

Abstract
Imaging of 300–500 mm mouse brain slices by laser photostimulation with flavoprotein autofluorescence (LFPA) allows the rapid and sensitive mapping of neuronal connectivity. It is accomplished using UV laser-based photo-uncaging of glutamate and imaging neuronal activation by capturing changes in green light (~520 nm) emitted under blue light (~460 nm) excitation. This fluorescence is generated by the oxidized form of flavoprotein and is a measure of metabolic activity. LPFA offers several advantages over imaging techniques that rely on dye loading. First, as flavoprotein imaging measures endogenous signals, it avoids the use of heterogeneously loaded and potentially cytotoxic dyes. Second, flavoprotein signals are large (1–20% above baseline), obviating the need for averaging. Third, the use of photostimulation ensures orthodromic neuronal activation and permits the rapid interrogation of multiple stimulation sites of the slice with a high degree of precision (~50 μm). Here we describe a step-by-step protocol for the incorporation of LPFA into virtually any slice rig, as well as how to do the experiment.
INTRODUCTION
In vitro functional imaging provides a powerful means to map neuronal connectivity. Imaging modalities that require the incorporation of external fluorophores (calcium and voltage-sensitive dye imaging) are limited by slow and heterogenous dye uptake, dye rundown over time and potential cytotoxicity1–3. Recently, a form of intrinsic imaging using flavoprotein autofluorescence (FA) has been adapted for use in the slice, and it yields an exceptionally strong signal-to-noise ratio for functional imaging of long-and short-range connections and retains stable responsiveness for hours4–7. FA imaging depends on the fluorescence of mitochondrial flavoproteins that fluoresce when oxidized, thereby providing an indicator of metabolic activity6,8. This signal is primarily related to postsynaptic activity, although a late phase may be related to glial activity5,9, and is of sufficient amplitude that it has a high signal-to-noise ratio and thus does not require one to average across multiple runs to yield reliable data. We recently combined laser photostimulation of caged glutamate with FA (LPFA) and demonstrated that LPFA can be used to rapidly (30 s) construct a map of connectivity between areas of a brain slice, with spatial resolution of 100–200 μm (ref. 10).
An extensively described technique10, laser scanning photostimulation, refers to a method in which focal areas of a brain slice that is bathed in caged, inactivated glutamate are stimulated with a laser that focally ‘uncages’ the chemical compound in a cylinder of tissue with a narrow (~50 μm) diameter. Essentially, the experimenter clicks on the area of the slice he or she is interested in stimulating, and glutamate is locally activated, only causing nearby cells to fire action potentials. As axons do not have glutamate receptors that would elicit an action potential, this technique avoids their activation, eliminating the possibility of stimulating ‘fibers of passage’, an issue that confuses electrical stimulation experiments. In addition, it enables rapid stimulation of multiple areas of the slice.
Thus, LPFA is the combination of techniques used to sequentially stimulate multiple 50-μm-diameter foci of a slice in a single run, while simultaneously imaging the resultant activation. This combination, which involves no mechanical perturbation of the slice, permits rapid stimulation and enables an experimenter to interrogate connectivity among many areas of an intact slice in a single experiment.
It is straightforward to adapt a rig equipped with an epifluorescence-capable microscope and a sideport for laser entry to perform LPFA; one needs only a camera, proper epifluorescence filters and a computer on which the images can be acquired and processed. Further, even without glutamate uncaging, FA imaging can be combined with other forms of stimulation to provide a rapid and economical way of assessing connectivity in a slice11. The technique has been used to investigate thalamocortical4,7, thalamoreticular, thalamostriatal and cortico-cortical connectivity5 in the juvenile and adult mouse brain slice. Given the flexibility of laser-based photo-stimulation and FA, both of which have been successfully applied across multiple brain preparations6,12–15, it is likely that LPFA can be applied outside of the thalamus and cortex. In addition, recent work demonstrating the utility of LPFA in human diseased cortical tissue16 suggests that this technique can be used to investigate disordered connectivity in human neuropathological states.
A potential disadvantage of LPFA is the protracted time course of the FA activation signal, which has a peak of 1–2 s, and takes approximately 10–15 s to return to baseline6,11,13. This may limit the ability to follow the detailed time course of neuronal activation. However, it should be noted that similar overall durations of activation are seen with other successful neuronal imaging techniques, such as blood-oxygen-level-dependent functional MRI signals17 and imaging of cytosolic calcium signals6. In addition, in its current state of development, LPFA has not been used for image activation at the individual neuronal level. Although flavoprotein signals in individual hepatocytes and cardiac myocytes have been reported using two-photon FA imaging techniques18,19, no such literature exists for neurons. Future work will clarify this issue.
Experiment design
An LPFA experiment involves several steps, which can be distilled down to the following general procedures: First, cut brain slices using procedures that are similar to those used in a standard in vitro electrophysiology experiment. Next, place a slice in a bath of oxygenated artificial cerebrospinal fluid (ACSF) containing caged glutamate. Determine the area in the slice you would like to stimulate and target it with the laser. Then expose the slice to epifluorescence illumination, adjust exposure time and begin imaging the slice. After collecting at least three or four frames (used for baseline), engage the laser stimulation. Continue collecting frames for the duration of the experiment, changing target sites and restimulating as desired (leave at least 4 s between each stimulation). After imaging is complete, disengage epifluorescence illumination and perform postacquisition processing. Wait for at least 2 min for the slice to recover before a new epifluorescence is run. These steps will be expanded upon in the PROCEDURE section.
To implement LPFA in the slice, one needs a minimum of the following equipment: an epifluorescence-capable microscope with a camera-mounting apparatus and sideport for laser entry, the necessary filter set, a source of illumination, a moderately high sensitivity camera, video capture software and a computer. As alluded to above, it is also possible to implement FA imaging without photo-stimulation techniques. Both setups will be described.
Standard techniques can be used to obtain brain tissue slices for FA imaging. As FA signals are derived from metabolic signaling pathways, we, similar to others5,6,11, take precautions to ensure the metabolic health of our tissue slices. For example, our experience suggests that perfusion of the animal with cold, oxygenated sucrose-based slicing solution immediately before slicing, and placing slices into a high-magnesium incubation medium after slicing, maximizes slice longevity. For anesthesia, 1 mg g−1 ketamine and 10 mg g−1 xylazine work well and avoid the mitochondrial suppression seen with barbiturates4. The incubation medium is based on standard ACSF with an increased ratio of magnesium to calcium (3:1), compared with normal ACSF (1:1, used during recordings), hereafter referred to as ‘incubation’ ACSF. A sucrose-based solution is used for perfusion and tissue slicing (slicing solution).
For laser photostimulation, nitroindolinyl-caged glutamate (Sigma-Aldrich) is dissolved in normal ACSF, with a final concentration of ~0.4 mM, and recirculated in the recording chamber for more than 4 min prior to photostimulation. Recirculation, although not ideal for slice health, is necessary because of the expense of caged glutamate; our experience suggests that slices are healthy for at least one hour in recirculated caged glutamate. This duration is more than enough to perform several connectivity maps of a mouse brain slice. Stimulation and recording in the caged glutamate solution typically lasts less than 20 min, with no evidence of run-down of the responsiveness of the tissue.
We did not have to use field potentials to determine the health of the slice. In fact, we have found that the best indicator of slice health is the ‘double-bubble’ finding described below, under ‘TROUBLESHOOTING.’ Further positive controls and procedures for situations in which no slice activation is seen are also described in the TROUBLESHOOTING section.
MATERIALS
REAGENTS
Chemicals for incubation of ACSF and imaging of ACSF
NaHCO3 (Sigma-Aldrich)
KCl (Sigma-Aldrich)
Glucose (Sigma-Aldrich)
NaCl (Sigma-Aldrich)
NaH2PO4·H2O (Sigma-Aldrich)
MgCl2·6H2O (Sigma-Aldrich)
CaCl2·2H2O (Sigma-Aldrich)
Chemicals for slicing solution
Sucrose (Sigma-Aldrich)
Glucose (Sigma-Aldrich)
NaHCO3 (Sigma-Aldrich)
KCl (Sigma-Aldrich)
NaH2PO4·H2O (Sigma-Aldrich)
MgCl2·6H2O (Sigma-Aldrich)
CaCl2·2H2O (Sigma-Aldrich)
Caged glutamate: Nitroindolinyl-caged glutamate (Sigma-Aldrich, cat. no. G3291)
Ketamine (Vetaket; Phoenix Scientific)
Xylazine (AnaSed; Lloyd Laboratories)
EQUIPMENT
Zeiss Axioskop FS2 + (Carl Zeiss)
QImaging Retiga-SRV (QImaging)
Streampix (Streampix 3; Norpix)
Dichroic mirror (Chroma)
Norprene (Saint-Gobain Performance Plastics)
Pulsed UV laser (DPSS Lasers)
UV-enhanced reflectors (Thorlabs)
Mirror galvanometers (Cambridge Technology)
Q-switch trigger (LS3-ZM2; Vincent Associates)
Neutral-density wheel (Edmund)
CorelDRAW X4 (Corel)
Pneumatic picopump (WPI)
ImageJ (available from the National Institutes of Health at http://rsbweb.nih.gov/ij/)
Matlab program (MathWorks)
REAGENT SETUP
Experimental tissue
BALB/c mice and human brain slice preparations have been used for these experiments; it is thus likely that preparations from a wide range of animals would yield LPFA signals. ! CAUTION Animal and human studies should be conducted in compliance with legislative and institutional requirements that pertain to the investigator.
Incubation ACSF
Incubation ACSF is composed of NaHCO3 (26 mM), KCl (2.5 mM), glucose (10 mM), NaCl (126 mM), NaH2PO4·H2O (1.25 mM), MgCl2·6H2O (3 mM) and CaCl2·2H2O (1.1 mM) dissolved into distilled water. We typically prepare a 10× concentrated solution (without the NaHCO3), which we store for several days at 4 °C and dilute it on the day of use. We add 20 ml of the stock solution to 100 ml of distilled H2O, then add 20 ml of a 10× NaHCO3 solution and 60 ml of distilled H2O to prepare a total of 200 ml of this solution. We begin oxygenating this solution with 95% O2 5% CO2 at least 30 min before adding the slices. ▲ CRITICAL Do not add concentrated stock solution to concentrated bicarbonate solution without first diluting with distilled water; some reagent may precipitate out.
Imaging ACSF
Imaging ACSF is composed of NaHCO3 (26 mM), KCl (2.5 mM), glucose (10 mM), NaCl (126 mM), NaH2PO4·H2O (1.25 mM) and MgCl2·6H2O (2 mM), CaCl2·2H2O (2 mM). We prepare 1,000 ml of this solution following the same protocol as the incubation ACSF (above), but with 100 ml of stock solution, 100 ml of concentrated bicarbonate solution and 800 ml of distilled water. We usually prepare this solution and begin oxygenating it just before slicing so that it is fully oxygenated before we begin the experiment. ▲ CRITICAL Do not add concentrated stock solution to concentrated bicarbonate solution without first diluting with distilled water; some reagent may precipitate out.
Slicing solution
Slicing solution is composed of the following reagents: sucrose (234 mM), glucose (11 mM), NaHCO3 (26 mM), KCl (2.5 mM), NaH2PO4·H2O (1.25 mM), MgCl2·6H2O (10 mM) and CaCl2·2H2O (0.5 mM). We prepare 200 ml of this solution for use in perfusing the animal in mouse brain-slice experiments, slicing tissue in both human and mouse experiments and for transporting human tissue slices from the operating room to the laboratories. We add 20 ml of stock solution to 40 ml of distilled water, then add 20 ml of 10× sucrose stock solution, followed by 40 ml of distilled water and 20 ml of 10× bicarbonate stock solution and dilute with 60 ml of distilled water to the final concentration. It should be chilled to ~4 °C and oxygenated in 95% O2 5% CO2 prior to use. ▲ CRITICAL Ensure that distilled water is added between each step to avoid reagent precipitation.
Caged glutamate
Dissolve a single 50 mg bottle of caged glutamate into 35 ml of ddH2O and aliquot into 0.7-ml aliquots and freeze at −20 °C prior to use; at the time of the experiment, dissolve the aliquot into ~7 ml of imaging ACSF resulting in a ~0.4-mM solution. ▲ CRITICAL The caged glutamate concentration is critical; we dissolved an aliquot into as many as 10 ml of normal ACSF, but do not recommend further dilution.
EQUIPMENT SETUP
Microscope
Any epifluorescence microscope can theoretically be utilized for FA imaging, provided that the optics pass all relevant wavelengths in the appropriate directions. We have had success with the Zeiss Axioskop FS2 + with an HBO 100 mercury lamp (Carl Zeiss) for epifluorescence illumination.
Image acquisition
We use a QImaging Retiga-SRV with Firewire interface to acquire images, although it is likely that lower sensitivity cameras could also be used with some success because of the large signals obtained.
Image capture
Streampix, a commercially available software package that enables rapid, low-overhead frame capture, is used to capture images to disk.
Computer
Our computer was built in-house. Although most new computers would perform well, we recommend at least 3 GB of RAM and the utilization of a hardware-based RAID-0 array with 128-kbit striping. This setup ensures rapid, real-time storage of data delivered to the computer from the camera. Slower systems, and/or those not using a zero-overhead software package, run the risk of introducing a write-to-disk delay that could affect the frame rate (and thus the temporal resolution) of acquired data.
Setup for FA
The optics for an FA imaging setup should deliver 460-nm light to the slice and 520-nm light to the camera. A band-reflectance dichroic mirror (transmission > 80% at 310–400 nm and 550 nm and above) is placed at a 45° angle between the horizontal excitation light beam and the vertically oriented path of light down to the slice (see Fig. 1). The dichroic mirror reflects the excitation wavelength (460 nm) down to the slice while also allowing the photostimulation laser light (355 nm) to pass down to the slice and enough of the fluorescent emission spectrum up from the slice to achieve robust FA imaging. A long-pass filter (>515 nm) is placed above the dichroic and laser port to ensure that other endogenous autofluorescence signals, including NADH autofluorescence (peak emission 450–480 nm (ref. 8)), are not imaged.
Figure 1.
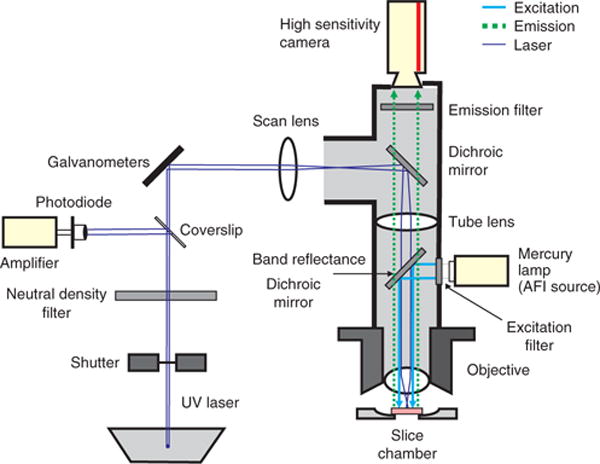
LPFA imaging setup. This schematic overview traces the light paths of the laser light required for photostimulation (purple), the excitation light (blue) and the autofluorescence (emission) light (green) from the slice. Figure is from Llano et al.5; used with permission.
Perfusion system
To maximize slice health, brain slices are placed onto a titanium mesh suspended ~1 mm above the bottom of the microscope chamber. This setup perfuses both sides of the slice, ensuring maximal slice health and, in practice, qualitatively improving the FA signal strength. To maximize oxygenation of the tissue, Norprene oxygen-impermeable tubing is used to deliver ACSF to the recording chamber.
UV laser
For photostimulation, a pulsed UV laser (355-nm wavelength, frequency-tripled Nd:YVO4, 100-kHz pulse repetition rate) is used. All laser stimuli are controlled by a Matlab program developed in the laboratory of Karel Svoboda. This program, called ‘ScanImage’, is available at http://www.neuroptikon.org/projects/display/ephus/ScanImage.
Optical setup
The laser beam is routed through a series of UV-enhanced reflectors, two mirror galvanometers and finally into a sideport of a Zeiss Axioskop FS2 + microscope. Angles of the galvanometers are controlled by ScanImage and determine the laser stimulation position. The optics generate a nearly cylindrical beam in the slice to keep the mapping two dimensional20. The Q-switch trigger of the laser and a shutter control the timing of the laser. A variable neutral-density wheel controls the intensity of the laser. A thin cover slip in the laser path reflects a small portion of the laser onto a photodiode. The current from this photodiode is amplified, acquired by the computer and used to monitor the laser intensity throughout the experiment.
Image analysis
To analyze collected images, we have developed several programs that are made to run on Matlab v. 7.04 in-house, although we primarily utilize just four. These include a program that reads images into Matlab, ‘imageproc_streampix.m’, one that reads images into Matlab while subtracting those that may interfere with the signal because of the stimulation method, ‘imageproc_laser.m’, and two more to generate ΔF/F (change in autofluorescence divided by baseline autofluorescence) images. These include ‘deltamarch.m’, which computes a running average difference in fluorescence by subtracting the average brightness values for each pixel of the first n (user-defined) images from the average of the subsequent n images, then divides by the average brightness value of the first n images. For example, if n = 3, the program will take the first three images, average their brightness values together and then subtract them from the average brightness values for images 4–6. The program then moves to the next set, subtracting the average of images 1–3 from the average of images 5–7, and dividing the result by the average of 1–3, then subtracting the average of 1–3 from the average of images 6–8 and so forth until reaching the end of the image sequence. This generates the classic ΔF/F commonly utilized throughout the imaging literature. Another program, ‘march.m’, moves both sets of images forward on each iteration, i.e., subtracting (the averages of) 1–3 from images 4–6, then 2–4 from 5–7, then 3–5 from 6–8 and so on, until reaching the end of the sequence. This reports that the slope of the brightness changes throughout the imaging run, lessening the effect of the drop-off in FA signal seen after the first stimulation run. The drop-off in signal is moderate (about 25–50%) and is typically recovered after 2–3 min of quiescence between runs. Thus, deltamarch.m is useful for shorter runs, whereas march.m is more appropriate for longer imaging runs. All Matlab programs are available in Supplementary Methods
ImageJ
Once images are generated by these programs, they are easily visualized in ImageJ by clicking ‘File’, ‘Import’, ‘Image Sequence’. The above procedures were used to generate the ΔF/F images that appear in all figures in this protocol. Further manipulation (letters and arrows) was carried out in CorelDRAW X4.
PROCEDURE
Slicing
1| Euthanize the mouse. Ketamine/xylazine is used rather than barbiturates, which have a depressive effect on mitochondria6,11,21.
-
2| Quickly perfuse the mouse transcardially with about 5–10 ml of cold (4 °C), oxygenated sucrose-based slicing solution. Once experienced, this step should take no more than 30 s after puncturing the diaphragm.
▲ CRITICAL STEP Although we have successfully imaged tissue from animals that we did not perfuse prior to an experiment, our subjective experience suggests that the tissue does not last as long and does not yield as robust a signal as tissue from perfused animals.
3| Rapidly remove the brain from the skull, block the brain at an appropriate angle (experiment-dependent) and mount it onto the slicer stage.
-
4| Slice the brain at desired thickness (300–500 μm is recommended). Here we focus on the mouse somatosensory thalamocortical slice, although the technique has been used in a number of other slice preparations4,5,7. Details on the preparation of the mouse somatosensory thalamocortical slice have been published previously22.
▲ CRITICAL STEP The angle and thickness of any given slice determines the connectivity that the slice has; if you are conducting experiments that involve grouping data from multiple animals, it is important to standardize slice thickness and slicing angle.
5| Place the slices into incubation ACSF (see REAGENT SETUP) at room temperature (19–21 °C) for 30–60 min.
Imaging
6| Remove the brain slice from the incubation ACSF and place it on a titanium mesh in a slice chamber under a microscope through which oxygenated imaging ACSF is flowing at ~3 ml/min.
7| Wait for 10 min for the slice to equilibrate with the imaging ACSF; while waiting ensure that all equipment is on and the imaging software is ready to acquire images. The fluorescent light source should be turned on at least 5 min prior to imaging to ensure brightness stability. We have conducted our experiments at room temperature.
-
8| Briefly switch over to fluorescence excitation (turn off bright-field illumination, move turret or open shutter to ‘turn on’ excitation light) and adjust exposure time such that the slice is readily visible but no part of the slice appears to be ‘bleached out’. This will determine the frame rate, which is typically between 10 and 20 Hz (exposure times range from 50–100 ms; see Fig. 2).
▲ CRITICAL STEP If the exposure time is long, the camera will become saturated, which will cause the signal to plateau. Although this is not important if one is simply interested in whether two structures in a slice are connected, it is critical that this is avoided in quantitative experiments.
-
9| Turn off the flow of imaging ACSF such that the fluid level in the chamber does not change.
▲ CRITICAL STEP This step is often necessary because, in our experience, any fluctuation of the ACSF level leads to very prominent stimulus artifacts. If the imaging ACSF level is held constant, it may be possible to perfuse the slice while imaging. Another way to mitigate this is to mount a glass cover slip onto two glass ‘feet’ that allow it to cover the slice without touching it. This provides a surface for the ACSF to wick onto, allowing continuous perfusion without a fluctuating ACSF level.
10| Immediately start imaging. If stimulating the slice (see Steps 15–17 for a description of laser stimulation, and Steps 18 and 19 for electrical or glutamate picospritzer-based stimulation), we recommend beginning the stimulation after 500 ms–1 s to allow at least five frames of baseline imaging before the stimulus. We also recommend imaging runs no longer than 30 s if not perfusing the slice.
11| After imaging, either turn the turret or close a shutter to prevent the excitation light from hitting the slice; this minimizes quenching.
-
12| Quickly turn the pump back on to perfuse the slice with fresh imaging ACSF.
■ PAUSE POINT Once this step is complete the slice can be allowed to recover for several minutes, thus allowing time for data analysis.
13| Use appropriate programs to analyze data (see EQUIPMENT SETUP section).
14| Wait at least 2 min after Step 16 before beginning another imaging run to allow the slice to fully recover.
Figure 2.
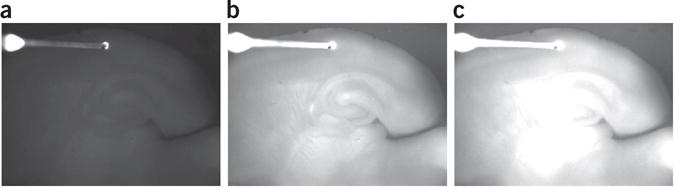
Brightness calibration example. (a) Image is too dark; pixel values are too close to 0, preventing detection of a negative-going signal. (b) Image is correct; pixel values in the area of interest are ~190, enabling a 34% signal increase without saturation. (c) Too bright; pixel values are too close to 255, so positive brightness changes will yield saturation (and thus no positive signal detection).
Laser stimulation
15| Thaw and dissolve an 0.7-ml aliquot of caged glutamate into about 7–10 ml of preoxygenated imaging ACSF, oxygenate it, and begin recirculation of the slice with caged glutamate solution.
-
16| We have had success in eliciting robust long-range activations using short trains of laser pulses of 0.5–1.0 ms and 10 mW of laser power. To stimulate the slice with minimal risk of photodamage from the laser, use short pulse trains (5 pulses, 20–40 ms interpulse interval, 1 ms pulses). Generally, in areas where strong connectivity exists, single trains are sufficient to elicit observable fluorescence and no averaging is needed. We use ScanImage software (see REAGENT SETUP), developed in Karel Svoboda’s laboratory, to control the photostimulation system.
? TROUBLESHOOTING
17| Imaging can occur continuously as the laser stimulates a series of loci. A flash artifact, time-locked to the laser pulse, may saturate the FA during the stimulus, and this flash can be used as a means to mark the stimulation time on the image sequence. Given the relatively long time course of the FA signal (1–2 s to peak, total duration of response ~10 s), the stimulus and neuronal response are separated enough in time to permit an unobscured view of the neuronal response.
Electrical or glutamate picospritzer-based stimulation
-
18| For electrical stimulation, place the electrode carefully on top of the tissue to be stimulated. For rapid exploration of tissue connectivity, we have used glass unipolar electrodes (5–10 μm in diameter) filled with ACSF. Stimulus strengths range from 5–120 μA, with 0.2- to 10-ms pulse durations, with pulses delivered at 20–40 pulses per second and pulse trains lasting 100–1,000 ms.
? TROUBLESHOOTING
-
19| For picospritzer application of glutamate, use micropipettes (5-μm diameter) filled with 2-mM L-glutamate dissolved in ACSF. Apply glutamate with a picospritzer (we have used a pneumatic picopump, WPI), with a pressure of 1 psi, 8-ms pulse duration and 1-s pulse train at 20 pulses per second.
? TROUBLESHOOTING
No remote ΔF/F signal using glutamate stimulation
If there is no remote ΔF/F signal using glutamate stimulation (Step 16 or 18), proceed with the following steps:
First, as a positive control, stimulate the tissue with an electrical stimulation probe (Step 18) with amplitudes of at least 25–50 μA, and plot the time course of pixels around stimulation site. If a positive signal is seen near the stimulus probe, and no other signal is seen elsewhere in the slice, it is likely that the slice does not have desired connectivity, or the health of the slice is poor. Move on to the next slice.
Second, to distinguish between poor slice connectivity versus poor slice preparation, the investigator may take a slice with known connectivity and attempt to visualize this connectivity using FA. One slice that has a high probability of retained connectivity is a 300-μm coronal slice containing neocortex. Electrical stimulation in the infragranular layer virtually always elicits a circular area of activation around the stimulus site, as well as an oblong-shaped locus of activation in layers 2/3 directly above the stimulation site. This ‘double-bubble’ (see Fig. 3) pattern is virtually always seen in healthy slices, and has also been reported by other investigators6. Absence of layer 2/3 activation in the presence of stimulus-site related activation would suggest that slice preparation should be optimized.
Figure 3.
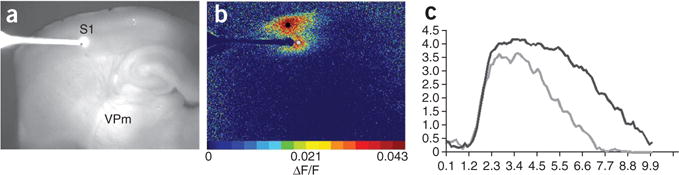
‘Double-bubble’ pattern. (a) Reference image for anatomy; slice is a thalamocortical preparation. (b) ΔF/F image after stimulation in layer 5; note the oblong activation in layers 2/3. (b,c) Gray circle in b corresponds to the stimulation site pixels analyzed in c; the black circle in b corresponds to the response site pixels analyzed in c. Color bar represents ΔF/F activation values (.043 = 4.3 percent). (c) ΔF/F traces for stimulation (gray) and response (black) sites. Stimulation parameters: 50 μA, 20 Hz, 10 pulses and 5 ms pulse width. Traces are averages of 5 × 5 pixels (277.9 μm2). Time in seconds is on the x axis and ΔF/F is on the y axis.
No signal even around the stimulus probe
If no signal is seen even around the electrical stimulation probe (assuming that the slice is not dead), this would suggest a technical malfunction of either the stimulation or imaging system.
Electrical stimulation or photostimulation malfunctions can be easily ruled out by using traditional electrical recording methods to record cellular activity during stimulation. To ensure that the stimulation method is working, one could use patch clamp recording or extracellular recording methods to measure neuronal activation near the stimulation site. Multiple examples of laser photostimulation and patch clamp recordings exist in the literature10,20.
Assuming slice health and stimulation are not the issues, the problem may lie in recording of FA signals or in signal processing. It is important that the tissue exposure to blue light be adequate to induce fluorescence and camera acquisition time be adequate to capture it. We typically use the highest light intensities that our mercury bulb-based illumination system is capable of at ×5 and ×2.5 magnification levels, and ~20% at ×40 magnification (higher magnification results in a greater concentration of light per unit area, eliciting rapid quenching). We then adjust exposure time such that the brightness in the area of interest is ~75% of the dynamic range of the camera (e.g., pixel values ~192 with 8-bit acquisition). This allows up to a 34% increase in fluorescence intensity (greater than the max fluorescence change observed so far) without saturating the camera.
The problem may also lie in image processing. FA carries the distinct advantage of having high signal-to-noise ratios, such that FA brightness changes can often be detected by merely looking at raw images; scroll back and forth through the image stack (load into ImageJ by clicking ‘File’, ‘Import’, ‘Image Sequence’). If a prominent signal is seen in the raw images, but not after processing, this would suggest that there is a bug in the image processing.
● TIMING
Steps 1–7: 6 min
Wait for 30 min to 1 h to allow slices to recover after slicing
Steps 8–10: ~10 min (per slice)
Steps 11–16: 10–30 s (per imaging run)
Step 17: 2 min (per imaging run)
Step 18: 10–30 s (per imaging run)
Step 19: 10–30 s (per imaging run)
ANTICIPATED RESULTS
The time course of activation is measured on the order of seconds. Plotting the time course of activation of pixels around the stimulation site and around the expected response site should give a characteristic time course with a positive peak at 1–2 s, a total duration of positive signal of approximately 5–10 s, and sometimes, a period of negative signal lasting another 5–10 s (see Fig. 4 for examples of thalamocortical and thalamoreticular activation using LPFA). If imaging runs and analyses were successful, the ΔF/F images should appear to be dark with bright spots that peak ~1–2 s after neuronal activity in areas of the slice that were active during imaging. At minimum, a bright spot should appear around the stimulation site. Although we have not quantitatively compared the results of electrical stimulation to photostimulation, the spatial patterns of long-range activation with both techniques appear similar5. As described below, one of the main utilities of electrical stimulation is to provide means to rule out a problem with the imaging apparatus.
Figure 4.
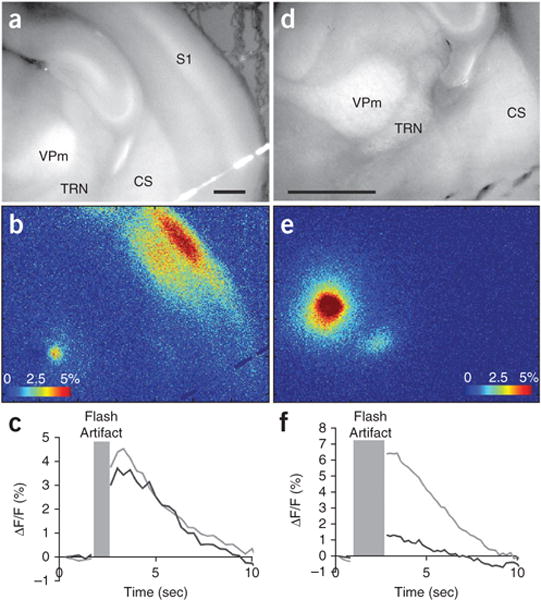
Examples of synaptic activation following photostimulation. (a–c) Correspond to thalamocortical activation at ×2.5 magnification after a single run of photostimulation in VPm, taken from the same set of experiments as Figure 7c of Llano et al5, but from a different run. (d–f) Correspond to thalamoreticular activation at ×5 magnification after a single run of photostimulation in VPm, taken from the same set of experiments as Figure 11 of Llano et al5, but from a different run from those illustrated in that publication. (a,d) Raw images for anatomical orientation. Scale bars, 1 mm. (b,e) ΔF/F images of activation. Color bars represent ΔF/F activation values in percentage. (c,f) ΔF/F traces for stimulation sites in the VPm and response site in S1 (c) or TRN (f). Light gray, stimulation site trace; black line, cortical (c) or thalamic reticular (f) response trace. Traces are averages of 10 × 10 pixels (555.8 μm2). Time, in seconds, is on the x axis and ΔF/F is on the y axis. Gray box corresponds to the time points that were excised because of a saturating flash artifact caused by the UV laser pulse. CS, corpus striatum, S1, primary somatosensory cortex, TRN, thalamic reticular nucleus and VPm, ventroposteriomedial thalamic nucleus.
Supplementary Material
Footnotes
Note: Supplementary information is available in the HTML version of this article.
AUTHOR CONTRIBUTIONS B.B.T. and D.A.L. generated data shown in the figures. B.B.T. generated a draft of the manuscript. All authors contributed to editing the manuscript and development of laser-scanning photostimulation.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Obaid AL, Koyano T, Lindstrom J, Sakai T, Salzberg BM. Spatiotemporal patterns of activity in an intact mammalian network with single-cell resolution: optical studies of nicotinic activity in an enteric plexus. J Neurosci. 1999;19:3073–3093. doi: 10.1523/JNEUROSCI.19-08-03073.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onimaru H, Homma I. A novel functional neuron group for respiratory rhythm generation in the ventral medulla. J Neurosci. 2003;23:1478–1486. doi: 10.1523/JNEUROSCI.23-04-01478.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fields RD, Shneider N, Mentis GZ, O’Donovan MJ. Imaging Nervous System Activity. John Wiley & Sons; 2001. [DOI] [PubMed] [Google Scholar]
- 4.Petrof I, Sherman SM. Synaptic properties of the mammillary and cortical afferents to the anterodorsal thalamic nucleus in the mouse. J Neurosci. 2009;29:7815–7819. doi: 10.1523/JNEUROSCI.1564-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llano DA, Theyel BB, Mallik AK, Sherman SM, Issa NP. Rapid and sensitive mapping of long-range connections in vitro using flavoprotein autofluorescence imaging combined with laser photostimulation. J Neurophysiol. 2009;101:3325–3340. doi: 10.1152/jn.91291.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shibuki K, et al. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theyel BB, Llano DA, Sherman SM. The corticothalamocortical circuit drives higher-order cortex in the mouse. Nat Neurosci. 2010;13:84–88. doi: 10.1038/nn.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinert KC, Dunbar RL, Gao W, Chen G, Ebner TJ. Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol. 2004;92:199–211. doi: 10.1152/jn.01275.2003. [DOI] [PubMed] [Google Scholar]
- 9.Reinert KC, Gao W, Chen G, Ebner TJ. Flavoprotein auto-fluorescence imaging in the cerebellar cortex in vivo. J Neurosci Res. 2007;85:3221–3232. doi: 10.1002/jnr.21348. [DOI] [PubMed] [Google Scholar]
- 10.Katz L, Dalva M. Scanning laser photostimulation: a new approach for analyzing brain circuits. J Neurosci Methods. 1994;54:205–218. doi: 10.1016/0165-0270(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 11.Shibuki K, Hishida R, Kitaura H, Takahashi K, Tohmi M. Handbook of Neurochemistry and Molecular Neurobiology. Springer; 2007. Coupling of brain function and metabolism: endogenous flavoprotein fluorescence imaging of neural activities by local changes in energy metabolism; pp. 321–342. Ch. 4.4. [Google Scholar]
- 12.Briggs F, Callaway EM. Laminar patterns of local excitatory input to layer 5 neurons in macaque primary visual cortex. Cerebral Cortex. 2005;15:479–488. doi: 10.1093/cercor/bhh154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Husson TR, Mallik AK, Zhang JX, Issa NP. Functional imaging of primary visual cortex using flavoprotein autofluorescence. J Neurosci. 2007;27:8665–8675. doi: 10.1523/JNEUROSCI.2156-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molnar P, Nadler JV. Mossy fiber-granule cell synapses in the normal and epileptic rat dentate gyrus studied with minimal laser photostimulation. J Neurophysiol. 1999;82:1883–1894. doi: 10.1152/jn.1999.82.4.1883. [DOI] [PubMed] [Google Scholar]
- 15.Reinert KC, Gao W, Chen G, Ebner TJ. Flavoprotein auto-fluorescence imaging in the cerebellar cortex in vivo. J Neurosci Res. 2007;85:3221–3232. doi: 10.1002/jnr.21348. [DOI] [PubMed] [Google Scholar]
- 16.Theyel BB, Kohrman MH, Frim DM, van Drongelen W. Network variability across human tissue samples in vitro: the problem and a solution. J Clin Neurophysiol. 2010;27:412–417. doi: 10.1097/WNP.0b013e3181fe0721. [DOI] [PubMed] [Google Scholar]
- 17.Logothetis NK, Wandell BA. Interpreting the BOLD signal. Annu Rev Physiol. 2004;66:735–769. doi: 10.1146/annurev.physiol.66.082602.092845. [DOI] [PubMed] [Google Scholar]
- 18.Gaspers LD, Thomas AP. Calcium-dependent activation of mitochondrial metabolism in mammalian cells. Methods. 2008;46:224–232. doi: 10.1016/j.ymeth.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82:2811–2825. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shepherd GMG, Pologruto TA, Svoboda K. Circuit analysis of experience-dependent plasticity in the developing rat barrel cortex. Neuron. 2003;38:277–289. doi: 10.1016/s0896-6273(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 21.Cohen PJ. Effect of anesthetics on mitochondrial function. Anesthesiology. 1973;39:153–164. doi: 10.1097/00000542-197308000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortexin vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.