

Abstract
BACKGROUND
The renin-angiotensin system is a complex regulatory hormonal network with a main biological peptide and therapeutic target, angiotensin (Ang) II (1–8). There are other potentially important Ang peptides that have not been well evaluated.
METHODS
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was used for concurrent evaluation of multiple Angs downstream of Ang I (1–10) and Ang II (1–8) in kidney and plasma from wild-type (WT) mice. Angiotensin converting enzyme 2 knockout (ACE2KO) was also used as a way to examine the Angs profile in the absence of ACE2, an enzyme that cleaves both Ang I (1–10) and Ang II (1–8).
RESULTS
In plasma from both WT and ACE2KO, levels of Ang I (1–10), Ang III (2–8), and Ang (2–10) were the highest of all the renin-angiotensin system (RAS) peptides. The latter two peptides are products of aminopeptidase A cleavage of Ang II (1–8) and Ang I (1–10), respectively. In contrast, plasma levels of Ang II (1–8), and Ang (1–7), the product of Ang II (1–8) cleavage by ACE2, were low. In kidney from both WT and ACE2KO, Ang II (1–8) levels were high as compared to plasma levels. In the ACE2KO mice, a significant increase in either Ang II (1–8) or a decrease in Ang (1–7) was not observed in plasma or in the kidney.
CONCLUSION
RAS-focused peptidomic approach revealed major differences in Ang peptides between mouse plasma and kidney. These Ang peptide profiles show the dominance of the aminopeptidase A/Ang (2–10) and aminopeptidase A/Ang III (2–8) pathways in the metabolism of Ang I (1–10) and Ang II (1–8) over the ACE2/Ang (1–7) axis. Ang III (2–8) and other peptides formed from aminopeptidase A cleavage may be important therapeutic RAS targets.
Keywords: ACE2, aminopeptidase A, angiotensin II, angiotensin III, angiotensin (1–7); blood pressure; hypertension.
The key effector peptide of the renin-angiotensin system (RAS), angiotensin (Ang) II (1–8), is an octapeptide hormone that influences, among others, water and salt homeostasis, blood pressure, inflammation, and fibrosis processes.1–9 Angiotensin converting enzyme 2 (ACE2) is a monocarboxypeptidase that hydrolyzes Ang II (1–8) and, to a lesser extent, Ang I (1–10) leading to Ang (1–7) and Ang (1–9) formation, respectively.10–15 The ACE2/Ang (1–7) axis has received increased attention because of multiple potentially beneficial effects of Ang (1–7).16 In previous studies our group showed that after the infusion of Ang II either recombinant human15 or murine ACE217 effectively lowered blood pressure in mice. This blood pressure-lowering effect of rACE2 in a model of acute Ang II-dependent hypertension was clearly attributable to a large increase in plasma ACE2 activity, which resulted in a more rapid fall in plasma Ang II (1–8) levels.15,17 However, when large doses of rACE2 were given to normotensive mice, the blood pressure was unaffected.15 We reasoned that other angiotensinases (or peptidases) that degrade Ang II (1–8) and other peptides within the RAS system, such as aminopeptidase A (APA),18,19 neprilysin,18 and others20 could significantly regulate Ang II levels as effectively or even more effectively than ACE2.
We hypothesized that the uncertainty related to the importance of ACE2 in normal metabolism of Ang peptides could be resolved by evaluation of the multiple peptides downstream of Ang I (1–10), the precursor peptide, and Ang II (1–8) for which there is a paucity of data regarding their abundance in plasma and particularly at the kidney level. A major limitation of research on Ang peptides has been the lack of direct methods to simultaneously measure multiple components of this cascade network with enough discriminative power and sensitivity. Because of the structural similarity of the several angiotensins, antibody-based detection methods, such as radioimmunoassay and enzyme immunoassay, suffer from limited discriminatory power.21 This shortcoming of antibody-based assays can be circumvented by combining radioimmunoassay or enzyme immunoassay assays with high-performance liquid chromatography.22,23 The process involved, however, is cumbersome and limited by the existing antibodies to only those angiotensins for which immunoassays are available.23 In this study, we aimed to examine concurrently circulating and kidney levels of several Ang peptides in mice using liquid chromatography–tandem mass spectrometry (LC-MS/MS) to generate a RAS-peptidomic profile.17,24 Our goal was to study kidney vs. plasma profiles of the RAS peptides as well as evaluate the various angiotensinases that regulate their formation and degradation both in the presence and the absence of endogenous ACE2.
METHODS
Mouse model and animal studies
All studies were conducted with the review and approval of the Institutional Animal Care and Use Committee.
Male wild-type (WT) and ACE2 knockout (ACE2KO) mice25,26 (C57BL6 genetic background) (breeding pairs donated by Dr S. Gurley, Duke University) were used to obtain kidneys and blood for plasma Ang peptides profiles, and to perform enzyme activity assays.
At 23–27 weeks of age WT and ACE2KO mice were given an overdose of Euthasol (390mg pentobarbitol-Na and 50mg phenytoin-Na per ml). To examine the potential effect of anesthesia on plasma Ang profiles, plasma RAS peptide profiles were also measured in a group of female WT mice (C57BL/6 genetic background) (n = 5) that were euthanized following Euthasol anesthesia as compared to a group of WT mice (n = 3) anesthetized with ketamine (200mg/kg of body weight). Under ketamine anesthesia, euthanasia was performed by cervical dislocation, and blood was rapidly drawn by cardiac puncture.15
Cardiac blood was immediately collected into tubes containing a cocktail of inhibitors of renin and RAS peptidases provided by Attoquant Diagnostics (Vienna, Austria). Kidneys after collection were placed immediately on dry ice and together with plasma stored at −80 °C. Plasma and kidney specimens were further processed by Attoquant Diagnostics before subjecting to LC-MS/MS analysis.
Liquid chromatography–tandem mass spectrometry (LC-MS/MS)
Extraction and detection of Ang peptides by LC-MS/MS24 are described in detail in Supplementary Methods online.
Enzymatic activities of angiotensinases
Activities of the following angiotensinases were evaluated: ACE2,17,27,28 ACE,29 APA,28 and neprilysin (details provided in Supplementary Methods).
Statistical analysis
Statistical analysis of 2 independent groups was done using unpaired t-test. Within-groups comparisons of 2 variables were performed using paired t-test. All tests were two-tailed. Significance was defined as P < 0.05. Data were expressed as mean ± SEM.
RESULTS
RAS peptides in plasma of WT and ACE2KO mice
Ang peptides were evaluated in individual plasma samples from 5 male WT mice and 4 ACE2KO mice that were euthanized under pentobarbital anesthesia. (Figure 1). In this figure, the levels of each peptide are given in order of decreasing concentrations. In WT mice, plasma Ang I (1–10) levels were more than 100 times higher than plasma levels of Ang II (1–8) (1,295±313 vs. 7.8±2.0 pg/ml, respectively, P < 0.01). Of note, the levels of plasma Ang III (2–8), a peptide that can be formed from Ang II (1–8) by APA cleavage18,19 and from Ang (2–10) by the action of ACE,30 were present at a much higher level (1,047±178 pg/ml) than Ang II (1–8). In fact, the levels of Ang III (2–8) were almost as high as those of Ang I (1–10) (1,295±313 pg/ml), the parent precursor of all the downstream Ang peptides. Plasma levels of Ang (2–10), a degradation product of Ang (1–10) by APA cleavage, were also relatively high (657±173 pg/ml) (Figure 1). By contrast, the plasma levels of Ang (1–7) and Ang (1–5) were very low and below the lower level of quantification in some of the samples. Ang (1–9) and Ang (3–7) levels were very low as well.
Figure 1.
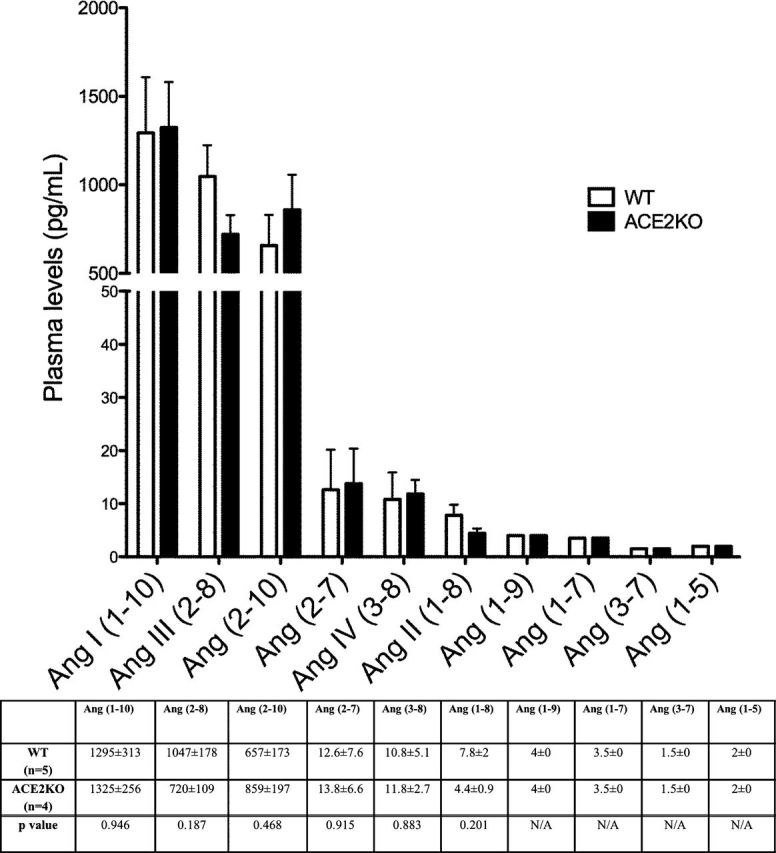
Plasma angiotensin (Ang) peptide levels in WT (□, n = 5) and ACE2KO (■, n = 4) mice represented in order of decreasing concentration (pg/ml). The bars and the values in the accompanying table below represent mean ± SE of each Ang peptide concentration in WT and ACE2KO mice. P values are from unpaired, two-tailed t-test. When concentrations were below the lower level of quantification (LLOQ), values were given as 0.5 × LLOQ.
To examine how a complete lack of ACE2 influences the levels of circulating RAS peptides, Ang peptides in ACE2KO mice were measured at the same time as in plasma collected from WT mice (all mice sacrificed at the same time). No significant differences were found in either Ang II (1–8) or Ang (1–7) (Figure 1). Similarly, there were no significant differences between WT and ACE2KO mice regarding plasma levels of any of the other 8 Ang peptides measured. In both, WT and ACE2KO mice, the Ang peptides found at the highest concentration downstream of Ang I (1–10) were Ang III (2–8) and Ang (2–10). In contrast, the levels of Ang II (1–8) and other downstream peptides were very low (Figure 1).
The precursor peptide, Ang I (1–10), is cleaved predominantly by ACE (and also by other enzymes, such as chymase, cathepsin D) to form Ang II (1–8)9 and by aminopeptidases, mainly APA, to form Ang (2–10).2,30 ACE2 cleaves Ang I (1–10) to form Ang (1–9) and Ang II (1–8) to form Ang (1–7).11,12,15 As depicted in Figure 2 (left panel), the concentration of peptides formed as a result of APA cleavage is much higher than the peptides formed as a result of ACE2 cleavage.
Figure 2.

The scheme depicts angiotensin peptides levels using the diameter of the circles to reflect their concentration in plasma (left) and kidney (right) in the WT (upper panels) and ACE2KO mice (lower panels). The enzymes known to cleave the various peptides are shown within the arrows. The peptides formed by ACE2 cleavage are in very low concentrations as opposed to those formed by APA cleavage (blue). Abbreviations: ACE, angiotensin I converting enzyme; ACE2, angiotensin converting enzyme 2; APA, aminopeptidase A; APN, aminopeptidase N; Apx, possible other aminopeptidases.
In both WT and ACE2KO mice, plasma levels of Ang (2–10) were markedly higher than those of Ang II (1–8), suggesting that Ang I (1–10) is used to form Ang (2–10) over Ang II (1–8). The known pathways of degradation of these peptides are shown in Figure 2. This “shift” toward Ang (2–10) formation could also be inferred from the ratio of plasma Ang (2–10)/Ang II (1–8) which in the ACE2KO was almost twice that of the WT mice (175±31 vs. 84±4, respectively, P < 0.01). This suggests that plasma Ang I (1–10) processing in ACE2 deficient mice is shifted toward Ang (2–10) formation even more efficiently than in the WT.
As expected, there was a positive correlation between Ang II (1–8) and Ang I (1–10) levels (Figure 3). This was observed in plasma from WT and ACE2KO (r = 0.872, P < 0.01 and r = 0.804, P < 0.01, respectively). In ACE2KO, this relationship was shifted downwards as compared to WT plasma (Figure 3). That Ang II (1–8) formation was reduced in plasma from the ACE2KO was evident also from the ratio of plasma Ang II (1–8)/Ang I (1–10) which was significantly lower in the ACE2KO as compared to WT (P < 0.01) (Figure 3). This shift in the formation of Ang II (1–8) from Ang I (1–10) was seen in the face of plasma Ang II (1–8) levels that were not significantly different between WT and ACE2KO mice (Figure 1). This decrease in Ang II (1–8) formation from Ang I (1–10) suggests that in this model of genetic ACE2 deficiency Ang II (1–8) accumulation is prevented, at least in part, by reduced formation when ACE2-mediated Ang II (1–8) degradation is absent.
Figure 3.

Correlation between plasma levels of Ang I (1–10) with Ang II (1–8) (left panel) and in WT (□, dotted trendline, n = 5) and ACE2KO mice (■, continuous trendline, n = 4). The ratio between Ang II (1–8) and Ang I (1–10) is shown in the right panel. There is a significant positive correlation between the levels of the precursor peptide Ang I (1–10) with the direct product of its degradation, Ang II (1–8), in WT and ACE2KO mice. In ACE2KO, the trend line for Ang II (1–8) formation from Ang I (1–10) is shifted downwards vs. WT plasma samples. The ratio between Ang II (1–8) and Ang I (1–10) is significantly lower in ACE2KO than WT mice plasma; **P < 0.01.
Serum activities of angiotensinases in WT and ACE2KO mice
In ACE2KO mice, ACE2 activity was at a level not significantly different from zero (0.2±0.1 relative fluorescence unit (RFU)/µl/h) (Table 1). In WT mice, serum ACE2 activity was low but detectable (0.8±0.2 RFU/µl/h) and significantly higher than in the ACE2KO (P < 0.01). The ACE2 activity in serum from WT mice (RFU/µl/h) corresponded to a concentration of 75 pg/ml of recombinant ACE2. Serum APA concentration in WT mice (RFU/µl/h) corresponded to a concentration of 801 pg/ml of recombinant APA and was 10 times higher than that of ACE2. ACE2 deletion was associated with a significantly higher level of APA activity as compared to WT mice (149±11, n = 11, vs. 94.9±9 RFU/µl/h, n = 17, respectively, P < 0.001). No significant differences were noted in serum ACE activity or neprilysin activity between ACE2KO and WT mice (Table 1).
Table 1.
Serum (RFU/µl/h) and kidney cortex enzyme activities (RFU/mg protein/h) of angiotensin processing enzymes measured in WT and ACE2KO mice
| Serum | Kidney | |||
|---|---|---|---|---|
| WT | ACE2KO | WT | ACE2KO | |
| ACE2 | 0.8±0.2 (n = 10) | 0.2±0.1** (n = 10) | 70,450±2,173 (n = 10) | 650±211*** (n = 10) |
| APA | 94.9±9 (n = 17) | 149±11*** (n = 11) | 1,084,521±43,032 (n = 10) | 868,908±29,919* (n = 10) |
| NEP | 0.74±0.11 (n = 8) | 0.86±0.11 (n = 8) | 733±61 (n = 10) | 700±55 (n = 10) |
| ACE | 59.7±2.9 (n = 10) | 66.1±2.9 (n = 10) | 1,890±100 (n = 10) | 2,179±143 (n = 10) |
Abbreviations: APA, aminopeptidase A; NEP, neprilysin; RFU, relative fluorescence unit; WT, wild-type.
*P < 0.05, **P < 0.01, ***P < 0.001 vs. WT.
Kidney levels of RAS peptides in WT and ACE2KO mice
The profiles of Ang peptides in kidney tissue differed from plasma in some important ways. Kidney Ang II (1–8) levels were almost 40 times higher than those measured in plasma (307±37 pg/g vs. 7.8±2 pg/ml, respectively). Ang (1–7) and Ang (1–9) were also higher in kidney than in plasma. In the WT, kidney levels of Ang I (1–10) were relatively high but lower than those measured in plasma obtained at the same time and from the same mice at the time of the sacrifice (468±96 pg/g vs. 1,295±313 pg/ml, respectively, n = 5). Ang (2–10) and Ang III (2–8) levels, by contrast, were markedly lower in kidneys as compared to plasma levels in the same animals (85±45 pg/g vs. 657±173 pg/ml, and 172±12 pg/g vs. 1,047±178 pg/ml, respectively). The differences between kidney and plasma levels of these peptides are very clear since 1g of kidney tissue is an approximate of 1ml of plasma.31 We did not report P values for these differences, however, because the units are not the same for plasma and kidney samples.
Kidney RAS peptides measured in WT mice were compared to those of ACE2 deficient mice (Figure 4). Kidney Ang II (1–8) levels were slightly higher in ACE2 deficient mice as compared to WT mice but the difference did not reach statistical significance (359±34 vs. 307±37 pg/g, P = NS, respectively; Figure 4). Ang (1–7) levels were also not significantly different between WT and ACE2KO kidneys. Using the kidney Ang (2–10)/Ang I (1–10) ratio as an index of Ang (2–10) formation from Ang I (1–10), a significant difference between ACE2KO and WT kidneys was found (0.385±0.074 vs. 0.158±0.047, P < 0.05, respectively). This difference suggests increased formation of Ang (2–10) in the ACE2KO kidney as compared to WT kidney. This way accumulation of Ang II (1–8) may be prevented owing to a shift toward Ang (2–10) formation from Ang I (1–10), which is mediated by APA cleavage (Figure 2). Unlike in plasma, the Ang II (1–8)/Ang I (1–10) ratio in ACE2KO kidneys taken as an index of Ang II (1–8) formation from Ang I (1–10) was not significantly different from that of the WT mice (0.677±0.130 vs. 0.735±0.125, respectively). There were no statistically significant differences in the levels of all other kidney peptides measured between ACE2KO and WT mice (Figure 4).
Figure 4.

Kidney Ang peptides levels in WT (□, n = 5) and ACE2KO (■, n = 5) mice represented in order of decreasing concentration (pg/g). The bars and the values in the accompanying table below represent mean ± SE of each angiotensin peptide concentration in WT and ACE2KO mice. P values are from unpaired, two-tailed t-test. When concentrations were below the lower level of quantification (LLOQ), values were given as 0.5 × LLOQ.
Kidney activities of angiotensinases in WT and ACE2KO mice
No significant differences were noted in kidney cortex activities for ACE and neprilysin between ACE2KO and WT mice (Table 1). APA activity in ACE2KO mice was slightly albeit significantly lower than in the WT mice (868,908±29,919, n = 10, vs. 1,084,521±43,032 RFU/mg/h, n = 10, respectively, P < 0.05). As anticipated, in ACE2KO, kidney ACE2 activity was not different from zero.
Plasma RAS peptides after pentobarbital and ketamine anesthesia
It has been previously documented that the use of pentobarbital anesthesia leads to plasma renin activation.32 We therefore examined whether anesthesia with pentobarbital could influence the RAS peptide profile.
Two groups of WT mice were compared: mice euthanized after pentobarbital anesthesia and mice euthanized after ketamine anesthesia followed by cervical dislocation (Figure 5). Ketamine anesthesia was associated with a directionally similar profile of plasma Ang peptides as pentobarbital anesthesia, namely the highest levels of plasma peptides were Ang I (1–10), Ang III (2–8), and Ang (2–10) (Figure 5). Concentrations of these 3 Angs were higher in mice after pentobarbital anesthesia than after ketamine anesthesia (Figure 5). Ang (2–7) levels were slightly but significantly higher in ketamine anesthetized mice as compared to mice after pentobarbital. No significant differences were noted in other plasma Ang peptides between these 2 anesthesia groups.
Figure 5.
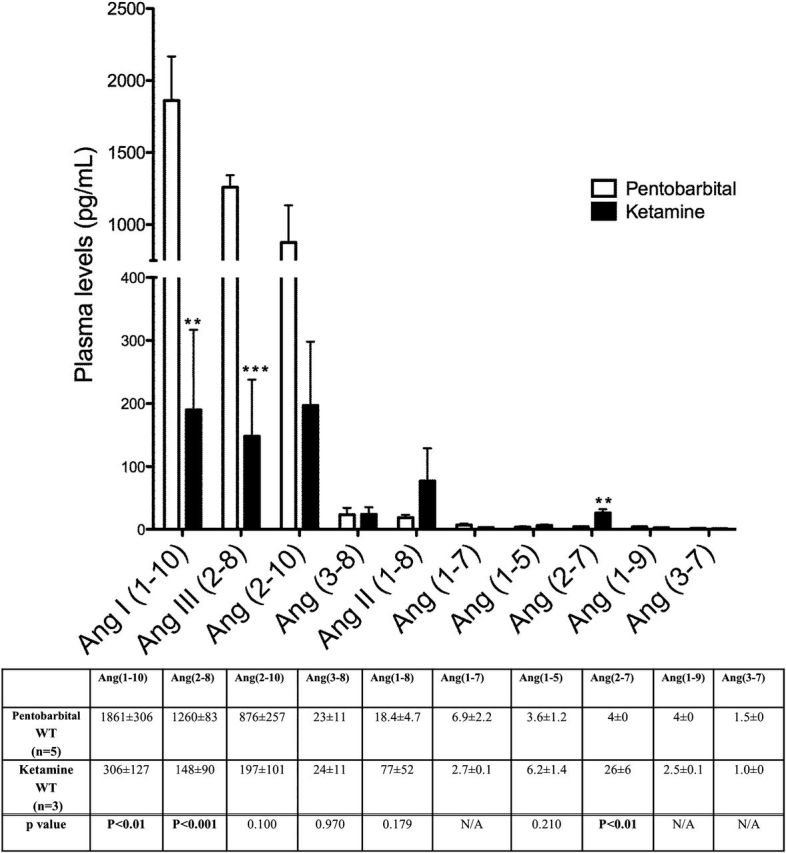
Plasma angiotensin (Ang) peptides levels represented in order of decreasing concentration (pg/ml) in female WT mice euthanized after pentobarbital (Euthasol) anesthesia (□, n = 5) and female WT mice euthanized after ketamine anesthesia (■, n = 3). The bars and the values in the accompanying table below represent mean ± SE of each Ang peptide concentration. P values are from unpaired, two-tailed t-test. When concentrations were below the lower level of quantification (LLOQ), values were given as 0.5 × LLOQ.
DISCUSSION
In this study, concurrent evaluation of 10 Ang peptides using an LC-MS/MS-based, RAS-centered peptidomic approach showed much higher levels of plasma Ang I (1–10), Ang III (2–8), and Ang (2–10) than those of Ang II (1–8), Ang (1–7), and Ang (1–5) which were very low (Figure 1). The observed profile of these peptides suggests that in plasma Ang I (1–10), the parent peptide, can be used to preferably form Ang (2–10), a process due largely to cleavage by APA33 and possibly other aminopeptidases34 (Figure 2). The degradation of Ang II (1–8) by ACE2 is not very active as judged by the low levels of Ang (1–7) and its downstream cleavage product [Ang (1–5)]. The degradation of Ang I (1–10) by ACE2 to form Ang (1–9) is also low as judged by the low levels of the latter peptide. Of note, the levels of Ang III (2–8) were the highest of the plasma peptides downstream of Ang I (1–10), likely as a reflection of high levels of APA activity in plasma. This angiotensinase also cleaves Ang I (1–10) to form Ang (2–10) which, in turn, is cleaved by ACE to form Ang III (2–8) (Figure 2). In keeping with this notion the relative enzymatic activities of APA and ACE in plasma were higher than those of ACE2 and neprilysin (Table 1). These enzyme differences are only relative because each one of these angiotensinases was evaluated with a different specific substrate. Notwithstanding this limitation, such large differences are consistent with the observed peptide products of cleavage by each of these angiotensinases (Figure 2).
Our findings in plasma from WT mice showing low levels of peptides formed from ACE2 cleavage were essentially similar to those of ACE2 deficient mice. This at first glance challenges the notion that ACE2 is a major Ang II (1–8) degrading enzyme.14,35,36 The latter concept is mainly supported by studies where recombinant (r) ACE2 was infused to dissipate exogenous Ang II (1–8).15,17 The effects of human rACE2 in vivo on the metabolism of Ang II were previously assessed by measuring the levels of Ang II (1–8) and Ang (1–7) by the enzyme-linked immunosorbent assay following the administration of Ang II (1–8) at a high dose.15 After administration of human rACE2, plasma Ang II (1–8) fell, whereas Ang-(1–7) increased.15 These in vivo findings replicated the effect of human rACE2 in vitro as assessed by high-performance liquid chromatography and demonstrated the importance of ACE2 in the metabolism of Ang II (1–8).15 Other studies also showed that ACE2 genetic ablation results in Ang II (1–8) accumulation, when this peptide is infused to achieve very high levels in plasma.15,25 In an ex vivo system, moreover, addition of recombinant ACE2 clearly enhances the formation of Ang (1–7).17 These clear effects of ACE2 as an Ang II (1–8)-processing angiotensinase, however, do not need to be manifested under conditions of relatively low Ang II (1–8) levels when possible differences might be difficult to detect and enzymes other than ACE2 can perfectly assume its degradation. Indeed, in our studies neither an increase in Ang II (1–8) nor a decrease in Ang (1–7) was seen in the ACE2KO mice under steady-state conditions.
An efficient circuit mechanism of enzymatic conversion of Ang I (1–10) to Ang (2–10), away from the conversion of Ang I (1–10) to Ang II (1–8), explains why the levels of RAS peptides metabolized by APA are high (Figure 2). APA is known to selectively hydrolyze N-terminal aspartyl residue from Ang II (1–8) to form Ang III (2–8).19,37 Circulating Ang III (2–8) and APA activity levels were both found to be high (Figure 1 and Table 1). Therefore, APA activity, which was found higher in serum of ACE2KO mice than in WT mice, can easily account, in part, for the lack of effect of ACE2 deficiency on plasma Ang II (1–8) levels. Additionally, the formation of Ang II (1–8) appears reduced in plasma from the ACE2KO as suggested by the lower levels of Ang II (1–8) for a given level of Ang I (1–10) (Figure 3). Thus, coupling of reduced formation and degradation may prevent Ang II (1–8) accumulation under conditions of ACE2 deficiency.
The inhibitors in the protease cocktail used in this study were present in the final sample at a vast molar excess to assure maximal inhibition of target enzymes, including renin. The delay between the injection of the anesthetic and the collection of samples was kept at the possible minimum. Despite of these precautions a possibility remains that renin inhibition was not complete which would explain the very high levels of Ang I (1–10) observed in this study (see below). An explanation as to why RAS peptides, such as Ang I (1–10), Ang (2–10), and Ang III (2–8) concentrations in plasma were much higher than those observed in previous studies23 could also be the use of pentobarbital anesthesia. This type of anesthesia is known to cause activation of renin.32,38 Increased plasma renin levels are mainly a compensatory reaction following a decline in arterial blood pressure due to sympathicolysis induced by anesthesia.38,39 In contrast to pentobarbital, ketamine stimulates sympathetic nerve system40 and in anesthetized mice using ketamine blood pressure remains largely unaffected.15,41 This might explain, in part, the very high levels of Ang I (1–10), Ang (2–10), and Ang III (2–8) observed after pentobarbital as compared to ketamine anesthesia. It is, however, important to note that independently of the anesthesia method used, in our study plasma concentrations of Ang (2–10) and Ang III (2–8) in mice remained the highest among the RAS peptides downstream of Ang I (1–10).
In the kidney, the levels of Ang II (1–8) were much higher than those observed in the plasma of the same animals. Previous studies by Navar and others have shown that renal content42–44 and kidney interstitial Ang I (1–10) and II (1–8) concentrations45 differ from circulating levels of these peptides. In agreement with these findings, we found markedly higher (~40 times) levels of Ang II (1–8) in the kidney than in plasma. Ang III (2–8) levels were also high in the kidney, albeit not as high as in the plasma. This peptide has been shown to markedly reduce sodium reabsorption in the presence of concurrent AT1 receptor blockade by translocating the AT2 receptor.19,46 High levels of Ang III (2–8) in plasma and in the kidney suggest that this peptide has important physiological actions. For instance, the known stimulatory effect of Ang II (1–8) on renal tubular Na+ reabsorption may be counterbalanced by an inhibitory effect of Ang III (2–8). It should be emphasized, however, that only when the AT1 receptor was blocked, Ang III (2–8) was shown to exert the natriuretic effect.19,46 Therefore, the significance of Ang III (2–8) in the day-to-day physiologic regulation of sodium excretion by the kidney is not clear at this time.
Ang (1–7) was higher in the kidney than in plasma in keeping with the fact that ACE2 is a tissue enzyme and thus is more involved in Ang (1–7) formation from Ang II (1–8) than in plasma where ACE2 levels are very low.27,29,47 In accord with other previous reports,25,26 we also did not detect Ang II (1–8) and Ang (1–7) differences between ACE2KO and WT in the kidney (Figure 4). Instead, in ACE2KO mice an almost 3-fold higher kidney concentration of Ang (2–10) and a significantly higher Ang (2–10)/AngI (1–10) ratio as compared to WT were found (Figure 3). This implies that in the face of ACE2 deficiency, the expected accumulation of Ang II (1–8) does not occur because of a shift to an increased formation of Ang (2–10) from Ang I (1–10), a process mediated by APA activity30 and possibly by other aminopeptidases.34 Since kidney APA activity was not increased, but rather decreased, in the ACE2KO mice an unknown peptidase rather than APA might be primarily involved.
In conclusion, LC-MS/MS evaluation of multiple RAS peptides revealed distinctive profiles of Ang peptides in plasma and in the kidney. The observed RAS peptides profiles demonstrate a dominance of the APA/Ang(2–10) and APA/Ang III (2–8) pathways in the metabolism of Ang I (1–10) and Ang II (1–8) over the ACE2/Ang(1–7) axis. Our findings also suggest that ACE2 deficiency does not result in a significant increase in Ang II (1–8) accumulation in plasma owing to a reduction in Ang II (1–8) formation from Ang I (1–10) which may be preferentially used to form Ang (2–10) by APA cleavage. The high levels of Ang III (2–8) and other peptides formed from APA cleavage observed in this study should invite further studies examining the function of those peptides and APA in health and disease. The physiology and pathophysiology of the RAS system could be better interpreted taking into account the complex interplay of all the changes in the various Ang peptides and the angiotensinases involved in their formation and degradation.
SUPPLEMENTARY MATERIALS
Supplementary materials are available at American Journal of Hypertension (http://ajh.oxfordjournals.org).
DISCLOSURE
The authors declared no conflict of interest.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institute of Diabetes and Digestive Kidney Diseases (grant 1R01DK080089–01A2) and a gift to Northwestern University by the Joseph and Bessie Feinberg Foundation.
REFERENCES
- 1. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59(3):251–287. [DOI] [PubMed] [Google Scholar]
- 2. Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, Dell'italia LJ. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond) 2014; 126:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Raij L. The pathophysiologic basis for blocking the renin-angiotensin system in hypertensive patients with renal disease. Am J Hypertens 2005; 18:95S–99S. [DOI] [PubMed] [Google Scholar]
- 4. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 2006; 103(47):17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baylis C, Brenner BM. Modulation by prostaglandin synthesis inhibitors of the action of exogenous angiotensin II on glomerular ultrafiltration in the rat. Circ Res 1978; 43:889–898. [DOI] [PubMed] [Google Scholar]
- 6. Schiffrin EL, Touyz RM. Inflammation and vascular hypertrophy induced by angiotensin II: role of NADPH oxidase-derived reactive oxygen species independently of blood pressure elevation? Arterioscler Thromb Vasc Biol 2003; 23(5):707–709. [DOI] [PubMed] [Google Scholar]
- 7. Ruiz-Ortega M, Egido J. Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int 1997; 52:1497–1510. [DOI] [PubMed] [Google Scholar]
- 8. Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 2011; 57:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park S, Bivona BJ, Ford SM, Jr, Xu S, Kobori H, de Garavilla L, Harrison-Bernard LM. Direct evidence for intrarenal chymase-dependent angiotensin II formation on the diabetic renal microvasculature. Hypertension 2013; 61:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002; 417:822–828. [DOI] [PubMed] [Google Scholar]
- 11. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000; 87:E1–E9. [DOI] [PubMed] [Google Scholar]
- 12. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000; 275:33238–33243. [DOI] [PubMed] [Google Scholar]
- 13. Ferrario CM, Varagic J. The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Renal Physiol 2010; 298:F1297–F1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Batlle D, Wysocki J, Soler MJ, Ranganath K. Angiotensin-converting enzyme 2: enhancing the degradation of angiotensin II as a potential therapy for diabetic nephropathy. Kidney Int 2012; 81:520–528. [DOI] [PubMed] [Google Scholar]
- 15. Wysocki J, Ye M, Rodriguez E, González-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension 2010; 55:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferreira AJ, Murça TM, Fraga-Silva RA, Castro CH, Raizada MK, Santos RA. New cardiovascular and pulmonary therapeutic strategies based on the Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas receptor axis. Int J Hypertens 2012; 2012:147825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye M, Wysocki J, Gonzalez-Pacheco FR, Salem M, Evora K, Garcia-Halpin L, Poglitsch M, Schuster M, Batlle D. Murine recombinant angiotensin-converting enzyme 2: effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension 2012; 60:730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Velez JC, Bland AM, Arthur JM, Raymond JR, Janech MG. Characterization of renin-angiotensin system enzyme activities in cultured mouse podocytes. Am J Physiol Renal Physiol 2007; 293:F398–F407. [DOI] [PubMed] [Google Scholar]
- 19. Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension 2008; 51:460–465. [DOI] [PubMed] [Google Scholar]
- 20. Grobe N, Weir NM, Leiva O, Ong FS, Bernstein KE, Schmaier AH, Morris M, Elased KM. Identification of prolyl carboxypeptidase as an alternative enzyme for processing of renal angiotensin II using mass spectrometry. Am J Physiol Cell Physiol 2013; 304(10):C945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volland H, Pradelles P, Ronco P, Azizi M, Simon D, Creminon C, Grassi J. A. solid-phase immobilized epitope immunoassay (SPIE-IA) permitting very sensitive and specific measurement of angiotensin II in plasma. J Immunol Methods 1999; 228(1–2):37–47. [DOI] [PubMed] [Google Scholar]
- 22. Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1–-7) metabolism in pulmonary and renal tissues. Am J Physiol Renal Physiol 2000; 279:F841–F850. [DOI] [PubMed] [Google Scholar]
- 23. Campbell DJ, Kladis A. Simultaneous radioimmunoassay of six angiotensin peptides in arterial and venous plasma of man. J Hypertens 1990; 8(2):165–172. [DOI] [PubMed] [Google Scholar]
- 24. Poglitsch M, Domenig O, Schwager C, Stranner S, Peball B, Janzek E, Wagner B, Jungwirth H, Loibner H, Schuster M. Recombinant Expression and Characterization of Human and Murine ACE2: Species-Specific Activation of the Alternative Renin-Angiotensin-System. Int J Hypertens 2012; 2012:428950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest 2006; 116(8):2218–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wysocki J, Ortiz-Melo DI, Mattocks NK, Xu K, Prescott J, Evora K, Ye M, Sparks MA, Haque SK, Batlle D, Gurley SB. ACE2 deficiency increases NADPH-mediated oxidative stress in the kidney. Physiol Rep 2014; 2(3):e00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wysocki J, Garcia-Halpin L, Ye M, Maier C, Sowers K, Burns KD, Batlle D. Regulation of urinary ACE2 in diabetic mice. Am J Physiol Renal Physiol 2013; 305:F600–F611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D. Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro . Hypertension 2014; 63:774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE, Coffman TM, Chen S, Batlle D. ACE and ACE2 activity in diabetic mice. Diabetes 2006; 55:2132–2139. [DOI] [PubMed] [Google Scholar]
- 30. Velez JC. The importance of the intrarenal renin-angiotensin system. Nat Clin Pract Nephrol 2009; 5:89–100. [DOI] [PubMed] [Google Scholar]
- 31. Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension 2002; 39:316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yun JC, Donahue JJ, Bartter FC, Kelly GD. Effect of pentobarbital anesthesia and laparotomy on plasma renin activity in the dog. Can J Physiol Pharmacol 1979; 57(4):412–416. [DOI] [PubMed] [Google Scholar]
- 33. Velez JC, Ryan KJ, Harbeson CE, Bland AM, Budisavljevic MN, Arthur JM, Fitzgibbon WR, Raymond JR, Janech MG. Angiotensin I is largely converted to angiotensin (1-7) and angiotensin (2-10) by isolated rat glomeruli. Hypertension 2009; 53:790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hildebrand D, Merkel P, Eggers LF, Schlüter H. Proteolytic processing of angiotensin-I in human blood plasma. PLoS One 2013; 8:e64027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J 2004; 383(Pt 1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 2002; 277:14838–14843. [DOI] [PubMed] [Google Scholar]
- 37. Kugler P. Aminopeptidase A is angiotensinase A. II. Biochemical studies on aminopeptidase A and M in rat kidney homogenate. Histochemistry 1982; 74:247–261. [DOI] [PubMed] [Google Scholar]
- 38. Janssen BJ, De Celle T, Debets JJ, Brouns AE, Callahan MF, Smith TL. Effects of anesthetics on systemic hemodynamics in mice. Am J Physiol Heart Circ Physiol 2004; 287(4):H1618–H1624. [DOI] [PubMed] [Google Scholar]
- 39. Schenk HD, Radke J, Ensink FB, Drobnik L, Kettler D, Sonntag H, Hellige G, Bretschneider HJ. Interactions between renal and general hemodynamics in fentanyl, droperidol, ketamine, thiopental and in peridural anesthesia--animal studies. Anaesthesiol Reanim 1995; 20(3):60–70. [PubMed] [Google Scholar]
- 40. Kitagawa H, Yamazaki T, Akiyama T, Mori H, Sunagawa K. Effects of ketamine on in vivo cardiac sympathetic nerve endings. J Cardiovasc Pharmacol 2001; 38(Suppl 1):S39–S42. [DOI] [PubMed] [Google Scholar]
- 41. Xu Q, Ming Z, Dart AM, Du XJ. Optimizing dosage of ketamine and xylazine in murine echocardiography. Clin Exp Pharmacol Physiol 2007; 34(5–6):499–507. [DOI] [PubMed] [Google Scholar]
- 42. Navar LG, Imig JD, Zou L, Wang CT. Intrarenal production of angiotensin II. Semin Neurol 1997; 17(5):412–422. [PubMed] [Google Scholar]
- 43. Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijn AJ. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension 1991; 18:763–773. [DOI] [PubMed] [Google Scholar]
- 44. van Kats JP, Schalekamp MA, Verdouw PD, Duncker DJ, Danser AH. Intrarenal angiotensin II: interstitial and cellular levels and site of production. Kidney Int 2001; 60:2311–2317. [DOI] [PubMed] [Google Scholar]
- 45. Siragy HM, Howell NL, Ragsdale NV, Carey RM. Renal interstitial fluid angiotensin. Modulation by anesthesia, epinephrine, sodium depletion, and renin inhibition. Hypertension 1995; 25:1021–1024. [DOI] [PubMed] [Google Scholar]
- 46. Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension 2009; 53:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006; 48:914–920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.