

Abstract
The alphabaculovirus Anticarsia gemmatalis multiple nucleopolyhedrovirus (AgMNPV) is the world’s most successful viral bioinsecticide. Through the 1980s and 1990s, this virus was extensively used for biological control of populations of Anticarsia gemmatalis (Velvetbean caterpillar) in soybean crops. During this period, genetic studies identified several variable loci in the AgMNPV; however, most of them were not characterized at the sequence level. In this study we report a full genome comparison among 17 wild-type isolates of AgMNPV. We found the pangenome of this virus to contain at least 167 hypothetical genes, 151 of which are shared by all genomes. The gene bro-a that might be involved in host specificity and carrying transporter is absent in some genomes, and new hypothetical genes were observed. Among these genes there is a unique rnf12-like gene, probably implicated in ubiquitination. Events of gene fission and fusion are common, as four genes have been observed as single or split open reading frames. Gains and losses of genomic fragments (from 20 to 900 bp) are observed within tandem repeats, such as in eight direct repeats and four homologous regions. Most AgMNPV genes present low nucleotide diversity, and variable genes are mainly located in a locus known to evolve through homologous recombination. The evolution of AgMNPV is mainly driven by small indels, substitutions, gain and loss of nucleotide stretches or entire coding sequences. These variations may cause relevant phenotypic alterations, which probably affect the infectivity of AgMNPV. This work provides novel information on genomic evolution of the AgMNPV in particular and of baculoviruses in general.
Keywords: baculovirus, evolution, wild isolates, horizontal gene transfer, deep sequencing, genetic diversity
Introduction
Baculoviruses are insect-specific viruses that are frequently restricted to one or a few related insect species, and thus became widely applied as biological controls against pests in agriculture and forestry (O'Reilly et al. 1993; Tanada and Kaya 1993; Moscardi 1999; Szewczyk et al. 2006). Baculoviruses have a circular, covalently closed, double-stranded DNA genome and, up to now, the most studied genera are the Alpha and Betabaculovirus. The Alphabaculovirus Anticarsia gemmatalis multiple nucleopolyhedrovirus (AgMNPV) was discovered in Peru, in 1962 (Steinhaus and Marsh 1962), and its first occurrence in Brazilian territory was registered in 1972 (Allen and Knell 1977). In Brazil, during the 1980s and 1990s, AgMNPV was extensively applied in soybean crops against populations of velvetbean caterpillars (A. gemmatalis), and these actions are recognized as the world’s largest biological control program using a virus as a bioinsecticide (Moscardi 1989, 1999). In nature, multiple viral particles of this virus are found embedded in paracrystalline protein structures called occlusion bodies (OBs) (Rohrmann 2011). When a caterpillar ingests these structures, the alkaline environment of the larvae midgut promotes OBs dissolution and occlusion-derived viruses (ODVs) are released, establishing primary infection. Within the first 2 hours the first budded viruses (BVs) are produced, and secondary infection takes place systemically, giving rise to new OBs (Friesen and Miller 2001). In most baculoviruses the infection cycle ends with insect disintegration (melting), a process that leads to the release of OBs into the environment. Interestingly, AgMNPV lacks the genes responsible for host cuticle rupture and tissue liquefaction (cathepsin and chitinase), and this fact favors the collection of dead insects, useful for future biological control application (Slack et al. 2004). In 2006, the isolate 2D was the first plaque-purified isolate of AgMNPV to have its whole genome sequenced (accession number DQ813662) (Oliveira et al. 2006). It contains 152 open-reading frames (ORFs), with short or absent intergenic regions, nine homologous regions (hrs), and shows high similarity to CfDefMNPV (Oliveira et al. 2006). Previous studies on AgMNPV diversity documented high levels of polymorphism in some genomic loci; however, the specific genetic elements affected by those changes, as well as their impact on AgMNPV evolution, are mostly unknown (Maruniak 1989; Maruniak et al. 1999). Wild populations of baculoviruses are commonly composed of different genotypes, which frequently recombine in a context of coinfection (Croizier and Ribeiro 1992). The genome evolution of baculoviruses involves gene horizontal gene transfer (HGT) (Hughes and Friedman 2003); insertion of noncoding sequences of cellular origin (transposons) (Blissard and Rohrmann 1990); and duplication/loss of genes or genomic fragments (Hayakawa et al. 2000), following a processes by which genomes tend to undergo net gain and losses of accessory genes, showing deviation from linkage disequilibrium among functional categories (Zanotto and Krakauer 2008). In this study, we describe the complete genome sequence and analyses of 17 naturally occurring South American isolates of AgMNPV. We report the main events outlining the AgMNPV genomic evolution, such as gene gain/loss, gene fusion/fission, among other structural variations. These analyses provide a better understanding of AgMNPV evolution and their molecular biology and encourage the search for new baculoviruses potentially able to be used in biological pest control.
Materials and Methods
Viral Samples, In Vivo Amplification, and DNA Extraction
The 17 wild-type samples were isolated from A. gemmatalis larvae. These insects had been collected from soybean crops with no previous intentional usage of baculoviruses as biological control agents. These samples were kindly provided by Dr Flavio Moscardi and Daniel Sosa-Gómez (EMBRAPA Soja, Londrina, Brazil). These isolates are wild-type genetically heterogeneous viral populations of AgMNPV, obtained from different geographical locations from 1980 to 1990, in southern Brazil, Argentina, and Uruguay (fig. 1 and table 1). The viral stocks were amplified separately in vivo (per os) in third/fourth-instar A. gemmatalis caterpillars fed on an artificial diet, as previously described (Greene et al. 1976; O'Reilly et al. 1993). The propagated polyhedra were purified from insect cadavers by steps of maceration, centrifugation and sucrose gradient ultracentrifugation, as previously described (Maruniak 1986). After purification, polyhedra were treated with DNase, to prevent any contamination with insect host genome. Virus particles were solubilized under alkaline conditions (Whitt and Manning 1987); genomic DNA was purified by successive phenol/chloroform/isoamyl alcohol extractions followed by ethanol precipitation and elution in TE buffer (Lo et al. 1996).
Fig. 1.—
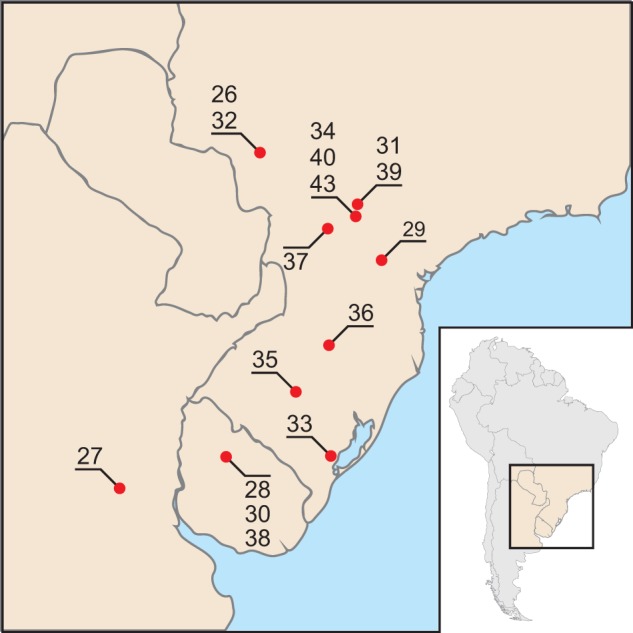
Geographic regions of original field sampling of the AgMNPV isolates (approximate coordinates).
Table 1.
Wild Isolates of AgMNPV
| Isolate | Accession Number | Country (Municipality, State) | Year |
|---|---|---|---|
| AgMNPV-26 | KR815455 | Brazil (Dourados, Mato Grosso do Sul) | 1984 |
| AgMNPV-27 | KR815456 | Argentina | 1982 |
| AgMNPV-28 | KR815457 | Uruguay | 1984 |
| AgMNPV-29 | KR815458 | Brazil (Ponta Grossa, Paraná) | 1985 |
| AgMNPV-30 | KR815459 | Uruguay | 1982 |
| AgMNPV-31 | KR815460 | Brazil (Sertanópolis, Paraná) | 1986 |
| AgMNPV-32 | KR815461 | Brazil (Dourados, Mato Grosso do Sul) | 1982 |
| AgMNPV-33 | KR815462 | Brazil (Pelotas, Rio Grande do Sul) | 1984 |
| AgMNPV-34 | KR815463 | Brazil (Londrina, Paraná) | 1990 |
| AgMNPV-35 | KR815464 | Brazil (Rio Grande do Sul) | 1984 |
| AgMNPV-36 | KR815465 | Brazil (Passo Fundo, Rio Grande do Sul) | 1982 |
| AgMNPV-37 | KR815466 | Brazil (Campo Mourão, Paraná) | 1982 |
| AgMNPV-38 | KR815467 | Uruguay | 1982 |
| AgMNPV-39 | KR815468 | Brazil (Sertanópolis, Paraná) | 1987 |
| AgMNPV-40 | KR815469 | Brazil (Londrina, Paraná) | N/Aa |
| AgMNPV-42 | KR815470 | N/Ab | 1987 |
| AgMNPV-43 | KR815471 | Brazil (Londrina, Paraná) | 1980 |
Note.—Isolate, location, year of collection and accession number are listed in the table.
Year of isolation unknown.
Geographic location of origin not specified.
454 Next-Generation Sequencing
First, to prepare viral DNA libraries, approximately 50 ng of genomic DNA from each sample were separately fragmented using the Roche Titanium-compatible Nextera DNA Sample Prep Kit (Epicentre Biotechnologies), according to the manufacturer’s protocol. In different reactions, viral DNA were MID-tagged (Roche 454 Life Sciences) during the Nextera amplification steps. The libraries were purified with Agencourt AMPure XP Beads (Beckman Coulter, Inc), at a 1:0.7 ratio (DNA:Beads) to eliminate fragments below 300 bp. The purified material was quantified and submitted to emPCR reactions, following the manufacturer’s instructions (emPCR Method Manual—Lib-L SV, Roche 454 Life Sciences). Finally, those pools were sequenced using a GS FLX Titanium platform (Roche 454 Life Sciences), in a single run, in accordance with manufacturer’s recommendations (Sequencing Method Manual, GS FLX Titanium Series, Roche 454 Life Sciences). The specific reads of each sample were segregated into 17 SFF files according to their MID tags, which were removed at this step.
Genome Assembly
There is considerable variability in wild-type, field isolates of natural organisms that did not experience significant, recent bottlenecks. Nevertheless, to facilitate comparisons, we aimed at the reconstruction of the predominant genotype of each viral population. This was done by de novo genome assemblies using the “Geneious” proprietary assembler implemented in Geneious 7.1.7 (Kearse et al. 2012). Summarizing, sequencing reads had their adapters and low quality regions trimmed before the assembly. The genomes were then assembled taking into account two parameters: Minimum overlap of 150 nt and 97% of identity among reads. The most frequent haplotypes reconstructed in large contigs were joined to obtain 17 full-length genomic scaffolds. 454-sequencing errors at homopolymeric regions were manually inspected and individually corrected. Information about total number of reads, average read length, and coverage can be found on supplementary figure S1, Supplementary Material online.
Genome Annotation and Sequence Data Analysis
Each genome was annotated using a pipeline generated by the EGene 2 platform (Durham et al. 2005). In this pipeline, coding regions were predicted ab initio using Glimmer 3.02 (Delcher et al. 1999), using all coding sequences of alphabaculoviruses Group I as a training set. All ORFs with at least 138 bp and presenting maximum overlapping of 148 bp between adjacent ORFs were annotated. All translated products were submitted to similarity searches with BLASTp against NCBI’s nr (nonredundant) database. Hits were considered positive when presenting e values below 10−6. Protein domains and families were identified through searches on InterPro (Mitchell et al. 2014) and CDD (Marchler-Bauer et al. 2007) databases. Gene Ontology (GO) terms of the three ontologies were assigned to each coding sequence (Binns et al. 2009). All translated products were also classified into orthologous groups according to eggNOG (Muller et al. 2010) and KEEG Orthology (KO) (Aoki-Kinoshita and Kanehisa 2007), and mapped onto KEGG pathways. Additional analyses included TMHMM 2.0 (Krogh et al. 2001), Phobius1.01 (Kall et al. 2004), and SignalP4.0 (Petersen et al. 2011) to predict signal peptides, protein motifs, and secondary structures (e.g., transmembrane helices, etc.). Finally, we also performed manual curation to include missing ORFs.
hrs were detected using Dotter (Sonnhammer and Durbin 1995), whereas Tandem Repeats Finder (Benson 1999) was applied to accurately locate hrs and direct repeats (drs) in the genomes. To analyze the genetic diversity present in the populations of geographical isolates of AgMNPV, we did alignments of gene coding regions with ClustalW (Thompson et al. 1994), which were further edited manually with Bioedit (Hall 1999). The genetic diversity was measured with DnaSP 5.0 (Librado and Rozas 2009) using the curated alignments. The DnaSP 5.0 software implements the Watterson’s estimator (θ = Ne.μ, assuming baculoviruses to be haploid) taking into account the number of segregating sites per gene (Watterson 1975). Moreover, a whole-genome alignment of all 17 AgMNPV genomes was performed using the “mauveAligner” algorithm implanted in Geneious 7.1.7 (Kearse et al. 2012), which in turn was applied to detect nonsynonymous changes. Both results of genetic diversity were plotted in a circular map using Circos (Krzywinski et al. 2009). Gene splicing in baculovirus appears to be rare and was reported in the ie-0/ie-1 genes of the AcMNPV (Chisholm and Henner et al. 1988; Chen et al. 2013). Nevertheless, similarly to what has been done for most baculovirus-annotated genomes available in GenBank to this date, we chose to not annotate splicing because RNA-Seq data were not available for the precise identification of intron boundaries, and the lack of sufficient comparative data would hamper the construction of reliable training sets for gene splicing prediction.
New Genes in the Genomes of the AgMNPV
We coupled our previous proteomics results and findings with new sequences obtained from the AgMNPV isolates. Briefly, to conduct our analysis we used the MASCOT 2.0 online software (MatrixSciense, Boston, MA) and the raw data files generated during the proteome study of AgMNPV (Braconi et al. 2014). We searched these data against the database containing the sequences of 17 wild-type genomes of AgMNPV. All parameters in the MASCOT software were set according to Braconi et al. (2014). In the end, the validated proteins had at least two independent spectra with a minimal length of ten amino acids, with greater than 99.0% probability estimated by the Peptide Prophet algorithm (Keller et al. 2002).
Results
Deep Sequencing of AgMNPV Genomes
The 17 AgMNPV viral populations were isolated from dead A. gemmatalis caterpillars and their genomes were sequenced using the next-generation technology Roche 454 GS-FLX Titanium. Each data set contained 26,474–78,766 reads, with an average length varying from 326 to 369 bp (supplementary fig. S1, Supplementary Material online). Following assembly, the size of the double-stranded circular DNA of AgMNPV genome varied, and we determined the similarities/differences at sequence level between all isolates. We also considered the fact that Roche/454 platform can induce erroneous base calls on homopolymeric regions, so all single nucleotide polymorphisms that mapped to those regions were ignored.
General Aspects of the AgMNPV Genomes
Compared with the AgMNPV-2D genome used as reference, the 17 wild-type genomes are from 0.04% to 1.31% smaller in length, their GC content varies from 44.5% to 44.6%, and their sequence similarity against the reference genome ranged from 98.9% to 99.5% (table 2). All AgMNPV isolates remained collinear to each other across their evolutionary history. No rearrangements, such as inversions and translocations, were detected, but insertions and deletions were common throughout the genomes (see below for more details). Most intergenic spacers in AgMNPV have not shown large modifications. The reference isolate (2D) has a highly compact genome, where 101 ORFs are separated from their neighboring ORFs by less than 50 nt, and 67 ORFs share from one to more than 140 nt with adjacent ORFs. This pattern of genome compaction is conserved in most isolates, and mutations in noncoding regions were mainly observed within loci of drs, hrs, and on regions involved in gain or loss of genes.
Table 2.
Genomic Features of AgMNPV Genotypes
| Isolate | Size (bp) | GC Content (%) | Number of ORFs | Number of hrs | Number of drs | Similarity with AgMNPV-2D (%) |
|---|---|---|---|---|---|---|
| 2D | 132,239 | 44.5 | 158 | 10 | 12 | — |
| 26 | 131,678 | 44.6 | 157 | 10 | 12 | 99.5 |
| 27 | 131,172 | 44.6 | 157 | 11 | 12 | 98.9 |
| 28 | 130,745 | 44.6 | 157 | 11 | 12 | 99.2 |
| 29 | 130,506 | 44.6 | 157 | 11 | 12 | 98.9 |
| 30 | 130,741 | 44.5 | 156 | 10 | 13 | 99.3 |
| 31 | 132,126 | 44.6 | 158 | 11 | 12 | 99.2 |
| 32 | 131,494 | 44.6 | 157 | 10 | 12 | 99.4 |
| 33 | 131,059 | 44.5 | 157 | 10 | 12 | 99.4 |
| 34 | 131,543 | 44.6 | 158 | 11 | 10 | 99.1 |
| 35 | 132,176 | 44.6 | 159 | 11 | 11 | 99.3 |
| 36 | 131,216 | 44.5 | 156 | 10 | 12 | 99.3 |
| 37 | 131,855 | 44.5 | 156 | 10 | 13 | 99.3 |
| 38 | 130,740 | 44.5 | 156 | 10 | 13 | 99.3 |
| 39 | 130,699 | 44.5 | 157 | 10 | 12 | 99.3 |
| 40 | 132,180 | 44.6 | 158 | 11 | 12 | 99.3 |
| 42 | 130,949 | 44.6 | 157 | 11 | 13 | 99.2 |
| 43 | 132,077 | 44.6 | 159 | 11 | 10 | 99.5 |
The aforementioned genomic features of AgMNPV were taken into account for genome annotation: Only ORFs of at least 138 bp in length (as observed in AgMNPV-2D, ORF010) and maximum overlap of 148 bp with adjacent ORFs (as in the anti-apoptotic 2, iap-2, ORF092) were annotated in the 17 AgMNPV wild-type isolates. Considering these parameters, a total of 167 ORFs were found in the pangenome of AgMNPV (fig. 2 and table 2). Among the 152 ORFs previously assigned to the AgMNPV-2D genome, 145 are shared by all isolates, but eight of them have size variations: 1629capsid (ORF002), pe-38-like-1 (ORF005), ie-2 (ORF011), ac91-like (ORF074), desmoplakin (ORF096), odv-e66 (ORF114), hisP (ORF136), and bro-b (ORF162). These ORFs were classified into distinct functional gene classes as follows: 1) Auxiliary, 2) structural proteins associated with BVs or ODVs, 3) packaging and assembly of new particles, 4) DNA replication, 5) viral transcription, 6) host interaction, and 7) genes with unknown function. Some of these genes code for auxiliary proteins: As pe-38-like-1 is a transcriptional transactivation factor, the ie-2 is a transactivator of early promoters and the desmoplakin is involved in the supercoiling process of DNA. Moreover, the 1629capsid codes for a structural protein at nucleocapsid base and odv-e66 codes for an ODV envelope protein. Due to structural polymorphisms leading to ORF fission, fusion or indels, some ORFs typically observed in AgMNPV-2D are not present in all genomes, namely pe-38-like-1 (ORF006), pe-38-like-2 (ORF007), he65-like-1 (ORF060), he65-like-2 (ORF061), ag144 (ORF144), bro-c (ORF156), and bro-a (ORF163). Completing the set of 167 ORFs, six additional ORFs present but not previously annotated in AgMNPV were found in all genomes (ORFs 027, 035, 042, 058, 112, and 135); and three new ORFs derived from indel events were observed in some isolates (ORFs 008, 012, and 139). To sum up, AgMNPV isolates have 151 genes in common, and besides coding sequences, a nonredundant set of 14 drs and 11 hrs were identified among all AgMNPV genomes (fig. 2 and table 2). For more details on presence or absence of ORFs, drs, and hrs, see supplementary tables S1 and S2, Supplementary Material online.
Fig. 2.—

Overview of the genomic organization of the AgMNPV pangenome. Red arrows represent ORFs previously annotated in the genome of AgMNPV-2D (Oliveira et al. 2006), whereas yellow ones are new ORFs not previously reported for AgMNPV-2D. These ORFs can be coding sequences absent in the isolate 2D; ORFs that were not annotated in that isolate; or ORFs derived from events of gene fusion/fusion. Green and blue rectangles stand for hrs and drs, respectively. Asterisks (*) indicate the genomic position of baculoviral core genes. The small diamonds between ORFs indicate different levels of genome compaction, such as overlaps between coding sequences (green, blue, red) and short intergenic spacers (gray diamonds).
Structural Variations in hrs and drs
In the pangenome of AgMNPV, most of the DNA length variations (indels) were observed within tandem repeats, such as hrs and drs. The 14 drs consist of two or more repeating units ranging in length from 14 to 28 bp. They are minisatellites found in both intra- and intergenic regions, and most indels in coding sequence were found within these regions, especially in 1629capsid (ORF002, dr1 and dr2), pe-38 (ORF005, dr3), and ac91-like (ORF074, dr6 and dr7), where the polymorphisms gave rise to different alleles varying in hundreds of base pairs. Only four drs were found in intergenic regions, being dr4 the longer and more variable of these elements, ranging from 344 to 525 bp.
A total of 11 hrs were identified in AgMNPV, and nine of them correspond to those previously found in AgMNPV-2D. Two of them correspond to new hrs composed of singleton palindromes (hr1 and hr2), which were observed in similar genomic positions as hr13 and hr12 in the CfDEFMNPV (Lauzon et al. 2005). Four hrs (hr3, hr9, hr10, and hr11) had size variation related to gain or loss of 28-bp core palindromes. Remarkable variations were observed in hr3 (former hr4 in AgMNPV-2D), which had four variants, ranging from 342 (5 × 30 bp imperfect palindromes) to 1,232 bp (19 × 30 bp imperfect palindromes) (supplementary table S2, Supplementary Material online).
Structural Variations in Coding Sequences
Some AgMNPV genes underwent insertions and deletions comprising stretches varying from dozens to hundreds of nucleotides, which caused structural variations, such as gene shortening and truncation. A large 295-bp indel generated a truncated form of the hisP (ORF136), a variation unique to the AgMNPV. This truncated allele potentially encodes a disrupted HAD-like domain, which shows a putative histidinol-phosphatase activity (Cohen et al. 2009), encoded by at least eight isolates: 26, 28, 29, 30, 36, 38, 39, and 42. A similar polymorphism was also observed in a disrupted form of the ODV envelope protein, odv-e66 (ORF114), which has a 327-bp indel in AgMNPV-42. Moreover, the 3′ region of pe-38 (ORF006) showed a remarkable pattern of gain and loss of genomic fragments that gave rise to at least four allelic variants (PE38-α, -β, -γ, -δ, and -ϵ). PE38-α is the variant found in AgMNPV-2D; PE38-ϵ is found in CfDEFMNPV; and the other three variants—β, γ, and δ—were observed, respectively, in (β) 26, 30, 32, 36, 37, 38; (γ) 27, 28, 29, 31, 34, 35, 40, 42 e 43; and (δ) 33, 39. The large variability observed in PE-38 was caused by the absence or duplication of Leucine Zipper motifs (Krappa and Knebel-Morsdorf 1991), which are structures composed mainly by leucine residues linked by glutamic acid, lysine, asparagine, and arginine (LEEKNRL), although other arrangements were also observed (fig. 3).
Fig. 3.—

Structural variations on the pe-38 locus. Each rectangle represents an amino acid block shared among different variants of PE38 (named from α to ϵ). The gray lines connecting each rectangle indicate the absence of amino acid blocks in some variants. The three blocks on the left are encoded by pe-38-like-2 (ORF007), where the block 2 is the RING Finger domain. The blocks from 4 to 15 are encoded by pe-38-like-1 (ORF006), and the Leucine Zipper motifs are located within the blocks 5, 6, and 7, which in turn correspond to a direct repeat (dr3). The scale above the diagram delimits intervals of ten amino acids.
Gene Fusion and Fission Events
In different isolates, genes pe-38, he65, ag144, and bro-c were observed as single ORFs or divided into two independent ORFs (supplementary fig. S2, Supplementary Material online). pe-38, he65, and ag144 had their coding sequences modified due to a 1-bp indel, a single nucleotide substitution, and a 10-bp indel, respectively. The gene pe-38 is originally split into two ORFs in AgMNPV-2D. The single ORF of he65 was encoded by most of the isolates, except AgMNPV-2D, -39, and -43. In these three isolates, the N and the C terminal proteins were encoded by he65-like-1 and he65-like-2, respectively. The same pattern was also observed for ag144 that was split into two short ORFs (ag145 and ag146) in most isolates, with the exception of AgMNPV-2D, -36, and -37. Finally, in bro-c, ORFs 157 and 158 were generated in at least two independent events of gene fission/fusion. One was probably mediated by slipped strand mispairing causing a 25-bp indel (isolate 32); and the second event derived from a 1-bp indel (isolates 35 and 36). The directionality of these ORF rearrangement events could not be precisely determined due to a lack of phylogenetic signal among the aforementioned haplotypes.
Other Structural Variations in Bro Genes
Proteins encoded bybro genes have high levels of plasticity and variability (Bideshi et al. 2003), and besides the structural variations involving bro-c, important changes affected two other bro genes. Alleles of bro are observed in multiple copies, varying from 1 to 16 copies (Ayres et al. 1994; Kuzio et al. 1999), although some baculoviruses lack these genes (Ahrens et al. 1997; Hyink et al. 2002). Among group I Alphabaculoviruses, isolate AgMNPV-2D has the higher number of bro genes: Seven copies (from bro-a to bro-g) (Oliveira et al. 2006). These seven copies of bro encode proteins with domains Bro-N (PF02498), DUF3627 (PF12299), and T5orf172 (PF10544), which can be covalently linked or separated in independent peptides. Interestingly, only bro-a (ORF163) is able to encode a protein with the T5orf172 domain. Interestingly, bro-a (ORF163) was absent inmost AgMNPV genomes, excluding AgMNPV-2D, -26, -31, -35, -40, and -43. The mechanisms that could explain bro-a (ORF163) absence are unknown and, based on sequence information, it was not possible to conclude whether this gene was lost or acquired during the recent evolutionary history of AgMNPV. Interestingly, bro-a and bro-b were contiguous in AgMNPV, and they encode proteins with contrasting domain architectures: Bro-N + T5orf172, and Bro-N + DUF3627, respectively. However, despite their divergent architecture, a fragment of 150 bp seems to be shared by these genes, and this observation gives us clues about the origin or loss of bro-a (fig. 4). Another important variation involving bro genes took place at the locus of bro-b. This gene showed at least three variants, which could be distinguished by the absence or presence of specific codons (from 1 to 7 in total), specifically located within their interdomain region, or inside the DUF3627 domain.
Fig. 4.—
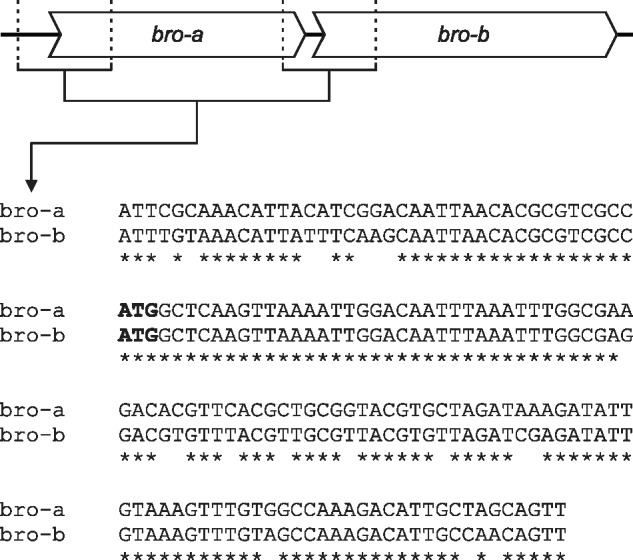
Sequence similarity between 5′ regions of bro-a and bro-b. The first nucleotide block corresponds to a 39-bp region upstream to the start codon of both genes (bold ATG in the second block). Asterisks highlight identical nucleotides (identity = 86%).
Further Genetic Novelties
Besides the unique ORFs 098 (ag98) and 132 (parp-like) observed in AgMNPV-2D (Oliveira et al. 2006), four other unique ORFs were found among the wild isolates: ORFs 008 (ag8), 012 (ag12), 135 (ag135), and 139 (rnf12-like). These ORFs are not present in all isolates, as indels disrupted their coding sequences in some genomes. Both ag8 and ag12 have small ORFs, and they are found inside the most variable region of AgMNPV genomes: The locus flanked by hr11 and hr2, where bro-a, pe-38, and ie-2 are located. Ag8 was present in nine isolates (27, 28, 29, 31, 34, 35, 40, 42, and 43), whereas ag12 was observed only in AgMNP-33 and -42.
The third new gene, rnf12-like (ORF139), potentially encodes a protein with a conserved RING-H2 Finger domain in its C-terminus. This domain is characterized by the motif C3H2C3, with a Hystidine replacing a Cysteine at the fourth position (Saurin et al. 1996), differing from other RING-Finger proteins of AgMNPV, which had a RING-HC Finger domain (C3HC4). A BLAST (Basic Local Alignment Search Tool) search revealed a significant match (e value = 8e-15) between this sequence and an RNF12-like protein from a lepidopteran (Bombyx mori), indicating a possible gene acquisition through an event of HGTs (fig. 5). The whole ORF of rnf12-like was found only in AgMNPV-34 and -37. In AgMNPV-2D and -32, rnf12-like was completely absent. All other isolates had a partial version encoding a small fragment of RING-H2 Finger (fig. 5). Seemingly, in this isolates this coding sequence was replaced and disrupted by an exogenous sequence composed mainly of a GT-dinucleotide repeat and a direct repeat (dr13; 4 × 15 bp).
Fig. 5.—

Structural variations observed in the locus of rnf12-like. The rectangles stand for nucleotide blocks located between ORFs 138 (v-ubi, upstream) and 140 (39K/pp31, downstream). Regions partially conserved are depicted as disrupted rectangles. Green and red regions encompass the rnf12-like coding sequence, where the red rectangle encodes a RING-H2 Finger domain. Regions on gray are nonconding regions, and the black region is an insertion that has a direct repeat (dr13).
Finally, a 45-aa polypeptide encoded by a nested ORF internal to fgf (ORF134), detected in the AgMNPV-2D proteome (Braconi et al. 2014), was found to be conserved in all AgMNPV genomes studied. As ORF135 (ag135) is located in opposite orientation to the fgf sequence, the protein it encodes does not match any region of FGF. The protein of this hypothetical gene is carried inside AgMNPV virions.
Genetic Diversity of AgMNPV Genes
The genetic diversity was calculated using both the Watterson’s genetic diversity estimator (θ) and the number of nonsynonymous changes (dN), which were obtained from multiple sequence alignments (MSA) of whole genomes and of 151 genes shared by all AgMNPV. The gene MSA was used to estimate the diversity as a function of the number of segregating sites (θ), where all genomes (including AgMNPV-2D) were assumed as genotypes of a hypothetical metapopulation of AgMNPV. Values of θ were divided into four categories, from A to D (in intervals of 0.01), where category A, in turn, was divided into four subcategories, from A1 to A4 (in intervals of 2.5 × 10−3) (fig. 6). The whole-genome MSA was used to detect nonsynonymous changes, and the total of polymorphisms per gene was plotted as absolute numbers in figure 6. Overall, at least 714 nonsynonymous changes were found, which are not evenly distributed along the genomes.
Fig. 6.—

Genetic diversity of the AgMNPV metapopulation. The external elements of this circular map represent ORFs in positive and negative sense, and the internal arrows highlight genes shared by all baculoviruses (core genes). The outer circle is a heat map of the number of nonsynonymous changes per gene. The inner circle shows genetic diversity measure based on the Watterson estimator (θ).
Among genes shared by all AgMNPV, 109 genes grouped within the subcategories A1 and A2, as they had low θ diversity, and most of them had (two or less) nonsynonymous changes. A total of 79 genes were observed within the subcategory A1, of which nine were 100% identical in all genomes, they were ac29-like, ac145-like, ac110-like, bro-a, bro-g, ctl-2, etm, iap3, and odv-e18. Contrastingly, only 17 genes were included within the other three categories (B–D). These genes carry more than 40% of all amino acids substitutions, and they are mainly observed in a highly variable region located between hr11 and hr2 (from 12 o’clock to 1 PM in the physical map) (fig. 6). A total of 12 genes with high diversity were located within or flanking this region: ie-2, ag10, pe-38, pk1, ag4, lef-2, ac4-like, bro-b, ptp-2, ac11-like, bro-c, and arif-1. These 12 genes accounted for 306 nonsynonymous changes (42% of the total).
Based on θ values of core and noncore (i.e., satellite) genes, the level of sequence variation between these two groups of genes was evaluated. Core genes constituted a set of 37 coding sequences shared by all baculoviruses, and in AgMNPV they were observed to have significantly low diversity (θ = 0.0031 ± 0.0028) when compared with noncore genes (θ = 0.0067 ± 0.0118) (fig. 7A). In terms of amino acid changes, the same pattern was observed, given that the average number of nonsynonymous changes in core genes was smaller than in noncore genes (table 3). Interestingly, from 19 core genes with very low genetic diversity (θ subcategory A1), 10 encoded structural proteins associated with BVs or ODVs (Braconi et al. 2014): 38k, p40/c42, vp1054, vp91/p95, p6.9, odv-e18, odv-e27, odv-ec43, pif-1, and pif-3. In general, core and satellite genes encoding structural proteins had the lowest average number of amino acid changes (3.14 ± 3.66). Some of these proteins had no changes, such as those encoded by p24, vp1054, odv-e18, pif-3, and polh (table 3). Interestingly, when protein size was plotted against number of nonsynonymous changes, we have found just a trend for positive correlation between these variables (r = 0.47), as some genes fell distant from the trend line (fig. 7B). These results gave us an interesting overview of the distribution of proteins in terms of polymorphisms along amino acid sites, where outliers, identified as those genes not included in the 95% CI in figure 7B, were either short proteins with high number of polymorphisms, or vice versa. Among these proteins, ten were short proteins (around 200–400 amino acids in length) that had a high number of changes (from 15 to 70). These proteins were encoded by ac11-like, ac91-like, bro-c, bro-b, ac34-like, pe-38-like-1, pk1, and ie-2 (fig. 7B). The last four genes in this list are known to be involved in DNA replication and/or transcription. This pattern was also observed for dnapol, which encodes the second largest protein of AgMNPV (991 aa), and was highly variable (22 changes). Conversely, not all long proteins appeared to have accumulated a high number of nonsynonymous changes. A high level of sequence conservation was observed in desmoplakin (886 aa) and helicase (1,221 aa), which had only two and six changes, respectively (fig. 7B).
Fig. 7.—
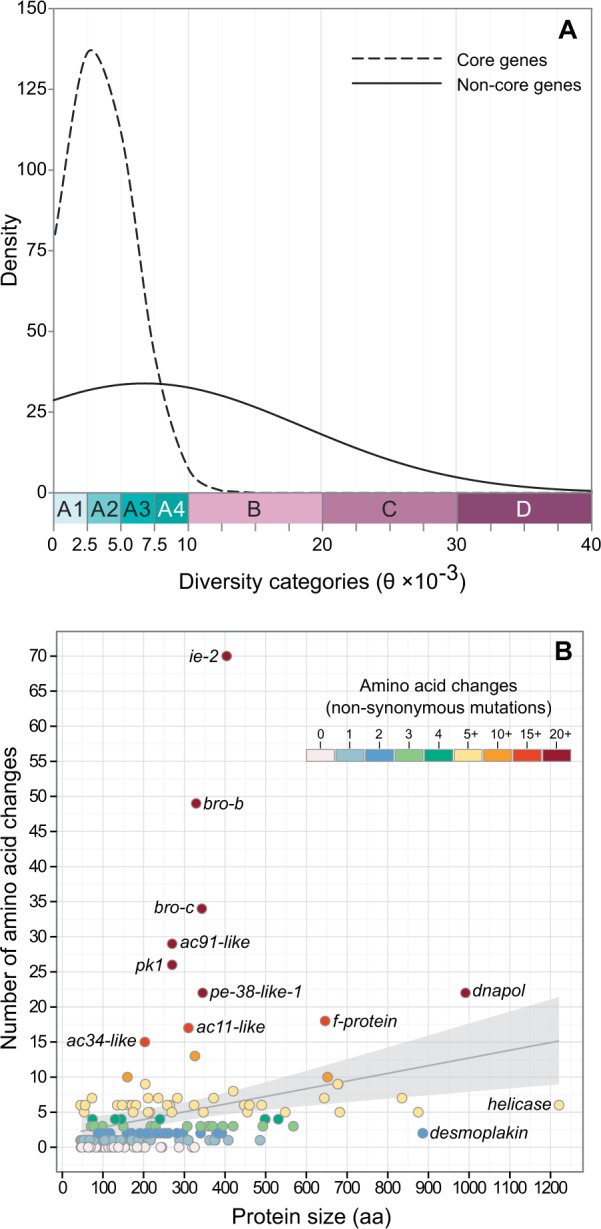
(A) Frequency distribution of core and noncore genes in the four categories and subcategories of θ diversity, as indicated by the intervals in the x-axis (A1–A4, B–D). Core genes show low genetic diversity when compared with satellite genes. (B) Scatter plot showing the number of amino acid changes (nonsynonymous changes) as a function of protein size (aa). A moderate correlation was observed between these variables (r = 0.47) and a 95% confidence interval is shown. Some outlier genes are shown for discussion.
Table 3.
Statistics of Nonsynonymous Changes (NSC) among Genes from Different Functional Categories
| Functional Category | Average NSC and Standard Deviation | Genes and Number of NSC |
|---|---|---|
| Auxiliary | 5.54 ± 11.01 | 25k fp (2); ac145-like (0); ac4-like (4); ac5-like (2); ag127 (1); ag128 (2); ag22 (1); bro-b (49); bro-c (34); bro-d (3); bro-e (0); bro-f (3); bro-g (0); cg30 (4); ctl-2 (0); desmoplakin (2); gp16 (0); he65 (5); hisP (6); ac141-like/exon0 (2); p48 (1); parp-like (3); pkip-1 (6); pnk/pnl (9); sod (3); v-trex (2) |
| Host modulation | 4.00 ± 4.24 | arif-1 (13); egt (3); fgf (0); iap-1 (2); iap-2 (2); iap-3 (0); p12 (0); p33 (6); ptp-1 (5); ptp-2 (10); v-ubi (3) |
| Replication | 7.73 ± 7.50 | alk-exo (8); dbp (3); dnapol (22); gta (4); helicase (6);lef-1 (2); lef-2 (9); lef-3 (1); lef-7 (2); me53 (6); pe-38-like-1 (22) |
| Transcription | 8.56 ± 17.47 | 39k/pp31 (2); ac38-like (2); ie-1 (3); ie-2 (70); lef-10 (1); lef-11 (0); lef-12 (1); lef-4 (5); lef-5 (6); lef-6 (1); lef-8 (5); lef-9 (6); met (5); p47 (3); pk1 (26); vlf-1 (1) |
| Capsid | 3.83 ± 5.25 | 1629capsid (18); 38k (1); ac53-like (1); p15 (1); p24 (0); p25 (3); p40 (3); p6.9 (1); p87 (9); vp1054 (0); vp39 capsid (2);vp91/p95 (7) |
| Virion | 2.78 ± 2.56 | ac108-like (1); ac150-like (1); ac68-like (1); ac81-like (2); f-protein (10); gp41 (2); gp64 (4); odv-e18 (0); odv-e25 (3); odv-e26 (5); odv-e27 (2); odv-e56 (7); odv-e66 (5);odv-ec43 (2); p10 (2); p49 (1); p74 (7); pif-1 (4); pif-2 (2); pif-3 (0); pif-4 (2); polh (0); pp34/pep (1) |
| Core genes | 3.65 ± 3.97 | See genes in bold above. |
| Noncore genes | 4.91 ± 9.27 | — |
| Total | 4.61 ± 8.32 | — |
Note.—Genes highlighted in bold correspond to core genes. Functional categories were assigned based on Cohen et al. (2009).
Discussion
The Genome Compaction of AgMNPV
As shown in figure 2, most of the AgMNPV ORFs have short or absent intergenic spacers, and this high level of genome compaction directly affects how viral genomes evolve (Chirico et al. 2010). Even cis-acting elements such as promoters, transcription starting sites, and polyadenylation sites can lie within adjacent coding sequences (Normark et al. 1983; Chen et al. 2013). Because mutations in these shared regions can impair more than one genetic element (whether coding or regulatory), these overlaps impose constrains on sequence evolution (Krakauer 2000). Moreover, as breaking points are likely to be deleterious when within overlaps, this high level of genome compaction could explain why genome collinearity was conserved among AgMNPV genotypes, as recently observed in other Alphabaculoviruses (Theze et al. 2014; Chateigner et al. 2015).
Polymorphisms in hrs
In previous studies involving other genotypes of AgMNPV, hrs were shown to be highly variable, differing from each other on their number of tandem repeats and core palindromes (Garcia-Maruniak et al. 1996; Maruniak et al. 1999). Among the 17 wild-type genotypes analyzed in this work, these copy-number variations were specially observed in large hrs, such as hr3, hr9, hr10, and hr11. Remarkably, the haplotypes flanked by thesehrs were the most variable in terms of gene content. Between hr11 and hr3 are variable genes as pe-38, 1629capsid, new genes as ag8 and ag12, as well as bro-a (absent in some isolates). On the other hand, the haplotype flanked by hr9 and hr10 encompassed the polymorphic gene hisP, and the parp-like (described previously in AgMNPV-2D) and rnf12-like (found as partial or full-length variants in some isolates) genes that are specific to the AgMNPV. Based on the repetitive nature of the hrs, and taking into account their roles as origins of replication (Habib and Hasnain 1997), the size variation observed within hrs could be the result of slipped strand mispairing during events of DNA replication (Levinson and Gutman 1987; Bzymek and Lovett 2001), and the genetic heterogeneity in the regions flanked by these variable hrs could be the result of homologous recombination between AgMNPV genotypes (Croizier and Ribeiro 1992; Crouch and Passarelli 2002). It is reasonable to expect that the decreased or increased number of palindromic sequences within variable hrs potentially affects regulation mechanisms mediated by these repetitive elements (Gemayel et al. 2010), as hrs work as enhancers for the expression of early genes (Olson et al. 2001; Landais et al. 2006). Size variations in hrs are common among alphabaculoviruses, such as SfMNPV (Simon et al. 2011), AcMNPV (Chateigner et al. 2015), and MacoNPV (Li et al. 2005), in which extensive indels were found.
The Dynamics of Gain and Loss of Bro Genes
In the AgMNPV, only bro-a (ORF163) encodes a polypeptide containing a T5orf172 domain. This gene is usually located adjacent to bro-b (ORF162), and flanked by ptp-1 and ac4-like (Oliveira et al. 2006). Gene content in this region is highly variable in different baculoviruses, especially due to partial or complete deletion of homologues of bro-a (supplementary fig. S3, Supplementary Material online). In AgMNPV, 12 isolates lacked bro-a, showing a haplotype similar to those observed in AcMNPV (Ayres et al. 1994) and CfDefMNPV (Lauzon et al. 2005). Moreover, the partial or complete lack of bro-a was recently suggested to be associated with multiple events of deletion in BmNPV isolates (Ardisson-Araujo et al. 2014). Both the structural variations and the similarity observed between bro-a and bro-b in AgMNPV (fig. 4), allowed us to hypothesize on the evolution of the bro-a/bro-b haplotype. Possibly the Bro-N domain encoded by bro-a could have been derived from a partial duplication of bro-b, followed by an independent acquisition of the T5orf172. Meanwhile, bro-a and bro-b could have been acquired together from a viral or a prokaryotic genome (Iyer et al. 2002). Moreover, the deletion of bro-a might have taken place during genome replication, whether due to intramolecular slipped-strand mispairing between bro-a and bro-b, or due to asymmetric homologous recombination between related genotypes (for details, see supplementary fig. 3, Supplementary Material online). In AgMNPV, the locus between hr11 and hr2 gathers at least one-third of the nonsynonymous changes that we detected. As shown in figure 6, this region encodes three bro genes: bro-a, -b, and -c. The latter two genes accumulated more than 11% of the amino acid changes reported for AgMNPV. Interestingly, this pattern was also observed in MacoNPV-A, in which gene gains/losses and high genetic diversity are observed in bro genes located specifically within a highly variable region (Li et al. 2005).
Gene Fusions/Fissions and Size Variations in Genes of AgMNPV
We presented at least four examples of gene fission and fusion in AgMNPV genes, events driven by point mutations (1-bp indels/substitutions) and short indels (10–25 bp). As observed in AcMNPV, the fusion of adjacent genes is a common mechanism of evolution among alphabaculoviruses (Chateigner et al. 2015). In the AcMNPV, at least four pairs of adjacent ORFs were found to be fused (Ac20/Ac21, Ac58/Ac59, Ac106/Ac107, and Ac112/Ac113), but these pairs do not correspond to those observed in our results (supplementary fig. S2, Supplementary Material online). Among cases of gene fission/fusion here reported, the one involving pe-38 attracts special attention. The protein encoded by pe-38 acts in the nucleus, where it stimulates DNA replication (Kool et al. 1994; Krappa et al. 1995). The fused version of pe-38 is known to possess internal late promoters (DTAAG motifs) located upstream its 3′ fragment, region that corresponds to the pe-38-like-1 (ORF006), and encodes a 20 kDa protein (Krappa et al. 1995). Remarkably, these promoters were conserved in the isolate 2D (that encoded both fragments individually), but their DTAAG motifs were mutated in most AgMNPV isolates (27, 28, 29, 31, 34, 35, 40, 42, and 43). As these isolates encoded the fused variant of pe-38, we hypothesize that this smaller version of the PE38 protein (∼20 kDa) could be expressed from its 3′ fragment (pe-38-like-1), probably using an alternative promoter (Krappa et al. 1995). Another gene that underwent fusion/fission was he65. The protein HE65 stimulates the formation of F-actin in the nucleus to promote viral particles production (Ohkawa et al. 2002). Interestingly, he65 was split into two independent ORFs in at least three isolates of AgMNPV (2D, 39, and 43), and differently of what was observed for pe-38, the internal early promoter of he65 (a TATA box followed by CANT) was conserved in all isolates. Interestingly, in AcMNPV, the he65 locus is known to encode at least two classes of transcripts with distinct sizes, and in different stages postinfection (Becker and Knebel-Morsdorf 1993). In the aforementioned cases of gene fusion/fission, the level of conservation of internal promoters suggested that small internal products may be expressed either from adjacent fused ORFs (pe38 and he65) or from their individual coding sequences derived from gene fission (pe38-like-1 and he65-like-2). These results indicated that the conservation of promoters determines how these ORF rearrangements impact the viral proteome, and consequently, on baculovirus infection.
A Nested Gene in AgMNPV
Small reading frames nested within large ORFs are common in viral, bacterial, and mitochondrial genomes (Normark et al. 1983; Krakauer 2000). Using mass spectrometry data recently obtained by our group, a 45-aa peptide was found to be encoded by an ORF located internally and in opposite orientation to fgf (ORF134). The short peptide encoded by this nested gene (ag135) seems to be expressed and carried along with AgMNPV virions (Braconi et al. 2014), and this might be the first report of a protein encoded by a nested gene in baculovirus. However, further studies are necessary to characterize the promoter motif, gene expression, and function of this new protein in AgMNPV.
Acquisition of an E3 ubiquitin-Ligase Gene
A new gene, rnf12-like (ORF139), was observed in at least two wild population of AgMNPV (34 and 37), and it encodes for a protein containing a RING-H2 Finger domain on its C-terminus. RING Finger domains are also encoded by other AgMNPV genes, such as cg30 (Passarelli and Miller 1994); the antiapoptotic genes, iap-1, iap-2, iap-3 (Clem and Miller 1994), ie-2 (Passarelli and Miller 1993), and pe-38 (Krappa et al. 1995), but their RING-HC Finger domains differ from the one encoded by rnf12-like (fig. 5). Proteins with any of these domains are known to belong to different functional categories (Freemont 2000). However, those with RING-H2 Finger are specially involved in the ubiquitin pathway, acting as E3 ubiquitin-protein ligases (Joazeiro and Weissman 2000). Among baculoviruses, three proteins with RING-HC Finger have already been pointed as E3 ubiquitin-protein ligases: IAP2, IE2, and PE38 (Imai et al. 2003). Almost all Alpha- and Betabaculoviruses express an additional component of the ubiquitin pathway: The viral ubiquitin, encoded by v-ubi (Guarino 1990). In AgMNPV, the gene v-ubi (ORF138) is located upstream of rnf12-like (ORF139), in the same orientation. As the ubiquitin–proteasome system has a central role targeting proteins for degradation (Katsuma et al. 2011), we hypothesize that the physical proximity of these viral genes could somehow be advantageous for their coexpression.
Based on the pattern of indels within the rnf12-like locus (fig. 5), the AgMNPV has probably acquired this gene through HGT, and then has gradually lost parts of this coding sequence in at least two moments over its evolutionary history. It is well known that the genomes of baculoviruses and other large DNA viruses evolve through subsequent events of HGT involving eukaryotic hosts, recombination with other viruses, and gene duplication followed by sequence divergence (Hughes and Friedman 2003; Shackelton and Holmes 2004; Zanotto and Krakauer 2008).
Most AgMNPV Genes Have Low Sequence Diversity
To analyze the genetic diversity in AgMNPV, we used two different metrics—θ and number of nonsynonymous (i.e., amino acid) changes. It is important to highlight that both measures of genetic diversity rely on nucleotide substitutions, but their results are slightly different, and are not necessarily proportional to each other. As these analyses were performed based on consensus genome sequences reconstructed from heterogeneous field isolates, a potential limitation of our results would be the assumption of clonality in AgMNPV populations, phenomenon uncommon for natural NPV isolates. Nevertheless, we preferred to summarize each population by its most frequent, master consensus sequence for expediency, which we assumed to be the predominant genotype of each viral population. Notwithstanding, our analyses revealed that 99 out of 167 AgMNPV genes had low sequence diversity (two or less polymorphisms), and core genes are on average less diverse than satellite genes, reinforcing the observations made by Oliveira et al. (2013). A total of 25 out of those 99 conserved genes encode structural proteins associated with viral envelope, nucleocapsid, or nucleosome formation (Braconi et al. 2014), and interactions among them or among these proteins with DNA are common (Rohrmann 1992). As changes at the sequence level can impair such interactions, we might expect that these proteins would be evolving slowly under purifying selection (Daugherty and Malik 2012), although other hypotheses to explain this observation are not discarded.
At the other extreme of diversity, a particular genomic region delimited by hr11 and hr2, approximately 18,000 bp grouped several genes with high sequence diversity (fig. 6). An increase in variability, such as the one around 12 o’clock in the physical map of the AgMNPV pangenome (fig. 6), was also observed in two isolates of MacoNPV, which have that region accumulating at least 50% of the total nucleotide changes, most of them located in two bro genes (Li et al. 2005). Nevertheless, this variable locus of MacoNPV does not match the one AgMNPV, as they differ in gene content. Interestingly, the variable locus at 12 o’clock in AgMNPV was shown to evolve through homologous recombinations, as reported in wild isolates studied by Croizier and Ribeiro (1992). In that work, different restriction sites were used as genomic markers to trace recombination between AgMNPV genotypes. One of these markers (named “ϵ”) is located exactly in the locus of ie-2, here presented as the most divergent gene, showing at least 70 polymorphisms. Curiously, this locus gathers several genes whose products act as trans-acting factors, namely: ac4-like, ac5-like (Lo et al. 2002), ac141-like/exon0, ie-1 (Kool et al. 1994), ie-2 (Prikhod’ko et al. 1999), and pe-38 (Krappa et al. 1995). This variable region also contains three bro genes (bro-a, -b, and -c), all of them encoding the domain Bro-N. This domain has been shown to have DNA-binding activity, and probably acts as transcriptional regulator of viral and host genes (Zemskov et al. 2000). The high level of polymorphism and the cluster of transcriptional regulators encoded there suggested that the variable region flanked by hr11 and hr2 may play a relevant role in determining the infectivity of AgMNPV. As this region frequently evolves through homologous recombination, it could facilitate gene exchange among AgMNPV genomes. Overall, the presence of variable regions and polymorphic alleles in baculoviral populations are important evolutionary advantages, once it is shown that these variable elements enable these viruses to respond rapidly to selective pressures (Li et al. 2005; Simon et al. 2011; Chateigner et al. 2015).
Conclusion
In this study, the complete genomic sequences of 17 wild isolates of Anticarsia gemmatalis MNPV were sequenced and characterized. A total of 167 predicted genes were observed in the pangenome of the AgMNPV. Among them, 151 are shared by all isolates, including the reference isolate 2D; six were implicated in events of gene fusion/fission; and other four were only found in a few genomes, including bro-a, and the new gene rnf12-like, which encodes for an E3 ubiquitin-ligase probably acquired through HGT. Gain and loss of small genome fragments were mainly observed in tandem repeats located inside or outside coding sequences. Most AgMNPV genes had low nucleotide diversity, and variable genes were mainly observed in a region previously described to be involved in homologous recombination. This study provides relevant information about AgMNPV genome evolution in particular and on baculovirus in general.
Supplementary Material
Supplementary figures S1–S3 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
A.F.B. and P.M.A.Z. thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, 2011/14537-0 and 2011/17120-3) for the financial support; C.T.B. holds a postdoctoral fellowship (FAPESP 2014/03911-7). A.G. and P.M.A.Z. hold a CNPq/PQ Scholarship. The authors declare that they have no competing interests. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Literature Cited
- Ahrens CH, et al. 1997. The sequence of the Orgyia pseudotsugata multinucleocapsid nuclear polyhedrosis virus genome. Virology 229(2):381–399. [DOI] [PubMed] [Google Scholar]
- Allen GE, Knell JD. 1977. A nuclear polyhedrosis virus of Anticarsia gemmatalis: I, ultrastructure, replication, and pathogenicity. Fla Entomol. 60(3):233–240. [Google Scholar]
- Aoki-Kinoshita KF, Kanehisa M. 2007. Gene annotation and pathway mapping in KEGG. Methods Mol Biol. 396:71–91. [DOI] [PubMed] [Google Scholar]
- Ardisson-Araujo DM, et al. 2014. Complete genome sequence of the first non-Asian isolate of Bombyx mori nucleopolyhedrovirus. Virus Genes 49(3):477–484. [DOI] [PubMed] [Google Scholar]
- Ayres MD, Howard SC, Kuzio J, Lopez-Ferber M, Possee RD. 1994. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 202(2):586–605. [DOI] [PubMed] [Google Scholar]
- Becker D, Knebel-Morsdorf D. 1993. Sequence and temporal appearance of the early transcribed baculovirus gene HE65. J Virol. 67:5867–5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. doi: gkc131 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bideshi DK, Renault S, Stasiak K, Federici BA, Bigot Y. 2003. Phylogenetic analysis and possible function of bro-like genes, a multigene family widespread among large double-stranded DNA viruses of invertebrates and bacteria. J Gen Virol. 84(Pt 9):2531–2544. [DOI] [PubMed] [Google Scholar]
- Binns D, et al. 2009. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics 25(22):3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blissard GW, Rohrmann GF. 1990. Baculovirus diversity and molecular biology. Annu Rev Entomol. 35:127–155. [DOI] [PubMed] [Google Scholar]
- Braconi CT, et al. 2014. Proteomic analyses of baculovirus Anticarsia gemmatalis multiple nucleopolyhedrovirus budded and occluded virus. J Gen Virol. 95(Pt 4):980–989. [DOI] [PubMed] [Google Scholar]
- Bzymek M, Lovett ST. 2001. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A. 98(15):8319–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateigner A, et al. 2015. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 7:3625–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, et al. 2013. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J Virol. 87(11):6391–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirico N, Vianelli A, Belshaw R. 2010. Why genes overlap in viruses. Proc Biol Sci. 277(1701):3809–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm GE, Henner DJ. 1988. Multiple early transcripts and splicing of the Autographa californica nuclear polyhedrosis virus IE-1 gene. J Virol. 62:3193–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RJ, Miller LK. 1994. Control of programmed cell death by the baculovirus genes p35 and iap. Mol Cell Biol.. 14(8):5212–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Marek M, Davies B, Vlak JM, van Oers M. 2009. Encyclopedia of Autographa californica nucleopolyhedrovirus genes. Virol Sin. 24:359–414. [Google Scholar]
- Croizier G, Ribeiro HCT. 1992. Recombination as a possible major cause of genetic heterogeneity in Anticarsia gemmatalis nuclear polyhedrosis virus wild populations. Virus Res.. 26:183–196. [Google Scholar]
- Crouch EA, Passarelli AL. 2002. Genetic requirements for homologous recombination in Autographa californica nucleopolyhedrovirus. J Virol. 76(18):9323–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Malik HS. 2012. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet.. 46:677–700. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27(23):4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham AM, et al. 2005. EGene: a configurable pipeline generation system for automated sequence analysis. Bioinformatics 21(12):2812–2813. [DOI] [PubMed] [Google Scholar]
- Freemont PS. 2000. Ubiquitination: RING for destruction? Curr Biol.. 10(2):R84–R87. [DOI] [PubMed] [Google Scholar]
- Friesen PD, Miller LK. 2001. Insect viruses In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Field’s virology. Philadelphia (PA): Lippincott Williams & Wilkins; p. 599–628. [Google Scholar]
- Garcia-Maruniak A, Pavan OH, Maruniak JE. 1996. A variable region of Anticarsia gemmatalis nuclear polyhedrosis virus contains tandemly repeated DNA sequences. Virus Res.. 41(2):123–132. [DOI] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. 2010. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet.. 44:445–477. [DOI] [PubMed] [Google Scholar]
- Greene GL, Leppla NC, Dickerson WA. 1976. Velvetbean caterpillar: a rearing procedure and artificial medium. J Econ Entomol. 69(4):487–488. [Google Scholar]
- Guarino LA. 1990. Identification of a viral gene encoding a ubiquitin-like protein. Proc Natl Acad Sci U S A.. 87(1):409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib S, Hasnain SE. 1997. A bifunctional baculovirus homologous region (hr1) sequence: enhancer and origin of replication functions reside within the same sequence element. Curr Sci.. 73(8):658–666. [Google Scholar]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. (41):95–98.10780396 [Google Scholar]
- Hayakawa T, Rohrmann GF, Hashimoto Y. 2000. Patterns of genome organization and content in lepidopteran baculoviruses. Virology 278(1):1–12. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. 2003. Genome-wide survey for genes horizontally transferred from cellular organisms to baculoviruses. Mol Biol Evol.. 20(6):979–987. [DOI] [PubMed] [Google Scholar]
- Hyink O, et al. Whole genome analysis of the Epiphyas postvittana nucleopolyhedrovirus. J Gen Virol. 83(Pt 4):957–971. [DOI] [PubMed] [Google Scholar]
- Imai N, et al. 2003. Ubiquitin ligase activities of Bombyx mori nucleopolyhedrovirus RING finger proteins. J Virol. 77(2):923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Koonin EV, Aravind L. 2002. Extensive domain shuffling in transcription regulators of DNA viruses and implications for the origin of fungal APSES transcription factors. Genome Biol.. 3(3):RESEARCH0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joazeiro CA, Weissman AM. 2000. RING finger proteins: mediators of ubiquitin ligase activity. Cell 102(5):549–552. [DOI] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. 2004. A combined transmembrane topology and signal peptide prediction method. J Mol Biol.. 338(5):1027–1036. [DOI] [PubMed] [Google Scholar]
- Katsuma S, Tsuchida A, Matsuda-Imai N, Kang W, Shimada T. 2011. Role of the ubiquitin-proteasome system in Bombyx mori nucleopolyhedrovirus infection. J Gen Virol. 92(Pt 3):699–705. [DOI] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem.. 74(20):5383–5392. [DOI] [PubMed] [Google Scholar]
- Kool M, Ahrens CH, Goldbach RW, Rohrmann GF, Vlak JM. 1994. Identification of genes involved in DNA replication of the Autographa californica baculovirus. Proc Natl Acad Sci U S A.. 91(23):11212–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer DC. 2000. Stability and evolution of overlapping genes. Evolution 54(3):731–739. [DOI] [PubMed] [Google Scholar]
- Krappa R, Knebel-Morsdorf D. 1991. Identification of the very early transcribed baculovirus gene PE-38. J Virol. 65(2):805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krappa R, Roncarati R, Knebel-Morsdorf D. 1995. Expression of PE38 and IE2, viral members of the C3HC4 finger family, during baculovirus infection: PE38 and IE2 localize to distinct nuclear regions. J Virol. 69(9):5287–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol.. 305(3):567–580. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, et al. 2009. Circos: an information aesthetic for comparative genomics. Genome Res.. 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzio J, et al. 1999. Sequence and analysis of the genome of a baculovirus pathogenic for Lymantria dispar. Virology 253(1):17–34. [DOI] [PubMed] [Google Scholar]
- Landais I, et al. 2006. Functional analysis of evolutionary conserved clustering of bZIP binding sites in the baculovirus homologous regions (hrs) suggests a cooperativity between host and viral transcription factors. Virology 344(2):421–431. [DOI] [PubMed] [Google Scholar]
- Lauzon HA, Jamieson PB, Krell PJ, Arif BM. 2005. Gene organization and sequencing of the Choristoneura fumiferana defective nucleopolyhedrovirus genome. J Gen Virol. 86(Pt 4):945–961. [DOI] [PubMed] [Google Scholar]
- Levinson G, Gutman GA. 1987. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol.. 4(3):203–221. [DOI] [PubMed] [Google Scholar]
- Li L, et al. 2005. Complete comparative genomic analysis of two field isolates of Mamestra configurata nucleopolyhedrovirus-A. J Gen Virol. 86:91–105. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. [DOI] [PubMed] [Google Scholar]
- Lo CF, et al. 1996. Detection of baculovirus associated with white spot syndrome (WSBV) in penaeid shrimps using polymerase chain reaction. Dis Aquat Org. 25(1–2):133–141. [Google Scholar]
- Lo HR, Chou CC, Wu TY, Yuen JP, Chao YC. 2002. Novel baculovirus DNA elements strongly stimulate activities of exogenous and endogenous promoters. J Biol Chem.. 277(7):5256–5264. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. 2007. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res.. 35(Database issue):D237–D240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruniak JE. 1986. Baculovirus structural proteins and protein synthesis In: Granados RR, Federici BA, editors. The biology of baculovirus. Vol. 1. Boca Raton, Florida: CRC Press; p. 129–146. [Google Scholar]
- Maruniak JE. 1989. Molecular biology of Anticarsia gemmatalis baculovirus. Mem Inst Oswaldo Cruz. 84(3):107–111. [Google Scholar]
- Maruniak JE, Garcia-Maruniak A, Souza ML, Zanotto PM, Moscardi F. 1999. Physical maps and virulence of Anticarsia gemmatalis nucleopolyhedrovirus genomic variants. Arch Virol. 144(10):1991–2006. [DOI] [PubMed] [Google Scholar]
- Mitchell A, et al. 2014. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43(Database issue):D213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscardi F. 1989. Use of viruses for pest control in Brazil: the case of the nuclear polyhedrosis virus of the soybean caterpillar, Anticarsia gemmatalis. Mem Inst Oswaldo Cruz 84:51–56. [Google Scholar]
- Moscardi F. 1999. Assessment of the application of baculoviruses for control of Lepidoptera. Annu Rev Entomol. 44:257–289. [DOI] [PubMed] [Google Scholar]
- Muller J, et al. 2010. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res.. 38(Database issue):D190–D195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark S, et al. 1983. Overlapping genes. Annu Rev Genet. 17:499–525. [DOI] [PubMed] [Google Scholar]
- O'Reilly DR, Miller LK, Luckow VA. 1993. Baculovirus expression vectors: a laboratory manual. New York: Oxford University Press. [Google Scholar]
- Ohkawa T, Rowe AR, Volkman LE. 2002. Identification of six Autographa californica multicapsid nucleopolyhedrovirus early genes that mediate nuclear localization of G-actin. J Virol. 76:12281–12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira JV, et al. 2006. Genome of the most widely used viral biopesticide: Anticarsia gemmatalis multiple nucleopolyhedrovirus. J Gen Virol. 87(Pt 11):3233–3250. [DOI] [PubMed] [Google Scholar]
- Oliveira JVC, et al. 2013. Modularity and evolutionary constraints in a baculovirus gene regulatory network. BMC Syst Biol.. 7(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson VA, Wetter JA, Friesen PD. 2001. Oligomerization mediated by a helix-loop-helix-like domain of baculovirus IE1 is required for early promoter transactivation. J Virol. 75(13):6042–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarelli AL, Miller LK. 1993. Three baculovirus genes involved in late and very late gene expression: ie-1, ie-n, and lef-2. J Virol. 67(4):2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarelli AL, Miller LK. 1994. In vivo and in vitro analyses of recombinant baculoviruses lacking a functional cg30 gene. J Virol. 68(2):1186–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8(10):785–786. [DOI] [PubMed] [Google Scholar]
- Prikhod’ko EA, Lu A, Wilson JA, Miller LK. 1999. In vivo and in vitro analysis of baculovirus ie-2 mutants. J Virol. 73(3):2460–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmann GF. 1992. Baculovirus structural proteins. J Gen Virol. 73 (Pt 4):749–761. [DOI] [PubMed] [Google Scholar]
- Rohrmann GF. 2011. Baculovirus molecular biology. Vol. 2012. Bethesda (MD): National Center for Biotechnology Information; Last accessed: 24 September 2013. Available from: http://www.ncbi.nlm.nih.gov/books/NBK49500/ [PubMed] [Google Scholar]
- Saurin AJ, Borden KL, Boddy MN, Freemont PS. 1996. Does this have a familiar RING? Trends Biochem Sci.. 21(6):208–214. [PubMed] [Google Scholar]
- Shackelton LA, Holmes EC. 2004. The evolution of large DNA viruses: combining genomic information of viruses and their hosts. Trends Microbiol.. 12(10):458–465. [DOI] [PubMed] [Google Scholar]
- Simon O, et al. 2011. Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: detecting positively selected genes. J Invertebr Pathol. 107:33–42. [DOI] [PubMed] [Google Scholar]
- Slack JM, Ribeiro BM, de Souza ML. 2004. The gp64 locus of Anticarsia gemmatalis multicapsid nucleopolyhedrovirus contains a 3′ repair exonuclease homologue and lacks v-cath and ChiA genes. J Gen Virol. 85(Pt 1):211–219. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Durbin R. 1962. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:GC1–10. [DOI] [PubMed] [Google Scholar]
- Steinhaus EA, Marsh GA. 1962. Report of diagnoses of diseased insects 1951-1961. Hilgardia 33:349–490. [Google Scholar]
- Szewczyk B, Hoyos-Carvajal L, Paluszek M, Skrzecz I, Lobo de Souza M. 2006. Baculoviruses—re-emerging biopesticides. Biotechnol Adv. 24(2):143–160. [DOI] [PubMed] [Google Scholar]
- Tanada Y, Kaya HK. 1993. Insect pathology. San Diego (CA): Academic Press. [Google Scholar]
- Theze J, et al. 2014. Genomic diversity in European Spodoptera exigua multiple nucleopolyhedrovirus isolates. J Gen Virol. 95:2297–2309. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res.. 22(22):4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 7(2):256–276. [DOI] [PubMed] [Google Scholar]
- Whitt MA, Manning JS. 1987. Role of chelating agents, monovalent anion and cation in the dissociation of Autograph calfornicu nuclear polyhedrosis virus occlusion body matrix by zinc chloride. J Invertebr Pathol. 49:61–69. [Google Scholar]
- Zanotto PMA, Krakauer DC. 2008. Complete genome viral phylogenies suggests the concerted evolution of regulatory cores and accessory satellites. PLoS One 3(10):e3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemskov EA, Kang W, Maeda S. 2000. Evidence for nucleic acid binding ability and nucleosome association of Bombyx mori nucleopolyhedrovirus BRO proteins. J Virol. 74(15):6784–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.