

Abstract
Importance
Cancer is caused by a diverse array of somatic and germline genomic aberrations. Advances in genomic sequencing technologies have improved the ability to detect these molecular aberrations with greater sensitivity. However, integrating them into clinical management in an individualized manner has proven challenging.
Objective
To evaluate the use of integrative clinical sequencing and genetic counseling in the assessment and treatment of children and young adults with cancer.
Design, Settings and Participants
An observational, consecutive case series (May 2012–October 2014) of 102 children and young adults (mean age, 10.6; median age, 11.5, range: 0–22 years) with relapsed, refractory, or rare cancer at a single major academic medical center.
Exposures
Each participant underwent integrative clinical exome (tumor and germline DNA) and transcriptome (tumor RNA) sequencing along with genetic counseling. Results were discussed in a multi-disciplinary Precision Medicine Tumor Board (PMTB) and recommendations were reported to treating physicians and families.
Main Outcomes and Measures
Proportion of patients with potentially actionable findings (PAF), results of clinical actions based on integrative clinical sequencing (ICS), and estimated proportion of patients or their families at risk for future cancer. PAF was defined as any genomic findings discovered during sequencing analysis that could lead to a 1) change in patient management by providing a targetable molecular aberration, 2) change in diagnosis or risk stratification or 3) provides cancer-related germline findings, which inform patients/families about a potential future risk of various cancers;
Results
We screened 104 patients and enrolled 102 patients of which 91 (89%) had adequate tumor tissue available to complete sequencing and only these patients were included in all subsequent calculations, including 28 (31%) with hematological malignancies and 63 (69%) with solid tumors. Overall, 42 (46%) patients had PAFs which changed patient management including, 54% (15/28) with hematological malignancies and 43% (27/63) with solid tumors. Overall, individualized actions were taken in 23 of the 91 (25%) patients and families based on actionable ICS findings, including change in treatment in 14 (15%) and genetic counseling for future cancer risk in 9 (10%) patients. 9/91 (10%) of these personalized clinical interventions resulted in ongoing partial clinical remission of 8–16 months duration or help sustain complete clinical remission of 6–21 months duration. All 9 (10%) patients and families with actionable incidental genetic findings agreed to formal genetic counseling and screening.
Conclusions and Relevance
In this single center case series of children and young adults with relapsed or refractory cancer, incorporation of data from integrative clinical sequencing into clinical management was feasible, revealed potentially actionable findings in 46% of patients, and was associated with change in treatment and family genetic counseling in a small proportion of patients. The lack of a control group limited our ability to judge whether better clinical outcomes were achieved compared to standard care.
Keywords: precision medicine, clinical sequencing, pediatric cancers, whole exome sequencing, transcriptome sequencing
INTRODUCTION
Outcomes of children and young adults with cancer have seen improvements, primarily due to an improved understanding of tumor biology and the clinical application of biological discoveries through multi-institutional clinical trials conducted by national consortia.1,2,3,4 However, survival for many pediatric oncology patients, including those with recurrent disease or metastatic disease, remains poor.5,6 To this end, integrative sequencing modalities offer a potentially useful platform to interrogate the individual cancer genome to identify actionable genomic alterations that can be matched to targeted therapies.7–9 As such, the concept of precision medicine, i.e., taking individual variability into account while designing therapy is not new, however, post-genome sequencing era discoveries provide renewed opportunities for personalizing care of individuals with cancer.10 In fact, “precision medicine” has been singled out as a priority initiative for the United States, with the goal of improving outcomes of hard-to-cure diseases, such as some pediatric cancers.11
Large-scale research projects, such as Therapeutically Applicable Research to Generate Effective Treatments (TARGET) and the Pediatric Cancer Genome Project (PCGP), are establishing the landscape of genomic alterations in common pediatric cancers.9,12–14 However, there are no prospective, pediatric studies demonstrating feasibility and utility of incorporating multiple comprehensive sequencing technologies (i.e., whole exome and transcriptome analysis) in the clinical management of children and young adults with cancer.
Following the establishment of a program called MiOncoSeq in 2011 to explore the feasibility of integrative clinical sequencing (ICS) in adult patients with advanced cancer15, we established Peds-MiOncoSeq in 2012, which is a prospective, observational clinical case series of relapsed, refractory or rare pediatric oncology patients, with an aim to study the feasibility and utility of ICS in the management of these patients. Our study also sought to identify limitations of this approach and barriers in translating sequencing findings into viable therapeutic options for pediatric oncology patients.
METHODS
Patients
Our study is a single center case series with prospective data collection, which enrolled patients at the University of Michigan C.S. Mott Children’s Hospital and was approved by our Institutional Review Board (see Supplementary Appendix Section I clinical protocol). Patients less than or equal to 25 years of age with a suspected diagnosis of a neoplastic disorder were eligible for the study. All patients were seen by a physician investigator and a genetic counselor. This study was initiated in May 2012 and is ongoing as of June 2015. All patients or parents/legal guardians provided informed consent (written assent if older than 10 years) and received mandatory pre-enrollment genetic counseling regarding the potential risks of incidental genetic findings (IGF) (Supplementary Appendix Section II consent documents). A “flexible default” consent model was employed which mandated disclosure of findings that directly influenced the current cancer management, but patients/parents could choose whether to receive incidental results, including those with possible significance for family members or conditions unrelated to the current cancer15,16. Once enrolled, a patient’s clinical course was updated quarterly in order to document clinical status and treatment decisions made by the primary team since last follow up (Supplementary Appendix Section I).
Integrative Clinical Sequencing
Board‐certified pathologists (R.R., L.P.K.) evaluated histologic sections for estimation of tumor content before submitting tissue for sequencing. Nucleic acid preparation and high-throughput sequencing were performed using standard protocols in our Clinical Laboratory Improvement Amendments (CLIA) compliant sequencing lab.17,18 Paired-end whole exome libraries from tumor samples and matched normal DNA, and transcriptome libraries from either poly-adenylated tumor RNA (PolyA+ transcriptome), or from total RNA captured by human all exon probes (capture transcriptome) were prepared and sequenced using the Illumina HiSeq 2000 and 2500 (Illumina Inc. San Diego, CA). Aligned exome and transcriptome sequencing reads were analyzed to detect putative somatic mutations, insertions/deletions (indels), copy-number alterations, gene fusions, and gene expression as described previously and detailed in Supplementary Methods.17,18 Summaries of sequencing depth and quality control parameters are presented in eTable 2.
Pathogenicity of germline variants were determined through review of the published literature, public databases including but not limited to ClinVar, Human Genome Mutation Database, and Leiden Open Variation Databases, and variant specific databases (e.g., International Agency for Research on Cancer TP53 Database, International Society for Gastrointestinal Hereditary Tumors mutation databases). Only variants that had been previously described as pathogenic were considered for disclosure. Variants with conflicting pathogenicity reports and variants not previously reported were considered to be of uncertain significance and were not considered for disclosure. Following disclosure, familial testing was recommended. Clinical relevance of somatic variants was investigated using an integrated approach incorporating technical considerations, (recurrence, variant allele fraction, expression levels, and predictive algorithms for pathogenicity), variant specific information (ClinVar, published literature, and curated gene specific resources), as well as published correlations of drug/variant sensitivity profiles. Considerations of tumor heterogeneity, including clonal versus subclonal mutations were addressed by comparing variant allele fractions and copy number estimates for each of the mutations to post-sequencing estimates of tumor content derived from SNV and copy number analyses. Variant allele fractions and tumor content estimates are shown in eTable 1. Each of the aberrations for which clinical action was based in this study were judged to be clonal.
Precision Medicine Tumor Board (PMTB) activity
A weekly, multi‐disciplinary PMTB interpreted and deliberated on sequencing results for each patient. PMTB participants included pediatric and adult oncologists, geneticists, pathologists, biologists, bioinformaticians, bioethicists, genetic counselors, study coordinators, and ad hoc expertise (see eFigure 1 for PMTB membership). Selected findings underwent additional independent CLIA-validated testing, and summarized results were disclosed to treating oncologists and families by the clinical sequencing team, board‐certified clinical geneticists, and/or counselors, as appropriate. A representative PMTB presentation is included in the Supplementary Appendix Section V.
For the purposes of this study potentially actionable findings (PAF) were defined as any genomic findings discovered during sequencing analysis that could lead to a 1) change in patient management by providing a targetable molecular aberration, 2) change in diagnosis or risk stratification or 3) provides cancer-related germline findings which inform patients/families about a potential future risk of various cancers.
RESULTS
Feasibility of Integrative Clinical Sequencing
We screened 104 patients and enrolled 102 patients (mean age 10.6, median age 11.5, age range 1–22 years) on the Peds-MiOncoSeq study between May 2012 to October 2014 (Figure 1). Two patients declined participation; for the first patient no tissue was available and the family declined research biopsy, while for second patient, cancer was in remission at the time of screening and family chose not to pursue the study using archived tissue. The patient population included common pediatric diagnoses including hematological malignancies, solid tumors and brain tumors (Table 1). Eighty one patients had refractory/relapsed disease at the time of enrollment who had exhausted all proven therapeutic options and were seen in clinic for experimental therapeutic options. The rest of the 21 patients were enrolled at the time of original diagnosis when they presented with either a very rare diagnosis or atypical presentation for pediatric age group (eTable1).
Figure 1. Overview of the Peds-MiOncoSeq Clinical Study.
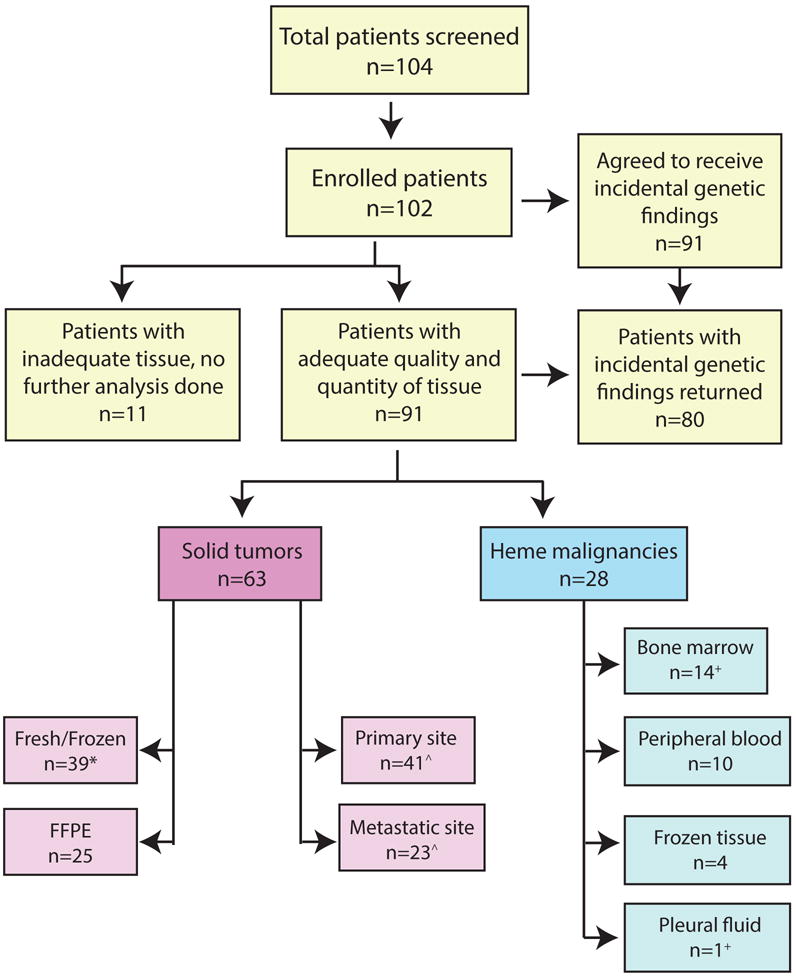
Flow chart of all patients who were screened and enrolled in the study. The number of patients with successful sequencing and details specifying the tumor type and tissue site are indicated. Two patients declined participation; for the first patient no tissue was available and the family declined research biopsy, while for second patient, cancer was in remission at the time of screening and family chose not to pursue the study using archived tissue. FFPE = Formalin fixed paraffin embedded. * One solid tumor patient was sequenced twice using frozen tissue, ˆ one solid tumor had both his primary and metastatic tumor site sequenced. + One leukemia patient had both bone marrow and malignant pleural fluid sequenced.
Table 1.
Patient demographics (102 patients)
| Diagnosis | Number of Patients | Gender (M/F) | Age (Range in Yr) | Age (Mean/Median in Yr) | Patients Sequenced at Relapse or Progression | Patients Sequenced at Diagnosis |
|---|---|---|---|---|---|---|
| All Patients | 102 | 52/50 | 0 – 22 | 10.6/11.5 | 81 | 21 |
| All Hematological Malignancies | 32 | 17/15 | 1 – 22 | 10.6/9 | 26 | 6 |
| Leukemia | 25 | 12/13 | 1 – 22 | 11.2/12 | 22 | 3 |
| Lymphoma | 4 | 3/1 | 2 – 20 | 11/11 | 3 | 1 |
| Other | 3 | 2/1 | 4 – 7 | 5/4 | 1 | 2 |
| All Solid Tumors | 70 | 35/35 | 0 – 22 | 10.6/12 | 55 | 15 |
| Brain Tumors | 8 | 4/4 | 1 – 13 | 7.1/7.5 | 6 | 2 |
| Neuroblastoma | 9 | 4/5 | 5 – 15 | 7.2/6 | 9 | 0 |
| Sarcoma | 29 | 14/15 | 0 – 22 | 11.1/12 | 21 | 8 |
| Renal Tumors | 3 | 2/1 | 18 – 22 | 20/20 | 1 | 2 |
| Liver Tumors | 7 | 4/3 | 0 – 17 | 7.3/5 | 5 | 2 |
| Ovarian Tumors | 3 | 0/3 | 9 – 13 | 11.7/13 | 2 | 1 |
| Other Solid Tumors | 11 | 7/4 | 0 – 18 | 13.7/15 | 10 | 1 |
Tumor tissue for sequencing was obtained by image-guided percutaneous core needle research biopsy in 7 patients performed by a study pediatric radiologist (J.R.D.), which was well tolerated and yielded adequate tissue. In 95 patients, tissue was obtained by standard of care diagnostic/therapeutic surgical procedures done either at the time of enrollment or from earlier procedures (eTable 1). Overall, in 91 cases (89%), we were able to obtain adequate quality and/or quantity of tumor to perform full sequencing analysis including, 28 hematological malignancies (31%) and 63 solid tumor cases (69%). These 91 patients were used as a denominator for all subsequent analyses (Figure 1). Details on the type of tissue used (i.e., frozen vs. formalin fixed paraffin embedded (FFPE)) as well as site of tissue collection (i.e., primary vs. metastasis), are summarized in Figure 1 and eTable 1.
Genetic counseling at study enrollment was well received by patients and families, with 91/102 (89%) enrolled in our study opting to receive optional IGFs. Overall, 80 (78%) actually received IGF results as these patients had both agreed to receive IGF and had adequate tumor to complete sequencing. The median turnaround time from study enrollment to presentation in PMTB for the overall cohort was 53 days, with a mean of 54 days (range: 15–114 days), which was longer than our anticipated 3–4 week timeline.15 The primary reasons for delay included waiting for bioinformatics analysis as well as wait times for the next PMTB which often delayed return of results by 2 weeks. However, over the course of the study, we were able to improve turnaround times for the first 51 patients (mean of 60 days) to the last 51 patients (mean of 48 days). We also assessed the actual costs of ICS from the time a sample was obtained and estimated the costs at approximately $6000 per patient including supplies, labor and bioinformatics analysis (eTable 3). However, patients/families were not charged for the sequencing and analysis.
Potentially Actionable Findings
One of the aims of our study was to estimate the prevalence of PAFs in children and young adults with cancer after completion of sequencing analysis. We identified 42 (46%) patients with PAFs (Tables 2,3 and eTable 1), including 9 (10%) patients with significant incidental germline findings (Table 3).
Table 2.
Summary of Hematological Malignancy PEDS-MIONCOSEQ Patients (14 Patients) with Potentially Actionable Findings
| Patient ID |
Diagnosis | Tissue Sequenced |
Informative and Actionable Genomic Findings |
Incidental Germline Findings |
Standard Therapy Without Sequencing |
Potentially Actionable Findings |
Potential Actions Based on Sequencing Results |
Action Taken |
Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Hematologic Malignancies | |||||||||
| 3 | Pre-B ALL | Bone Marrow† Pleural Fluid† |
Homozygous CDKN2A deletion; ETV6-ABL1 fusion | Palliative cytotoxic therapy for relapsed ALL or Phase-I clinical trials | Homozygous CDKN2A deletion; ETV6-ABL1 fusion | Imatinib targeting ABL1, CDK inhibitors (NCT01037790) | Yes | Sustained clinical remission for 21 months on imatinib. | |
| 9 | MDS, AML | Bone Marrow† | N-Ras G13R Mutation; Loss at Chr6q and Chr20q | Palliative cytotoxic therapy or Azacitidine or Phase-I clinical trials | N-Ras G13R Mutation | MEK inhibitor (NCT01907815) | No | Patient died of progressive disease; no MEK inhibitor available for clinical trial at the time and no dosing information for off-label use in pediatrics. | |
| 14 | Pre-B ALL | Bone Marrow† | TP53 (p.R196*), NRAS (p.Q61H), MED12 (p.R1467*), MLL2 (p.S4042fs); CDKN2A/2B homozygous deletion, chr7p one copy loss; CRLF2 overexpression | Palliative cytotoxic therapy or Phase-I clinical trials | NRAS (p.Q61H), CDKN2A/2B homozygous deletion | CDK inhibitors (NCT01037790), MEK inhibitor (NCT01907815) | No | Patient died of progressive disease; No MEK or CDK inhibitor available for clinical trial and no dosing information for off-label use in pediatrics. | |
| 30 | AML | Bone Marrow† | CSF3R p.T640N and p.Q768* point mutations, EIF4A2-MECOM fusion; chr7q copy losses of EPHA1, EPHB6, EZH2, MLL3, MNX1, RHEB, SHH, BRAF, CREB3L2, GRM8, PRSS1, SMO, chr3q copy gain, chr21 copy gain | Palliative cytotoxic therapy or Azacitidine or Phase-I clinical trials | CSF3R p.T640N and p.Q768* point mutations | Targeting CSF3R p.T640N and p.Q768* point mutations with JAK2 inhibitor ruxolitinib | No | Patient could not be treated on ruxolitinib on clinical trial or off label due to rapid progression before availability of results. | |
| 38 | T-ALL | Peripheral Blood† | CDKN2A and CDKN2B deletions, PRSS1 deletion, NTRK1 overexpression; | Palliative cytotoxic therapy or Phase-I clinical trials | CDKN2A and CDKN2B deletions | CDK inhibitors (NCT01037790) | No | Patient died of rapid progressive disease; no CDK inhibitors available for clinical trial and no dosing information for off-label use in pediatrics. | |
| 41 | ETP-ALL | Peripheral Blood† | FLT3 ITD mutation; Chr16p gain, Chr16q loss; FLT3 overexpression | Allogenic BMT, No adjuvant therapy following BMT | FLT3 ITD mutation, FLT3 overexpression | FLT3 inhibitor | Yes | Patient in clinical remission post-BMT, on sorafenib post-BMT for 15 months. | |
| 49 | Pre-B ALL | Peripheral Blood† | FLT3 deletion; BLK and FLT3 overexpression | Allogenic BMT for relapsed ALL. No adjuvant therapy following BMT | FLT3 deletion, FLT3 overexpression | FLT3 inhibitor | Yes | Patient in clinical remission for 9 months post-BMT, on sorafenib for 6 months. | |
| 53 | LCH | Axillary LN† (M) | BRAF V600D mutation | Steroids, Vinblastine for 6 months | BRAF V600D mutation | BRAF inhibitor | None required | Patient in clinical remission after chemotherapy. Eligible for BRAF inhibitors in case of post-treatment. | |
| 54 | AML | Bone Marrow† | NF1 (Y333*) mutation, NF1 frame-shift deletion, TSC2 stop-gain SNV insertion (truncated after a.a.646; FL=1807 a.a.); CBFB-MYH11 fusion | Allogenic BMT for AML, No adjuvant therapy following BMT | NF1 (Y333*) mutation, NF1 frame-shift deletion | MEK inhibitors (NCT02049801) | None required | Patient in clinical remission following BMT. Eligible for MEK inhibitor in case of post-treatment. | |
| 55 | AML | Bone Marrow† | CDK6 overexpression, FLT3 (D835Y) mutation | No adjuvant therapy following Donor Leukocyte Infusion | FLT3 (D835Y) mutation | Next generation FLT inhibitor (NCT02039726) | None required | Patient in clinical remission following DLI. Eligible for next generation FLT inhibitor in case of post-treatment. | |
| 65 | AML | Bone Marrow† Bone Marrow‡ |
9q loss, WT1, NF1 frame-shift indels, BIRC6-CEBPZ inactivating fusion, PTPT11 E76Q mutation | Palliative cytotoxic therapy or Phase-I clinical trials | NF1 frame-shift | MEK inhibitor (NCT02049801) or mTOR inhibitors | No | Patient died quickly following Allogeneic BMT. | |
| 66 | JMML | Bone Marrow‡ | Somatic CBL (p.Y371H), NRAS (p.G12D), NRAS (p.G13D), PTPN11 (p.D61Y) | Chemotherapy, 13-Cis Retinoic acid followed by Allogenic BMT | NRAS (p.G12D), NRAS (p.G13D) | MEK inhibitor (NCT01907815) | None required | Patient clinically stable and experiencing spontaneous regression without therapy. MEK inhibitors in case fails standard therapy. | |
| 76 | T/My Bi-Phenotypic Leukemia | Peripheral Blood† | NRAS (G60E), TP53 (P72R), PHF6 (R320*) mutations, SPI1 frame-shift insertion (Q78fs, somatic); ASXL1 frame-shift insertion, CBLC frame-shift insertion, JAK3 (M511I) activating mutation; JAK3 overexpression | Cytotoxic chemotherapy for relapsed leukemia, Phase-I clinical trials | NRAS (G60E), JAK3 (M511I) activating mutation; JAK3 overexpression | JAK3 inhibitor or MEK inhibitor (NCT01907815) | Yes | Patient treated with JAK3 inhibitor tofacitinib but could not tolerate full dose due to GI toxicity, died of progressive disease. | |
| 92 | ALL | Bone Marrow† | Rearrangement of T-cell receptors and immunoglobulins detected; ZCCHC7-PAX5, EBF1-PDGFRB fusions | Allogenic BMT for elevated MRD | EBF1-PDGFRB fusion | PDGFRB inhibitors (imatinib) | None so far | Patient in clinical remission post-BMT, treating physician in process of getting approval for Imatinib | |
| 98 | Pre-B ALL | Peripheral Blood‡ | Rearrangement B and T cell antigen receptors; BCR-ABL1 fusion, RUNX1-MSH6 (loss of RUNX1 function), MSH6-RUNX1 (reciprocal; functional RUNX1), HBS1L-MYB (out-of-frame) fusions; FLT3 overexpression | Imatinib therapy post Allogenic BMT | BCR-ABL1 fusion | Imatinib, dasatinib targeting BCR-ABL1 fusion | No | Patient already on imatinib post-BMT. | |
Tissues are fresh frozen unless indicated as FFPE= Formalin Fixed Paraffin Embedded Tissue; Tissue type abbreviations: P=Primary, M=Metastatic; gender abbreviations: F=Female, M=Male.
Bold Genomic findings: Potentially Actionable Findings (PAF), which can potentially impact patient’s diagnosis, risk stratification, management or informs patient/family of future risk for serious health condition including cancers.
Abbreviations: ALL=Acute Lymphoblastic Leukemia, MDS=Myelodysplastic Syndrome, AML=Acute Myeloid Leukemia, ETP-ALL=Early T-cell Precursor Acute Lymphoblastic Leukemia, LCH= Langerhan’s Cell Histiocytosis, JMML= Juvenile Myelomonocytic Leukemia, LN=Lymph Node, indel=insertion/deletion, BMT=Bone Marrow Transplant, DLI= Donor Lymphocyte Infusion, MRD= Minimal Residual Disease.
DNA paragroups are indicated by an asterisk (*) placed after the main haplogroup.
Tissue used for sequencing was after relapse or refractory or progressive disease following treatment with surgery, chemotherapy, radiation or biologic therapy
Tissue used for sequencing was prior to starting any treatment
Table 3.
Summary of Solid Tumor PEDS-MIONCOSEQ Patients (27 Patients) with Potentially Actionable Findings
| Patient ID |
Diagnosis | Tissue Sequenced |
Informative and Actionable Genomic Findings |
Incidental Germline Findings |
Traditional Therapy Options Without Sequencing |
Potentially Actionable Findings |
Potential Actions Based on Sequencing Results |
Action Taken |
Clinical Actions and Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Solid Tumors | |||||||||
| 5 | SS | Thigh† (P) FFPE Lung† (M) FFPE |
NOTCH1 T1997M | Available Phase-I clinical trial for relapsed solid tumors | NOTCH1 T1997M | NOTCH1 inhibitor (NCT01154452) | No | Patient eligible for adult NOTCH1 inhibitor trial, but in hospice with progressive disease. | |
| 13 | PEComa | Peritoneal Mass† (P) | SFPQ-TFE3 fusion | Available Phase-I clinical trial for relapsed solid tumors mTOR or VEGF inhibitors | SFPQ-TFE3 fusion | VEGF or mTOR inhibitors | Yes | Patient on pazopanib therapy for Post-treatment with 90% tumor reduction for longer than 16 months. | |
| 15 | WT | Paraspinal Mass† (M) | WTX1 AMER1 deletion, MYC P44L, MAX R60Q | HOXB13 (p.G84E) mutation | Available Phase-I clinical trial for relapsed solid tumors, no genetic counseling | MYC P44L, Germline HOXB13 (p.G84E) mutation | mTOR and VEGF inhibitors, family counseling for prostate cancer risk | Yes | Patient on VEGF2 inhibitor (XL-184) with a partial response for longer than 15 months. Family referred to genetics clinic. |
| 21 | Melanoma | Lymph Node‡ (M) FFPE | BRAF V600E | BAP1 (p.D567X) | No adjuvant therapy | BRAF V600E, Germline BAP1 (p.D567X) | BRAF inhibitor (NCT01677741), family genetic counseling for melanoma and other cancers | Yes | Family in genetics clinic for counseling. BRAF inhibitor as available option for relapse. |
| 23 | NBL | Adrenal mass‡ (P) | Many CNAs, ATRX deletion | BARD1 fs insertion (p.E139fs) | Available Phase-I clinical trial for relapsed NBL, no genetic counseling | Germline BARD1 fs insertion (p.E139fs) | Genetic counseling for familial cancer | Yes | Family in genetics clinic for counseling. |
| 24 | Colorectal Carcinoma | Colon Mass† (P) Lymph Node‡ (M) FFPE |
TP53 (C135F), GNAS (R201H), SFPQ-TFE3 fusion | Available Phase-I clinical trial for relapsed solid tumors or VEGF inhibitors | SFPQ-TFE3 fusion | VEGF or mTOR inhibitors | Yes | Action taken; Patient progressed on pazopanib therapy. | |
| 31 | ACC | Adrenal Mass‡ (P) FFPE | STAG2 W706G; Chr12q copy gain, Chr22 copy loss (CHEK2), MDM2 amplification and overexpression | Mitotane or Available Phase-I clinical trial for relapsed solid tumors | MDM2 amplification and overexpression | MDM2 inhibitors (NCT01901172) | No | Patient eligible for MDM2 inhibitor but decided to go on other investigational therapy. | |
| 33 | ARMS | Hepatic Lobe† (M) | Chr3p, 4q, 12q, 14p, 14q, and 16q copy losses, MYCN copy gain; TP53 (R248W), PAX3-FOXO1 fusion; MYCN, OLIG2 overexpression FGF8 amplification and overexpression |
Available Phase-I clinical trial for relapsed solid tumors or palliative cytotoxic chemotherapy | FGF8 amplification and overexpression | FGFR4 inhibitor (NCT01976741 or NCT01703481) | Yes | Action taken; patient received FGFR inhibitor ponatinib but discontinued due to skin toxicity. | |
| 37 | ERMS | Peritoneal Fluid† (M) | MDM2, YEATS4 amplification; MAFB, CSF1R, SPI1 overexpression; FGFR4 V550E | Available Phase-I clinical trial for relapsed solid tumors | FGFR4 V550E | FGFR4 inhibitor (NCT01976741 or NCT01703481) | No | No FGFR inhibitor available in clinical trials or dosing information available for off label use for pediatric patients. | |
| 39 | RMS | Oropharynx Mass† (P) | FANCD2 frame-shift deletion; ATIC-ALK fusion | Adjuvant cytotoxic chemotherapy | ATIC-ALK fusion | ALK inhibitor in combination with cytotoxic therapy | None required | Patient in clinical on cytotoxic chemotherapy. ALK inhibitor is an option for post-treatment. | |
| 43 | IFS | Forearm Mass† (P) | Chr3q copy loss, chr16 copy gain; STAG2 Y355F mutation, IL-3 indel; Homozygous deletion CDKN2A, CDKN2B; LMNA-NTRK1 fusion; NTRK1, LMNA overexpression | Available Phase-I clinical trial for relapsed solid tumors | Homozygous deletion CDKN2A, CDKN2B; LMNA-NTRK1 fusion | Change in diagnosis, NTRK inhibitors or CDK inhibitors (NCT01037790) | Yes | 50% reduction in lung masses on crizotinib therapy for NTRK1 inhibition, on therapy for 8 months. | |
| 44 | OS | Leg Mass† (P) FFPE | Homozygous deletions of NF-1, PTEN, FAS, P53; frame-shift insertion of ATRX; WNT5B, WNT16 overexpression | Available Phase-I clinical trial for relapsed solid tumors or palliative cytotoxic chemotherapy | Homozygous deletions of NF-1 | MEK inhibitors (NCT01725100) | Yes | Action taken; patient treated with MEK inhibitor trametinib but progressed after 2 months. | |
| 45 | Epithelioid Sarcoma | Lymph Node† (M) | KRAS, ALAS1, BIRC3, WNT8A overexpression | Available Phase-I clinical trial for relapsed solid tumors | KRAS overexpression | MEK inhibitors (NCT01725100) | Yes | Patient started on MEK inhibitor in combination with mTOR inhibitor but progressed in 4 weeks. | |
| 51 | Cholangio-carcinoma | Liver Mass† (P) | IDH1 (p.R132C), TP53 splice site mutation; KRAS amplification, KRAS overexpression | Available Phase-I clinical trial for relapsed solid tumors | IDH1 (p.R132C) mutation, KRAS amplification, KRAS overexpression | IDH inhibitors or MEK inhibitors (NCT01725100) | No | Patient rapidly progressed and died. No IDH or MEK inhibitor in clinical trials and no dosing information for off label use. | |
| 57 | RMS | Cerebellar Tumor‡ (P) FFPE Cerebellar Tumor† (P) |
Chr1q, 5p copy gain; chr7p, 16q copy loss; DES overexpression; overexpression of FGF8, FGF9, FGFR4, ALK; PAX3-NCOA2 fusion; MYOG, MYOD1 overexpression (RMS markers) | Available Phase-I clinical trial for brain tumors or cytotoxic regimen directed at Medulloblastoma | Overexpression of FGF8, FGF9, FGFR4, ALK; PAX3-NCOA2 fusion; MYOG, MYOD1 overexpression (RMS markers) | Change in diagnosis, treatment plan | Yes | Change in treatment after sequencing to RMS therapy, remained in remission 6 months following change in management before progressing. | |
| 58 | PPB | Mediastinal Mass‡ (P) | Rearranged genome; TP53 homozygous deletion; MLL3 (G315S) mutation, CTNNB1 frame-shift deletion; moderate FGFR1, FGFR4 overexpression Somatic DICER1 (G1809R) point mutation- near hotspots |
DICER1 (p.E1788X) | Cytotoxic chemotherapy and genetic counseling | Germline DICER1 (p.E1788X) | Genetic counseling for DICER1 family of tumors | Yes | Family seen in genetics clinic for counseling for DICER1 family of tumors. |
| 68 | ATRT | Posterior Fossa‡ (P) FFPE | SMARCB1 frameshift deletion, (deletion of exon 2), LOH at SMARCB1 | No additional therapy | SMARCB1 frameshift deletion, (deletion of exon 2), LOH at SMARCB1 | CDK4/6 inhibitor (NCT01747876) | None required | Patient in clinical remission following chemotherapy. | |
| 69 | MBL | Cerebellum‡ (P) FFPE | Overexpression of PTCH1, PTCH2, GLI1, GLI2, MYCN (SHH subtype markers); overexpression of ERBB4, NTRK1, NTRK3; Sonic Hedgehog Pathway (SHH) activation | Palliative cytotoxic chemotherapy or Available Phase-I clinical trial for relapsed brain tumor | Sonic Hedgehog Pathway (SHH) activation | SHH inhibitor | No | No SHH inhibitors in clinical trial and no dosing information available for off label use in children. | |
| 70 | Ovarian Small Cell Carcinoma | Ovarian Tumor‡ (P) | SMARCA4 (p.T858K); WT1 overexpression | SMARCA4 (p.R979X) | Cytotoxic chemotherapy and genetic counseling | SMARCA4 (p.T858K), Germline SMARCA4 (p.R979X) | Genetic counseling for family members for ovarian tumors | Yes | Family seen in genetics clinic for counseling for ovarian tumors. |
| 81 | NBL | Right Kidney† (M) | Chr7, Chr17q copy gains; Chr11q, Chr1p copy losses; MYCN single copy gain; RHD, GSTM1 homozygous deletion; CCND1, NTRK1 overexpression; ALK (F1174L) hotspot mutation | Available Phase-I clinical trial for relapsed NBL | ALK (F1174L) hotspot mutation | ALK inhibitor (crizotinib) | Yes | Action taken; patient was treated with crizotinib but progressed after 2 months. | |
| 86 | IMFM | Neck Mass† (M) | NOTCH3, PDGFRB overexpression; Somatic PDGFRB (p.N666K) | PDGFRB (p.R561C) | No adjuvant therapy, no genetic counseling | Germline PDGFRB (p.R561C) | PDGFRB inhibitors (imatinib), family counseling | Yes | Family referred to genetics clinic for counseling. No actions at present on patient, in clinical remission following chemotherapy. Eligible for PDGFRB inhibitors in case of post-treatment. |
| 89 | High Grade Glioma | Brain Tumor‡ (P) | PDGFRA, MYC, PVT1, CHIC2, RBPJ, FGF2, ING4, ZNF384 amplification; LRP6-ETV6 fusion; PDGFRA, MYC, PVT1, CHIC2, RBPJ, FGF2, ING4, ZNF384 overexpression | Available Phase-I clinical trial for relapsed brain tumors | PDGFRA amplification | Pazopanib targeting PDGFRA | No | Patient died before starting targeted therapy. | |
| 91 | RCC | Left Renal Mass‡ (P) | CDKN2A/2B one copy loss; PPM1D frame-shift insertion (p.T506fs); ASPSCR1-TFE3 fusion | Sunitinib or Sorafennib or Pazopanib | ASPSCR1-TFE3 fusion | Pazopanib targeting TFE3 fusion | Yes | Patient on pazopanib with SD for 10 months. | |
| 93 | Omental mass | Panniculitis with infiltrate‡ (P) FFPE | DICER1, FGF7 overexpression | MITF (p.E318K), GJB1 (p.C179Y) | No adjuvant therapy or no genetic counseling | Germline MITF (p.E318K), | Genetic counseling for melanoma risk | Yes | Family genetic counseling for melanoma risk. Diagnosis of X-linked CMT confirmed |
| 94 | ERMS | Labia Mass‡ (P) | SMARCB1, BCR, UGT2B17 homozygous deletion; EZH2 copy loss; CDK8, FGF11 overexpression | TP53 (p.Y236X) | Palliative cytotoxic chemotherapy or Available Phase-I clinical trial for ERMS, Genetic counseling | SMARCB1 homozygous deletion, Germline TP53 (p.Y236X) | CDK4/6 inhibitor (NCT01747876), family genetic counseling for Li-Fraumani family tumors | Yes | Family genetic counseling confirmed Li-Fraumani syndrome. No actions could be taken; no CDK4/6 inhibitor available for ERMS clinical trials in children. |
| 95 | Ovarian Small Cell Carcinoma | Left Ovarian Mass‡ (P) FFPE | SMARCA4 p.K835fs, somatic | SMARCA4 p.Q415fs | Genetic counseling for family members for ovarian tumors | Germline SMARCA4 p.Q415fs | Genetic counseling for family members for ovarian tumors | Yes | Family seen in genetics clinic for counseling for ovarian tumors confirming SMARCA4mutation. |
| 102 | NPCA | Nasopharyngeal Mass‡ (P) FFPE | KRAS p.G12D mutation, BRAF p.G469E mutation | Palliative cytotoxic chemotherapy or Available Phase-I clinical trial for relapsed solid tumors | KRAS p.G12D mutation, BRAF p.G469E mutation | RAF or MEK inhibitor for BRAF p.G469E mutation | Yes | Patient on adjuvant RAF inhibitor for 6 months with no evaluable disease. | |
Tissues are fresh frozen unless indicated as FFPE= Formalin Fixed Paraffin Embedded Tissue; Tissue type abbreviations: P=Primary, M=Metastatic; gender abbreviations: F=Female, M=Male.
Bold Genomic findings: Potentially Actionable Findings (PAF), which can potentially impact patient’s diagnosis, risk stratification, management or informs patient/family of future risk for serious health condition including cancers.
Abbreviations: SS=Synovial Cell Sarcoma, PEcoma=Perivascular Epithelioid cell tumor, WT=Wilms’ Tumor, NBL=Neuroblastoma, ACC=Adrenocortical Carcinoma, ARMS=Alveolar Rhabdomyosarcoma, ERMS=Embronal Rhabdomyosarcoma, RMS=Rhabdomyosarcoma, IFS=Infantile Fibrosarcoma, OS=Osteosarcoma, PPB=Pleuropulmonaryblastoma, ATRT= Atypical Teratoid Rhabdoid Tumor of Brain, MBL=Medulloblastoma, IMFM=Infantile Myofibromatosis, RCC=Renal Cell Carcinoma, NPCA=Nasopharyngeal Carcinoma, LN=Lymph Node, indel=insertion/deletion, CNA=Copy Number Alterations
DNA paragroups are indicated by an asterisk (*) placed after the main haplogroup.
SD=Stable Disease, PR= Partial Remission. Patient disease status for solid tumors is evaluated by RECIST 1.1 (Response Evaluation Criteria in Solid Tumors).
Tissue used for sequencing was after relapse or refractory or progressive disease following treatment with surgery, chemotherapy, radiation or biologic therapy
Tissue used for sequencing was prior to starting any treatment
Actionable findings in pediatric hematological malignancies
Potentially actionable findings were identified in 54% (15/28) of patients with hematological malignancies (Table 2). In patient 3, a 9 year-old girl with precursor-B acute lymphoblastic leukemia (pre-B ALL) (Table-2, eTable 1), RNA sequencing revealed an actionable, cryptic gene fusion involving ETV6 and ABL1 (eFigure 2C) that was not detected by other standard diagnostic tests including cytogenetics and Fluorescence In Situ Hybridization (FISH) for BCR-ABL. As predicted19,20, pre-clinical in vitro assays on this patient’s primary leukemia cells demonstrated their sensitivity to imatinib, a tyrosine kinase inhibitor (eFigure 2E, F). With patient having failed all standard therapeutic options, she was started on imatinib and chemotherapy. She was unable to tolerate ongoing cytotoxic chemotherapy with imatinib and was treated with imatinib alone for most of her course. She maintained morphological, cytogenetic and molecular remission for 21 months with excellent quality of life on imatinib (Table 2, eTable 1).
Additional hematological malignancy patients with potentially actionable findings and clinical course are discussed in Table 2 and eTable 1, including a cryptic, actionable EBF1-PDGFRB fusion in a patient with refractory pre-B ALL and three patients with hematologic malignancies who all had actionable alterations in the FLT3 kinase detected by sequencing. Sorafenib has shown activity in patients with refractory leukemia with FLT3 alterations.21
Actionable ICS findings in pediatric solid tumors
We identified potentially actionable findings in 43% (27/63) patients with pediatric solid tumors (Table 3). Patient 43 is a 3 year-old girl originally diagnosed as infantile myofibromatosis and subsequently (by sequencing) as high-grade spindle cell sarcoma negative for the ETV6-NTRK3 fusion (Supplementary Appendix section IV, V).22 Her transcriptome analysis identified a novel in-frame fusion of LMNA-NTRK1, which preserves the functional tyrosine kinase domain of NTRK1 (eFigure 3B). While almost 90% of Infantile Fibrosarcoma (IFS) patients have the canonical ETV6-NTRK3 fusion, the LMNA-NTRK1 fusion reported in this index patient is functionally analogous. NTRK1 fusions in other cancers, including lung cancers, have shown sensitivity to crizotinib, an ALK and c-MET inhibitor.23,24 Discovery of the LMNA-NTRK1 fusion in our patient suggested the diagnosis of IFS and changed the management of this patient to oral crizotinib. Within six weeks of starting therapy, she achieved a partial remission and has since maintained a favorable response on crizotinib for greater than 8 months without major toxicity (Table 3, eFigure 3C, D).
The second solid tumor case example features patient 57, a 4 year-old girl who was diagnosed as medulloblastoma, and enrolled on the study at the time of her relapse. Our analysis identified a cryptic fusion between the PAX3 gene and NCOA2 gene (eFigure 3G) suggestive of a diagnosis of rhabdomyosarcoma (RMS).25,26,27 Intracranial RMS is an extremely rare diagnosis (≤0.1% of all intracranial tumors) with a poor prognosis.28 The diagnosis of RMS was confirmed by using RNA-Seq to evaluate the expression of genes associated with all four molecular subgroups of medulloblastoma29 and genes associated with RMS (i.e., myogenin, desmin, FGFR4). We detected extremely high expression of genes associated RMS and low expression for most medulloblastoma-lineage genes (eFigure 3H and supplementary Appendix section IV). Furthermore, the tumor stained strongly positive for myogenin confirming the diagnosis of RMS. The change in diagnosis for this patient resulted in a change of management as well (Table 3). Other solid tumors with PAFs are summarized in Table 3 and Appendix section IV.
Cancer-related incidental germline findings (IGF)
By default, patients enrolled in our study received information with regards to cancer-related IGF unless they opted out. Nine patients (10%) had significant incidental germline findings in our cohort, potentially impacting patients and other family members (Table 3). In four of these families, the history was unremarkable for a familial cancer syndrome, and they would never have been otherwise referred for cancer genetics counseling. All nine patients and families have since undergone formal counseling and genetic screening in our cancer genetics clinic. Specific mutations identified are listed in Table 2. These included mutations associated with established syndromes (DICER1 syndrome, Infantile Myofibromatosis, Li-Fraumeni syndrome, and SMARCA4 Related Small Cell Ovarian Cancer Hypercalcemic Type) and in more recently described cancer risk genes (BAP1, BARD1, HOXB13, and MITF) where cancer risk is less clearly defined. A case example of actionable germline findings was patient 21, a 17 year-old female with relapsed metastatic melanoma in whom the canonical BRAFV600E mutation was identified, as well as a germline truncation of the BRCA1-associated protein 1 (BAP1, pD567X) (Table 3 and Supplementary Appendix Section IV). BAP1 is a tumor suppressor gene implicated in proper BRCA1 function, and germline BAP1 mutations are implicated in cancer predisposition for malignant mesothelioma, atypical melanocytic tumors, uveal melanoma, and cutaneous melanoma.30 This patient had a family history of cancer, including in her mother who had ovarian cancer at 44 years of age. However, she was already seen in the cancer genetics clinic and was screened negative for BRCA gene mutations. The patient and her family agreed to be seen in our cancer genetics clinic again, this time for counseling and further testing for the BAP1 gene in family members.
Clinical actions based on integrative sequencing
Overall, our study revealed potentially actionable findings in tumor or germline in 46% (n=42) patients. Among patients with PAFs, we were able to act upon results in 23 of the 91 (25%) patients and families, including change in treatment in 14 (15%), genetic counseling for future cancer risk in 9 (10%) patients and both in 1(1%) patient. In 9/91 (10%) of these personalized clinical interventions resulted in ongoing partial clinical remission of 8–16 months duration or help sustain complete clinical remission for 6–21 months duration. while in 5 (5%) patients they were unsuccessful. All 9 (10%) patients and families with actionable incidental genetic findings (IGFs) agreed to formal genetic counseling and genetic screening. The primary reasons for not being able to act upon PAFs included a) patients in clinical remission on current therapy and the role of genomically informed adjuvant treatment to prevent relapse in these settings is not well established, and b) the treating physician felt no additional therapy was necessary. Other major reasons for not being able to take clinical actions based on molecular findings included, limited access to drugs, family/physician preference, or results being available too late in the clinical course to act (Table 2,3).
Overall landscape of molecular alterations in the cohort
As expected, recurrent driver gene fusions were more prevalent in the hematologic malignancies (57%) as compared to solid tumors (27%); and amongst solid tumors they were most prevalent in sarcomas (Figure 2, eFigure 7). All functional fusions discovered in our study are listed in eTable 5, 6 and eFigure 8. An overview of all the classes of aberrations identified in our cohort is shown in eTable 6. As 36% of patients exhibited a driving gene fusion, this indicates a potential role of including RNA-seq (i.e., transcriptome sequencing), in addition to whole exome analysis, in the work-up of individuals with cancer. Furthermore, the presence of actionable germline findings in 10% of patients suggests a role for matched normal sequencing and mandatory genetic counseling in the management children and young adults with cancer.
Figure 2. Summary results of the Peds-MiOncoSeq study.

A matrix representation of selected informative findings from the sequencing results from the Peds-MiOncoSeq cohort. Patients are characterized on the Y axis according to disease type. Molecular aberrations are indicated on the X axis and grouped according to type. The presence of specific mutations, insertion/deletions, amplification/deletions, and gene fusions are indicated by colored blocks. Color-coding of the blocks is indicated in the legend. Data represented in this figure are derived from all 91 patients with completed whole exome as well as transcriptome sequencing of tumors and exome sequencing of germline DNA. Only sequencing findings with biological significance are included. SNV, single nucleotide variant; indel: insertion/deletion.
DISCUSSION
To our knowledge, Peds-MiOncoSeq is the first prospective, observational case series exploring the feasibility of integrative clinical sequencing and its potential influence in clinical decision-making as well as in management of children and young adults with cancer. Through our study, we were able to identify actionable findings in 46% of patients and furthermore, we were able to take clinical actions in 25% of these patients. Overall, 10% of patients showed durable clinical responses and in another 10% of patients and their families, their care was influenced by germline results. We hope our experience will guide other clinical sequencing efforts and therefore have made our clinical protocols and consent documents available as part of this study (Supplementary Appendix).
Our approach facilitated clinical decision-making and enabled discovery opportunities. In order to balance the cost of sequencing, bioinformatics analysis, and likelihood of finding clinically actionable information, we chose to perform both exome and transcriptome sequencing but not whole genome sequencing. We generally achieved >150X coverage by whole exome analysis which allowed us to detect sub-clonal populations of approximately 10%. All of the PAFs we reported for this case series, we believe are clonal events. Higher depths of sequencing will be required to detect minor sub-clones which may impact disease progression and resistance mechanisms. Pediatric cancers have a well-known paucity of recurrent point mutations compared to adult tumors and RNA-seq provided valuable insights in our patients’ cancers, including structural variations leading to a new diagnosis (PAX3-NCOA2), novel gene fusions (NTRK1), and new treatment options (ETV6-ABL1, TFE3, and ALK).31–34 RNA-seq discoveries alone accounted for almost 20% of the actionable findings in our study, which would have been missed otherwise.
Our study also used germline sequencing, which led to about 10% of patients and families receiving formal genetic screening for familial cancer syndromes based on significant actionable incidental findings revealed by our study. Many of these families had no significant family history and would likely have not been referred to genetic counseling under routine clinical care. A majority of patients/families opted for disclosure of incidental genetic results (89%), which is consistent with other studies examining parent preferences for return of research results.35 We mandated genetic counseling as an integral part of the study, along with follow up in our cancer genetics clinic for significant IGFs. This was well received by our participants and will be important moving forward, as how to optimally inform families of the risks of clinical sequencing, is receiving increased attention from both bioethicists and empirical researchers.36–38 Pediatric cancers, particularly leukemias post-allogenic transplant, have the added challenge of deciphering somatic alterations in the background of donor-derived cells, best exemplified by patient 3 in our study.
A key feature of our study is that we employed multidisciplinary PMTBs, which discussed, critiqued, and deliberated on genomic findings as well as assessed the feasibility of pursuing actionable findings when applicable. We believe that the unique expertise assembled in our PMTB allowed it to not only deliberate on the scientific merit of actionable genomic findings, but also discuss possible logistical and ethical issues before sharing the existence of candidate clinical trials, potential off-label use of approved agents, and age-dependent dosing of agents with the treating team.
Finally, our study identified several findings, which warrant further characterization and may, in some cases, suggest novel directions for research in translational science and experimental therapeutics. Among the observations of interest were an ALK fusion in rhabdomyosarcoma, a new NTRK1 fusion in IFS, and a novel YAP-MAML2 fusion in meningioma. Our study also identified several patients with disruption of SWI/SWNF chromatin modifiers (ARID1A/B, SMARCB1, SMARCA4) and tumor suppressors CDKN2A/B and CDKN1B/C implicating these genes in the pathogenesis of a wide variety of pediatric tumors.
Our study had several limitations, many of which were inherent to the study design. A major limitation was the observational nature of the study without a control group, at a single academic institution, which limited our ability to ascertain if the study actually improved clinical outcome as compared to standard of care. In addition, several of the patients were sequenced at the time of relapse using original diagnostic material, which we realize is not ideal, as evolution of tumor genome in response to therapy, is well documented. Another limitation of our study was the non-availability of drugs for the pediatric population, either through clinical trials or for off-label use. This was especially true in very young patients, where formulations and dosing uncertainty created an additional barrier in using off-label agents. While this is not surprising given the smaller number of investigational agents and clinical trials available for pediatric patients, mostly available through major consortium, it nonetheless prohibited several patients from potentially benefiting from actionable sequencing findings for which there are drugs available for adults. In addition, in many of our patients, we identified aberrations in multiple pathways, which will likely require combining multiple targeted agents (+/− chemotherapy) in order to have a meaningful effect on clinical outcome.39 Finally, longer than expected sequencing turn-around time also limited our ability to take clinical actions in many cases. Improvements in turnaround time can be anticipated in the future through incorporation of rapid sequencing modes, newer streamlined library preparation and capture protocols, and the use of cloud based computing resources for higher throughput analyses. Together, these improvements should reduce turnaround time to two weeks or less.
CONCLUSION
In this single center case series of children and young adults with relapsed or refractory cancer, incorporation of data from integrative clinical sequencing into clinical management was feasible, revealed potentially actionable findings in 46% of patients, and was associated with change in treatment and family genetic counseling in a small proportion of patients. The lack of a control group limited our ability to judge whether better clinical outcomes were achieved compared to standard care.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by NIH Clinical Sequencing Exploratory Research (CSER) Award NIH 1UM1HG006508, the Prostate Cancer Foundation, Mr. Tim Wadham, Good Charity, Inc., and the Raymond and Eva Shapiro family. None of the sponsors played a role in the design; data collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. Funds from NIH Clinical Sequencing Exploratory Research (CSER) Award NIH 1UM1HG006508, were utilized for conduct of the study. A.M.C. is a Howard Hughes Medical Institute Investigator, an A. Alfred Taubman Scholar, and an American Cancer Society Professor.
Additional Contributions: We would like to acknowledge Rashmi Chugh, M.D., David Smith, M.D., Steven Pipe, M.D., Laurence Boxer, M.D., Hugh Garton, M.D., Elizabeth Lawlor, M.D., Ph.D., Erika Newman, M.D., James Geiger, M.D., Peter Ehrlich, M.D., Jyoti Athanikar, Ph.D., Rhonda Mcdougall, P.N.P., Marcia Leonard, P.N.P., Javed Siddiqui, M.S., Courtney Oliver, M.S., Ashley Carpenter, M.P.H., Lynda Hodges, M.A., Angela Stovall, M.A., Christine Brennan, B.S., Erica Rabban, B.S., Terrence Barrette, Christine Betts, Karen Giles, Pallavi Mohapatra, Xiaoxuan Dong for their help in the conduct of this study as well as in preparing this manuscript. All acknowledged above are affiliated with University of Michigan. Most importantly, the authors would like to recognize the enormous generosity and kindness of the pediatric oncology patients and families at C.S. Mott Children’s Hospital for participating in this study.
Footnotes
Author Contributions
Drs. Mody and Chinnaiyan had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Mody, Wu, Robinson, Chinnaiyan
Acquisition, analysis, and interpretation of the data: Mody, Wu, Lonigro, Cao, Vats, Asangani, Palanisamy, Dillman, Rabah, Kunju, Everett, Raymond, Frank, M.S. Ning, Wang, Stoffel, Innis, Roberts, Robertson, Yanik, Chamdin, Connelly, Choi, Harris, Kitko, Jasty Rao, and Chinnaiyan
Drafting of the manuscript: Mody, Wu, Roychowdhury, Prensner, Robinson, Chinnaiyan
Critical revision of the manuscript for important intellectual content: Mody, Roberts, Levine, Castle, Hutchinson, Talpaz, Wu, Roychowdhury, Prensner, Robinson, Chinnaiyan
Administrative, technical, or material support: Cao, Frank, Vats, Wu, Lonigro, Talpaz, Robinson, Chinnaiyan
Study supervision: Mody, Wu, Robinson, Chinnaiyan
Conflict of Interest: A.M.C. serves on the scientific advisory board of Paradigm Diagnostics which is a non-profit tumor sequencing company of the University of Michigan. Paradigm was not involved with the conduct of this study.
References
- 1.Gaynon PS, Angiolillo AL, Carroll WL, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a Children’s Oncology Group Report. Leukemia. 2010 Feb;24(2):285–297. doi: 10.1038/leu.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moroz V, Machin D, Faldum A, et al. Changes over three decades in outcome and the prognostic influence of age-at-diagnosis in young patients with neuroblastoma: a report from the International Neuroblastoma Risk Group Project. European journal of cancer. 2011 Mar;47(4):561–571. doi: 10.1016/j.ejca.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 3.Thiele CJ, Cohn SL. Genetically InFormed therapies–a “GIFT” for children with cancer. Clin Cancer Res. 2012 May 15;18(10):2735–2739. doi: 10.1158/1078-0432.CCR-11-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011 Feb 10;29(5):551–565. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oberlin O, Rey A, Lyden E, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008 May 10;26(14):2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.London WB, Castel V, Monclair T, et al. Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011 Aug 20;29(24):3286–3292. doi: 10.1200/JCO.2010.34.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012 Aug 14;22(2):153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010 Oct;11(10):685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 9.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009 Nov;41(11):1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington, DC: The National Academies Press; 2011. [PubMed] [Google Scholar]
- 11.Collins FS, Varmus H. A new initiative on precision medicine. The New England journal of medicine. 2015 Feb 26;372(9):793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Benavente CA, McEvoy J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012 Jan 19;481(7381):329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012 Mar;44(3):251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welch JS, Westervelt P, Ding L, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011 Apr 20;305(15):1577–1584. doi: 10.1001/jama.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011 Nov 30;3(111):111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. 07//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013 Dec;45(12):1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer discovery. 2013 Jun;3(6):636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien SG, Vieira SA, Connors S, et al. Transient response to imatinib mesylate (STI571) in a patient with the ETV6-ABL t(9;12) translocation. Blood. 2002 May 1;99(9):3465–3467. doi: 10.1182/blood.v99.9.3465. [DOI] [PubMed] [Google Scholar]
- 20.Nand R, Bryke C, Kroft SH, Divgi A, Bredeson C, Atallah E. Myeloproliferative disorder with eosinophilia and ETV6-ABL gene rearrangement: efficacy of second-generation tyrosine kinase inhibitors. Leuk Res. 2009 Aug;33(8):1144–1146. doi: 10.1016/j.leukres.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Widemann BC, Kim A, Fox E, et al. A phase I trial and pharmacokinetic study of sorafenib in children with refractory solid tumors or leukemias: a Children’s Oncology Group Phase I Consortium report. Clin Cancer Res. 2012 Nov 1;18(21):6011–6022. doi: 10.1158/1078-0432.CCR-11-3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourgeois JM, Knezevich SR, Mathers JA, Sorensen PH. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. The American journal of surgical pathology. 2000 Jul;24(7):937–946. doi: 10.1097/00000478-200007000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nature medicine. 2013 Nov;19(11):1469–1472. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui JJ, Tran-Dubeì M, Shen H, et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK) Journal of Medicinal Chemistry. 2011;54(18):6342–6363. doi: 10.1021/jm2007613. 2011/09/22. [DOI] [PubMed] [Google Scholar]
- 25.Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2002 Jun 1;20(11):2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 26.Mosquera JM, Sboner A, Zhang L, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes, chromosomes & cancer. 2013 Jun;52(6):538–550. doi: 10.1002/gcc.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sumegi J, Streblow R, Frayer RW, et al. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes, chromosomes & cancer. 2010 Mar;49(3):224–236. doi: 10.1002/gcc.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford R, Crockard HA, Isaacson PG. Primary rhabdomyosarcoma of the central nervous system: case report. Neurosurgery. 1985 Jul;17(1):101–104. doi: 10.1227/00006123-198507000-00019. [DOI] [PubMed] [Google Scholar]
- 29.Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011 Apr 10;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carbone M, Ferris LK, Baumann F, et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. Journal of translational medicine. 2012;10:179. doi: 10.1186/1479-5876-10-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011 Jan 28;331(6016):435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janeway KA, Place AE, Kieran MW, Harris MH. Future of clinical genomics in pediatric oncology. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013 May 20;31(15):1893–1903. doi: 10.1200/JCO.2012.46.8470. [DOI] [PubMed] [Google Scholar]
- 33.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013 Jul 11;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014 Jan 23;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez CV, Gao J, Strahlendorf C, et al. Providing research results to participants: attitudes and needs of adolescents and parents of children with cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009 Feb 20;27(6):878–883. doi: 10.1200/JCO.2008.18.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tabor HK, Brazg T, Crouch J, et al. Parent perspectives on pediatric genetic research and implications for genotype-driven research recruitment. Journal of empirical research on human research ethics: JERHRE. 2011 Dec;6(4):41–52. doi: 10.1525/jer.2011.6.4.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haga SB, O’Daniel JM, Tindall GM, Lipkus IR, Agans R. Public attitudes toward ancillary information revealed by pharmacogenetic testing under limited information conditions. Genet Med. 2011 Aug;13(8):723–728. doi: 10.1097/GIM.0b013e31821afcc0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilfond BS, Carpenter KJ. Incidental findings in pediatric research. The Journal of law, medicine & ethics: a journal of the American Society of Law, Medicine & Ethics. 2008;36(2):332–340. 213. doi: 10.1111/j.1748-720X.2008.00277.x. Summer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goltsov A, Langdon SP, Goltsov G, Harrison DJ, Bown J. Customizing the therapeutic response of signaling networks to promote antitumor responses by drug combinations. Front Oncol. 2014;4:13. doi: 10.3389/fonc.2014.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.