS, Khan M, Mahmood R, et al. Mesenchymal stem cells conditioned with glucose depletion augments their ability to repair infarcted myocardium. J Cell Mol Med. 2012;16:2518–29. doi: 10.1111/j.1582-4934.2012.01568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buravkova LB, Rylova YV, Andreeva ER, et al. Low atp level is sufficient to maintain the uncommitted state of multipotent mesenchymal stem cells. Biochim Biophys Acta. 2013;1830:4418–4425. doi: 10.1016/j.bbagen.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 19.Estrada JC, Albo C, Benguria A, et al. Culture of human mesenchymal stem cells at low oxygen tension improves growth and genetic stability by activating glycolysis. Cell Death Differ. 2012;19:743–755. doi: 10.1038/cdd.2011.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grayson WL, Zhao F, Bunnell B, et al. Hypoxia enhances proliferation and tissue formation of human mesenchymal stem cells. Biochem Biophys Res Commun. 2007;358:948–953. doi: 10.1016/j.bbrc.2007.05.054. [DOI] [PubMed] [Google Scholar]
- 21.Mylotte LA, Duffy AM, Murphy M, et al. Metabolic flexibility permits mesenchymal stem cell survival in an ischemic environment. Stem Cells. 2008;26:1325–1336. doi: 10.1634/stemcells.2007-1072. [DOI] [PubMed] [Google Scholar]
- 22.Chen CT, Shih YRV, Kuo TK, et al. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2008;26:960–968. doi: 10.1634/stemcells.2007-0509. [DOI] [PubMed] [Google Scholar]
- 23.Pattappa G, Heywood HK, De Bruijn JD, et al. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J Cell Physiol. 2011;226:2562–2570. doi: 10.1002/jcp.22605. [DOI] [PubMed] [Google Scholar]
- 24.Yoon DS, Kim YH, Jung HS, et al. Importance of Sox2 in maintenance of cell proliferation and multipotency of mesenchymal stem cells in low-density culture. Cell Prolif. 2011;44:428–440. doi: 10.1111/j.1365-2184.2011.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao F, Ma T. Perfusion bioreactor system for human mesenchymal stem cell tissue engineering: Dynamic cell seeding and construct development. Biotechnol Bioeng. 2005;91:482–493. doi: 10.1002/bit.20532. [DOI] [PubMed] [Google Scholar]
- 26.Grayson WL, Ma T, Bunnell B. Human mesenchymal stem cells tissue development in 3d pet matrices. Biotechnol Prog. 2004;20:905–912. doi: 10.1021/bp034296z. [DOI] [PubMed] [Google Scholar]
- 27.Munoz N, Kim J, Liu Y, et al. Gas chromatography-mass spectrometry analysis of human mesenchymal stem cell metabolism during proliferation and osteogenic differentiation under different oxygen tensions. J Biotechnol. 2014;169:95–102. doi: 10.1016/j.jbiotec.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Ma T. Bioreactor strategy in bone tissue engineering: Pre-culture and osteogenic differentiation under two flow configurations. Tissue Eng Part A. 2012;18:2354–2364. doi: 10.1089/ten.tea.2011.0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lv FJ, Tuan RS, Cheung KM, et al. The surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408–1419. doi: 10.1002/stem.1681. [DOI] [PubMed] [Google Scholar]
- 30.Rafalski VA, Mancini E, Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012;125:5597–5608. doi: 10.1242/jcs.114827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu XL, Duan SL, Yi F, et al. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013;18:325–332. doi: 10.1016/j.cmet.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian WN, Braunstein LD, Pang JD, et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998;273:10609–10617. doi: 10.1074/jbc.273.17.10609. [DOI] [PubMed] [Google Scholar]
- 35.Stroncek DF, Sabatino M, Ren J, et al. Establishing a bone marrow stromal cell transplant program at the National Institutes of Health Clinical Center. Tissue Eng Part B Rev. 2014;20:200–205. doi: 10.1089/ten.teb.2013.0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colter DC, Class R, DiGirolamo CM, et al. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc Natl Acad Sci USA. 2000;97:3213–3218. doi: 10.1073/pnas.070034097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiGirolamo CM, Stokes D, Colter D, et al. Propagation and senescence of human marrow stromal cells in culture: A simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br J Haematol. 1999;107:275–281. doi: 10.1046/j.1365-2141.1999.01715.x. [DOI] [PubMed] [Google Scholar]
- 38.Jingting Li, Brendan Jones, Ying Zhang, et al. Low-density expansion protects human synovium-derived stem cells from replicative senescence: a preliminary study. Drug Deliv Transl Res. 2012;2:363–374. doi: 10.1007/s13346-012-0094-y. [DOI] [PubMed] [Google Scholar]
- 39.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuznetsov SA, Mankani MH, Bianco P, et al. Enumeration of the colony-forming units-fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res. 2009;2:83–94. doi: 10.1016/j.scr.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorrentino A, Ferracin M, Castelli G, et al. Isolation and characterization of cd146(+) multipotent mesenchymal stromal cells. Exp Hematol. 2008;36:1035–1046. doi: 10.1016/j.exphem.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Sacchetti B, Funari A, Michienzi S, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 43.Jones EA, Kinsey SE, English A, et al. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheuma. 2002;46:3349–3360. doi: 10.1002/art.10696. [DOI] [PubMed] [Google Scholar]
- 44.Boiani M, Scholer HR. Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Bio. 2005;6:872–884. doi: 10.1038/nrm1744. [DOI] [PubMed] [Google Scholar]
- 45.Kleijn RJ, van Winden WA, van Gulik WM, et al. Revisiting the C-13-label distribution of the non-oxidative branch of the pentose phosphate pathway based upon kinetic and genetic evidence. FEBS J. 2005;272:4970–4982. doi: 10.1111/j.1742-4658.2005.04907.x. [DOI] [PubMed] [Google Scholar]
- 46.Brekke EMF, Walls AB, Schousboe A, et al. Quantitative importance of the pentose phosphate pathway determined by incorporation of C-13 from [2-C-13]- and [3-C-13]glucose into TCA cycle intermediates and neurotransmitter amino acids in functionally intact neurons. J Cereb Blood Flow Metab. 2012;32:1788–1799. doi: 10.1038/jcbfm.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dusick JR, Glenn TC, Lee WP, et al. Increased pentose phosphate pathway flux after clinical traumatic brain injury: A [1,2-C-13(2)]glucose labeling study in humans. J Cereb Blood Flow Metab. 2007;27:1593–1602. doi: 10.1038/sj.jcbfm.9600458. [DOI] [PubMed] [Google Scholar]
- 48.Brandl A, Meyer M, Bechmann V, et al. Oxidative stress induces senescence in human mesenchymal stem cells. Exp Cell Res. 2011;317:1541–1547. doi: 10.1016/j.yexcr.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Van Blerkom J. Mitochondria in early mammalian development. Semin Cell Dev Biol. 2009;20:354–364. doi: 10.1016/j.semcdb.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Schieke SM, Ma MC, Cao L, et al. Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J Biol Chem. 2008;283:28506–28512. doi: 10.1074/jbc.M802763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simsek T, Kocabas F, Zheng JK, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arranz L, Urbano-Ispizua A, Mendez-Ferrer S. Mitochondria underlie different metabolism of hematopoietic stem and progenitor cells. Haematologica. 2013;98:993–995. doi: 10.3324/haematol.2013.084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romero-Moya D, Bueno C, Montes R, et al. Cord blood-derived CD34(+) hematopoietic cells with low mitochondrial mass are enriched in hematopoietic repopulating stem cell function. Haematologica. 2013;98:1022–1029. doi: 10.3324/haematol.2012.079244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lonergan T, Brenner C, Bavister B. Differentiation-related changes in mitochondrial properties as indicators of stem cell competence. J Cell Physiol. 2006;208:149–153. doi: 10.1002/jcp.20641. [DOI] [PubMed] [Google Scholar]
- 55.Chen CT, Hsu SH, Wei YH. Upregulation of mitochondrial function and antioxidant defense in the differentiation of stem cells. Biochim Biophys Acta. 2010;1800:257–263. doi: 10.1016/j.bbagen.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 56.Ma T, Grayson WL, Frohlich M, et al. Hypoxia and stem cell-based engineering of mesenchymal tissues. Biotechnol Prog. 2009;25:32–42. doi: 10.1002/btpr.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palomaki S, Pietila M, Laitinen S, et al. HIF-1 alpha is upregulated in human mesenchymal stem cells. Stem cells. 2013;31:1902–1909. doi: 10.1002/stem.1435. [DOI] [PubMed] [Google Scholar]
- 58.Fink BD, Hong YS, Mathahs MM, et al. UCP2-dependent proton leak in isolated mammalian mitochondria. J Biol Chem. 2002;277:3918–3925. doi: 10.1074/jbc.M107955200. [DOI] [PubMed] [Google Scholar]
- 59.Krauss S, Zhang CY, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci USA. 2002;99:118–122. doi: 10.1073/pnas.012410699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Khvorostov I, Hong JS, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860–4873. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rombouts WJC, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–170. doi: 10.1038/sj.leu.2402763. [DOI] [PubMed] [Google Scholar]
- 62.Li WY, Choi YJ, Lee PH, et al. Mesenchymal stem cells for ischemic stroke: Changes in effects after ex vivo culturing. Cell Transplant. 2008;17:1045–1059. doi: 10.3727/096368908786991551. [DOI] [PubMed] [Google Scholar]
- 63.McMurray RJ, Gadegaard N, Tsimbouri PM, et al. Nanoscale surfaces for the longterm maintenance of mesenchymal stem cell phenotype and multipotency. Nat Mater. 2011;10:637–644. doi: 10.1038/nmat3058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.