

Abstract
Multiple sclerosis (MS) is a heterogeneous disease that develops as an interplay between the immune system and environmental stimuli in genetically susceptible individuals. There is increasing evidence that viruses may play a role in MS pathogenesis acting as these environmental triggers. However, it is not known if any single virus is causal, or rather several viruses can act as triggers in disease development. Here, we review the association of different viruses to MS with an emphasis on two herpesviruses, Epstein-Barr virus (EBV) and human herpesvirus 6 (HHV-6). These two agents have generated the most impact during recent years as possible co-factors in MS disease development. The strongest argument for association of EBV with MS comes from the link between symptomatic infectious mononucleosis and MS and from seroepidemiological studies. In contrast to EBV, HHV-6 has been found significantly more often in MS plaques than in MS normal appearing white matter or non-MS brains and HHV-6 re-activation has been reported during MS clinical relapses. In this review we also suggest new strategies, including the development of new infectious animal models of MS and antiviral MS clinical trials, to elucidate roles of different viruses in the pathogenesis of this disease. Furthermore, we introduce the idea of using unbiased sequence-independent pathogen discovery methodologies, such as next generation sequencing, to study MS brain tissue or body fluids for detection of known viral sequences or potential novel viral agents.
Keywords: Multiple sclerosis, human herpesvirus 6, Epstein-Barr virus
INTRODUCTION
The infectious etiology of multiple sclerosis (MS) has been suspected for well over one hundred years and a large number of viruses as well as other infectious agents have been associated with the disease during this time. Association of a disease with a virus certainly does not imply causation and indeed most associated agents have failed to stand the test of time. For example, measles virus was tightly linked to MS in the past, but the introduction of measles vaccination had no apparent effect on the prevalence of MS [1]. However, accumulating evidence still suggests a role for environmental factors, such as viruses, in MS pathogenesis. There has been a re-emergence of the infectious theory in MS during recent years- not as a sole factor but as a part of a more complex etiology- in which both genes and environment play a role. The findings that genetic or epigenetic factors cannot fully explain the MS disease discordance in monozygotic twins [2], has again pointed to the importance of environmental factors in MS pathogenesis. The largest body of evidence during the last few years has accumulated around Epstein-Barr virus (EBV) and human herpesvirus 6 (HHV-6). Other associated agents include varicella-zoster virus (VZV), and human endogenous retroviruses (HERVs) to name a few. Still to date, none of the associated agents have proven to be causative.
The more we understand the mechanisms of MS disease pathogenesis, the clearer it has become that MS is truly a complex disease with genetic, environmental and immunological components. The extent of interplay between these different factors is under rigorous investigation. The heterogeneity of the disease might also suggest that MS is not caused or triggered by only one virus, but rather a more complex set of viral infections could act as triggers in genetically susceptible individuals [3]. Some of the questions that need to be explored include the extent of the interplay between MS risk genes, vitamin D levels, UV-light exposure, tobacco smoking and viruses- are there risk factors that when present can amplify the risk of MS or are all these risk factors independent? Answering these questions might lead to a greater understanding of possible mechanisms viruses could induce in MS. In addition, population based studies on the prevalence of different MS associated viruses throughout the world might reveal similarities with prevalence of the disease itself in certain regions, strengthening an association between suspected viruses and MS.
The purpose of this report is to review the latest research on infectious agents in MS, the main focus being EBV and HHV-6. The most important finding associating EBV to MS is a higher seroprevalence and higher titers of EBV antibodies in MS patients compared to age-matched controls. The findings in support of EBV are rather uniform in the literature while for HHV-6 there has been more variability in studies demonstrating higher HHV-6 antibody titers or prevalence in MS patients than controls. This probably reflects the lack of reliable, validated, and commercially available HHV-6 serological tests. Indeed, most of the serological data suggesting association between HHV-6 and MS has been produced using “home-brewed” methods that are more difficult to reproduce and require knowledge in assay development. By contrast, HHV-6 DNA, protein or RNA can be found from different body fluids and brain tissue from patients with MS in higher prevalence than in tissue from healthy controls, a finding that has not been consistently seen in EBV.
In addition to a review of viral association with MS, we want to suggest new strategies to evaluate the role of infectious agents in MS. These new strategies include development of new infectious animal models of MS, clinical trials with effective anti-viral drugs and highly sensitive and unbiased sequence identification strategies (e.g. panmicrobial microarrays and next generation sequencing methods to identify novel pathogens or confirm the presence of previously associated pathogens in MS patients).
POSSIBLE MECHANISMS ASSOCIATING MS WITH VIRUSES
Most of the data linking MS to virus(es) has been generated by studying either the presence of part of a virus, such as DNA, RNA or proteins, in the body fluids or tissue or the immune response to a virus. The immune response to viruses includes mainly studies of antibodies in serum or CSF in patients with MS compared to findings from those in appropriate control groups. T cell responses to different viruses have also been studied, although less intensively. The control patient groups used in different studies vary and healthy subjects are frequently used. However, healthy subjects might not be the most informative control group, but rather more closely related patient populations such as other inflammatory neurological disorders (OIND) should be considered. These studies have produced large numbers of MS-associated agents, but little is known about how viruses could actually could trigger or modulate the MS disease process. Animal models for MS have been developed, including experimental autoimmune encephalitis (EAE) and models for virus-induced demyelination such as Theiler’s murine encephalomyelitis virus induced demyelinating disease in mice. Animal models have helped research understand some mechanisms, like molecular mimicry and bystander activation, that might play a role in a possible virus-induced pathology in MS. Unfortunately, animal models for human viruses, e.g. EBV and HHV-6, which have been suggested to play a role in the pathogenesis of MS are not available. Most of the herpesviruses are strictly species specific and therefore the development of animal models to study these viruses has been difficult to advance.
DIRECT MECHANISM
Although the current view of MS pathogenesis highlights the role of immune cells, including T- and B cells, the direct death of myelin-producing oligodendrocytes or direct toxic effects of active infection to CNS cells cannot be ruled out. It is possible that immune activation and lymphocyte infiltration in the MS plaque is a secondary effect due to active viral infection in oligodendrocytes or possibly in other resident CNS cells. Direct viral infection of oligodendrocytes can indeed cause cell death and demyelination. JC virus, the causative agent of progressive multifocal leukoencephalopathy (PML), is a known example of human virus capable of infecting oligodendrocytes and causing demyelination. JC virus is a ubiquitous virus that is usually acquired in childhood. It is not entirely known why it causes PML in some individuals, although immunosuppression is a common factor among all patients with PML. Of note, a small fraction of MS patients treated with natalizumab, an anti-α4-integrin monoclonal antibody, has developed PML. The biology of JC virus latency and reactivation in PML is not well known.
A chronic viral infection of the CNS can cause disease and demyelination. In subacute sclerosing pancephalitis (SSPE), a chronic slow infection of the CNS by measles virus leads to inflammatory disease of the both gray and white matter. The measles virus has been identified in neurons, glial and immune cells in the CNS of patients with SSPE [4]. Viral infection of CNS cells other than oligodendrocytes can also cause demyelination, as in the case of canine distemper virus, which preferentially infects astrocytes in white matter [5] and infection of oligodendrocytes is actually a rare finding in canine distemper. HHV-6 has been found in MS plaques significantly more frequently than in control brains or MS normal appearing white matter (NAWM) [6, 7]. The expression of HHV-6 antigen has been observed mainly in oligodendrocytes and to some extent in astrocytes, but whether this is a cause or a consequence of MS plaque formation, is not known.
MOLECULAR MIMICRY
Molecular mimicry, when some of the pathogen proteins have homologous amino acid sequences with self proteins, has been suggested to be a possible mechanism for viral-induced autoimmunity [8]. Several viral sequences have been shown to be homologous to myelin proteins by homology searches [8]. Later, by searching structural requirements for T cell receptor recognition of MHC-bound peptides, several viral peptides that could not have been identified by homology searches were found to have structural similarities and were able to activate autoreactive T cells [9]. Although it is known that viral peptides mimicking self proteins can induce a disease [8], for a long time it was not known if viral infection can induce a disease through molecular mimicry. This was addressed by Zhao et al. [10] in a mouse model of autoimmune herpes stromal keratitis. The authors showed that autoreactive T cells recognized an HSV-1 protein, UL-6. Furthermore, the mutant virus lacking the epitope responsible for T cell recognition was not able to induce disease [10]. This was a clear demonstration that a viral infection was able to induce autoimmunity through a molecular mimicry mechanism.
An increased lymphoproliferative response to HHV-6A antigen has been suggested to occur in patients with MS as compared to controls [11]. The response to HHV-6B was similar in patients and controls. Furthermore, HHV-6 protein U24 shares a proline rich amino acid stretch (PRTPPPS) with myelin basic protein that could act as a molecular mimic. It was shown that more than 50% of the T cells recognizing MBP peptide cross-reacted and could be activated by HHV-6 U24 peptide in MS patients, therefore suggesting molecular mimicry [12]. The frequency of the subpopulation of T cells recognizing both peptides was significantly increased in patients with MS compared to healthy controls. There have also been reports of increased CD4+ and CD8+ T cell responses to EBV, particularly to EBNA-1, in patients with MS compared to controls [13–15]. A fraction of the EBNA-1 specific CD4+ T cells also recognized myelin antigens, suggesting molecular mimicry [16].
In addition to cross-reactive T cell responses between viral antigens and myelin antigens, cross-reactive antibodies between viruses and myelin antigens have been studied, although less intensively. In one report investigating CSF antibody specificity to over 37,000 tagged proteins, the two most frequently detected antigens were demonstrated to be EBV latency associated proteins, EBNA-1 and BRRF2 [13]. In another study, using combinatorial libraries expressing single-chain variable fragments cloned from PBMCs from patients with MS, Gabibov et al. [17] were able to identify cross-reactive antibodies that recognized MBP and EBV latent membrane protein 1 (LMP1). By contrast, others have produced monoclonal antibodies from CSF B cells of MS patients and have not found any putative autoantigens or infectious antigens [18].
BYSTANDER ACTIVATION AND EPITOPE SPREADING
The term epitope spreading was first used to describe a process in which a diverse immune response is generated against several epitopes of a single protein, although the initial immune response was initiated against a single dominant epitope of that protein [19]. In addition, many proteins contain cryptic epitopes that are hidden from immune system within intact protein, but can be revealed to the immune cells after dissociation. In chronic tissue damage as a result of viral infection, graft rejection, or autoimmune process, the specificity of the immune response spreads to include self epitopes different than the initial protein that initiated the inflammatory process [20]. Epitope spreading can include both self-reactive T and B cells, but is mostly studied in the context of autoreactive T cells.
In addition to their role as antibody producing cells, B cells can act as antigen presenting cells (APC). As a response to viral infection, APCs capture viral antigen and present viral peptides through MHC class II molecules to T cell receptors. B cell – CD4 T cell interaction is, in general, needed for abundant antibody production. During active infection, B cells present viral antigens to T cell receptors in the context of MHC class II. An additional interaction between B cell CD40 and T cell CD40L is needed for production of interleukin 2, 4 and 5 by T cells, which in turn activates B cells to proliferate and maturate into antibody producing plasma cells. It is possible that B cells in the CNS of MS patients might process and present infectious agents, and subsequently parts of infected cells or damaged surrounding tissue, as a result of epitope spreading to T cells (Fig. 1). In this way, autoimmunity might be promoted at both the B- and T cell level. In addition, if this B cell maturation is compartment specific i.e. only occurs in CNS, it could explain the presence of oligoclonal bands in MS. In MS, oligoclonal bands are mostly seen in CSF only, although in some MS cases the banding pattern includes serum bands as well. However, epitope spreading is difficult to study in humans, and in disorders such as MS the initiating antigen is usually not known. Thus, most data on epitope spreading comes from mouse models such as EAE and Theiler’s murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD).
Fig. 1.
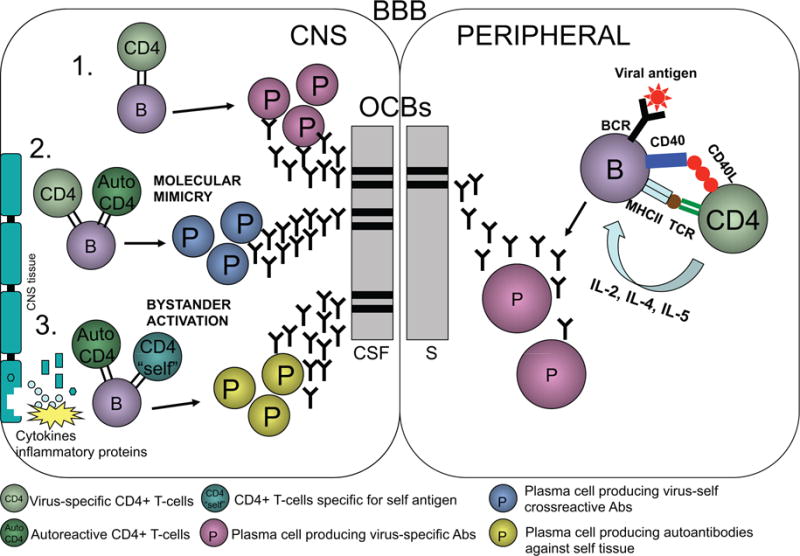
As a response to viral infection B cells can process and present viral peptides to T cells leading to activation of T cells and maturation of B cells into antibody producing plasma cells. These plasma cells can produce clonal IgG molecules that are present in serum during the active infection of the periphery. If virus invades the CNS, B cells can activate and maturate into plasma cells and produce antibodies in a mannar similar to in the periphery (1). The viral antigens might resemble myelin antigens and induce T cell as well as B cell responses including cross-reactive antibodies to viral as well as autoantigens through molecular mimicry mechanisms (2). Chronic viral infection can also cause local tissue damage leading to presentation of autoantigens, which are then recognized by autoreactive T or B cells (3). This leads to production of novel clones of T cells and plasma cells that recognize only autoantigens but not viral antigens that may have initiated the process. Any of these events occurring in the CNS could produce antibodies that are not seen in peripheral blood and therefore would be present in CSF as oligoclonal bands.
Using different myelin peptides as antigens it has been shown that epitope spreading, both intramolecular and intermolecular, is instrumental in disease development in mouse EAE. For example, SJL mice immunized with proteolipid protein (PLP) peptide 139–151 develop a relapsing remitting disease in which CD4+ T cells specific for PLP 139–151 appear in three days. Later, during the first relapse, CD4+ T cells specific for PLP 179–191 can be detected. During a second relapse, CD4+ T cell responses against MBP 84–104 can be demonstrated, representing both intramolecular and intermolecular epitope spreading [21]. Epitope spreading also plays a role in TMEV-IDD. Myelin destruction is initiated by myelin specific T cells that attack persistent virus [22]. After demyelination starts, CD4+ T cell responses to PLP 139–151 can be detected. Later in disease, CD4+ T cells specific for less encephalitogenic myelin peptides such as PLP 178–191, PLP 56–70 and MOG 92–106 are seen [23]. In MS, immune responses including B cell and T cell components could first be directed against an active or chronic viral infection and later diversify through molecular mimicry, bystander activation and/or epitope spreading against self tissue.
COMPLEX GENOME-VIROME INTERACTION
The severity of the viral infection depends on many different factors. Possibly most important is the interplay between virus and host immune mechanisms. Host immune mechanisms, like all traits, are influenced by genetics and may sometimes have an impact on the development of symptomatic disease as a response to infectious agents. As our knowledge of common microbial flora has grown, it has become clear that we have not only a common flora of bacteria but also a common flora of viruses, called the virome [24]. Our virome consists of common viruses such as endogenous retroviruses and herpesviruses. The role of commensal bacteria as part of human physiology is well established, but the role of the virome in human physiology and pathophysiology is poorly understood. It is known that microbes are needed to challenge and shape the immune system and more recently the role of ubiquitous viruses has been suggested to play a role in this process [24].
Although some believe that MS could be caused by a single virus [25], the clinical heterogeneity of MS and the diversity of MS plaques in the CNS [26] suggests that there might be more than one infectious agent in the pathogenesis of this disease [27]. Furthermore, the recognition of more than one infectious trigger in MS would, at least partially, explain the long list of viruses that have been associated with this disease (Table 1). MS is not the only complex human disease that has been suggested to develop as a result of genome-virome interaction. Similarly, genome-virome interactions have been suggested to play a role in type 1 diabetes, inflammatory bowel disease and asthma. These diseases could be induced by several different viral infections associated with host-virus interactions in a model in which common frequent viral infections may or may not cause symptoms [28].
Table 1.
Partial List of Viruses and the Year(s) they were Reported to be Associated with MS (Adopted from Johnson, 1994 [118])
| Virus | Years |
|---|---|
| Rabies | 1946, 1964 |
| Herpes simplex | 1964 |
| Scrapie agent | 1965 |
| MS-associated agent | 1962 |
| Parainfluenza virus 1 | 1972 |
| Measles | 1972 |
| Simian virus 5 | 1978 |
| Chimpanzee cytomegalovirus | 1979 |
| Coronavirus | 1980 |
| SIMON-like virus | 1982 |
| Tick borne encephalitis virus | 1982 |
| HTLV-1 | 1986 |
| MSRV (HERV-W) | 1989, 1997 |
| HSV-1 | 1989 |
| MS1533 (retrovirus) | 1994 |
| HHV-6 | 1993, 1995 |
| Borna virus | 1998 |
| EBV | 1998, 2003, 2007 |
| VZV | 2004 |
| Torque Teno virus | 2005 |
HERPESVIRUSES AND MULTIPLE SCLEROSIS
Several features of some common herpesviruses (EBV, HHV-6 and possibly VZV) make them interesting candidates to act as triggers in MS. These three viruses are ubiquitous, infecting almost all individuals in early years of life. Large epidemiological studies of human migration have suggested that MS susceptibility is acquired during infancy or childhood-favoring an early infection of a ubiquitous virus as a trigger. Herpesviruses remain in the body in an inactive latent form with periodic reactivations. All these viruses are neurotropic, but also neurovirulent i.e. they can infect CNS cells, and also cause CNS disease. Although it is possible that a virus triggering MS directly lyses cells in the CNS, it is more likely that MS is associated with an immunopathogenic host-immune response to the virus.
EPSTEIN – BARR VIRUS (EBV) AND MS
EBV (human herpesvirus 4) was isolated from lymphoblasts from patients with lymphoma. The primary infection of EBV can be asymptomatic or can cause mild disease. EBV is the causative agent of infectious mononucleosis. Close to 100% of the population in Western countries is infected with EBV and it remains latent in B cells [29] after primary infection. Although the early studies of EBV in MS date back to the early 70’s [30], the association between MS and EBV infection has strengthened during the last few years. Large-scale population based studies on EBV antibodies have shown consistently higher seroprevalence in patients with MS as compared to controls.
IMMUNE RESPONSE TO EBV IN MS
Both higher seroprevalence and higher EBV antibody titers have been reported in patients with MS compared to controls. There are three different major antigens that are generally used in EBV antibody studies: viral capsid antigen (VCA), EBV nuclear antigen (EBNA) complex (or part of it e.g. EBNA-1) and early antigen (EA). There is some general variation in results depending on which antigen was used. In a recent meta-analysis, Santiago et al. [31] reviewed the results from MS case-control studies of antibodies to different antigens separately and found odds ratios of 5.5 for VCA, 5.4 for EBNA complex, 12.1 for EBNA-1 and 1.3 for EA. Therefore, it seems that EBNA-1 is the most relevant marker of EBV infection in MS. In all of the studies presented in Table 2, MS patients had a higher seroprevalence than controls. In all but one study, the prevalence was close to 90–100% of MS patients and lower in controls. There are only two studies for anti-EBNA-1 antibodies in CSF that have reported higher prevalence of EBNA-1 antibodies. Although the prevalence of serum antibodies to VCA shows more variation than antibodies to EBNA-1 (Table 3), a higher VCA seroprevalence is still seen in patients with MS. In summary, the increased prevalence of antibodies to different EBV antigens in MS patients compared to controls is some of the most convincing evidence supporting the association of EBV with MS.
Table 2.
Prevalence of EBNA-1 Antibodies in Patients with MS Compared to Control Patients
| Sample | Reference | MS (%) | Control (%) |
|---|---|---|---|
| Serum IgG | |||
| Larsen et al., 1985 [119] | 100 | 83.9 | |
| Wandinger et al., 2000 [120] | 100 | 90.2 | |
| Munch et al., 1998 [121] | 99.3 | 89.9 | |
| Pohl et al., 2006 [122] | 92.5 | 57.5 | |
| Banwell et al., 2007 [123] | 57.9 | 56.3 | |
| Haahr et al., 2004 [124] | 100 | 94.3 | |
| Wagner et al., 2000 [125] | 100 | 90.2 | |
| Sundstrom et al., 2004 [126] | 99.6 | 95.3 | |
| Lindsey et al., 2010 [127] | 97.5 | 92.5 | |
| Ingram et al., 2010 [128] | 94.7 | 93.3 | |
| Villegas et al., 2011 [129] | 86.8 | 82.7 | |
| CSF IgG | |||
| Bray et al., 1992 [130] | 79.7 | 14.5 | |
| Villegas et al., 2011 [129] | 82 | 69 | |
Table 3.
Prevalence of EBV Viral Capsid Antigen (VCA) Antibodies in MS and Controls
| Sample | Reference | MS (%) | Control (%) |
|---|---|---|---|
| Serum IgG | |||
| Banwell et al., 2007 [123] | 85.7 | 63.5 | |
| Pohl et al., 2006 [122] | 98.6 | 72.1 | |
| Zivadinov et al., 2006 [131] | 95 | 100 | |
| Ponsonby et al., 2005 [132] | 100 | 96.5 | |
| Alotaibi et al., 2004 [133] | 83.3 | 56.7 | |
| Sundstrom et al., 2004 [126] | 100 | 98.7 | |
| Ascherio et al., 2001 [134] | 99.3 | 93.4 | |
| Myhr et al., 1998 [135] | 97.9 | 81.1 | |
| Shirodaria et al., 1987 [136] | 100 | 92.3 | |
| Bray et al., 1992 [130] | 57.7 | 24.8 | |
| Bray et al., 1983 [137] | 98.7 | 89.4 | |
| Sumaya et al., 1980 [138] | 98.7 | 93.8 | |
| Sumaya et al., 1976 [139] | 98.6 | 94.6 | |
IS EBV PRESENT IN MS BRAINS?
In addition to the increased seroprevalance of EBV in MS patients, some reports have also shown that the immune response, at least in a subset of patients, could be intrathecal. This suggests virus-specific responses within the CNS and thus indirectly suggests the presence of virus in this compartment [32]. This intrathecal response, however, has also been suggested to be part of the polyspecific response in MS [33]. Whether EBV is present in MS brains and particularly in the MS plaques is an important issue with conflicting reports in the literature. The data demonstrating the presence or absence of EBV in MS brains was reviewed and attempts to explain the reasons for the different published results were addressed by the NeuroproMiSe EBV Working Group [34]. Although the Group concluded that most of the controversies are due to the different specificities and sensitivities of the methods used in different studies, there are still controversial results in different reports using the same methods, i.e. in situ hybridization for EBER RNA [35, 36]. The small non-coding RNAs, EBERs, abundantly expressed in every latently infected cell serve as the gold standard for detection of EBV infection in situ. There is an agreement that EBER RNA can be detected in situ from EBV abundant tumor samples. The difference comes when looking at brain tissue samples that have low numbers, if any, of infected B cells. However, current data suggests that EBV is typically not present in MS brain plaques.
INFECTIOUS MONONUCLEOSIS AND MS
Primary EBV infection can be asymptomatic or present as infectious mononucleosis. In a recent meta-analysis [37], including 18 population-based or case-control studies, the role of infectious mononucleosis as a risk factor in MS was studied. The total number of MS cases included in the meta-analysis was 19,390, and 16,007 controls were studied as well. The relative risk determined in this meta-analysis was 2.17 (95% CI 1.97 – 2.39; p<10−54) confirming the results (RR 2.3) from previous meta-analysis done with less patients and controls [38]. It has been suggested that infectious mononucleosis increases the risk of MS and the risk persists for at least 30 years after infection [39]. It is poorly understood why symptomatic EBV infection increases the risk of MS compared to those with asymptomatic infection. One feasible explanation could be the hygiene theory [40]; populations in Western countries are in contact with less germs and therefore the immune system might start responding to self tissues that may lead to autoimmune disease. The hygiene hypothesis has been used to explain the recent increase in allergies and autoimmune diseases in developed countries. In fact, infectious mononucleosis is more common if EBV primary infection occurs later in life. This might suggest that individuals who acquire EBV later in life have also had less contact with other microbes and therefore, according to the hygiene theory, may be more prone to autoimmune diseases such as MS.
PRESENCE OF EBV DNA IN MULTIPLE SCLEROSIS
The presence of EBV DNA in different MS samples is summarized in Table 4. In general, EBV DNA is a rare finding in cell-free body fluids such as plasma or CSF. No differences have been reported in published studies on the prevalence of EBV DNA in plasma between MS patients and controls [41, 42]. Because sample sizes in these studies were small, it is hard to draw any definitive conclusions based on these reports. In addition, presence of EBV DNA in CSF is highly unusual and no differences are found between MS and control CSF. As EBV resides in B cells during latency, DNA is detected more often in PBMCs. Some investigators have found significant differences in the prevalence of EBV DNA in PBMCs between MS and controls [43], while others have not [44–46]. In conclusion, the findings of EBV DNA, other than in latent form in PBMCs, is rare and there seems to be no significant differences in the prevalence of EBV DNA detected by PCR between MS patients and controls.
Table 4.
Prevalence of EBV DNA in Patients with MS Compared to Controls
| Sample | Reference | MS (%) | Control (%) |
|---|---|---|---|
| Plasma DNA | |||
| Höllsberg et al., 2005 [42] | 15.2 | 5.5 | |
| Wagner et al., 2004 [41] | 29 | 16.1 | |
| PBMC DNA | |||
| Alvarez et al., 2000 [44] | 32.2 | 35.3 | |
| Ferrante et al., 2000 [45] | 50 | 38.9 | |
| Hay and Tenser, 2000 [46] | 100 | 100 | |
| Sotelo et al., 2007 [43] | 84.2 | 63.1 | |
| Brain DNA | |||
| Morré et al., 2001 [140] | 0 | 0 | |
| Sanders et al., 1996 [141] | 27 | 37.8 | |
| CSF DNA | |||
| Alvarez-Lafuente et al., 2008 [142] | 2.1 | 0 | |
| Denne et al., 2007 [143] | 0 | 0 | |
| Mancuso et al., 2007 [144] | 2.5 | 0 | |
| Martin et al., 1997 [145] | 0 | 0 | |
| Morre et al., 2001 [140] | 0 | 0 | |
HUMAN HERPESVIRUS 6 (HHV-6)
HHV-6 was first isolated from patients with AIDS and immunoproliferative syndromes in 1986 [47]. Two different types of HHV-6, HHV-6A and HHV-6B, have been identified and they differ from each other in biological, immunological and clinical features [48]. HHV-6B is acquired early in life, usually before the age of two or three. After primary infection, which can be asymptomatic or present as exanthema subitum (roseola infantum), the virus becomes latent and is primarily found in PBMCs for life. HHV-6B virus is a ubiquitous virus with seroprevalency approaching 100% in Western countries. Due to the lack of serological assays for detection of HHV-6A, the prevalence and acquisition time of HHV-6A is not known. The cross-reactive nature of HHV-6A and HHV-6B antibodies, which in vivo are not cross-protective since double infections can be found, have hindered the serological studies of HHV-6A. However, HHV-6A has been suggested to be more neurotropic than HHV-6B, based on more prevalent detection of HHV-6A in CSF than in PBMCs [49].
HHV-6 AND MS
Soon after its discovery in 1986 [47] preliminary reports suggested a possible association of HHV-6 in the pathogenesis of MS. Sola et al. [50] investigated serum antibody titers by IFA and viral DNA in PBMCs by PCR from 126 MS patients and 500 controls. Significantly higher serum antibody titers were found from patients with MS. In contrast, HHV-6 DNA was found only rarely from MS patients or control PBMCs and therefore it was concluded that high serum HHV-6 antibody titers might be a consequence of immune impairment rather than HHV-6 reactivation of a latent HHV-6 infection. In another study [51], HHV-6 DNA was detected in three CSF samples of 21 MS patients (14.3%), but not in patients with other neurological diseases (OND) including myalgic encephalitis, meningitis and chronic fatigue syndrome or in healthy controls. In this study, HHV-6 serum antibody titers as measured by ELISA were also higher in sera of patients with MS compared to OND or healthy controls, supporting the possible role of HHV-6 in the pathogenesis of MS. Increased antibody titers in serum to HHV-6 reported by Sola et al. [50] was confirmed by other investigators soon after the initial reports [51]. However, the later studies have shown far more variation in HHV-6 viral titers and prevalence of HHV-6 antibodies in MS and controls (Table 5). This may be explained by different patient and control populations, but also differences in serological assays used.
Table 5.
HHV-6 Antibody Findings in MS
| Sample | Reference | MS (%) | Control (%) |
|---|---|---|---|
| Serum IgG | Sola et al., 1993 [50] | 71 | 41 |
| Liedtke et al., 1995 [146] | 39 | 18 | |
| Soldan et al., 1997 [147] | 85 | 72 | |
| Ablashi et al., 1998 [148] | 69 | 28 | |
| Enbom et al., 1999 [149] | 100 | 100 | |
| Ablashi et al., 2000 [150] | 90 | 75 | |
| Taus et al., 2000 [151] | 30 | 25 | |
| Derfuss et al., 2005 [152] | 84 | 88 | |
| Virtanen et al., 2007 [153] | 100 | 69 | |
| Kuusisto et al., 2008 [154] | 88 | 86 | |
| Behzad-Behbahani et al., 2011 [155] | 100 | 73 | |
| Serum IgM | Liedtke et al., 1995 [146] | 3 | 2 |
| Soldan et al., 1997 [147] | 73 | 18 | |
| Ablashi et al., 1998 [148] | 56 | 19 | |
| Friedman et al., 1999 [56] | 80 | 16 | |
| Ablashi et al., 2000 [150] | 71 | 15 | |
| Enbom et al., 2000 [156] | 2 | NT | |
| Taus et al., 2000 [151] | 0 | 0 | |
| Riverol et al., 2007 [157] | 35 | 34 | |
| Kuusisto et al., 2008 [154] | 6 | 0 | |
| CSF IgG | Sola et al., 1993 [50] | 7 | NT |
| Wilborn et al., 1994 [51] | 0 | 0 | |
| Ablashi et al., 1998 [148] | 39 | 7 | |
| Friedman et al., 1999 [56] | 94 | 100 | |
| Ongradi et al., 1999 [158] | 43 (6A) 87 (6B) | 17 (6A) 0 (6B) | |
| Ablashi et al., 2000 [150] | 4 | NT | |
| Derfuss et al., 2005 [152] | 34 | 12 | |
| Virtanen et al., 2007 [153] | 15 | 0 | |
| Kuusisto et al., 2008 [154] | 0 | 0 |
The first direct evidence for the involvement of HHV-6 in the pathogenesis of MS was reported in 1995 [7]. Challoner and co-workers used representational difference analysis (RDA), introduced by Lisitsyn et al. [52]. RDA is a subtractive hybridization method that can be used to identify nucleic acid sequences that are unique to, or present in greater numbers in diseased compared to healthy tissue. The DNA content from MS brain tissue was compared to DNA from peripheral blood leukocytes of healthy donors. By RDA they were able to identify a DNA sequence of 341 bp from one out of five patients that was essentially identical to the HHV-6 gene encoding HHV-6 major DNA binding protein. Challoner extended these findings by detecting the presence of HHV-6 DNA in brain samples by nested PCR and found that HHV-6 DNA was present in 78% and 74% of MS cases and controls, respectively. Although the authors of the paper concluded that HHV-6, especially variant B is a commensal virus in the human brain, they also demonstrated HHV-6B antigen expression in MS plaques in oligodendrocytes, but not in control brains or in non-plaque regions in MS brains. Since the destruction of oligodendrocytes (leading to degradation of myelin) is a hallmark of MS, the studies suggested an association of HHV-6 with the etiology or pathogenesis of MS. To extend these observations further, Cermelli and others [53] conducted a study in which MS plaques were isolated by laser microdissection from brain samples and DNA was purified and used for detection of HHV-6 DNA by nested PCR. Controls included brain samples from normal appearing white matter (NAWM) from the same patients with MS and brain samples from patients with other neurological diseases and patients with other inflammatory non-neurological disorders. While the rate of HHV-6 DNA was similar in NAWM samples of MS patients and in control brains, the frequency of detection of HHV-6 DNA was significantly higher in MS plaques. Others have studied HHV-6 antigen expression in MS brain samples as well. Carrigan and others [54, 55] demonstrated HHV-6 antigen in eight out of 11 brain samples from patients with MS but not in any of the seven control brain samples. The result was confirmed by others [6, 56, 57], although Coates and Bell [58] were not able to identify HHV-6 antigen in any of the 23 brain samples from patients with MS (though they did successfully identify HHV-6 antigen in salivary gland tissue). Collectively, these studies suggest that while HHV-6 is found in human brains, it is found significantly more often within MS plaques. Whether this suggests that HHV-6 is a cause or consequence of MS plaque development is an unanswered question that needs to be addressed.
HHV-6 DNA findings in different sample materials from patients with MS compared to control groups also vary between studies (Table 6). In serum, DNA detection rates range from 0 to 83% and 0 to 53% in MS cases and controls, respectively. In CSF, the detection rates are from 0 to 78% and 0 to 20% in cases and controls, respectively. Few studies have reached a statistically significant difference between groups. In brain samples, the detection rates are higher than in serum or CSF and may suggest that HHV-6 activation is tissue-restricted and HHV-6 does not usually present as free viral particles in body fluids or does so only a short time during viral activation possibly at the time of relapse.
Table 6.
Detection of HHV-6 DNA in Patients with MS Compared to Control Patients
| Sample | Reference | MS (%) | Control (%) |
|---|---|---|---|
| Serum DNA | Wilborn et al., 1994 [51] | 0 | 0 |
| Martin et al., 1997 [145] | 0 | NT | |
| Soldan et al., 1997 [147] | 30 | 0 | |
| Fillet et al., 1998 [159] | 6 | 0 | |
| Goldberg et al., 1999 [160] | 4 | 0 | |
| Mirandola et al., 1999 [161] | 0 | 0 | |
| Tejada-Simon et al., 2002 [162] | 67 | 33 | |
| Al-Shammari et al., 2003 [163] | 0 | 0 | |
| Tejada-Simon et al., 2003 [12] | 83 | 55 | |
| Alvarez-Lafuente et al., 2006 [164] | 25 | 0 | |
| Virtanen, et al., 2007 [153] | 0 | 0 | |
| Kuusisto et al., 2008 [154] | 0 | 0 | |
| Ahram et al., 2009 [165] | 27 | 24 | |
| Behzad-Behbahani et al., 2011 [155] | 33 | 5 | |
| PBMC DNA | Sola et al., 1993 [50] | 3 | 4 |
| Torelli et al., 1995 [166] | 3 | 22 | |
| Merelli et al., 1997 [167] | 5 | 0 | |
| Mayne et al., 1998 [168] | 25 | 24 | |
| Rotola et al., 1999 [169] | 41 | 29 | |
| Ablashi et al., 2000 [150] | 75 | 60 | |
| Hay and Tenser, 2000 [46] | 7 | 14 | |
| Kim et al., 2000 [170] | 21 | 0 | |
| Rotola et al., 2000 [171] | 40 | 37 | |
| Taus et al., 2000 [151] | 14 | 0 | |
| Alvarez-Lafuente et al., 2002 [172] | 49 | 22 | |
| Alvarez-Lafuente et al., 2002 [173] | 53 | 30 | |
| Chapenko et al., 2003 [66] | 62 | 29 | |
| Alvarez-Lafuente et al., 2006 [164] | 81 | 30 | |
| Alvarez-Lafuente et al., 2007 [174] | RRMS 54 SPMS 38 |
30 | |
| CSF DNA | Wilborn et al., 1994 [51] | 14 | 0 |
| Liedtke et al., 1995 [146] | 11 | 5 | |
| Martin et al., 1997 [145] | 0 | 0 | |
| Ablashi et al., 1998 [148] | 17 | 0 | |
| Fillet et al., 1998 [159] | 6 | 0 | |
| Enbom et al., 1999 [149] | 6 | 6 | |
| Goldberg et al., 1999 [160] | 0 | 0 | |
| Mirandola et al., 1999 [161] | 0 | 0 | |
| Taus et al., 2000 [151] | 0 | 0 | |
| Tejada-Simon et al., 2002 [162] | 47 | 20 | |
| Cirone et al., 2002 [175] | 78 | NT | |
| Virtanen, et al., 2007 [153] | 0 | 0 | |
| Kuusisto et al., 2008 [154] | 0 | 0 | |
| Alvarez-Lafuente et al., 2008 [142] | 10 | 0 | |
| Mancuso et al., 2010 [176] | 2 | 0 | |
| Brain DNA | Challoner et al., 1995 [7] | 78 | 74 |
| Sanders et al., 1996 [141] | 57 | 38 | |
| Merelli et al., 1997 [167] | 0 | 50 | |
| Friedman et al., 1999 [56] | 36 | 14 | |
| Cermelli et al., 2003 [53] | 58 | 27 |
HHV-6 ACTIVATION DURING RELAPSES
The idea that viruses can cause MS relapses or that they are activated as an epiphenomenon during relapses has been known for a long time. Several common viruses have been associated with relapses and indeed as much as one third of all MS relapses have been associated with common transmissible pathogens [59–62]. Interestingly, vaccinations are not a risk factor for MS relapses or for disease development [63, 64] suggesting that immune activation alone does not affect the risk of relapses in MS.
HHV-6 active replication can be most reliably measured by detection of viral mRNA from infected cells. However, the presence of viral DNA in cell-free body fluids, such as serum, centrifuged CSF, and urine, is also indicative of viral replication, although HHV-6 DNA can be present in apparently cell-free material from lysed latently infected cells. In order to examine if HHV-6 is associated with exacerbations in MS, Berti et al. [65] collected 215 serum samples from 59 MS patients. Presence of HHV-6 DNA was studied using sensitive nested PCR techniques. HHV-6 DNA was found more frequently in serum samples from MS patients in clinical exacerbation compared to patients in clinical remission. Chapenko et al. [66] also found HHV-6 genomes by PCR in PBMCs of 61.5% patients with MS, which was significantly higher than that of patients with other neurological disorders (28.6%) or normal blood donors (28.7%). These studies suggested that there might be an active infection in some of the patients in relapse. In later studies [67] assessing 105 RRMS patients and 49 normal blood donors, viral RNA transcripts of U16/U17, U89/U90 and U94 genes were found from PBMCs of 17 patients with RRMS (16%), but not from controls. U94 gene RNA, the only known HHV-6 gene transcribed in latency [68], was found from nine additional MS patients and 3 controls suggesting latent virus in PBMCs. When these patients were further analyzed, seven out of 32 (22%) patients that had relapses were found to have active HHV-6 infection (U16/U17+, U89/U90+, U94+) and only one (3%) patient had latent HHV-6 infection (U16/U17−, U89/U90−, U94+). These results further support the role of HHV6 infection in relapses in at least a subset of MS patients.
VZV AND MS
The primary infection of VZV causes varicella (chicken pox) and reactivation in later life causes zoster (shingles). By the age of 15, 95% of people in developed countries have acquired the infection [69]. Marrie and Wolfson [70] analyzed published data on association of VZV and MS in a meta-analysis in 2001. Most of the seroepidemiological and case-control studies reviewed in the meta-analysis failed to show any correlation between VZV infection or varicella and MS. Some of the studies, however, have shown higher seroprevalence of VZV antibodies in CSF of MS patients [71, 72]. This probably reflects a polyspecific response to several neurotropic viruses, such as measles, rubella and VZV in MS. In addition, VZV has been suggested to be present in PBMCs of patients during clinical relapse by PCR [43, 73]. VZV viral particles were also observed by electron microscopy in CSF during relapse [74] although this was not confirmed by others [75]. In a nationwide, population-based study conducted in Taiwan, Kang et al., [76] followed 315,550 patients with herpes zoster and 946,650 randomly selected control patients for one year. The risk for development of MS was found to be 3.63-fold greater in the group with herpes zoster. This finding has to be confirmed in other populations. Although the association of VZV with MS is not as strong as in the case of EBV or HHV-6, this large-scale study might suggest that VZV reactivation could act also as a trigger in MS disease onset and warrants further studies to explore this association.
ENDOGENOUS RETROVIRUSES AND MS
Endogenous retroviruses entered into the human genome millions of years ago. In total, up to 8% of the whole human genome can consist of endogenous retroviral sequences. It is therefore not surprising from an evolutionary standpoint that these retroviral sequences, and possible proteins that are translated from these viral transcripts, are associated with both health and disease. Some endogenous viral sequences have the capability to affect host gene transcription or even transactivate other viruses. In contrast, several viruses have been shown to regulate or activate the transcription of endogenous retroviral genes, such as env gene. Furthermore, it has been suggested that several herpesviruses, such as VZV, HSV-1, EBV and HHV-6, might activate HERV-W retroviral elements [77].
Reverse transcriptase activity and retroviral particles were identified in leptomeningeal cell lines from patients with MS [78]. It was first thought to be related to, but distinct from known T cell lymphotropic viruses (HTLV), thus explaining the similarities between HTLV-1 associated myelopathy / tropical spastic paraparesis (HAM/TSP) and MS [79]. Later characterization of the viral elements, however, proved it to be endogenous, not a known exogenous retrovirus [80–82], and was first called MS associated retroviral element (MSRV). Later the virus was identified as a new family of HERVs, HERV-W [83]. The presence of HERV-W is more prevalent in patients with MS than in controls [84] and the presence of HERV-W RNA in CSF of patients with MS has been suggested to be a short-term [85] as well as long-term [86] clinical prognostic marker of the disease.
HERVS AND NEUROINFLAMMATION
How could HERVs then play a role in the pathogenesis of MS? HERV-W env gene encodes a protein termed syncytin-1 that has been more often expressed in MS than in control brains [87]. Syncytin-1 is found in astrocytes, perivascular macrophages and activated microglia [88]. It promotes cytokine expression and release of reactive oxygen species in astrocytes that leads to oligodendrocyte damage. It also might induce innate immunity through Toll-like receptor 4 (TLR-4), which leads to the release of proinflammatory cytokines [89]. Syncytin-1 can also cause endoplasmic reticulum stress in astrocytes [90]. These events initiated by syncytin-1 (possibly increased from over expression of HERV-W in MS compared to controls) could possibly promote neuroinflammation in developing MS plaques.
MEASLES IN MS
Both measles virus (paramyxovirus) and rubella virus (togavirus) can cause demyelinating disease of the CNS. Measles is the cause of subacute sclerosing panencephalitis (SSPE) and rubella is the cause of progressive rubella panencephalitis. Increased measles antibody levels in serum and higher frequency of antibodies in CSF was detected more often in patients with MS than controls [91]. Attemps to detect measles RNA in MS blood [92] and brain tissue [93] have been unsuccessful. Although MS patients vaccinated against measles virus have measles antibodies in CSF, MS patients with natural measles infection have higher serum and CSF measles antibodies than those that have been vaccinated [94].
INTRATHECAL ANTIBODY RESPONSE TO VIRUSES – SPECIFIC OR POLYSPECIFIC
The only laboratory marker in the clinical diagnosis of MS is abnormal intrathecal IgG production. There are two methods that have been used for detection of intrathecal IgG antibody in MS; measurement of IgG index and detection of oligoclonal bands. The IgG index is calculated by determining CSF and serum IgG ratios that have been corrected for albumin concentrations in CSF and serum. Oligoclonal bands are detected using isoelectric focusing methods to separate different clones of IgG in CSF and serum. Separated IgG molecules are then detected by immunofixation or immunoblotting. Matched CSF and serum samples are run in parallel and positive findings are defined by two or more distinct IgG bands found in CSF but not in serum. Neither of these techniques is specific for MS and can be demonstrated in CSF from other inflammatory diseases of the CNS. The IgG index measurement is quantitative while detection of oligoclonal bands is qualitative. In general, polyspecific IgG production within the CNS increases IgG index, but does not affect the quantity of oligoclonal bands. Oligoclonal bands are considered more of a marker of clonal antigen-specific activation of different B cell clones. Therefore, it is reasonable to suggest that CSF oligoclonal responses should target the disease relevant antigen.
As mentioned, oligoclonal bands are not specific for MS and are seen in other inflammatory, mostly infectious, diseases of the CNS. Importantly, in these diseases it has been shown that oligoclonal bands are specific for the causative virus. These diseases include, but are not restricted to; SSPE, in which the oligoclonal bands are specific for measles virus, HTLV-1 associated neurological disease (HAM/TSP), in which the oligoclonal bands are specific for HTLV-1, and herpes simplex encephalitis, in which the oligoclonal bands are specific for HSV-1. Knowing that oligoclonal bands are typically found from patients with infectious diseases of the CNS, it is tempting to suggest that oligoclonal bands may be a biomarker for an infectious trigger. Gilden has suggested: “if EBV or any other virus causes MS, it should be possible to demonstrate that MS OGBs contain antibody directed against the suspected agent” [95]. Several attempts have been made to identify infectious agents responsible for oligoclonal band formation in MS. For example, it has been shown that some of the oligoclonal bands are specific for Chlamydia pneumoniae [96, 97], EBV [13, 98] and HHV-6 [99].
In our recent studies, we also found that a proportion (approximately one third) of patients with MS had either EBV or HHV-6 specific oligoclonal bands in CSF (Manuscript submitted). One intriguing finding is the observation that oligoclonal bands in MS remain the same during MS disease development and progression, even when the B cells are depleted by anti-CD20 (rituximab) treatment [100, 101]. In our study we also assessed longitudinal CSF samples and have demonstrated that HHV-6-specific OCBs, similar to total IgG OCBs, remained the same over time. In addition, we found that patients with herpesvirus specific oligoclonal bands, either EBV or HHV-6, had fewer contrast enhancing MRI lesions. This finding might suggest that constant intrathecal antibody production against herpesviruses, presenting as virus-specific OCBs in CSF, may control the virus within the CNS. Patients who do not have a strong intrathecal antibody response to virus might fail to control virus, which could lead to viral activation and increased CNS damage reflecting increased number of contrast enhancing lesions. This is consistent with the finding that patients in relapse are found to have more active HHV-6 infections [65–67].
INFECTIOUS ANIMAL MODELS OF MS
Experimental autoimmune encephalomyelitis (EAE) is the most commonly used animal model of MS. It is induced, most commonly in mice, by the use of myelin antigens or myelin peptides with the aid of adjuvant. The disease varies with the animal strain and myelin epitopes being used. The hallmark of the disease is infiltration of myelin specific Th1 CD4+ T cells into the CNS. The presence of Th1 CD4+ T cells has been shown to be an important feature in the disease induction since adoptive transfer of myelin-specific CD4+ T cells is sufficient to induce the disease [102]. In monophasic EAE, the clearing of inflammatory infiltrates from the CNS is associated with disease recovery. First attempts to create an infectious demyelinating mouse model induced by molecular mimicry included a recombinant vaccinia virus expressing proteolipid protein [103]. Although disease was not induced by infection with this virus, infected animals later developed a more severe EAE when encephalitogenic myelin peptides were introduced to animals. It is obvious that much has been learned from EAE about how autoimmunity could play a part in MS and EAE has served as a useful preclinical model for MS clinical therapies. However, if an infectious component plays a role in MS, then EAE may not be appropriate. In addition to EAE, there are some infectious mouse models of demyelination that have been studied.
THEILER’S MURINE ENCEPHALOMYELITIS VIRUS-INDUCED DEMYELINATING DISEASE OF THE CNS
TMEV, a mouse picorna virus, is a natural pathogen of mice. It usually infects the gastrointestinal tract, but is also capable of causing demyelination within the CNS. The ability to induce a demyelinating disease depends on the virus strain and genetic background of the mouse. Two groups of TMEV have been identified; one causing acute encephalitis and the other causing chronic progressive demyelinating disease primarily in the spinal cord. Intracerebral infection with viruses that cause demyelinating disease leads to persistent infection of the CNS. The main cause of the demyelinating disease, however, is the infiltration of immune cells into the CNS. In general, TMEV induced demyelinating disease progresses more slowly than EAE. Also, the humoral response seems to be much stronger in TMEV-induced demyelination and in MS than in EAE [104].
JAPANESE MACAQUE ENCEPHALOMYELITIS
Recently, an interesting report of spontaneous CNS disease resembling MS in Japanese macaques was published [105]. The disease, Japanese macaque encephalomyelitis (JME), appeared in the Oregon National Primate Research Center 21 years after the establishment of the colony. Axthelm et al. [105] reported that the disease has typically affected 1 to 3 % of the colony each year since the initial case was observed. The clinical symptoms of JME include paralysis, ataxia, and ocular motor paresis. Generally the disease appears in young adult primates, but cases of juvenile and older animals have been seen as well. Although the initial onset of the disease is often severe and JME animals usually fail to recover, a few animals have recovered and been placed back in the colony. All of the replaced animals have since had relapses. Pathologically, animals with JME have multifocal areas of demyelination and oligodendrogliosis together with some axonal loss. In affected areas including, cerebrum, cerebellum, brainstem and spinal cord, cellular infiltration of macrophages and lymphocytes was observed.
Interestingly, when white matter lesions from animals with JME were cultured over primary rhesus fibroblasts, an infectious agent was identified. In electron microscopic analysis it appeared as a herpesvirus and was further identified in sequence analysis to be a novel gamma-2 herpesvirus, called JM rhadinovirus (JMRV). It is most closely related to a rhesus macaque rhadinovirus (RRV). To prove causation, further research is needed. Intriguingly, JMRV has been isolated from active CNS lesions in 5 different macaques with JME, but not from normal appearing white matter from macaques with or without JME. Although JMRV is associated with JME, nothing is yet known of the prevalence of JMRV and its biology or possible pathogenetic mechanisms associating this virus to JME. In summary, JME as a model of MS has great potential to shed new light on the possible role herpesviruses may have in the pathogenesis of MS and other demyelinating diseases of the CNS.
MARMOSET AS A MODEL OF HHV-6 INDUCED NEUROLOGICAL DISEASE
Mouse EAE has several differences compared to human MS. Common marmosets are more closely related to humans in terms of genetics and immunology [106]. Another major advantage of using common marmosets in EAE studies is that brain lesions in EAE marmosets can be visualized with similar MRI techniques as used in the clinical setting. EAE can be induced with white matter homogenate, myelin proteins, or myelin peptides together with adjuvant.
HHV-6 uses CD46 as a cellular receptor for viral entry into the cell. Since mice lack this molecule, HHV-6 does not establish infection in mice. Transgenic mice expressing CD46 have had difficulties supporting HHV-6 infection, which might indicate that there are other co-receptors not expressed in mice as well. However, common marmosets express CD46 and are able to support HHV-6 infection. The possible role of HHV-6, not just in MS but more generally in neurological diseases such as temporal lobe epilepsy [107, 108] and encephalitis [109], reinforces the need for an animal model for this virus. As mentioned, the marmoset will also be an ideal animal model since MR imaging can monitor their brains. HHV-6A but not HHV-6B has been reported to cause a neurological disease in marmosets (Genain et al., 2008, Baltimore, 6th International Conference on HHV-6 & 7). The route of HHV-6 infection has also been shown to have differential effects on the immune response and viral persistence in marmosets (Leibovitch et al., 2011, Amsterdam, ECTRIMS). We are optimistic that developments in this field will help to clarify the role of HHV-6 in disease as well as understand the basic biology and immunology involved in HHV-6 infection.
CLINICAL TRIALS WITH ANTIVIRALS IN MS
It is clear that biomarkers will be needed to monitor MS disease progression and MS patient selection for medical therapy. The disease course of MS is unpredictable and varying disease progressions between patients make clinical trial design challenging. Therefore, it is clear that better patient stratification methods are needed. For example, it might be that only a subset of patients might benefit from antiviral therapy, which highlights the role of monitoring viral markers before, during, and after anti-viral therapy in MS in addition to clinical markers such as MRI and occurrence of relapses. Furthermore, it is possible that associated viral infection occurs years before the clinical onset of the disease and thus anti-viral therapy is not beneficial in MS. However, if viral infection is associated with relapses, anti-viral therapy might reduce the relapse rate.
There have been a few antiviral clinical trials in MS using anti-herpesvirus drugs. Studies reported by Bech et al., [110] in Scandinavia and Friedman et al., [111] in the USA evaluated the effect of valacyclovir therapy, an oral form of acyclovir, in randomized, double-blind, placebo-controlled trials in MS. The primary endpoint in the Scandinavian study was the number of new active MRI-evident lesions over 24 weeks of treatment and progression of the disease in the American study. Both studies failed to meet the primary endpoints. However, Bech and others [110] found in subgroup analysis that valacyclovir treatment was associated with reduced amount of new active lesions in patients with high MRI activity, determined by more than one active lesion at baseline. Friedman and others [111] concluded that there were trends (but not statistically significant) toward drug effect over placebo in the severe clinical category. It is noteworthy to mention that acyclovir is not effective against HHV-6 in vitro [112] or in vivo [42]. If active HHV6 was indeed present only in a subset of MS patients, then acyclovir would not be the drug of choice. Antiviral clinical trials might be the best (only) way to address the question of whether or not viruses play a role in MS, if the virus is active during MS disease course. However, to prove or disprove this hypothesis we need well-designed effective, antiviral clinical trials with safe, efficient and CNS-penetrable antiviral drugs. Future studies should include virologic and immunologic stratification of patients before therapy to assess clinical and laboratory outcomes [113].
PATHOGEN DISCOVERY IN MS
Until recently, the majority of surveillance and pathogen discovery efforts have relied heavily upon prior knowledge of the nucleotide sequence of known agents [114]. Prior techniques have amplified target sequences using known primer sequences with competitive PCR and subsequent microarray analysis, which then directs the search for a specific agent in question [115]. This strategy adopts an obvious level of bias when looking for pathogens as the causative agents for disease and fails to identify new pathogens and those that have diverged significantly from their related ancestors due to the lack of annealing of the specific primer sequence [114].
During the past few decades, sequence independent nucleic acid amplifications have been developed. The goal of these methods is to amplify DNA or RNA sequences that are found only from diseased samples, or in much higher levels from diseased samples compared to healthy tissue, and to further identify these sequences. Indeed, HHV-6 sequences were found from the MS brain tissue using an unbiased technique called representational differential analysis that is a hybridization-based subtraction method for identification of differential DNA fragments between samples [7]. This is one way of identifying “new” infectious agents that might be associated with MS. Other approaches to identify new infectious agents in MS include the use of pathogen-microarrays [116] or sequencing by next generation sequencing platforms [117]. Both of these methods have been highly successful in identifying new viruses in healthy and diseased samples. Despite the obvious power of these novel techniques in identification of new pathogens, the identification of novel viruses has been more successful in acute diseases upon active replication of the virus such as in hemorrhagic or upper respiratory infections. By contrast, MS is a chronic disease of the CNS that has significant limitations; (i) if viral infection is a trigger of MS it is most likely a low level persistent (or latent) infection (ii) the selection of appropriate sample material to be used in pathogen discovery in MS is difficult since brain tissue cannot usually be obtained in early disease. Attempts to identify possible triggers should include active lesions where the triggering agent most likely would still be present. Along these lines (and taking into account the unbiased nature of these novel approaches) the pathogen discovery approach to identify novel agents and/or confirm the presence of previously associated agents in MS brains and body fluids is promising.
FINAL REMARKS
In this review we have discussed the role of infectious agents, mainly viruses, in MS. Despite evidence of the association between MS and several viruses, no virus to date has been proven to be the cause of this neurological disease. Recently, compelling evidence has focused on members of the herpesvirus family, namely EBV and HHV-6. Since these viruses are ubiquitous, it presents unique challenges in establishing causation with this (or any) disease. The isolation of the presumptive agent from MS disease tissue such as active plaques within the CNS and increased humoral and cellular immune responses to these viruses in peripheral blood are strong arguments in support of these viruses as triggers in the MS disease process. Our recent detection of herpesvirus-specific OCBs from MS patients and the establishment of a novel HHV-6 non human primate model of infection also lends support that these viruses may play a role in the pathogenesis of this disease. Ultimately, only through well-controlled antiviral treatment trials can the causative versus consequential impact that these viruses have in MS ever be established.
Acknowledgments
We would like to thank Anna Abrams for her assistance in preparation of this review.
ABBREVIATIONS
- APC
Antigen presenting cell
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- EA
Early antigen
- EAE
Experimental autoimmune encephalitis
- EBER
EBV encoded RNA
- EBNA
EBV nuclear antigen
- EBV
Epstein – Barr virus
- HAM/TSP
HTLV-1 associated myelopathy / tropical spastic paraparesis
- HERV
Human endogenous retrovirus
- HHV-6
Human herpesvirus 6
- HSV-1
Herpes simplex virus 1
- HTLV
Human T-cell lymphotropic virus
- JME
Japanese macaque encephalomyelitis
- JMRV
Japanese macaque rhadinovirus
- LMP-1
Latent membrane protein 1
- MHC
Major histocampatibility complex
- MBP
Myelin basic protein
- MOG
Myelin oligodendrocyte glycoprotein
- MSRV
MS associated retroviral element
- NAWM
Normal appearing white matter
- OIND
Other inflammatory neurological disease
- OND
Other neurological disease
- PBMC
Peripheral blood mononuclear cell
- PLP
Proteolipid protein
- PML
Progressive multifocal leukoencephalopathy
- RDA
Representational difference analysis
- RRV
Rhesus macaque rhadinovirus
- SSPE
Subacute sclerosing panencephalitis
- TLR
Toll-like receptor
- TMEV-IDD
Theiler’s murine encephalomyelitis virus induced demyelinating disease
- VCA
Viral capsid antigen
- VZV
Varicella – Zoster virus
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
References
- 1.Ahlgren C, Oden A, Toren K, Andersen O. Multiple sclerosis incidence in the era of measles-mumps-rubella mass vaccinations. Acta Neurol Scand. 2009;119:313–320. doi: 10.1111/j.1600-0404.2008.01131.x. [DOI] [PubMed] [Google Scholar]
- 2.Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, Zhang L, Farmer AD, Bell CJ, Kim RW, May GD, Woodward JE, Caillier SJ, McElroy JP, Gomez R, Pando MJ, Clendenen LE, Ganusova EE, Schilkey FD, Ramaraj T, Khan OA, Huntley JJ, Luo S, Kwok PY, Wu TD, Schroth GP, Oksenberg JR, Hauser SL, Kingsmore SF. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krone B, Grange JM. Multiple sclerosis: are protective immune mechanisms compromised by a complex infectious background? Autoimmune Dis. 2011;2011:708–750. doi: 10.4061/2011/708750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isaacson SH, Asher DM, Godec MS, Gibbs CJ, Jr, Gajdusek DC. Widespread, restricted low-level measles virus infection of brain in a case of subacute sclerosing panencephalitis. Acta Neuropathol. 1996;91:135–139. doi: 10.1007/s004010050404. [DOI] [PubMed] [Google Scholar]
- 5.Mutinelli F, Vandevelde M, Griot C, Richard A. Astrocytic infection in canine distemper virus-induced demyelination. Acta Neuropathol. 1989;77:333–335. doi: 10.1007/BF00687587. [DOI] [PubMed] [Google Scholar]
- 6.Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J Infect Dis. 2003;187:1365–1376. doi: 10.1086/368172. [DOI] [PubMed] [Google Scholar]
- 7.Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci USA. 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 9.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao ZS, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344–1347. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 11.Soldan SS, Leist TP, Juhng KN, McFarland HF, Jacobson S. Increased lymphoproliferative response to human herpesvirus type 6A variant in multiple sclerosis patients. Ann Neurol. 2000;47:306–313. [PubMed] [Google Scholar]
- 12.Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol. 2003;53:189–197. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- 13.Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Bussow K, Sommer N, Hemmer B. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115:1352–1360. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Munz C, Martin R. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain. 2006;129:1493–1506. doi: 10.1093/brain/awl067. [DOI] [PubMed] [Google Scholar]
- 15.Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, Kleeberg J, Le Goff G, Pantaleo G, Du Pasquier RA. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain. 2008;131:1712–1721. doi: 10.1093/brain/awn108. [DOI] [PubMed] [Google Scholar]
- 16.Lunemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, Münz C. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. 2008;205:1763–1773. doi: 10.1084/jem.20072397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabibov AG, Belogurov AA, Jr, Lomakin YA, Zakharova MY, Avakyan ME, Dubrovskaya VV, Smirnov IV, Ivanov AS, Molnar AA, Gurtsevitch VE, Diduk SV, Smirnova KV, Avalle B, Sharanova SN, Tramontano A, Friboulet A, Boyko AN, Ponomarenko NA, Tikunova NV. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J. 2011;25(12):4211–4221. doi: 10.1096/fj.11-190769. [DOI] [PubMed] [Google Scholar]
- 18.Yu X, Gilden DH, Ritchie AM, Burgoon MP, Keays KM, Owens GP. Specificity of recombinant antibodies generated from multiple sclerosis cerebrospinal fluid probed with a random peptide library. J Neuroimmunol. 2006;172:121–131. doi: 10.1016/j.jneuroim.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 20.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 21.McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karpus WJ, Pope JG, Peterson JD, Dal Canto MC, Miller SD. Inhibition of Theiler’s virus-mediated demyelination by peripheral immune tolerance induction. J Immunol. 1995;155:947–957. [PubMed] [Google Scholar]
- 23.Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, Katz-Levy Y, Carrizosa A, Kim BS. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 24.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 25.Lipton HL, Liang Z, Hertzler S, Son KN. A specific viral cause of multiple sclerosis: one virus, one disease. Ann Neurol. 2007;61:514–523. doi: 10.1002/ana.21116. [DOI] [PubMed] [Google Scholar]
- 26.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 27.Steiner I, Sriram S. The “one virus, one disease” model of multiple sclerosis is too constraining. Ann Neurol. 2007;62:529. doi: 10.1002/ana.21233. author reply 529. [DOI] [PubMed] [Google Scholar]
- 28.Foxman EF, Iwasaki A. Genome-virome interactions: examining the role of common viral infections in complex disease. Nat Rev Microbiol. 2011;9:254–264. doi: 10.1038/nrmicro2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 30.Nikoskelainen J, Panelius M, Salmi A. E.B. virus and multiple sclerosis. Br Med J. 1972;4:111. doi: 10.1136/bmj.4.5832.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santiago O, Gutierrez J, Sorlozano A, de Dios Luna J, Villegas E, Fernandez O. Relation between Epstein-Barr virus and multiple sclerosis: analytic study of scientific production. Eur J Clin Microbiol Infect Dis. 2010;29:857–866. doi: 10.1007/s10096-010-0940-0. [DOI] [PubMed] [Google Scholar]
- 32.Castellazzi M, Tamborino C, Cani A, Negri E, Baldi E, Seraceni S, Tola MR, Granieri E, Contini C, Fainardi E. Epstein-Barr virus-specific antibody response in cerebrospinal fluid and serum of patients with multiple sclerosis. Mult Scler. 2010;16:883–887. doi: 10.1177/1352458510368051. [DOI] [PubMed] [Google Scholar]
- 33.Otto C, Oltmann A, Stein A, Frenzel K, Schroeter J, Habbel P, Gärtner B, Hofmann J, Ruprecht K. Intrathecal EBV antibodies are part of the polyspecific immune response in multiple sclerosis. Neurology. 2011;76:1316–1321. doi: 10.1212/WNL.0b013e318215286d. [DOI] [PubMed] [Google Scholar]
- 34.Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue–report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011;134:2772–2786. doi: 10.1093/brain/awr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, Andreoni L, Trivedi P, Salvetti M, Faggioni A, Aloisi F. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, Roughan JE, Almendinger SE, Blewett MM, Brück W, Hafler DA, O’Connor KC. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. 2009;132:3318–3328. doi: 10.1093/brain/awp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One. 2010:5. doi: 10.1371/journal.pone.0012496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nielsen TR, Rostgaard K, Askling J, Steffensen R, Oturai A, Jersild C, Koch-Henriksen N, Sørensen PS, Hjalgrim H. Effects of infectious mononucleosis and HLA-DRB1*15 in multiple sclerosis. Mult Scler. 2009;15:431–436. doi: 10.1177/1352458508100037. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen TR, Rostgaard K, Nielsen NM, Koch-Henriksen N, Haahr S, Sorensen PS, Hjalgrim H. Multiple sclerosis after infectious mononucleosis. Arch Neurol. 2007;64:72–75. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 40.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 41.Wagner HJ, Munger KL, Ascherio A. Plasma viral load of Epstein-Barr virus and risk of multiple sclerosis. Eur J Neurol. 2004;11:833–834. doi: 10.1111/j.1468-1331.2004.00871.x. [DOI] [PubMed] [Google Scholar]
- 42.Hollsberg P, Kusk M, Bech E, Hansen HJ, Jakobsen J, Haahr S. Presence of Epstein-Barr virus and human herpesvirus 6B DNA in multiple sclerosis patients: associations with disease activity. Acta Neurol Scand. 2005;112:395–402. doi: 10.1111/j.1600-0404.2005.00516.x. [DOI] [PubMed] [Google Scholar]
- 43.Sotelo J, Ordonez G, Pineda B. Varicella-zoster virus at relapses of multiple sclerosis. J Neurol. 2007;254:493–500. doi: 10.1007/s00415-006-0402-x. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez R, Cour I, Kanaan A, Benedicto M, Martin-Estefania C, Arroyo R, Varela de Seijas E, Picazo JJ. Detection of viral genomes of the Herpesviridae family in multiple sclerosis patients by means of the polymerase chain reaction (PCR) Enferm Infec Microbiol Clin. 2000;18:223–228. [PubMed] [Google Scholar]
- 45.Ferrante P, Mancuso R, Pagani E, Guerini FR, Calvo MG, Saresella M, Speciale L, Caputo D. Molecular evidences for a role of HSV-1 in multiple sclerosis clinical acute attack. J Neurovirol. 2000;6(Suppl 2):S109–S114. [PubMed] [Google Scholar]
- 46.Hay KA, Tenser RB. Leukotropic herpesviruses in multiple sclerosis. Mult Scler. 2000;6:66–68. doi: 10.1177/135245850000600202. [DOI] [PubMed] [Google Scholar]
- 47.Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Kramarsky B, Markham PD, Salahuddin SZ, Sturzenegger S, Wong-Staal F. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science. 1986;234:596–601. doi: 10.1126/science.2876520. [DOI] [PubMed] [Google Scholar]
- 48.De Bolle L, Naesens L, De Clercq E. Update on human herpesvirus 6 biology, clinical features, and therapy. Clin Microbiol Rev. 2005;18:217–245. doi: 10.1128/CMR.18.1.217-245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hall CB, Caserta MT, Schnabel KC, Long C, Epstein LG, Insel RA, Dewhurst S. Persistence of human herpesvirus 6 according to site and variant: possible greater neurotropism of variant A. Clin Infect Dis. 1998;26:132–137. doi: 10.1086/516280. [DOI] [PubMed] [Google Scholar]
- 50.Sola P, Merelli E, Marasca R, Poggi M, Luppi M, Montorsi M, Torelli G. Human herpesvirus 6 and multiple sclerosis: survey of anti-HHV-6 antibodies by immunofluorescence analysis and of viral sequences by polymerase chain reaction. J Neurol Neurosurg Psychiatry. 1993;56:917–919. doi: 10.1136/jnnp.56.8.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilborn F, Schmidt CA, Brinkmann V, Jendroska K, Oettle H, Siegert W. A potential role for human herpesvirus type 6 in nervous system disease. J Neuroimmunol. 1994;49:213–214. doi: 10.1016/0165-5728(94)90198-8. [DOI] [PubMed] [Google Scholar]
- 52.Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- 53.Cermelli C, Berti R, Soldan SS, Mayne M, D’Ambrosia JM, Ludwin SK, Jacobson S. High frequency of human herpesvirus 6 DNA in multiple sclerosis plaques isolated by laser microdissection. J Infect Dis. 2003;187:1377–1387. doi: 10.1086/368166. [DOI] [PubMed] [Google Scholar]
- 54.Carrigan DR, Knox KK. Human herpesvirus six and multiple sclerosis. Mult Scler. 1997;3:390–394. doi: 10.1177/135245859700300607. [DOI] [PubMed] [Google Scholar]
- 55.Knox KK, Brewer JH, Henry JM, Harrington DJ, Carrigan DR. Human herpesvirus 6 and multiple sclerosis: systemic active infections in patients with early disease. Clin Infect Dis. 2000;31:894–903. doi: 10.1086/318141. [DOI] [PubMed] [Google Scholar]
- 56.Friedman JE, Lyons MJ, Cu G, Ablashl DV, Whitman JE, Edgar M, Koskiniemi M, Vaheri A, Zabriskie JB. The association of the human herpesvirus-6 and MS. Mult Scler. 1999;5:355–362. doi: 10.1177/135245859900500509. [DOI] [PubMed] [Google Scholar]
- 57.Virtanen JO, Zabriskie JB, Siren V, Friedman JE, Lyons MJ, Edgar M, Vaheri A, Koskiniemi M. Co-localization of human herpes virus 6 and tissue plasminogen activator in multiple sclerosis brain tissue. Med Sci Monit. 2005;11:BR84–87. [PubMed] [Google Scholar]
- 58.Coates AR, Bell J. HHV-6 and multiple sclerosis. Nat Med. 1998;4:537–538. doi: 10.1038/nm0598-537b. [DOI] [PubMed] [Google Scholar]
- 59.Edwards S, Zvartau M, Clarke H, Irving W, Blumhardt LD. Clinical relapses and disease activity on magnetic resonance imaging associated with viral upper respiratory tract infections in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;64:736–741. doi: 10.1136/jnnp.64.6.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oikonen M, Laaksonen M, Aalto V, Ilonen J, Salonen R, Eralinna JP, Panelius M, Salmi A. Temporal relationship between environmental influenza A and Epstein-Barr viral infections and high multiple sclerosis relapse occurrence. Mult Scler. 2011;17:672–680. doi: 10.1177/1352458510394397. [DOI] [PubMed] [Google Scholar]
- 61.Buljevac D, Flach HZ, Hop WC, Hijdra D, Laman JD, Savelkoul HF, van Der Meché FG, van Doorn PA, Hintzen RQ. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002;125:952–960. doi: 10.1093/brain/awf098. [DOI] [PubMed] [Google Scholar]
- 62.Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–1315. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Confavreux C, Suissa S, Saddier P, Bourdes V, Vukusic S. Vaccinations and the risk of relapse in multiple sclerosis. Vaccines in Multiple Sclerosis Study Group. N Engl J Med. 2001;344:319–326. doi: 10.1056/NEJM200102013440501. [DOI] [PubMed] [Google Scholar]
- 64.Farez MF, Correale J. Immunizations and risk of multiple sclerosis: systematic review and meta-analysis. J Neurol. 2011;258:1197–1206. doi: 10.1007/s00415-011-5984-2. [DOI] [PubMed] [Google Scholar]
- 65.Berti R, Brennan MB, Soldan SS, Ohayon JM, Casareto L, McFarland HF, Jacobson S. Increased detection of serum HHV-6 DNA sequences during multiple sclerosis (MS) exacerbations and correlation with parameters of MS disease progression. J Neurovirol. 2002;8:250–256. doi: 10.1080/13550280290049615-1. [DOI] [PubMed] [Google Scholar]
- 66.Chapenko S, Millers A, Nora Z, Logina I, Kukaine R, Murovska M. Correlation between HHV-6 reactivation and multiple sclerosis disease activity. J Med Virol. 2003;69:111–117. doi: 10.1002/jmv.10258. [DOI] [PubMed] [Google Scholar]
- 67.Alvarez-Lafuente R, De las Heras V, Bartolome M, Picazo JJ, Arroyo R. Relapsing-remitting multiple sclerosis and human herpesvirus 6 active infection. Arch Neurol. 2004;61:1523–1527. doi: 10.1001/archneur.61.10.1523. [DOI] [PubMed] [Google Scholar]
- 68.Rotola A, Ravaioli T, Gonelli A, Dewhurst S, Cassai E, Di Luca D. U94 of human herpesvirus 6 is expressed in latently infected peripheral blood mononuclear cells and blocks viral gene expression in transformed lymphocytes in culture. Proc Natl Acad Sci USA. 1998;95:13911–13916. doi: 10.1073/pnas.95.23.13911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnes DW, Whitley RJ. CNS diseases associated with varicella zoster virus and herpes simplex virus infection. Pathogenesis and current therapy. Neurol Clin. 1986;4:265–283. [PubMed] [Google Scholar]
- 70.Marrie RA, Wolfson C. Multiple sclerosis and varicella zoster virus infection: a review. Epidemiol Infect. 2001;127:315–325. doi: 10.1017/s0950268801005891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leinikki P, Shekarchi I, Iivanainen M, Taskinen E, Holmes KV, Madden D, Sever JL. Virus antibodies in the cerebrospinal fluid of multiple sclerosis patients detected with ELISA tests. J Neurol Sci. 1982;57:249–255. doi: 10.1016/0022-510X(82)90031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Forghani B, Cremer NE, Johnson KP, Fein G, Likosky WH. Comprehensive viral immunology of multiple sclerosis. III. Analysis of CSF antibodies by radioimmunoassay. Arch Neurol. 1980;37:616–619. doi: 10.1001/archneur.1980.00500590040004. [DOI] [PubMed] [Google Scholar]
- 73.Ordonez G, Pineda B, Garcia-Navarrete R, Sotelo J. Brief presence of varicella-zoster vral DNA in mononuclear cells during relapses of multiple sclerosis. Arch Neurol. 2004;61:529–532. doi: 10.1001/archneur.61.4.529. [DOI] [PubMed] [Google Scholar]
- 74.Sotelo J, Martinez-Palomo A, Ordonez G, Pineda B. Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann Neurol. 2008;63:303–311. doi: 10.1002/ana.21316. [DOI] [PubMed] [Google Scholar]
- 75.Burgoon MP, Cohrs RJ, Bennett JL, Anderson SW, Ritchie AM, Cepok S, Hemmer B, Gilden D, Owens GP. Varicella zoster virus is not a disease-relevant antigen in multiple sclerosis. Ann Neurol. 2009;65:474–479. doi: 10.1002/ana.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kang JH, Sheu JJ, Kao S, Lin HC. Increased risk of multiple sclerosis following herpes zoster: a nationwide, population-based study. J Infect Dis. 2011;204:188–192. doi: 10.1093/infdis/jir239. [DOI] [PubMed] [Google Scholar]
- 77.Perron H, Bernard C, Bertrand JB, Lang AB, Popa I, Sanhadji K, Portoukalian J. Endogenous retroviral genes, Herpesviruses and gender in Multiple Sclerosis. J Neurol Sci. 2009;286:65–72. doi: 10.1016/j.jns.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 78.Perron H, Geny C, Laurent A, Mouriquand C, Pellat J, Perret J, Seigneurin JM. Leptomeningeal cell line from multiple sclerosis with reverse transcriptase activity and viral particles. Res Virol. 1989;140:551–561. doi: 10.1016/s0923-2516(89)80141-4. [DOI] [PubMed] [Google Scholar]
- 79.Koprowski H, DeFreitas EC, Harper ME, Sandberg-Wollheim M, Sheremata WA, Robert-Guroff M, Saxinger CW, Feinberg MB, Wong-Staal F, Gallo RC. Multiple sclerosis and human T-cell lymphotropic retroviruses. Nature. 1985;318:154–160. doi: 10.1038/318154a0. [DOI] [PubMed] [Google Scholar]
- 80.Haahr S, Sommerlund M, Christensen T, Jensen AW, Hansen HJ, Moller-Larsen A. A putative new retrovirus associated with multiple sclerosis and the possible involvement of Epstein-Barr virus in this disease. Ann NY Acad Sci. 1994;724:148–156. doi: 10.1111/j.1749-6632.1994.tb38903.x. [DOI] [PubMed] [Google Scholar]
- 81.Perron H, Gratacap B, Lalande B, Genoulaz O, Laurent A, Geny C, Mallaret M, Innocenti P, Schuller E, Stoebner P, Seignerurin JM. In vitro transmission and antigenicity of a retrovirus isolated from a multiple sclerosis patient. Res Virol. 1992;143:337–350. doi: 10.1016/s0923-2516(06)80122-6. [DOI] [PubMed] [Google Scholar]
- 82.Perron H, Garson JA, Bedin F, Beseme F, Paranhos-Baccala G, Komurian-Pradel F, Mallet F, Tuke PW, Voisset C, Blond JL, Lalande B, Seigneurin JM, Mandrand B. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci USA. 1997;94:7583–7588. doi: 10.1073/pnas.94.14.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blond JL, Beseme F, Duret L, Bouton O, Bedin F, Perron H, Mandrand B, Mallet F. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J Virol. 1999;73:1175–1185. doi: 10.1128/jvi.73.2.1175-1185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garson JA, Tuke PW, Giraud P, Paranhos-Baccala G, Perron H. Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis. Lancet. 1998;351:33. doi: 10.1016/s0140-6736(98)24001-3. [DOI] [PubMed] [Google Scholar]
- 85.Sotgiu S, Serra C, Mameli G, Pugliatti M, Rosati G, Arru G, Dolei A. Multiple sclerosis-associated retrovirus and MS prognosis: an observational study. Neurology. 2002;59:1071–1073. doi: 10.1212/wnl.59.7.1071. [DOI] [PubMed] [Google Scholar]
- 86.Sotgiu S, Mameli G, Serra C, Zarbo IR, Arru G, Dolei A. Multiple sclerosis-associated retrovirus and progressive disability of multiple sclerosis. Mult Scler. 2010;16:1248–1251. doi: 10.1177/1352458510376956. [DOI] [PubMed] [Google Scholar]
- 87.Bhat RK, Ellestad KK, Wheatley BM, Warren R, Holt RA, Power C. Age- and disease-dependent HERV-W envelope allelic variation in brain: association with neuroimmune gene expression. PLoS One. 2011;6:e19176. doi: 10.1371/journal.pone.0019176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Antony JM, van Marle G, Opii W, Butterfield DA, Mallet F, Yong VW, Wallace JL, Deacon RM, Warren K, Power C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- 89.Rolland A, Jouvin-Marche E, Viret C, Faure M, Perron H, Marche PN. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J Immunol. 2006;176:7636–7644. doi: 10.4049/jimmunol.176.12.7636. [DOI] [PubMed] [Google Scholar]
- 90.Antony JM, Ellestad KK, Hammond R, Imaizumi K, Mallet F, Warren KG, Power C. The human endogenous retrovirus envelope glycoprotein, syncytin-1, regulates neuroinflammation and its receptor expression in multiple sclerosis: a role for endoplasmic reticulum chaperones in astrocytes. J Immunol. 2007;179:1210–1224. doi: 10.4049/jimmunol.179.2.1210. [DOI] [PubMed] [Google Scholar]
- 91.Adams JM. Measles antibodies in patients with multiple sclerosis. Neurology. 1967;17:707. doi: 10.1212/wnl.17.7.707. passim. [DOI] [PubMed] [Google Scholar]
- 92.Brankin B, Osman M, Herlihy L, Hawkins SA, Cosby SL. Failure to detect measles virus RNA, by reverse transcription-polymerase chain reaction, in peripheral blood leucocytes of patients with multiple sclerosis. Mult Scler. 1996;1:204–206. [PubMed] [Google Scholar]
- 93.Geeraedts F, Wilczak N, van Binnendijk R, De Keyser J. Search for morbillivirus proteins in multiple sclerosis brain tissue. Neuroreport. 2004;15:27–32. doi: 10.1097/00001756-200401190-00007. [DOI] [PubMed] [Google Scholar]
- 94.Ahlgren C, Oden A, Haghighi S, Andersen O, Bergstrom T, Lycke J. The effect of live, attenuated measles vaccine and measles infection on measles antibody levels in serum and CSF of patients with multiple sclerosis or clinically isolated syndrome. J Neuroimmunol. 2011;235:98–103. doi: 10.1016/j.jneuroim.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 95.Gilden DH. Viruses and multiple sclerosis. JAMA. 2001;286:3127–3129. doi: 10.1001/jama.286.24.3127. [DOI] [PubMed] [Google Scholar]
- 96.Yao SY, Stratton CW, Mitchell WM, Sriram S. CSF oligoclonal bands in MS include antibodies against Chlamydophila antigens. Neurology. 2001;56:1168–1176. doi: 10.1212/wnl.56.9.1168. [DOI] [PubMed] [Google Scholar]
- 97.Fainardi E, Castellazzi M, Tamborino C, Seraceni S, Tola MR, Granieri E, Contini C. Chlamydia pneumoniae-specific intrathecal oligoclonal antibody response is predominantly detected in a subset of multiple sclerosis patients with progressive forms. J Neurovirol. 2009;15:425–433. doi: 10.3109/13550280903475580. [DOI] [PubMed] [Google Scholar]
- 98.Franciotta D, Di Stefano AL, Jarius S, Zardini E, Tavazzi E, Ballerini C, Marchioni E, Bergamaschi R, Ceroni M. Cerebrospinal BAFF and Epstein-Barr virus-specific oligoclonal bands in multiple sclerosis and other inflammatory demyelinating neurological diseases. J Neuroimmunol. 2011;230:160–163. doi: 10.1016/j.jneuroim.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 99.Virtanen JO, Pietilainen-Nicklen J, Uotila L, Farkkila M, Vaheri A, Koskiniemi M. Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J Neuroimmunol. 2011;237:93–97. doi: 10.1016/j.jneuroim.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 100.Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, Cross AH. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol. 2010;67:707–714. doi: 10.1001/archneurol.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, HERMES Trial Group B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 102.McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD. Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38:229–240. doi: 10.1016/0165-5728(92)90016-e. [DOI] [PubMed] [Google Scholar]
- 103.Barnett LA, Whitton JL, Wada Y, Fujinami RS. Enhancement of autoimmune disease using recombinant vaccinia virus encoding myelin proteolipid protein. J Neuroimmunol. 1993;44:15–25. doi: 10.1016/0165-5728(93)90263-x. [DOI] [PubMed] [Google Scholar]
- 104.Pachner AR. Experimental models of multiple sclerosis. Curr Opin Neurol. 2011;24:291–299. doi: 10.1097/WCO.0b013e328346c226. [DOI] [PubMed] [Google Scholar]
- 105.Axthelm MK, Bourdette DN, Marracci GH, Su W, Mullaney ET, Manoharan M, Kohama SG, Pollaro J, Witkowski E, Wang P, Rooney WD, Sherman LS, Wong SW. Japanese macaque encephalomyelitis: a spontaneous multiple sclerosis-like disease in a nonhuman primate. Ann Neurol. 2011;70:362–373. doi: 10.1002/ana.22449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.t Hart BA, Laman JD, Bauer J, Blezer E, van Kooyk Y, Hintzen RQ. Modelling of multiple sclerosis: lessons learned in a non-human primate. Lancet Neurol. 2004;3:588–597. doi: 10.1016/S1474-4422(04)00879-8. [DOI] [PubMed] [Google Scholar]
- 107.Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, Heiss JD, Cogen P, Gaillard WD, Sato S, Theodore WH, Jacobson S. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology. 2003;61:1405–1411. doi: 10.1212/01.wnl.0000094357.10782.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fotheringham J, Donati D, Akhyani N, Fogdell-Hahn A, Vortmeyer A, Heiss JD, Williams E, Weinstein S, Bruce DA, Gaillard WD, Sato S, Theodore WH, Jacobson S. Association of human herpesvirus-6B with mesial temporal lobe epilepsy. PLoS Med. 2007;4:e180. doi: 10.1371/journal.pmed.0040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yao K, Honarmand S, Espinosa A, Akhyani N, Glaser C, Jacobson S. Detection of human herpesvirus-6 in cerebrospinal fluid of patients with encephalitis. Ann Neurol. 2009;65:257–267. doi: 10.1002/ana.21611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bech E, Lycke J, Gadeberg P, Hansen HJ, Malmestrom C, Andersen O, Christensen T, Ekholm S, Haahr S, Höllsberg P, Bergström T, Svennerholm B, Jakobsen J. A randomized, double-blind, placebo-controlled MRI study of anti-herpes virus therapy in MS. Neurology. 2002;58:31–36. doi: 10.1212/wnl.58.1.31. [DOI] [PubMed] [Google Scholar]
- 111.Friedman JE, Zabriskie JB, Plank C, Ablashi D, Whitman J, Shahan B, Edgell R, Shieh M, Rapalino O, Zimmerman R, Sheng D. A randomized clinical trial of valacyclovir in multiple sclerosis. Mult Scler. 2005;11:286–295. doi: 10.1191/1352458505ms1185oa. [DOI] [PubMed] [Google Scholar]
- 112.Reymen D, Naesens L, Balzarini J, Holy A, Dvorakova H, De Clercq E. Antiviral activity of selected acyclic nucleoside analogues against human herpesvirus 6. Antiviral Res. 1995;28:343–357. doi: 10.1016/0166-3542(95)00058-5. [DOI] [PubMed] [Google Scholar]
- 113.Goodman AD, Miller DH. Infections and MS: clinical trials move to center stage. Neurology. 2002;58:7–8. doi: 10.1212/wnl.58.1.7. [DOI] [PubMed] [Google Scholar]
- 114.Whitley R. The new age of molecular diagnostics for microbial agents. N Engl J Med. 2008;358:988–989. doi: 10.1056/NEJMp0708085. [DOI] [PubMed] [Google Scholar]
- 115.Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL. Microarray-based detection and genotyping of viral pathogens. Proc Natl Acad Sci USA. 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Palacios G, Quan PL, Jabado OJ, Conlan S, Hirschberg DL, Liu Y, Zhai J, Renwick N, Hui J, Hegyi H, Grolla A, Strong JE, Towner JS, Geisbert TW, Jahrling PB, Büchen-Osmond C, Ellerbrok H, Sanchez-Seco MP, Lussier Y, Formenty P, Nichol MS, Feldmann H, Briese T, Lipkin WI. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg Infect Dis. 2007;13:73–81. doi: 10.3201/eid1301.060837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- 118.Johnson RT. The virology of demyelinating diseases. Ann Neurol. 1994;36(Suppl):S54–S60. doi: 10.1002/ana.410360715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Larsen PD, Bloomer LC, Bray PF. Epstein-Barr nuclear antigen and viral capsid antigen antibody titers in multiple sclerosis. Neurology. 1985;35:435–438. doi: 10.1212/wnl.35.3.435. [DOI] [PubMed] [Google Scholar]
- 120.Wandinger K, Jabs W, Siekhaus A, Bubel S, Trillenberg P, Wagner H, Wessel K, Kirchner H, Hennig H. Association between clinical disease activity and Epstein-Barr virus reactivation in MS. Neurology. 2000;55:178–184. doi: 10.1212/wnl.55.2.178. [DOI] [PubMed] [Google Scholar]
- 121.Munch M, Riisom K, Christensen T, Moller-Larsen A, Haahr S. The significance of Epstein-Barr virus seropositivity in multiple sclerosis patients? Acta Neurol Scand. 1998;97:171–174. doi: 10.1111/j.1600-0404.1998.tb00632.x. [DOI] [PubMed] [Google Scholar]
- 122.Pohl D, Krone B, Rostasy K, Kahler E, Brunner E, Lehnert M, Wagner HJ, Gärtner J, Hanefeld F. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology. 2006;67:2063–2065. doi: 10.1212/01.wnl.0000247665.94088.8d. [DOI] [PubMed] [Google Scholar]
- 123.Banwell B, Krupp L, Kennedy J, Tellier R, Tenembaum S, Ness J, Belman A, Boiko A, Bykova O, Waubant E, Mah JK, Stoian C, Kremenchutzky M, Bardini MR, Ruggieri M, Rensel M, Hahn J, Weinstock-Guttman B, Yeh EA, Farrell K, Freedman M, Iivanainen M, Sevon M, Bhan V, Dilenge ME, Stephens D, Bar-Or A. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol. 2007;6:773–781. doi: 10.1016/S1474-4422(07)70196-5. [DOI] [PubMed] [Google Scholar]
- 124.Haahr S, Plesner AM, Vestergaard BF, Hollsberg P. A role of late Epstein-Barr virus infection in multiple sclerosis. Acta Neurol Scand. 2004;109:270–275. doi: 10.1046/j.1600-0404.2003.00221.x. [DOI] [PubMed] [Google Scholar]
- 125.Wagner HJ, Hennig H, Jabs WJ, Siekhaus A, Wessel K, Wandinger KP. Altered prevalence and reactivity of anti-Epstein-Barr virus antibodies in patients with multiple sclerosis. Viral Immunol. 2000;13:497–502. doi: 10.1089/vim.2000.13.497. [DOI] [PubMed] [Google Scholar]
- 126.Sundstrom P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nystrom L, Dillner J, Forsgren L. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. 2004;62:2277–2282. doi: 10.1212/01.wnl.0000130496.51156.d7. [DOI] [PubMed] [Google Scholar]
- 127.Lindsey JW, Hatfield LM, Vu T. Epstein-Barr virus neutralizing and early antigen antibodies in multiple sclerosis. Eur J Neurol. 2010;17:1263–1269. doi: 10.1111/j.1468-1331.2010.03005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ingram G, Bugert JJ, Loveless S, Robertson NP. Anti-EBNA-1 IgG is not a reliable marker of multiple sclerosis clinical disease activity. Eur J Neurol. 17:1386–1389. doi: 10.1111/j.1468-1331.2010.03083.x. [DOI] [PubMed] [Google Scholar]
- 129.Villegas E, Santiago O, Carrillo JA, Sorlozano A, Guerrero M, Fernandez O, Gutiérrez J. Low intrathecal immune response of anti-EBNA-1 antibodies and EBV DNA from multiple sclerosis patients. Diagn Microbiol Infect Dis. 70:85–90. doi: 10.1016/j.diagmicrobio.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 130.Bray PF, Luka J, Bray PF, Culp KW, Schlight JP. Antibodies against Epstein-Barr nuclear antigen (EBNA) in multiple sclerosis CSF, and two pentapeptide sequence identities between EBNA and myelin basic protein. Neurology. 1992;42:1798–1804. doi: 10.1212/wnl.42.9.1798. [DOI] [PubMed] [Google Scholar]
- 131.Zivadinov R, Nasuelli D, Tommasi MA, Serafin M, Bratina A, Ukmar M, Pirko I, Johnson AJ, Furlan C, Pozzi-Mucelli RS, Monti-Bragadin L, Grop A, Zambon M, Antonello RM, Cazzato G, Zorzon M. Positivity of cytomegalovirus antibodies predicts a better clinical and radiological outcome in multiple sclerosis patients. Neurol Res. 2006;28:262–269. doi: 10.1179/016164106X98134. [DOI] [PubMed] [Google Scholar]
- 132.Ponsonby AL, van der Mei I, Dwyer T, Blizzard L, Taylor B, Kemp A, Simmons R, Kilpatrick T. Exposure to infant siblings during early life and risk of multiple sclerosis. JAMA. 2005;293:463–469. doi: 10.1001/jama.293.4.463. [DOI] [PubMed] [Google Scholar]
- 133.Alotaibi S, Kennedy J, Tellier R, Stephens D, Banwell B. Epstein-Barr virus in pediatric multiple sclerosis. JAMA. 2004;291:1875–1879. doi: 10.1001/jama.291.15.1875. [DOI] [PubMed] [Google Scholar]
- 134.Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ, Hankinson SE, Hunter DJ. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA. 2001;286:3083–3088. doi: 10.1001/jama.286.24.3083. [DOI] [PubMed] [Google Scholar]
- 135.Myhr KM, Riise T, Barrett-Connor E, Myrmel H, Vedeler C, Gronning M, Kalvenes MB, Nyland H. Altered antibody pattern to Epstein-Barr virus but not to other herpesviruses in multiple sclerosis: a population based case-control study from western Norway. J Neurol Neurosurg Psychiatry. 1998;64:539–542. doi: 10.1136/jnnp.64.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shirodaria PV, Haire M, Fleming E, Merrett JD, Hawkins SA, Roberts SD. Viral antibody titers. Comparison in patients with multiple sclerosis and rheumatoid arthritis. Arch Neurol. 1987;44:1237–1241. doi: 10.1001/archneur.1987.00520240019006. [DOI] [PubMed] [Google Scholar]
- 137.Bray PF, Bloomer LC, Salmon VC, Bagley MH, Larsen PD. Epstein-Barr virus infection and antibody synthesis in patients with multiple sclerosis. Arch Neurol. 1983;40:406–408. doi: 10.1001/archneur.1983.04050070036006. [DOI] [PubMed] [Google Scholar]
- 138.Sumaya CV, Myers LW, Ellison GW. Epstein-Barr virus antibodies in multiple sclerosis. Arch Neurol. 1980;37:94–96. doi: 10.1001/archneur.1980.00500510052009. [DOI] [PubMed] [Google Scholar]
- 139.Sumaya CV, Myers L, Ellison GW. Epstein-Barr virus antibodies in multiple sclerosis. Trans Am Neurol Assoc. 1976;101:300–302. [PubMed] [Google Scholar]
- 140.Morre SA, van Beek J, De Groot CJ, Killestein J, Meijer CJ, Polman CH, van der Valk P, Middeldorp JM, van Den Brule AJ. Is Epstein-Barr virus present in the CNS of patients with MS? Neurology. 2001;56:692. doi: 10.1212/wnl.56.5.692. [DOI] [PubMed] [Google Scholar]
- 141.Sanders VJ, Felisan S, Waddell A, Tourtellotte WW. Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol. 1996;2:249–258. doi: 10.3109/13550289609146888. [DOI] [PubMed] [Google Scholar]
- 142.Alvarez-Lafuente R, Garcia-Montojo M, De Las Heras V, Dominguez-Mozo MI, Bartolome M, Benito-Martin MS, Arroyo R. Herpesviruses and human endogenous retroviral sequences in the cerebrospinal fluid of multiple sclerosis patients. Mult Scler. 2008;14:595–601. doi: 10.1177/1352458507086425. [DOI] [PubMed] [Google Scholar]
- 143.Denne C, Kleines M, Dieckhofer A, Ritter K, Scheithauer S, Merz U, Häusler M. Intrathecal synthesis of anti-viral antibodies in pediatric patients. Eur J Paediatr Neurol. 2007;11:29–34. doi: 10.1016/j.ejpn.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 144.Mancuso R, Delbue S, Borghi E, Pagani E, Calvo MG, Caputo D, Granieri E, Ferrante P. Increased prevalence of varicella zoster virus DNA in cerebrospinal fluid from patients with multiple sclerosis. J Med Virol. 2007;79:192–199. doi: 10.1002/jmv.20777. [DOI] [PubMed] [Google Scholar]
- 145.Martin C, Enbom M, Soderstrom M, Fredrikson S, Dahl H, Lycke J, Bergström T, Linde A. Absence of seven human herpesviruses, including HHV-6, by polymerase chain reaction in CSF and blood from patients with multiple sclerosis and optic neuritis. Acta Neurol Scand. 1997;95:280–283. doi: 10.1111/j.1600-0404.1997.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 146.Liedtke W, Malessa R, Faustmann PM, Eis-Hubinger AM. Human herpesvirus 6 polymerase chain reaction findings in human immunodeficiency virus associated neurological disease and multiple sclerosis. J Neurovirol. 1995;1:253–258. doi: 10.3109/13550289509114021. [DOI] [PubMed] [Google Scholar]
- 147.Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, Brennan MB, Maloni HW, McFarland HF, Lin HC, Patnaik M, Jacobson S. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA. Nat Med. 1997;3:1394–1397. doi: 10.1038/nm1297-1394. [DOI] [PubMed] [Google Scholar]
- 148.Ablashi DV, Lapps W, Kaplan M, Whitman JE, Richert JR, Pearson GR. Human Herpesvirus-6 (HHV-6) infection in multiple sclerosis: a preliminary report. Mult Scler. 1998;4:490–496. doi: 10.1177/135245859800400606. [DOI] [PubMed] [Google Scholar]
- 149.Enbom M, Wang FZ, Fredrikson S, Martin C, Dahl H, Linde A. Similar humoral and cellular immunological reactivities to human herpesvirus 6 in patients with multiple sclerosis and controls. Clin Diagn Lab Immunol. 1999;6:545–549. doi: 10.1128/cdli.6.4.545-549.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ablashi DV, Eastman HB, Owen CB, Roman MM, Friedman J, Zabriskie JB, Peterson DL, Pearson GR, Whitman JE. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J Clin Virol. 2000;16:179–191. doi: 10.1016/s1386-6532(99)00079-7. [DOI] [PubMed] [Google Scholar]
- 151.Taus C, Pucci E, Cartechini E, Fie A, Giuliani G, Clementi M, Menzo S. Absence of HHV-6 and HHV-7 in cerebrospinal fluid in relapsing-remitting multiple sclerosis. Acta Neurol Scand. 2000;101:224–228. doi: 10.1034/j.1600-0404.2000.101004224.x. [DOI] [PubMed] [Google Scholar]
- 152.Derfuss T, Hohlfeld R, Meinl E. Intrathecal antibody (IgG) production against human herpesvirus type 6 occurs in about 20% of multiple sclerosis patients and might be linked to a polyspecific B-cell response. J Neurol. 2005;252:968–971. doi: 10.1007/s00415-005-0794-z. [DOI] [PubMed] [Google Scholar]
- 153.Virtanen JO, Farkkila M, Multanen J, Uotila L, Jaaskelainen AJ, Vaheri A, Koskiniemi M. Evidence for human herpesvirus 6 variant A antibodies in multiple sclerosis: diagnostic and therapeutic implications. J Neurovirol. 2007;13:347–352. doi: 10.1080/13550280701381332. [DOI] [PubMed] [Google Scholar]
- 154.Kuusisto H, Hyoty H, Kares S, Kinnunen E, Elovaara I. Human herpes virus 6 and multiple sclerosis: a Finnish twin study. Mult Scler. 2008;14:54–58. doi: 10.1177/1352458507080063. [DOI] [PubMed] [Google Scholar]
- 155.Behzad-Behbahani A, Mikaeili MH, Entezam M, Mojiri A, Pour GY, Arasteh MM, Rahsaz M, Banihashemi M, Khadang B, Moaddeb A, Nematollahi Z, Azarpira N. Human herpesvirus-6 viral load and antibody titer in serum samples of patients with multiple sclerosis. J Microbiol Immunol Infect. 2011;44:247–251. doi: 10.1016/j.jmii.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 156.Enbom M, Linde A, Evengard B. No evidence of active infection with human herpesvirus 6 (HHV-6) or HHV-8 in chronic fatigue syndrome. J Clin Microbiol. 2000;38:2457. doi: 10.1128/jcm.38.6.2457-2457.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Riverol M, Sepulcre J, Fernandez-Diez B, Villoslada P, Fernandez-Alonso M, Rubio M, Rodriguez A, Uccelli A, Brieva L. Antibodies against Epstein-Barr virus and herpesvirus type 6 are associated with the early phases of multiple sclerosis. J Neuroimmunol. 2007;192:184–185. doi: 10.1016/j.jneuroim.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 158.Ongradi J, Rajda C, Marodi CL, Csiszar A, Vecsei L. A pilot study on the antibodies to HHV-6 variants and HHV-7 in CSF of MS patients. J Neurovirol. 1999;5:529–532. doi: 10.3109/13550289909045382. [DOI] [PubMed] [Google Scholar]
- 159.Fillet AM, Lozeron P, Agut H, Lyon-Caen O, Liblau R. HHV-6 and multiple sclerosis. Nat Med. 1998;4:537. doi: 10.1038/nm0598-537a. author reply 538. [DOI] [PubMed] [Google Scholar]
- 160.Goldberg SH, Albright AV, Lisak RP, Gonzalez-Scarano F. Polymerase chain reaction analysis of human herpesvirus-6 sequences in the sera and cerebrospinal fluid of patients with multiple sclerosis. J Neurovirol. 1999;5:134–139. doi: 10.3109/13550289909021995. [DOI] [PubMed] [Google Scholar]
- 161.Mirandola P, Stefan A, Brambilla E, Campadelli-Fiume G, Grimaldi LM. Absence of human herpesvirus 6 and 7 from spinal fluid and serum of multiple sclerosis patients. Neurology. 1999;53:1367–1368. doi: 10.1212/wnl.53.6.1367-a. [DOI] [PubMed] [Google Scholar]
- 162.Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Killian JM, Zhang JZ. Detection of viral DNA and immune responses to the human herpesvirus 6 101-kilodalton virion protein in patients with multiple sclerosis and in controls. J Virol. 2002;76:6147–6154. doi: 10.1128/JVI.76.12.6147-6154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Al-Shammari S, Nelson RF, Voevodin A. HHV-6 DNAaemia in patients with multiple sclerosis in Kuwait. Acta Neurol Scand. 2003;107:122–124. doi: 10.1034/j.1600-0404.2003.01234.x. [DOI] [PubMed] [Google Scholar]
- 164.Alvarez-Lafuente R, De Las Heras V, Bartolome M, Garcia-Montojo M, Arroyo R. Human herpesvirus 6 and multiple sclerosis: a one-year follow-up study. Brain Pathol. 2006;16:20–27. doi: 10.1111/j.1750-3639.2006.tb00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ahram M, El-Omar A, Baho Y, Lubad MA. Association between human herpesvirus 6 and occurrence of multiple sclerosis among Jordanian patients. Acta Neurol Scand. 2009;120:430–435. doi: 10.1111/j.1600-0404.2009.01187.x. [DOI] [PubMed] [Google Scholar]
- 166.Torelli G, Barozzi P, Marasca R, Cocconcelli P, Merelli E, Ceccherini-Nelli L, Ferrari S, Luppi M. Targeted integration of human herpesvirus 6 in the p arm of chromosome 17 of human peripheral blood mononuclear cells in vivo. J Med Virol. 1995;46:178–188. doi: 10.1002/jmv.1890460303. [DOI] [PubMed] [Google Scholar]
- 167.Merelli E, Bedin R, Sola P, Barozzi P, Mancardi GL, Ficarra G, Franchini G. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J Neurol. 1997;244:450–454. doi: 10.1007/s004150050121. [DOI] [PubMed] [Google Scholar]
- 168.Mayne M, Krishnan J, Metz L, Nath A, Auty A, Sahai BM, Power C. Infrequent detection of human herpesvirus 6 DNA in peripheral blood mononuclear cells from multiple sclerosis patients. Ann Neurol. 1998;44:391–394. doi: 10.1002/ana.410440317. [DOI] [PubMed] [Google Scholar]
- 169.Rotola A, Cassai E, Tola MR, Granieri E, Di Luca D. Human herpesvirus 6 is latent in peripheral blood of patients with relapsing-remitting multiple sclerosis. J Neurol Neurosurg Psychiatry. 1999;67:529–531. doi: 10.1136/jnnp.67.4.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kim JS, Lee KS, Park JH, Kim MY, Shin WS. Detection of human herpesvirus 6 variant A in peripheral blood mononuclear cells from multiple sclerosis patients. Eur Neurol. 2000;43:170–173. doi: 10.1159/000008158. [DOI] [PubMed] [Google Scholar]
- 171.Rotola A, Caselli E, Cassai E, Tola MR, Granieri E, Luca DD. Novel human herpesviruses and multiple sclerosis. J Neurovirol. 2000;6(Suppl 2):S88–S891. [PubMed] [Google Scholar]
- 172.Alvarez-Lafuente R, Martin-Estefania C, de las Heras V, Castrillo C, Cour I, Picazo JJ, Varela De Seijas E, Arroyo R. Prevalence of herpesvirus DNA in MS patients and healthy blood donors. Acta Neurol Scand. 2002;105:95–99. doi: 10.1034/j.1600-0404.2002.1o050.x. [DOI] [PubMed] [Google Scholar]
- 173.Alvarez-Lafuente R, Martin-Estefania C, de Las Heras V, Castrillo C, Picazo JJ, Varela de Seijas E, González RA. Active human herpesvirus 6 infection in patients with multiple sclerosis. Arch Neurol. 2002;59:929–933. doi: 10.1001/archneur.59.6.929. [DOI] [PubMed] [Google Scholar]
- 174.Alvarez-Lafuente R, de las Heras V, Garcia-Montojo M, Bartolome M, Arroyo R. Human herpesvirus-6 and multiple sclerosis: relapsing-remitting versus secondary progressive. Mult Scler. 2007;13:578–583. doi: 10.1177/1352458506072667. [DOI] [PubMed] [Google Scholar]
- 175.Cirone M, Cuomo L, Zompetta C, Ruggieri S, Frati L, Faggioni A, Ragona G. Human herpesvirus 6 and multiple sclerosis: a study of T cell cross-reactivity to viral and myelin basic protein antigens. J Med Virol. 2002;68:268–272. doi: 10.1002/jmv.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mancuso R, Hernis A, Cavarretta R, Caputo D, Calabrese E, Nemni R, Ferrante P, Delbue S, Clerici M. Detection of viral DNA sequences in the cerebrospinal fluid of patients with multiple sclerosis. J Med Virol. 2010;82:1051–1057. doi: 10.1002/jmv.21764. [DOI] [PubMed] [Google Scholar]