

Abstract
p38 mitogen-activated protein kinases (p38 MAPKs) are a group of serine/threonine protein kinases that together with ERK (extracellular signal-regulated kinases) and JNK (c-Jun N-terminal kinases) MAPKs act to convert different extracellular signals into specific cellular responses through interacting with and phosphorylating downstream targets. In contrast to the mitogenic ERK pathway, mammalian p38 MAPK family proteins (alpha, beta, gamma, and delta), with and without JNK participation, predominantly regulate inflammatory and stress response. Recent emerging evidence suggests that the p38 stress MAPK pathway may function as a tumor suppressor through regulating Ras-dependent and -independent proliferation, transformation, invasion and cell death by isoform-specific mechanisms. A selective activation of a stress pathway to block tumorigenesis may be a novel strategy to control human malignancies.
Keywords: The p38 MAPK pathway, Ras, Tumor Suppressor, isoform-specific, Oncogenesis, Review
2. INTRODUCTION
MAPKs (mitogen-activated protein kinases) consist of ERK (extracellular signal-regulated kinase), JNK (c-Jun N-terminal kinase), and p38 cascades (1, 2). Classically, MAPKs function by phosphorylating substrates containing consensus sequence Ser/Thr-Pro (3) after they are phosphorylated and activated by upstream kinases (MAPK kinases). Each of these MAPK pathways has several family members but all share the same conservative Thr-Xaa-Tyr phosphorylation motif (where Xaa is any amino-acid) (1, 2, 4). The ERK activity is mostly frequently activated by mitogens and required for cell proliferation, differentiation and/or transformation (5, 6). JNK and p38 pathways, on the other hand, are predominantly responsive to stress and cytokine signaling and play an in important role in regulating stress response and inflammation (7, 8). MAPKs can be activated almost by all type of stimuli and are consequently involved in many critical biological processes such as proliferation, differentiation, cell death and transformation through regulating downstream gene expression and/or interacting with other signaling cascades.
The p38 upstream activators include MAPK kinase 6 (MKK6) and MKK3. Downstream effectors consist of kinases such as MK2 (MAPK-activating protein kinase 2) and PRAK (p38-related/activated protein kinase) as well as transcription factors including ATF-2 (activating transcription factor-2), MEF2 (myocyte enhancement factor 2), and c-Jun (4, 9). The mammalian p38 family consists of four isoform proteins (alpha, beta, gamma, and delta), with p38alpha and p38beta 75% identical in their amino-acid sequence, and p38gamma and p38delta about 60% identical to p38alpha (4, 10). p38alpha and p38beta are susceptible to inhibition by SB drugs (SB203580 and SB202190) whereas p38gamma and p38delta activity is unaffected by these compounds due to their differences in ATP binding pocket (11, 12). While all p38 family proteins can be phosphorylated by MKK6/MKK3 and share many substrates, p38alpha and p38beta have a higher activity to phosphorylate MK2/MK3, whereas p38gamma and p38delta appear to have a selective effect on a microtubule-associated protein Tau and scaffold proteins SAP90 and SAP97 (4, 10). The in vivo signaling selectivity of p38 family proteins, however, remain mostly unknown, which may be critical for understanding broad activities of p38 MAPK activation.
p38alpha {also called p38} was originally identified in study of protein tyrosine phosphorylation in response to stress (13, 14). Later on, p38beta (15), p38gamma (16) {also called SAPK3 (17) and ERK6 (18)}, and p38delta (19) {also called SAPK4 (20)} were cloned and characterized. p38alpha is ubiquitously expressed, whereas other p38 proteins are detectable in a tissue-specific manner (4). p38alpha knockout turns out to be lethal as a result of extraembryonic effects (21, 22), whereas mice with p38beta (23) or p38gamma and/or p38delta knockout (24) are viable and fertile. Although studies of a p38-interacting protein suggest a redundant role of p38 family proteins in development (25), most published results about the p38 pathway are from analyzing p38alpha (4, 26) and specific effects of other p38 family proteins are just emerging (4, 10). Signaling through the p38 pathway has been shown to be involved in regulating inflammation, cytokine synthesis, cell death and cell differentiation, which has been recently reviewed in detail (4, 9, 10). Below we will review recent studies about roles of the p38 pathway activation in Ras-dependent and -independent malignancies and these results together suggest that a tumor suppressing role of the p38 stress pathway may act by isoform-specific and/or tissue-specific mechanisms. A review about specific effects of p38alpha in tumorigenesis has also been recently published (26).
3. RAS ONCOGENE AND THE ERK/JNK MAPK PATHWAYS
Ras proteins play central roles in the control of normal and transformed cell growth, and are among the most frequently mutated and activated genes in human cancers; 30% of Ras mutations occur in codons 12, 13, and 61 (27–29). There are three members of Ras proteins, i.e., H-Ras, K-Ras and N-Ras, and experiments with antisense and knockout studies showed that only K-Ras is required for normal cell growth and/or mouse development (30, 31). In human cancer, 85% of these mutations involve K-Ras whereas less than 15% are from H-Ras (27). K-Ras mutations occur frequently in pancreatic, colon, lung and thyroid cancers, whereas H-Ras mutations mostly arise from bladder and kidney tumors (27–29). In addition to mutations, high levels of normal Ras protein expression also contribute to breast cancer progression downstream of activated membrane tyrosine kinase receptors (29, 32). Targeting Ras oncogene has been therefore an intensive research for cancer therapeutic development for several decades (27, 28, 33).
Ras oncogene signals through multiple pathways or effectors to induce a malignant phenotype in which MAPKs are among the most critical cascades to mediate and modify its activity. The ERK pathway acts downstream of Ras/Raf/MEK to provide a common route by which signals from different growth factor receptors and oncogene Ras converge to activate major transcription factors (34). The ERK MAPK pathway has been shown to be required for Ras-induced transformation (6) (Figure 1). Inhibition of the ERK pathway has consequently become an important tool to screen for novel therapeutics to control Ras-related malignancies (35–37). Activation of the ERK MAPK pathway alone is also known to be sufficient to transform cells, as demonstrated with Raf (38, 39) and constitutively active MEK (5, 40, 41). Recent studies further revealed frequent mutations/activations of B-Raf proteins in human melanoma (42–45). Although normal cells also require the ERK activity for proliferation and differentiation in response to growth factors, a selective inhibition of constitutively active proliferative signaling from Ras/Raf/MEK/ERK cascades remains a major research direction for cancer therapeutic developments.
Figure 1.
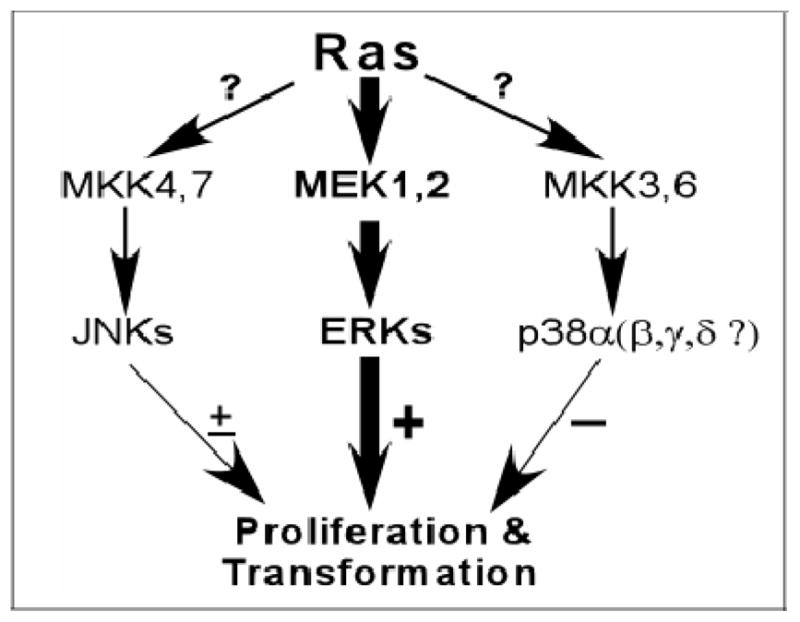
An opposing role of the ERK and p38 pathways in Ras transformation. The ERK activity is required for Ras transformation and the ERK activation alone is sufficient to transform cells. The p38 MAPK pathway, as demonstrated with p38alpha, on the other hand, is inhibitory to Ras oncogene signaling. Thus, the ERK and p38 MAPK pathways oppose each other in determining Ras transforming activity. The inconsistent effect of the JNK pathway on Ras transformation was illustrated with the “+” sign, whereas “?” indicates that p38beta, gamma and delta proteins may have a distinct role in regulating Ras oncogene activity.
The JNK/MAPK pathway has inconsistent roles in Ras transformation (Figure 1). Early studies using a dominant negative JNK kinase (SEK) showed a required role of the JNK pathway in Ras transformation (46). This observation is further confirmed by Ras transformation experiments in mouse fibroblasts with genetic disrupting c-Jun gene (47) or stably expressing a dominant negative c-Jun protein (Tam67) (48). Additional experiments showed that JNK2 is required for Ras transformation independent of c-Jun/AP1 regulation (49). This conclusion, however, is challenged by an observation in which increased tumor growth was demonstrated when Ras-transformed JNK1/2 null cells were injected into mice as compared to the wild-type cells administration, suggesting a suppressive role of the JNK pathway in Ras transformation (50). Additional studies by application of a specific JNK inhibitor SP600125, however, revealed that the JNK inhibition has no substantial effects on Ras-induced transformion in rat intestine epithelial cells (51). This discrepancy is not clearly understood at the present time and may relate to differences in mouse species or cell lines used resulting in context-dependent effects.
4. THE p38ALPHA PATHWAY FUNCTIONS AS A RAS ONCOGENE SUPPRESSOR
The p38alpha (p38) pathway generally acts as a suppressor for Ras signaling (Figure 1) (see the review (26, 52). An inhibitory role of the p38 pathway in Ras activity was first suggested by an observation that p38 activation inhibits gene expression of cyclin D1 (53), an essential downstream effector of Ras proliferative signaling (54). Furthermore, application of a dominant negative MKK6 or the SB inhibitor was shown to reverse MEKK3 (an activator of several MAPK pathways (55)) induced cyclin D1 down-regulation and/or cell-growth inhibition (56). Later on, several studies showed that endogenous normal Ras activity is required for the p38 activation by growth factors. For example, treatment of cells with GM-CSF (granulocyte/macrophage colony-stimulating factor) and PDGF (platelet-derived growth factor) requires Ras to activate p38, leading to an increased proliferation (57) or cell motility (58). Furthermore, the Src oncoprotein requires Ras/p38/JNK pathways to induce STAT3 transcriptional activity (59). These results together suggest that endogenous Ras may act upstream of the p38 pathway to regulate cell growth and gene expression.
The first systemic analyses about the role of p38 pathways in Ras oncogene activity was reported in 2000 (Figure 2A and Table 1) (60). In this study, transient expression of Ras oncogene in NIH 3T3 cells stimulated the kinase activity of MKK6, p38alpha, and its downstream kinases PRAK/MK2. This stimulation was blocked by co-expressing their respective upstream dominant negative and/or the non-phosphorable ERK, indicating an activation of the entire p38 cascade downstream of the ERK pathway. Moreover, dominant negative forms of each of these molecules inhibit Ras-induced proliferation and gene expression, indicating a negative feedback property of the p38 pathway activation (60). Additional analyses revealed that this suppressive effect of the p38 pathway may occur through its inhibitory activity on Ras-induced JNK activation. Importantly, the p38 activation was only growth-inhibitory in human bladder cancer cells that harbor a mutated but not a normal Ras gene. These results together suggest that the entire p38 pathway from MKK6 through p38alpha to PRAK/MK2 may function as a Ras suppressor by negative feedback and its activation may inhibit Ras-dependent malignant growth.
Figure 2.

p38 pathways regulate Ras oncogene activity by negative feedback (A) and signaling integration (B). Along the p38 pathway, MKK6, p38alpha and MK2/PRAK have been shown to be activated by Ras oncogene through MEK/ERK pathways and in turn suppress Ras activity by negative feedback (A) (60). The p38gamma expression, on the other hand, is induced by Ras that is required for Ras transformation (72). Phosphorylated p38alpha was recently shown to down-regulate p38gamma protein expression by ubiquitin/proteasome pathways (74). In a given system, therefore, the Ras transforming activity will be determined by integrated signaling from the Ras-suppressor p38alpha and the Ras-effector p38gamma (B) (72, 74). Solid lines were used for elucidative purpose and detail pathways for these regulations remain to be established.
Table 1.
Studies showing a regulatory role of p38 pathways in tumorigenesis
| Year | Authors | Results | Ref |
|---|---|---|---|
| 2000 | Chen et al | The p38 pathway MKK6/p38alpha/PRAK/MK2 inhibits Ras oncogene-induced proliferation by a negative feedback | 60 |
| 2002 | Pruitt et al | Increased Ras transformation in cell culture by the p38 inhibitor SB203580 | 51 |
| 2002 | Bulavin et al | Increased tumor growth of Ras transformed p38alpha−/− MEFs in mice | 131 |
| 2003 | Brancho et al | Increased tumor growth of Ras transformed MKK3/6−/− MEFs in mice | 63 |
| 20031 | Elenitoba- Johnson et al | Decreased tumor growth of human lymphoma by the p38 inhibitor SB203580 in mice | 85 |
| 2004 | Bulavin et al | Increased mammary tumor formation by SB203580 in PPM1D null mice carrying MMTV-Erbb2 transgene | 61 |
| 2005 | Tang et al | Decreased Ras transformation by p38gamma depletion or p38alpha phosphorylation | 72 |
| 2005 | Timofeev et al | Decreased tumor growth in nude mice by injecting inducible MKK6-expressing cells | 62 |
| 20061 | Matsuo et al | Decreased lung metastasis of murine tumors without growth alterations in p38alpha+/− mice | 118 |
| 2007 | Sun et al | Increased Ras transformation in PRAK null MEFs and increased DMBA-induced skin tumors in PRAK null mice | 65 |
| 2007 | Hui et al | Increased carcinogen-induced liver tumors in mice with liver-specific depletion of p38alpha gene | 132 |
| 2007 | Demidov et al | Decreased mammary tumor formation in MMTV-ErB2/PPM1D and MMTV-MKK6 transgenic mice | 66 |
The tumor suppressive activity of the MKK6/p38alpha/PRAK/MK2 pathway was demonstrated in most of these studies except two occasions (1) in which the opposite effect was observed likely as a result of involvement of tumor/host/tissue-specific factors and/or other p38 family proteins.
The inhibitory effect of the p38 pathway on Ras oncogene activity was further confirmed and extended by a series of in vitro and in vivo studies (Table 1). First, inhibition of the p38 pathway by SB203580 increases Ras-induced soft agar growth whereas JNK inhibition with SP600125 does not have substantial effect, indicating a suppressive activity of endogenous p38 in Ras transformation (51). The suppressive effect of the p38 on tumorigenesis was further established in mice by a SB203580-mediated increase (61) and MKK6-induced decrease in tumor growth (62). Furthermore, knockout of several endogenous genes along the p38 pathway was shown to increase Ras-induced transformation and resultant tumor growth in mice. These include MKK3/6 (63), p38alpha (61, 64), and PRAK (65). In these studies, wild-type and knockout mouse embryonic fibroblasts were typically expressed with Ras oncogene and resultant tumorigenesis was assessed by in vitro transformation and/or by in vivo tumor growth following their injection into mice. Studies by Sun et al further showed that PRAK knockout also renders mice prone to chemically induced skin tumors (65). In addition, the MMTV-MKK6 transgene reduces the mammary tumor formation in MMTV-ErbB2/PPM1D transgenic mice (66). These studies together establish an inhibitory role of the p38 pathway in Ras transformation in experimental cancers.
5. THE p38 PATHWAY REGULATES RAS ACTIVITY BY ISOFORM-SPECIFIC MECHANISMS
The mammalian p38 pathway consists of four isoform proteins with distinct biological activities that can act cooperatively or antagonistically in executing various functions. p38gamma, but not its other family proteins, was first shown to bind and phosphorylate alpha1-syntrophin through its unique C-terminal PDZ-binding motif (67). Further studies showed that p38beta increases whereas p38gamma and p38delta decrease or have no effects on stress-induced AP-1 transcriptional activity in human breast cancer cells (68). In another separate study using rat primary hepatocytes, however, p38alpha, p38beta, and p38delta decreased while p38gamma increased the gene expression of HO-1 (heme oxygenase-1) (69). In mouse mesangial cells, on the other hand, TGF-beta1 was shown to signal through p38alpha and p38delta, but not p38beta, downstream of MKK3 (70). Moreover, in human keratinocytes, p38alpha increases whereas p38delta decreases MKK6-induced gene expression (71). These results together indicate that p38 family proteins may play a distinct role in regulating gene expressions by a cell- or tissue-specific mechanism. Most of these analyses, however, have been performed by over-expressing p38 proteins and roles of endogenous p38 MAPKs in these regulations remain mostly un-established.
In rat intestinal epithelial cells (IEC-6), Ras oncogene was found to increase p38gamma RNA and protein expression with concurrently stimulated p38alpha phosphorylation and decreased p38gamma phosphorylation in which p38beta and p38delta proteins were undetectable (72). Further analysis using the p38alpha inhibitor SB and p38gamma siRNA showed that induced phospho-p38alpha inhibits while resultant elevated p38gamma proteins promote Ras transformation, indicating an opposite role of two p38 family proteins in Ras transformation (Figure 2B). More importantly, p38gamma mRNA was unanimously increased in a group of primary human colon cancer tissues as compared to the matched normal tissues, pointing to its potential role in colon cancer development. Ras also increases p38gamma protein expression (but not p38alpha phosphorylation) in human breast cancer (73). In this case, p38gamma protein acts to mediate Ras-invasive signaling without affecting its proliferative activity, which was inhibited by estrogen receptor alpha (ER) through their direct interactions. These results together indicate that increased p38gamma gene expression is required for Ras oncogenic activity. Recent studies further showed that phospho-p38alpha can down-regulate p38gamma protein expression through c-Jun dependent ubiquitin/proteasome pathways (74). Because p38alpha is ubiquitously expressed and its activating signals are more abundant, stress signaling may act through both stimulating p38alpha phosphorylation and p38gamma down-regulation to inhibit Ras oncogene activity. However, an increased p38gamma gene expression in human colon and breast cancers suggests its potential role in Ras-dependent and perhaps also in Ras-independent human malignancies.
6. THE ROLES OF THE p38 PATHWAY IN REGULATING MALIGNANT GROWTH
In normal tissues, p38 activation is known to lead to an inhibition of liver cell proliferation, as demonstrated by decreased growth by MKK6 that was reversed by the SB inhibitor (75). Moreover, studies with the organ-specific p38alpha knockout and over-expression also showed that p38alpha is inhibitory to fetal cardiomyocyte proliferation (76). In macrophages, however, p38 activation is involved in promoting cell proliferation in response to GM-CSF (77) and inhibiting cell death (78). This distinct p38 activity may relate to its regulatory effects on the biosynthesis as well as the stability of pro-inflammatory molecules, which may play an important role in regulating cancer initiation and progression {see the recent review (10, 79)}.
A moderate activation of the p38 pathway also frequently leads to growth-inhibitory response in cancer. Studies from Pramanik et al, for example, showed that the MKK6 expression inhibits DNA synthesis in human MCF-7 breast cancer cells and this inhibitory effect is enhanced by the co-expressed p38gamma but decreased by the p38beta, indicating isoform-specific mechanisms involved (68). The p38 activation was further shown to inhibit tumor growth in vivo, as adenoviral-mediated MKK6 over-expression completely abolished the tumor formation of human breast cancer in nude mice, although the involved p38 family protein(s) was not identified in this study (80). In addition, p38 activation with MKK6 and inhibition with SB 203580 were found to decrease and increase tumor growth in chick embryos (81). The p38 activation has also been reported to inhibit proliferation of K-Ras mutated pancreatic cancer cells (82) and is required for the growth inhibition by activin, a member of the TGFbeta family proteins (83). In some cases, however, the p38 activity can be growth-stimulatory: inhibition of p38alpha/beta activity with SB203580 suppresses DNA synthesis in human thyroid carcinoma cells in vitro (84) and blocks the growth of human lymphoma in vitro and in mice (85). Whether these different effects result from specific tumor lines or are due to non-specific activities of SB and/or different contributions of p38alpha versus p38beta proteins remains unknown (Table 1).
The growth inhibitory activity of the p38 MAPK may be associated with its role in promoting differentiation. The best example for this is that activation of the p38 pathway by MKK6 induces terminal differentiation of human rhabdomyosarcoma cells that couples with a reduced proliferation (86). Further studies also showed that p38 activity is required for neuronal differentiation in PC12 cells (87, 88). Moreover, p38 activity is required for muscle differentiation (89) and knockout studies revealed that myoblasts lacking p38alpha, but not p38beta and p38delta, are deficient in differentiation that couples with a continuous proliferation (90). Studies from Uddin et al further showed that there is a differentiation-associated expression of p38alpha and p38gamma in primary human erythroid cells (91). Moreover, p38gamma and p38delta were also demonstrated to be involved in myoblasts (18) and keratinocytes differentiation respectively (92). These results together indicate that the p38 pathway may induce cellular differentiation through isoform- and -tissue specific mechanisms, which could contribute to its growth-inhibitory activity.
7. THE ROLES OF THE p38 PATHWAY IN REGULATING CELL DEATH
The p38 pathway activity is usually pro-apoptotic. An inducible expression of ASK1 (apoptosis signal-regulating kinase 1), an upstream kinase that activates both the JNK and p38 pathways, was first shown to induce cell death (93). Later studies showed that the p38 activation by adenovirus-mediated MKK6 gene delivery or a chemical stimulus arsenite induces cell death in estrogen receptor negative (ER−) but not in ER+ breast cancer cells through c-Jun activation (80). In human colon cancer cells, however, the p38 activation only leads to a cell-death response when K-Ras gene is mutated, and this selective K-Ras dependent apoptotic effect was shown to be due to a down-regulation of anti-apoptotic vitamin D receptor (VDR) protein expression (94). Knockout studies further revealed a pro-apoptotic property of the p38 pathway by demonstrating a decreased cell death in cells lacking MKK6 (95), p38alpha (64, 96) and MK2 (97). These results together suggest a sufficient role of the p38 pathway in inducing cell death. As previously demonstrated (80, 85), the pro-apoptotic activity of the p38 pathway may be an important component of its tumor-inhibitory activity in vitro and in vivo.
Additional studies with genetic and/or chemical inhibitors showed that the p38 pathway can mediate various upstream signaling to induce cell death. For example, activation of p38 and/or JNK stress MAPK pathways has been shown to be required for a cell-death response after treatment with cancer therapeutic agents such as adriamycin (98), etoposide (99), cisplatin (100), UV radiation (7, 101), gamma-irradiation (102) and even ligand epidermal growth factor (EGF) (103) in various systems. p38 activation, however, was also found to be anti-apoptotic in melanoma (104) and glioma cells (105) in response to UV radiation. In addition to cell-types, some of these effects may be due to involvements of distinct p38 family proteins in this process. Indeed, recent evidence suggests that in contrast to p38alpha, p38beta appears to be anti-apoptotic in some systems (106–109). Furthermore, p38gamma, by both over-expression and depletion, was recently shown to be anti-apoptotic through antagonizing the pro-apoptotic p38alpha signaling (74). The pro-apoptotic activity of p38alpha is likely integrated by anti-apoptotic effects of its family proteins, leading to either a cell-survival or cell-death response by a cell-type and/or stimuli specific mechanism.
Multiple mechanisms have been shown to be involved in p38 regulating cell death. Early studies showed that that p38 activation may be required for UV-induced cell death through phosphorylating p53 protein at Ser33/46 and thereby increasing its stability and pro-apoptotic activity (101). A serine/threonine specific protein phosphatase Wip1 (PPM1D) of the PP2C family was later found to be induced by a p38-dependent and p53-dependent pathway and in turn dephosphorylate p38, thereby preventing p53-mediated apoptosis (110). Recent studies further showed that p38 can promote apoptosis by directly phosphorylating Bcl-2 family proteins: it phosphorylates BimEL protein at Ser65 to enhances its pro-apoptotic functions (111) but phosphorylates Bcl-2 at Thr56 and Ser87 to inhibits its anti-apoptotic potential (112). Additional experiments showed that the p38 pathway can increase cell death through activating a downstream target p18Hamlet, a transcriptional co-activator (113), or crosstalking with the PI3K pathway (102, 114). Therefore, dissecting signaling interactions of the p38 pathway with different pro-apoptotic and anti-apoptotic cascades may additionally contribute to understanding its pleiotropic cell-death regulatory activities.
8. THE ROLES OF THE p38 PATHWAY IN REGULATING CELL INVASION AND CANCER METASTASIS
p38 activity has been shown to increase cancer cell invasion/migration in many systems. Levels of phospho-p38 proteins, for example, are positively correlated with breast cancer invasive activity and its inhibition with the SB compound was shown to decrease the invasion likely through decreasing the mRNA stability of uPA (urokinase plasminogen activator) and its receptor uPAR (115). Inhibition of the p38 activity with different dominant negative mutants further showed that it is p38alpha, but not p38beta, activation that is responsible for increasing the uPA and uPAR RNA expression. Additional studies revealed that the p38 acts downstream of MKK3 and upstream of MK2 to maintain the uPA stability and the invasive phenotype (116). Of interest, in human umbilical vein endothelial cells (HUVECs) expression of dominant negative forms of p38alpha and p38gamma as well as its downstream kinase MK2, but not p38beta or p38delta, blocks VEGF (vascular endothelial growth factor) induced endothelial migration (117). However, a recent study showed a resistance of p38alpha+/− mice to experimental lung metastasis as compared to the normal mice following an intravenous injection of mouse melanoma F10 and Lewis lung carcinoma cells (118). These results indicate that p38alpha activity in tumor versus host may play an opposite role in regulating tumor invasion and metastasis.
In experimental cancer, p38 activity was shown to be required for H-Ras, but not N-Ras, induced cell invasion in breast epithelial MCF10A cells (119). This effect may involve Rac1/PI3K activation and up-regulation of MMP-2/9 (matrix metalloproteinase 2 and 9) (120). Additional studies also showed that p38 phosphorylation is required for K-Ras dependent invasion in pancreatic cancer (121) as well as for prostate (122) and lung cancer invasion (123). Although studies with over-expressed dominant negative p38 proteins showed that only p38alpha (but not other isoforms) is required for H-Ras-induced invasion in 3T3 cells (124), p38gamma proteins were recently shown to promote breast cancer invasion downstream of Ras (73). Furthermore, p38alpha, but not p38beta, can phosphorylate pro-invasive EGFR (epidermal growth factor receptor) at Y1045 and T669, leading to its internalization and destruction (125–127). Although the relationship between the p38-mediated EGFR down-regulation and the p38 invasion-stimulatory effect remains to be established further, outcomes of the p38 pathway activation on cancer invasion and/or metastasis will likely be determined by the integrated signaling from its associated pro-invasive and anti-invasive pathway activities.
9. CONCLUSION REMARKS
Activation of the entire p38 stress MAPK pathway (MKK6-p38alpha-PRAK) has been systemically demonstrated to suppress Ras-dependent and -independent malignant growth through inhibition of cell proliferation, induction of differentiation and/or cell-death. It was further shown that the tumor-suppressive and/or cell-death promoting effect of p38alpha couples with its activity to disrupt its antagonistic family protein p38gamma. These results together indicate a general tumor suppressor activity of the p38 MAPK stress pathway. It should be pointed out that the p38 pathway activation by phosphorylation may only act as a tumor suppressor during early stages of tumorigenesis, with the final outcomes likely different in different types of cancers and different with the systemic versus the neoplastic p38 activation by isoform-specific mechanisms. It is therefore not surprising to note that different than other tumor suppressors there exists an inversed correlation between levels of phospho-p38 proteins (and JNK) and breast cancer patient survival (128, 129). Perhaps, an altered expression of p38 family proteins may be more relevant to clinical cancer progression, as demonstrated with an increased p38gamma expression in human colon and breast cancers (72), increased p38beta transcripts in human lymphomas (85) and increased p38alpha expression and phosphorylation in thyroid cancers (84). Since a high rate of Ras mutations and/or over-expression has been observed in all these clinical malignancies (27, 85), studies of Ras activation and p38 pathway regulations may provide a unique angle to understand roles of this stress pathway in tumorigenesis. Moreover, with the well established role of the p38 MAPKs in regulating cytokine signaling and inflammatory response that can affect tumorigenesis through multiple pathways (130), investigating systemic effects of the p38 MAPK activation may be particularly needed for understanding its roles in malignant progression.
Acknowledgments
The work was supported by grants from National Institutes of Health (2R01 CA91576), Department of Veterans Affair (Merit Review), Advancing a Healthier Wisconsin (AHW) fund (to GC).
Abbreviations
- p38 MAPKs
p38 mitogen-activated protein kinases
- ERK
extracellular signal-regulated kinases
- JNK
c-Jun N-terminal kinases
- MAPKs
mitogen-activated protein kinases
- MKK
MAPK kinase
- MK
MAPK-activating protein kinase
- PRAK
p38-related/activated protein kinase
- ATF-2
activating transcription factor-2
- MEF2
myocyte enhancement factor 2
- MEK
MAP kinase/ERK kinase
- SEK
dominant negative JNK kinase
- GM-CSF
granulocyte/macrophage colony-stimulating factor
- PDGF
platelet-derived growth factor
- HO-1
heme oxygenase-1
- ER
estrogen receptor alpha
- ASK1
apoptosis signal-regulating kinase 1
- VDR
vitamin D receptor
- uPA
urokinase plasminogen activator
- HUVECs
human umbilical embryonic cells
- VEGF
vascular endothelial growth factor
- MMP
matrix metalloproteinase
- EGFR
epidermal growth factor receptor
References
- 1.Whitmarsh AJ, Davis RJ. A central control for cell growth. Nature. 2000;403:255–256. doi: 10.1038/35002220. [DOI] [PubMed] [Google Scholar]
- 2.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends in Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends in Cell Biology. 1997;7:353–361. doi: 10.1016/S0962-8924(97)01105-7. [DOI] [PubMed] [Google Scholar]
- 4.Ono K, Han J. The p38 signal transduction pathway. Activation and function. Cell Sign. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 5.Mansour S, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 6.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 7.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 8.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 9.Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. TIBS. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 10.Cuenda A, Rousseau S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Kuma Y, Sabio G, Bain J, Shpiro N, Marquesz R, Cuenda A. RIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J Biol Chem. 2005;280:19472–19479. doi: 10.1074/jbc.M414221200. [DOI] [PubMed] [Google Scholar]
- 12.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 13.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 14.Lee JC, Laydon JT, McDonnel PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38γ: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- 17.Mertens S, Craxton M, Goedert M. SAP kinase-3, a new member of the family of mammalian stress-activated protein kinases. FEBS Lett. 1996;383:273–276. doi: 10.1016/0014-5793(96)00255-4. [DOI] [PubMed] [Google Scholar]
- 18.Lechner C, Zahalka MA, Giot J, Moller NP, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Pro Natl Acad Sci USA. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Padova FD, Ulevitch RJ, Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinase, p38δ. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 20.Goedert M, Cuenda A, Craxton M, Jakes R, Cohen P. Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6): comparison of its substrate specificity with that of other SAP kinases. EMBO J. 1997;16:3563–3571. doi: 10.1093/emboj/16.12.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda AR. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- 22.Tamura T, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38α in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 23.Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, McIlrath J, Carr JM, Armit LJ, Clacher C, Malone L, Kollias G, Arthur C. Generation and characterization of p38β (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–10464. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabio G, Simon J, Arthur C, Kuma Y, Peggie M, Carr J, Murray-Tait V, Centeno F, Goebeler M, Morrice N, Cuenda A. p38γ regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005;24:1134–1145. doi: 10.1038/sj.emboj.7600578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zohn I, Li Y, Skolnik EY, Anderson KV, Han J, Niswander L. p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell. 2006;125:957–969. doi: 10.1016/j.cell.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 26.Han J, Sun P. The pathways to tumor supression via route p38. Trends Biochem Sci. 2007;32:364–371. doi: 10.1016/j.tibs.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Downward J. Targeting Ras signalling pathways in cancer therapy. Nature Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 28.Malumbres M, Barbacid M. Ras oncogenes: the first 30 years. Nature Rev Cancer. 2003;3:7–13. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 29.Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Sem Cancer Biol. 2004;14:105–114. doi: 10.1016/j.semcancer.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 30.Chen G, Oh S, Monia BP, Stacey DW. Antisense oligonucleotides demonstrate a dominant role of c-Ki-RAS proteins in regulating the proliferation of diploid human fibroblasts. J Biol Chem. 1996;271:28259–28265. doi: 10.1074/jbc.271.45.28259. [DOI] [PubMed] [Google Scholar]
- 31.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson RT, Umanoff H, Edelmann W, Kucherlapati R, Jacks T. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes & Dev. 1997;11:2648–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE, Boss GR. Ras activation in human breast cancer. Breast Cancer Res Treat. 2000;62:51–62. doi: 10.1023/a:1006491619920. [DOI] [PubMed] [Google Scholar]
- 33.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 34.Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch. Nature Reviews. 2002;3:30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- 35.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van DD, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 36.Kato-Stankiewicz J, Hakimi I, Zhi G, Zhang J, Serebriiskii I, Guo L, Edamatsu H, Koide H, Menon S, Eckl R, Sakamuri S, Lu Y, Chen Q, Agarwal S, Baumbach WR, Golemis EA, Tamanoi F, Khazak V. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotype in human cancer cells. Pro Natl Acad Sci USA. 2002;99:14398–14403. doi: 10.1073/pnas.222222699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 38.Rosario M, Paterson H, Marshall C. Activation of the Raf/MAP kinase cascade by the Ras-related protein TC21 is required for the TC21-mediated transformation of NIH 3T3 cells. EMBO J. 1999;18:1270–1279. doi: 10.1093/emboj/18.5.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vale T, Nge T, White MA, Linsky PE. Raf-induced transformation requires an interleukin 1 autocine loop. Cancer Res. 2001;61:602–607. [PubMed] [Google Scholar]
- 40.Robinson M, Stippec SA, Goldsmith E, White MA, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol. 1998;8:1141–1150. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- 41.Govindarajan B, Bai X, Cohen C, Zhong H, Kilroy S, Louis G, Moses M, Arbiser JL. Malignant transformation of melanocytes to melanoma by constitutive activation of mitogen-activated protein kinase kinase (MAPKK) signaling. J Biol Chem. 2003;278:9790–9795. doi: 10.1074/jbc.M212929200. [DOI] [PubMed] [Google Scholar]
- 42.Dong J, Phelps RG, Qiao R, Yao S, Benard O, Ronai Z, Aaronson SA. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63:3883–3885. [PubMed] [Google Scholar]
- 43.Wellbrock C, Ogilvie L, Hedley D, Karasardes M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 44.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934–936. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 45.Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Project CG, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 46.Clark GJ, Westwick JK, Der CJ. p120 GAP modulates Ras activation of Jun kinases and transformation. J Biol Chem. 1997;272:1677–1681. doi: 10.1074/jbc.272.3.1677. [DOI] [PubMed] [Google Scholar]
- 47.Johnson R, Spiegelman B, Hanahan D, Wisdom R. Cellular transformation and malignancy induced by ras require c-jun. Mol Cell Biol. 1996;16:4504–4511. doi: 10.1128/mcb.16.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim S, Brown PH, Birrer M. The inhibitory activity of a transdominant c-jun mutant fused to the ligand binding domain of the estrogen receptor. Oncogene. 1996;12:1043–1053. [PubMed] [Google Scholar]
- 49.Nielsen C, Thastrup J, Bottzauw T, Jaattela M, Kallunki T. c-Jun NH2-terminal kinase 2 is required for Ras transformation independently of activator protein 1. Cancer Res. 2007;67:175–185. doi: 10.1158/0008-5472.CAN-06-2801. [DOI] [PubMed] [Google Scholar]
- 50.Kennedy NJ, Sluss HK, Jones SN, Bar-Sagi D, Flavell R, Davis RJ. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes & Dev. 2003;17:629–637. doi: 10.1101/gad.1062903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J Biol Chem. 2002;277:31808–31817. doi: 10.1074/jbc.M203964200. [DOI] [PubMed] [Google Scholar]
- 52.Kennedy NJ, Cellurale C, Davis RJ. A radical role for p38 MAPK in tumor initiation. Cancer Cell. 2007;11:101–103. doi: 10.1016/j.ccr.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Lavoie J, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 54.Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the cdk inhibitor p27kip1. Mol Cell Biol. 1997;17:3850–3857. doi: 10.1128/mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uhlik MT, Abell AM, Cuevas BD, Nakamura K, Johnson GL. Wiring diagrams of MAPK regulation by MEKK1, 2, and 3. Biochem Cell Biol. 2004;82:658–663. doi: 10.1139/o04-114. [DOI] [PubMed] [Google Scholar]
- 56.Ellinger-Ziegelbauer H, Kelley K, Siebenlist U. Cell cycle arrest and reversion of ras-induced transformation by a conditionally activated form of mitogen-activated protein kinase kinase kinase 3. Mol Cell Biol. 1999;19:3857–3868. doi: 10.1128/mcb.19.5.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rausch O, Marshall CJ. Cooperation of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways during granulocyte colony-stimulating factor-induced hemopoietic cell proliferation. J Biol Chem. 1999;274:4096–4105. doi: 10.1074/jbc.274.7.4096. [DOI] [PubMed] [Google Scholar]
- 58.Matsumoto T, Yokote K, Tamura K, Takemoto M, Ueno H, Saito Y, Mori S. Platelet-derived growth factor activates p38 mitogen-activated protein kinase through a ras-dependent pathway that is important for actin reorganization and cell migration. J Biol Chem. 1999;274:13954–13960. doi: 10.1074/jbc.274.20.13954. [DOI] [PubMed] [Google Scholar]
- 59.Turkson J, Bowman T, Adnane J, Zhang Y, Djeu JY, Sekharam M, Frank DA, Holzman LB, Wu J, Sebti S, Jove R. Requirement for Ras/Rac-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol Cell Biol. 1999;19:7519–7528. doi: 10.1128/mcb.19.11.7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback to Ras proliferative signaling. J Biol Chem. 2000;275:38973–38980. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 61.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CM, Appella E, Fornace JA. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19Arf pathway. Nature Genet. 2004;36:343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 62.Timofeev O, Lee TY, Bulavin DV. A subtle change in p38 MAPK activity is sufficient to suppress in vivo tumorigenesis. Cell Cycle. 2005;4:118–120. doi: 10.4161/cc.4.1.1342. [DOI] [PubMed] [Google Scholar]
- 63.Brancho D, Tanaka N, Jaeschke A, Ventura J, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell R, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes & Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 65.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout M, Dong M-Q, Frangou CG, Yates JR, III, Wright PE, Han J. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 66.Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E, Bulavin DV. The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene. 2007;26:2502–2506. doi: 10.1038/sj.onc.1210032. [DOI] [PubMed] [Google Scholar]
- 67.Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, Goedert M. Stress-activated protein kinase-3 interacts with the PDZ domain of α1-syntrophin: a mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- 68.Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, Han J, Chen G. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun: The determinant role of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 2003;278:4831–4839. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- 69.Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxgenase-1 gene expression by MAP kinase of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem. 2003;278:17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Ma R, Flavell R, Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38α and p38δ MAPK isoforms by TGF-β1 in murine mesangial cells. J Biol Chem. 2002;277:47257–47262. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 71.Dashti S, Efimova T, Eckert RL. MEK6 regulates human involucrin gene expression via a p38α- and p38δ-dependent mechanism. J Biol Chem. 2001;276:27214–27220. doi: 10.1074/jbc.M100465200. [DOI] [PubMed] [Google Scholar]
- 72.Tang J, Qi X, Mercola D, Han J, Chen G. Essential role of p38γ in K-Ras transformation independent of phosphorylation. J Biol Chem. 2005;280:23910–23917. doi: 10.1074/jbc.M500699200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qi X, Tang J, Loesch M, Pohl N, Alkan S, Chen G. p38γ MAPK integrates signaling cross-talk between Ras and estrogen receptor to increase breast cancer invasion. Cancer Res. 2006;66:7540–7547. doi: 10.1158/0008-5472.CAN-05-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qi X, Pohl NM, Loesch M, Hou S, Li R, Qin JZ, Cuenda A, Chen G. p38α antagonizes p38γ activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response. J Biol Chem. 2007;282:31398–31408. doi: 10.1074/jbc.M703857200. [DOI] [PubMed] [Google Scholar]
- 75.Awad MM, Enslen H, Boylan JM, Davis RJ, Gruppuso PA. Growth regulation via p38 mitogen-activated protein kinase in developing liver. J Biol Chem. 2000;275:38716–38721. doi: 10.1074/jbc.M008040200. [DOI] [PubMed] [Google Scholar]
- 76.Engel FB, Schebesta M, Duong MT, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes & Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Senokuchi T, Matsumura T, Sakai M, Yano M, Taguchi T, Matsuo T, Sonoda K, Kukidome D, Imoto K, Nishikawa T, Kim-Mitsuyama S, Takuwa Y, Araki E. Statins suppress osidized low density lipoprotein-induced macrophage proliferation by inactivation of the samll G protein-p38 MAPK pathway. J Biol Chem. 2005;280:6627–6633. doi: 10.1074/jbc.M412531200. [DOI] [PubMed] [Google Scholar]
- 78.Park JM, Greten FR, Li Z, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 79.Li W-W, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qi X, Borowicz S, Pramanik R, Schultz RM, Han J, Chen G. Estrogen receptor inhibits c-Jun-dependent stress-induced cell death by binding and modifying c-Jun activity in human breast cancer cells. J Biol Chem. 2004;279:6769–6777. doi: 10.1074/jbc.M311492200. [DOI] [PubMed] [Google Scholar]
- 81.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERKMAPK activity as a determinant of tumor growth and dormancy; regulation by p38SAPK. Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- 82.Ding X, Adrian TE. MEK/ERK-mediated proliferation is negatively regulated by p38 MAP kinase in the human pancreatic cancer cell line, PANC-1. Biochem Biophy Res Comm. 2001;282:447–453. doi: 10.1006/bbrc.2001.4595. [DOI] [PubMed] [Google Scholar]
- 83.Cocolakis E, Lemay S, Ali S, Lebrun J. The p38 MAPK pathway is required for cell growth inhibition of human breast cancer cells in response to activin. J Biol Chem. 2001;276:18430–18436. doi: 10.1074/jbc.M010768200. [DOI] [PubMed] [Google Scholar]
- 84.Pomerance M, Quillard J, Chantoux F, Blondeau JP. High-level expression, activation, and subcellular localization of p38-MAP kinase in thyroid neoplasms. J Pathol. 2007;209:298–306. doi: 10.1002/path.1975. [DOI] [PubMed] [Google Scholar]
- 85.Elenitoba-Johnson KSJ, Jenson SD, Abbott RT, Palais RA, Bohling SD, Lin Z, Tripp S, Shami PJ, Wang LY, Coupland RW, Buckstein R, Perez-Ordonez B, Perkins SL, Lim MS. Involvement of multiple signaling pathways in follicular lymphoma transformation: p38-mitogen-activated protein kinase as a target for therapy. Pro Natl Acad Sci USA. 2003;100:7259–7264. doi: 10.1073/pnas.1137463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, Feramisco JR, Karin M, Wang JYJ. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes & Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 87.Morooka T, Nishida E. Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J Biol Chem. 1998;273:24285–24288. doi: 10.1074/jbc.273.38.24285. [DOI] [PubMed] [Google Scholar]
- 88.Iwasaki S, Iguchi M, Watanabe K, Hoshino R, Tsujimoto M, Kohno M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogetic protein-2. J Biol Chem. 1999;274:26503–26510. doi: 10.1074/jbc.274.37.26503. [DOI] [PubMed] [Google Scholar]
- 89.Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco J, Karin M, Wang J, Puri PL. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perdiguero E, Ruiz-Bonilla V, Gresh L, Hui L, Ballestar E, Sousa-Victor P, Baeza-Raja B, Jardi M, Bosch-Comas A, Esteller M, Caelles C, Esteller M, Caelles C, Serrano AL, Wagner EF, Munoz-Canoves P. Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38α in abrogating myoblast proliferation. EMBO J. 2007;26:1245–1256. doi: 10.1038/sj.emboj.7601587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Uddin S, Ah-Kang J, Ulaszek J, Mahmud D, Wickrema A. Differentiation stage-specific activation of p38 mitogen-activated protein kinase isoforms in primary human erythroid cells. Pro Natl Acad Sci USA. 2004;101:147–152. doi: 10.1073/pnas.0307075101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Efimova T, Broome A, Eckert RL. A regulatory role for p38δ MAPK in keratinocyte differentiation Evidence for p38δ-ERK1/2 complex formation. J Biol Chem. 2003;278:34277–34285. doi: 10.1074/jbc.M302759200. [DOI] [PubMed] [Google Scholar]
- 93.Ichijo H, Nishida E, Irie K, Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK?JNK and p38 signaling pathway. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 94.Qi X, Tang J, Pramanik R, Schultz RM, Shirasawa S, Sasazuki T, Han J, Chen G. p38 MAPK activation selectively induces cell death in K-ras mutated human colon cancer cells through regulation of vitamin D receptor. J Biol Chem. 2004;279:22138–22144. doi: 10.1074/jbc.M313964200. [DOI] [PubMed] [Google Scholar]
- 95.Tanaka H, Kamanaka M, Enslen H, Dong C, Wysk M, Davis RJ, Flavell RA. Differential involvment of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Reports. 2002;3:785–791. doi: 10.1093/embo-reports/kvf153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Porras A, Zuluaga S, Black E, Valladares A, Alvarez AM, Ambrosino C, Benito M, Nebreda AR. p38α mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol Biol Cell. 2004;15:922–933. doi: 10.1091/mbc.E03-08-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vertii A, Hakim C, Kotlyarov A, Gaestel M. Analysis of properties of small heat shock protein Hsp25 in MAPK-activated protein kinase 2 (MK2)-deficient cells. MK2-dependent insolubilization of Hsp25 oligomers correlates with susceptibility to stress. J Biol Chem. 2006;281:26966–26975. doi: 10.1074/jbc.M602134200. [DOI] [PubMed] [Google Scholar]
- 98.Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol. 2005;25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Juo P, Kuo CJ, Reynolds SE, Konz RF, Raingeaud J, Davis RJ, Biemann H, Blenis J. Fas activation of the p38 mitogen-activated protein kinase signaling pathway requires ICE/CED-3 family proteases. Mol Cell Biol. 1997;17:24–35. doi: 10.1128/mcb.17.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 101.Bulavin D, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, Fornace A., Jr Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–6854. doi: 10.1093/emboj/18.23.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kumar P, Miller AI, Polverini PJ. p38 MAPK mediates γ-irradiation-induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol Chem. 2004;279:43352–43360. doi: 10.1074/jbc.M405777200. [DOI] [PubMed] [Google Scholar]
- 103.Tikhomirov O, Carpenter G. Ligand-induced, p38-dependent apoptosis in cells expressing high levels of epidermal growth factor receptor and ErbB-2. J Biol Chem. 2004;279:12988–12996. doi: 10.1074/jbc.M311655200. [DOI] [PubMed] [Google Scholar]
- 104.Ivanov VN, Ronai Z. p38 protects human melanoma cells from UV-induced apoptosis through down-regulation of NF-kappaB activity and Fas expression. Oncogene. 2000;19:3003–3012. doi: 10.1038/sj.onc.1203602. [DOI] [PubMed] [Google Scholar]
- 105.Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003;23:8306–8315. doi: 10.1128/MCB.23.22.8306-8315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guo Y, Kang B, Han J, Williamson JR. p38β MAP kinase protects rat mesangial cells from TNF-α-induced apoptosis. J Cell Biochem. 2001;82:556–565. doi: 10.1002/jcb.1180. [DOI] [PubMed] [Google Scholar]
- 107.Nemoto S, Xiang J, Huang S, Lin A. Induction of apoptosis by SB202190 through inhibition of p38β mitogen-activated protein kinase. J Biol Chem. 1998;273:16415–16420. doi: 10.1074/jbc.273.26.16415. [DOI] [PubMed] [Google Scholar]
- 108.Silva G, Cunha A, Gregoire IP, Seldon MP, Soares MP. The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38α MAPK isoform. J Immunol. 2006;177:1894–1903. doi: 10.4049/jimmunol.177.3.1894. [DOI] [PubMed] [Google Scholar]
- 109.Verma A, Mohindru M, Deb DK, Sassano A, Kambbampati S, Ravandi F, Minucci S, Kalvakolanu DV, Platanias LC. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to arsenic trioxide. J Biol Chem. 2002;277:44988–44995. doi: 10.1074/jbc.M207176200. [DOI] [PubMed] [Google Scholar]
- 110.Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19:6517–6526. doi: 10.1093/emboj/19.23.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J Biol Chem. 2006;281:25215–25222. doi: 10.1074/jbc.M512627200. [DOI] [PubMed] [Google Scholar]
- 112.Chiara GD, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodel S, Palamara AT, Russo T, Garaci E, Cozzolino F. Bcl-2 phosphorylation by p38. Identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 113.Cuadrado A, Lafarga V, Cheung PCF, Dolado I, Llanos S, Cohen P, Nebrada A. A new p38 MAP kinase-regulated transcriptional coactivator that stimulates p53-dependent apoptosis. EMBO J. 2007;26:2115–2126. doi: 10.1038/sj.emboj.7601657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem. 2007;282:21598–21608. doi: 10.1074/jbc.M611316200. [DOI] [PubMed] [Google Scholar]
- 115.Huang S, New L, Pan Z, Han J, Nemerow GR. Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38α mitogen-activated protein kinase activity. J Biol Chem. 2000;275:12266–12272. doi: 10.1074/jbc.275.16.12266. [DOI] [PubMed] [Google Scholar]
- 116.Han Q, Leng J, Bian D, Mahanivong C, Carpenter KA, Pan ZK, Han J, Huang S. Rac1-MKK3-p38-MAPKAPK2 pathway promotes urokinase plasminogen activator mRNA stability in invasive breast cancer cells. J Biol Chem. 2002;277:48379–48385. doi: 10.1074/jbc.M209542200. [DOI] [PubMed] [Google Scholar]
- 117.Yu J, Bian D, Mahanivong C, Cheng R, Zhou W, Huang S. p38 mitogen-activated protein kinase regulation of endothelial cell migration depends on urokinase plasminogen activator expression. J Biol Chem. 2004;279:50446–50454. doi: 10.1074/jbc.M409221200. [DOI] [PubMed] [Google Scholar]
- 118.Matsuo Y, Amano S, Furuya M, Namiki K, Sakurai K, Nishiyama M, Sudo T, Tatsumi K, Kuriyama T, Kimura S, Kasuya Y. Involvement of p38α mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem. 2006;281:36767–36775. doi: 10.1074/jbc.M604371200. [DOI] [PubMed] [Google Scholar]
- 119.Kim MS, Lee EJ, Kim HRC, Moon A. p38 kinase is a key moleculae for H-Ras-induced cell motility and invasive phenotype in human breast epethelial cells. Cancer Res. 2003;63:5454–5461. [PubMed] [Google Scholar]
- 120.Shin I, Kim S, Song H, Kim H-RC, Moon A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway. Its critical role in invasion and migration of breast epithelial cells. J Biol Chem. 2005;280:14675–14683. doi: 10.1074/jbc.M411625200. [DOI] [PubMed] [Google Scholar]
- 121.Dreissigacker U, Mueller MS, Unger M, Siegert P, Genze F, Gierschik P, Giehl K. Oncogenic K-Ras down-regulates Rac and RhoA and enhances migration and invasion of pancreatic carcinoma cells through activation of p38. Cellular Signalling. 2006;18:1156–1168. doi: 10.1016/j.cellsig.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 122.Chen LXS, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. 2006;25:2987–2998. doi: 10.1038/sj.onc.1209337. [DOI] [PubMed] [Google Scholar]
- 123.Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, Hung MC, Kuo ML. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell. 2006;9:209–223. doi: 10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 124.Behren A, Binder K, Vucelic G, Herberhold S, Hirt B, Loewenheim H, Preyer S, Zenner HP, Simon C. The p38 pathway is required for Ha-ras induced in vitro invasion of NIH3T3 cells. Exp Cell Res. 2005;303:321–330. doi: 10.1016/j.yexcr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 125.Frey M, Dise RS, Edelblum KL, Polk DB. p38 kinase regulates epidermal growth factor receptor downregulation and cellular migration. EMBO J. 2006;25:1–10. doi: 10.1038/sj.emboj.7601457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zwang Y, Yarden Y. p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J. 2006;25:4195–4206. doi: 10.1038/sj.emboj.7601297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Winograd-Katz SE, Levitzki A. Cisplatin induces PKB/Akt activation and p38 (MAPK) phosphorylation of the EGF receptor. Oncogene. 2006;25:7381–7390. doi: 10.1038/sj.onc.1209737. [DOI] [PubMed] [Google Scholar]
- 128.Yeh YT, Hou MF, Chung YF, Chen YJ, Yang SF, Chen DC, Su JH, Yuan SS. Decreased expression of phosphorylated JNK in breast infiltrating ductal carcinoma is associated with a better overal survival. Int J Cancer. 2006;118:2678–2684. doi: 10.1002/ijc.21707. [DOI] [PubMed] [Google Scholar]
- 129.Davidson B, Konstantinovsky S, Kleinberg L, Nguyen MT, Bassarova A, Kvalheim G, Nesland JM, Reish R. The mitogen-activated protein kinases (MAPK) p38 and JNK are markers of tumor progression in breast carcinoma. Gynecol Oncol. 2007;102:453–461. doi: 10.1016/j.ygyno.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 130.Lin W-W, Karin M. A cytokine-mediated link between innate immunity, inflamation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace JA, Appella E. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nature Genet. 2002;31:210–215. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 132.Hui L, Bakri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Kommenovic V, Scheuch H, Beug H, Wagner EF. p38α suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]