

Abstract
Penicillium digitatum and Penicillium expansum are two closely related fungal plant pathogens causing green and blue mold in harvested fruit, respectively. The two species differ in their host specificity, being P. digitatum restricted to citrus fruits and P. expansum able to infect a wide range of fruits after harvest. Although host-specific Penicillium species have been found to have a smaller gene content, it is so far unclear whether these different host specificities impact genome variation at the intraspecific level. Here we assessed genome variation across four P. digitatum and seven P. expansum isolates from geographically distant regions. Our results show very high similarity (average 0.06 SNPs [single nucleotide polymorphism] per kb) between globally distributed isolates of P. digitatum pointing to a recent expansion of a single lineage. This low level of genetic variation found in our samples contrasts with the higher genetic variability observed in the similarly distributed P. expansum isolates (2.44 SNPs per kb). Patterns of polymorphism in P. expansum indicate that recombination exists between genetically diverged strains. Consistent with the existence of sexual recombination and heterothallism, which was unknown for this species, we identified the two alternative mating types in different P. expansum isolates. Patterns of polymorphism in P. digitatum indicate a recent clonal population expansion of a single lineage that has reached worldwide distribution. We suggest that the contrasting patterns of genomic variation between the two species reflect underlying differences in population dynamics related with host specificities and related agricultural practices. It should be noted, however, that this results should be confirmed with a larger sampling of strains, as new strains may broaden the diversity so far found in P. digitatum.
Keywords: Penicillium, genomics, genome variation
Introduction
Worldwide, green mold, caused by Penicillium digitatum (Pers: Fr.), and blue mold, caused by Penicillium expansum (Link) Thom, are the two most damaging postharvest diseases of fruits (Marcet-Houben et al. 2012; Ballester et al. 2014). Penicillium digitatum infects citrus fruits exclusively (Marcet-Houben et al. 2012) whereas P. expansum can infect and develop decay in a large range of fruit crops, among which apples (Buron-Moles et al. 2015), pears (Paster et al. 2009), peaches (Yang et al. 2011), hazel (Yang et al. 2014), and others (Judet-Correia et al. 2010; Cao et al. 2012). Infection of the fruits by both pathogens takes place through surface injuries inflicted in the peel of the fruit during harvest and subsequent handling in the packhouse. Spores present in these injuries become active, germinate, colonize the injured tissue, and produce disease. Besides the economic impact of these fungi for the fruit industry, P. expansum is also a potential threat to public health due to its ability to produce patulin, a toxic mycotoxin, which can contaminate infected fruits and their derived products (Morales et al. 2007; Ballester et al. 2014). Other Penicillium species are also able to synthesize a wide array of toxins which has increased the interest for these fungi. As a result, the number of sequenced genomes has also increased, with the number of Penicillium genome projects reaching more than 40 (NCBI database [Geer et al. 2010]). Sequencing at the intraspecies level has also received increased attention, due to interest in unveiling the genetic bases of phenotypic variations observed between different strains. For instance, the three P. expansum strains sequenced by Ballester et al. (2014) presented different levels of Patulin production, with one strain being unable to produce the toxin. Despite its importance, the genomic dynamics of different strains of Penicillium species are still poorly understood.
Studies of genetic polymorphism in other species from the Trichocomaceae family are scarce, but available data point to the existence of several globally distributed lineages, so that isolates from distant locations can show high similarity while, at the same time, isolates from nearby locations may show larger differences. For instance, a comparison between two strains of Penicillium chrysogenum from China and United States revealed a very low level of sequence divergence between the two strains measured as the density of single nucleotide polymorphisms (SNP) between the two strains (∼0.08 SNPs/kb; Wang et al. 2014). In contrast, a comparison of the P. chrysogenum type strain to P. chrysogenum KF-25 showed a high level of protein sequence divergence where proteins shared only 75.1% similarity on average, suggesting they are actually different species (Peng et al. 2014). In Aspergillus fumigatus, the comparison of two strains, A1163, a derivative strain of CBS144-89/CEA10 isolated from a patient in Paris, and Af293 isolated from a patient in United Kingdom, showed an identity of 99.8% at nucleotide level between the core genes (Fedorova et al. 2008). A genome-wide nucleotide diversity analysis showed that the genetic diversity of the eight Aspergillus oryzae strains from Japan (1.05 SNPs/kb) is approximately 25% of that found in eight Aspergillus flavus strains from different parts in the world (2.06 SNPs/kb) (Gibbons et al. 2012). In Aspergillus niger, the type strain CBS513.88, from The Netherlands, was compared with A. niger SH2 from China, which showed a high level of similarity to the type strain with a SNP density of 0.051 SNPs/kb (Yin et al. 2014) while the second strain (ATCC1015 from United States) had a more than hundred higher average SNP density of 8 SNPs/kb (Andersen et al. 2011). Comparison between this latter strain and a fourth A. niger strain (ATCC9029, also from United States) showed an intermediate level of divergence (2 SNPs/kb) (Andersen et al. 2011). Remarkably, in all cases, SNPs were shown to cluster in hypervariable regions in the genome rather than being uniformly distributed. In the strain A. niger SH2 these hypervariable regions were located in regions where gene order was not conserved with respect to other Aspergillus species. This effect was first observed in A. oryzae where the comparison of two strains showed that proteins with mutations were more often found in genes whose synteny was not conserved among other Aspergillus species (Umemura et al. 2012). These regions were often subtelomeric and were enriched in genes encoding secondary metabolism clusters, hydrolases, and transporters.
In addition, the increase of genetic variability can be linked to sexual recombination between diverged strains (Horn et al. 2009; Böhm et al. 2015; Teixeira et al. 2015). Until some decades ago the fungal kingdom was considered to comprise a high proportion of asexual species. Now, a growing number of studies in diverse species previously thought to be asexual, show that they are indeed able to reproduce sexually at some stage of their life cycle, or under some specific conditions (Ropars et al. 2012; Böhm et al. 2015; Taylor et al. 2015). Sex in fungi is controlled by the MAT loci, which encode key regulators of mating, production of pheromones and pheromone receptors (Fowler et al. 1999). The fungal kingdom includes outbreeding (heterothallism) systems with at least two and up to thousands of different mating types and inbreeding (homothallism) systems (Ni et al. 2011). Specifically, in Ascomycetes, MAT regions can vary in size from 1 to 6 kb and contain between one and three MAT genes, which encode homeodomain, α-domain, and/or high mobility group-domain transcription factors (Dyer 2008). Sexual reproduction is driven by mating compatibility and is mainly regulated by two MAT idiomorphs (dissimilar sequences occupying the same locus on the chromosome). Heterothallic individuals require the interaction of two partners of opposite mating types with compatible MAT idiomorphs in order to reproduce. Homothallic organisms are self-fertile and can present primary or secondary homothallism (pseudohomothallism), in the first one both idiomorphs are present in the same individual genome, either fused together or in different genomic regions; in the second one, an individual that has several mating types in the same genome is able to switch between them in order to reproduce with an individual of an opposite mating type (Ni et al. 2011; Billiard et al. 2012; Almeida et al. 2015).
Of note, the above mentioned studies focus on generalist species that inhabit a variety of niches. Indeed A. niger can be found in mesophilic environments such as decaying vegetation or soil and plants (Schuster et al. 2002) and P. chrysogenum is found in moist soil in habitats ranging from degraded forests (Jha et al. 1992) to arctic subglacial ice (Sonjak et al. 2006). Both species can colonize decaying fruits of a wide variety of species, likely resulting from their opportunistic saprophytic lifestyle. The initial sequencing of the P. digitatum genome revealed that strains isolated from Spain and China had nearly identical mitochondrial sequences, which was suggestive of a recent global expansion of this species (Marcet-Houben et al. 2012). On the other hand, P. expansum presented a higher degree of variability in the three sequenced strains (Ballester et al. 2014). The authors suggested that these differences in genome variability were correlated with smaller gene content and higher host-specificity (Ballester et al. 2014). Currently, there are four sequenced strains for P. digitatum (Marcet-Houben et al. 2012; Sun et al. 2013) and seven for P. expansum (Ballester et al. 2014; Nordberg et al. 2014; Yang et al. 2014; Yu et al. 2014; Li et al. 2015) which doubles the number of genomes used in previous analyses. Here, we assessed patterns of intraspecific variation in the two species, which extended previous observation of contrasting levels of variability. In addition, we reconstructed the evolutionary history of the sequenced lineages and assessed the contribution of recombination among divergent strains to patterns of genome variability.
Materials and Methods
Fungal Strains and Genome Data
Four strains of P. digitatum (table 1) and seven strains of P. expansum (table 2) were used in this study. These strains were isolated in different parts of the world and from different substrates (fig. 1A). The two Spanish strains of P. digitatum were isolated from orange and grapefruit (Marcet-Houben et al. 2012), the Chinese strain from citrus (Sun et al. 2013), and the Israeli one was isolated from decayed grapefruit and was sequenced within this study (see below). The Chinese, Israeli, Spanish and three American strains of P. expansum were isolated from decayed apples (Ballester et al. 2014; Yang et al. 2014; Yu et al. 2014;Li et al. 2015), while the additional American strain was isolated from hazel “Corylus avellana L.” and was initially misidentified as Penicillium aurantiogriseum (Yang et al. 2014), and corrected later (Ballester et al. 2014). Based on their higher quality, the Spanish P. digitatum (PHI26) and the Israeli P. expansum (PEXP) strains were used as reference for their respective species. Due to the lack of publicly stored data for two strains of P. expansum (R19 and T01) we simulated sequencing paired-end reads (70 bases read length, and 100 bases insert size) based on their genome assembly using wgsim 0.3.1-r13 tool (Li 2012) setting the error parameter to zero (rRXe = 0).
Table 1.
Summary of the Main Assembly and Annotation Features of the Genomes of the Four P. digitatum Strains Used for This Study
| P. digitatum Strains | PHI26 | Pd1 | Pd01-ZJU | PDC 102 |
|---|---|---|---|---|
| Origin | Spain | Spain | China | Israel |
| Sample | Orange | Grapefruit | Citrus | Grapefruit |
| Sequencing technology | Illumina (HiSeq 2000) | Roche 454 | Roche 454, Illumina (GAII) | Illumina (HiSeq 2000) |
| Genome size (Mb) | 25.6 | 24.9 | 26 | 24.9 |
| Sequencing coverage | 83× | 24× | 300× | 500× |
| Number of contigs | 287 | 544 | 1,817 | 826 |
| Number of scaffolds | 102 | 54 | 76 | ND |
| Number of large scaffolds (>100 kb) | 36 | 26 | 35 | ND |
| N50 (base pairs) | 878,909 | 1,533,507 | 1,047,382 | 70,904 |
| GC content (%) | 48.9 | 48.9 | 48.4 | 48.9% |
| Number of protein-coding genes | 9,153 | 8,969 | 9,006 | 8,070 |
Table 2.
Summary of the Main Assembly and Annotation Features of the Genomes of the Seven P. expansum Strains Used for This Study
| P. expansum Strains | PEXP | PEX1 | PEX2 | NRRL 62431 | ATCC 24692 | R19 | T01 |
|---|---|---|---|---|---|---|---|
| Origin | Israel | Spain | United States | United States | United States | United States | China |
| Sample | Apple | Apple | Apple | Nuts | Apple | Apple | Apple |
| Sequencing technology | Illumina (HiSeq 2000) | Illumina (HiSeq 2000) | Illumina (HiSeq 2000) | Illumina GAIIx | Illumina (HiSeq 2000) | Illumina (MiSeq Benchtop) | Illumina (HiSeq 2000) |
| Genome size (Mb) | 32.1 | 31.1 | 32.4 | 32.7 | 32.5 | 31.4 | 33.5 |
| Sequencing coverage | 605× | 73× | 473× | 50× | 119× | 27× | 527× |
| Number of contigs | 269 | 1,723 | 377 | 4,775 | 136 | 1,231 | 527 |
| Number of scaffolds | 249 | 1,723 | 345 | ND | 12 | ND | 108 |
| Number of large scaffolds (>100 kb) | 80 | 9 | 89 | ND | 8 | ND | ND |
| N50 (base pairs) | 422,053 | 31,189 | 376,691 | 17,305 | 5,330,000 | 48,518 | 5,031,090 |
| GC content (%) | 47.6 | 48.2 | 47.6 | 48.5 | ND | 48.24 | 47.1 |
| Number of protein-coding genes | 11,048 | 10,683 | 11,070 | 11,476 | 1,830 | 10,554 | 11,770 |
Fig. 1.—

Approximate geographic locations and host species where the four strains of P. digitatum and the seven strains of P. expansum analyzed in this study were isolated (A). Venn diagrams indicating shared SNPs in (B) P. digitatum using the PHI26 strain as a reference for the other three strains (Pd1, Pd01-ZJU, PDC 102), (C) P. expansum using the PEXP strain as a reference for the other six strains (PEX1, PEX2, NRRL 62431, ATCC 24692, R19, T01).
Genome Sequencing of P. digitatum Strain PDC 102
Here, we report the genome sequence of P. digitatum strain PDC 102, originally isolated from decayed grapefruit. The genome was sequenced using Illumina HiSeq 2000 at the high throughput sequencing unit, Technion—Israel Institute of Technology, Haifa, Israel. A total of 83,360,197 paired-end reads with a length of 100 nt were generated with genome coverage of approximately 500-fold. The paired-end reads were assembled into 883 contigs with an N50 contig length of 70,904 bp by the ABySS (Simpson et al. 2009) software followed by SSPACE (Boetzer et al. 2011) and GapFiller (Boetzer and Pirovano 2012) software to create longer scaffolds with paired-end data. This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under accession No. LHSL00000000. The version described in this article is the first version (accession No. LHSL00000000). Gene prediction and annotation were done using the maker website pipeline (Cantarel et al. 2008) and the Augustus software pipeline (Stanke et al. 2004).
Detection of SNPs
To improve the reference genomes, we mapped their reads onto their respective reference genome using BWA 0.7.6a-r433 (Li and Durbin 2009) and we identified SNPs with GATK (McKenna et al. 2010), using thresholds for mapping quality (>40) and read depth (>20) as filters. Then we substituted the reference nucleotides with the highly reliable SNPs, as they are most likely the result of errors in the assembly sequenced. Then, sequence reads from each strain were mapped onto the respective improved reference genome for the SNP calling, using the same tools described before (BWA and GATK), but this time, lowering the read depth to 10. To study patterns of SNP distribution along the genomes, we plotted Venn diagrams using InteractiVenn tool (Heberle et al. 2015) and for each strain, we plotted the SNP density per contig for nonoverlapping windows of 1 kb.
Population Genetics Parameters
We estimated the Nei’s gene diversity index (Nei 1973) in each species. To investigate the variation of nonsynonymous and synonymous SNPs in the coding regions per strain, we compared nonsynonymous changes per nonsynonymous site (πN) to synonymous changes per synonymous site (πS) by assuming that three-fourth of all sites are nonsynonymous.
Phylogenetic Analysis
A species tree was built using a gene concatenation approach. We used Penicillium camemberti as an outgroup. We selected the orthologous proteins predicted during the phylome reconstruction of P. expansum (Ballester et al. 2014). Genes that had one-to-one orthologs in the three species of interest (P. camemberti, P. digitatum, and P. expansum; Gabaldón 2008) were selected. After discarding genes with no SNP in any of the three species we kept 5,892 protein-coding genes for further analyses. We first did a protein alignment using the pipeline described by Huerta-Cepas et al. (2011). Briefly, this alignment strategy consists of doing six alignments using three different programs (MUSCLE v3.8.31 [Edgar 2004], MAFFT v6.861b [Katoh et al. 2002], Kalign v2.04 [Lassmann and Sonnhammer 2005]) and applying them in forward and reverse sequence orientation. The six resulting alignments are then combined using M-COFFEE as implemented in T-COFEE v8.80 (Notredame et al. 2000). The alignment is then back-translated to coding sequence. After concatenating all the alignments, the resulting nucleotide alignment was trimmed using trimAl v1.4 (Capella-Gutiérrez et al. 2009) to remove all positions with gaps. The final alignment had 9321774 positions. A phylogenetic tree was then reconstructed from this alignment using PhyML (Guindon and Gascuel 2003) using the general time reversible model and other options as default. 100 bootstrap repetitions were calculated.
Genomic Recombination
To compute the number of recombination events we selected the four strains of P. digitatum and four strains of P. expansum (PEXP, PEX1, NRRL 62431, and ATCC 24692). LDhat v. 2.2 (Auton and McVean 2007) was used to infer the minimum number of recombination events (Rmin) per species. In addition, for P. expansum, we used RDP4 (Martin and Rybicki 2000) on the largest contigs (>500 kb) to analyze the recombination breakpoints and recalculate the SNP density without recombination tracks (Martin and Rybicki 2000).
MAT Locus Search
In order to search the presence of MAT idiomorph regions in P. digitatum and P. expansum, we performed a blast search of the two idiomorphs available in NCBI database (Geer et al. 2010) in the species closely related to P. digitatum and P. expansum (supplementary table S1, Supplementary Material online) against each genome.
Results and Discussion
Genome Sequence of P. digitatum PDC 102
To increase the genomic information of P. digitatum, we undertook the sequencing of an additional strain of this species: PDC 102, isolated from decayed grapefruit in Israel. This genome was sequenced using Illumina HiSeq 2000 pair-end approach (see Materials and Methods) and the final assembly resulted in a genome size (24.9 Mb) similar to other sequenced strains of P. digitatum (Marcet-Houben et al. 2012). The assembly includes 883 contigs with an N50 contig length of 70,904 bp and a coverage of 500× (table 1). The genome has an average G+C content of 48.9%. A total of 8,070 predicted proteins (minimum length of 44 amino acids, maximum length of 6,923 amino acids) were identified in this strain.
Patterns of Polymorphism in P. expansum and P. digitatum
Including the newly sequenced P. digitatum PDC 102 genome we have a total of four P. digitatum genomes and seven P. expansum genomes isolated from different geographical locations (fig. 1A). This allowed us to derive maps of genome variation in both species (see Materials and Methods). For each species we mapped the read data to the chosen reference assembly of the same species to detect SNPs (see Materials and Methods). These analyses showed that in P. digitatum there were few polymorphic sites, with an average of 0.06 SNPs per kb (table 3 and fig. 1B). In contrast, the same analysis in P. expansum showed a 40-fold larger amount of genetic variation, with an average of 2.44 SNPs per kb (table 3 and fig. 1C). These differences were also apparent when considering Nei’s index of genetic diversity (Hs) which was 47 times higher in P. expansum as compared with P. digitatum (table 3). These results are consistent with previous comparisons done with the two Spanish strains of P. digitatum, and supported by the comparison of the mitochondrial sequence of the Chinese strain (Marcet-Houben et al. 2012) and three completely sequenced strains of P. expansum (Ballester et al. 2014). Interestingly, for P. expansum we found no correlation between the number of SNPs shared between the strains and their geographical distribution. For instance, the strains that shared more SNPs are from United States (ATCC 24692) and China (T01), whereas the strains that shared less SNPs are both from United States (PEX2 and R19). In P. digitatum, the strains from China (Pd01-ZJU) and Israel (PDC 102) shared more SNPs (fig. 1B).
Table 3.
Number of SNPs (Total, Synonymous, and Nonsynonymous SNPs), Density of SNPs (SNPs/kb), πN/πS Ratio, Nei Index (Hs), and Minimum Number of Recombination Events (Rmin) per kb of the P. digitatum and P. expansum Genome Strains Using as a Reference PHI26 and PEXP Strain, Respectively
|
P. expansum Strains |
P. digitatum Strains |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PEX1 | PEX2 | NRRL 62431 | ATCC 24692 | R19 | T01 | Pd1 | Pd01-ZJU | PDC 102 | ||
| SNPs | 42,559 | 56,969 | 41,750 | 231,312 | 40,706 | 61,445 | 312 | 1,395 | 3,155 | |
| SNPs syn | 8,665 | 10,020 | 9,831 | 53,033 | 9,092 | 10,556 | 23 | 135 | 319 | |
| SNPs nonsyn | 6,392 | 7,124 | 7,126 | 30,544 | 6,698 | 7,410 | 51 | 271 | 542 | |
| πN/πS | 0.24 | 0.24 | 0.24 | 0.19 | 0.25 | 0.23 | 0.73 | 0.67 | 0.57 | |
| SNPs/kb | 1.368 | 1.758 | 1.277 | 7.128 | 1.296 | 1.833 | 0.013 | 0.054 | 0.127 | |
| SNPs/kb average | 2.44 | 0.06 | ||||||||
| Hs | 27.89 × 10-4 | 0.59 × 10-4 | ||||||||
| Rmin/kba | 0.036 | 0.004 | ||||||||
In P. expamsum, the Rmin was obtained using the strains: PEXP, PEX1, NRRL 62431, and ATCC 24692.
Patterns of Synonymous and Nonsynonymous SNPs
We next analyzed the distribution of SNPs present within protein-coding exons and observed that the number of nonsynonymous SNPs was higher than the number of synonymous SNPs in all the strains of P. digitatum, whereas in P. expansum all strains displayed an excess of synonymous SNPs (table 3). We calculated per site nonsynonymous and synonymous nucleotide diversity by assuming that three-fourth of all sites are nonsynonymous and computed the ratio between these values (table 3). Our results indicate an excess in the rate of nonsynonymous to synonymous substitutions in P. digitatum (0.57–0.73) as compared with P. expansum (0.19–0.25). These results indicate a relaxation of purifying selection in P. digitatum relative to P. expansum. Considering that this excess is distributed along the entire genome and is present in all sampled isolates, an underlying common demographic history of the population should provide an explanation to our observation. A possible explanation is that the excess of nonsynonymous SNPs in P. digitatum results, at least in part, from a population bottleneck followed by a recent population expansion. Earlier studies based on simulations (Travis et al. 2007; Lohmueller et al. 2008) showed that during a population bottleneck the proportion of nonsynonymous SNPs increases. When the population expanded, the fraction of nonsynonymous SNPs increases because the increase in population size results in many more mutations, of which most are nonsynonymous because of the genetic code. This scenario would also imply that the population growth was recent enough so that purifying selection has not had enough time to reduce the fraction of slightly deleterious nonsynonymous SNPs to the equilibrium value expected for a larger population.
Evolutionary Relationships Based on SNP Data
We used SNP data to reconstruct the phylogenetic relationships among the strains considered, using P. camemberti as an outgroup. We limited the analysis to coding regions that had one to one orthologs in all three species and that had at least one SNP in any P. expansum or P. digitatum strains. In total, 5,892 single copy genes fulfilled these requirements. These genes were concatenated and a species tree was reconstructed using a maximum likelihood approach (see Materials and Methods). As seen in figure 2, there was no correlation between the phylogenetic position and the geographical origins of P. expansum and P. digitatum. Neither did we find a correlation with the host specificity in P. expansum. Branch lengths from the common ancestor to P. digitatum were longer than the lengths to P. expansum. At the strain level, however, branches in P. digitatum were shorter, in consonance with the smaller amount of genomic variation observed (table 3). Collectively these data indicate that the time to coalescence is longer in P. expansum strains as compared with P. digitatum strains (i.e.,P. expansum strains started to diverge earlier).
Fig. 2.—
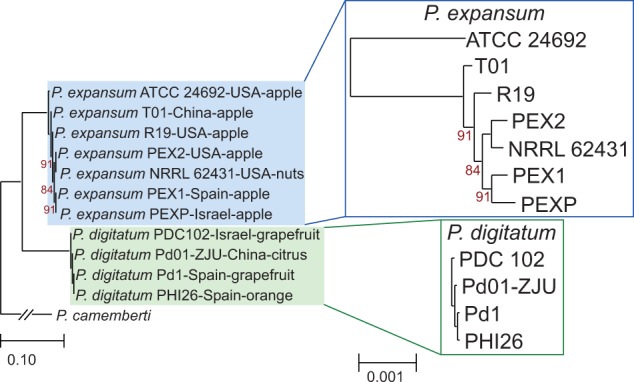
Maximum likelihood species tree derived from the SNPs found in 5,892 protein-coding genes present in all species considered. All bootstrap values that are not maximal (100%) are indicated in the figure. Scales below indicate substitutions per site. Insets show detail of the subtrees containing P. expansum and P. digitatum lineages at a larger scale, to better appreciate the differences in branch lengths.
Signatures of Recombination and Heterothallism in the P. expansum Genome
Interestingly, we found that the SNPs were not equally distributed along the P. expansum genome, but rather they formed clusters (see fig. 3 for an example). Such pattern is reminiscent of possible recombination events, in which a given region of the genome has been introgressed from a genetically different strain. To test this possibility we searched for recombination events in both species (see Materials and Methods). Our results show a much larger number of predicted recombination events in P. expansum (0.036 Rmin per kb, where Rmin is the minimum number of recombination events) versus the 0.004 Rmin per kb found in P. digitatum (table 3). To assess to what extent recombination may have contributed to the observed larger diversity in P. expansum we compared the SNPs density in a subset of the longest 13 contigs, comprising 10.4 Mb, before and after excluding recombination tracks (see Materials and Methods). In the evaluated regions, SNP density decreased from 7.68 to 7.49 SNPs/kb, indicating that roughly 2.5% of the variation may be attributed to recombination tracks between the strains compared. This result shows some contribution to genetic variability in P. expansum of recombination among diverged lineages, although this process can only explain a small part of the larger genetic variability when compared with that found in P. digitatum (0.06 SNPs/kb). The low level of recombination events detected in P. digitatum (0.004 Rmin per kb) (table 3) is not necessarily indicative of a low prevalence of recombination, as recombination events between very closely related strains are very difficult to detect.
Fig. 3.—

(A) Visualization with the integrative genome viewer of the mapping results of a selected 32 kb region (coordinates with respect to the reference: contig 129:71,272–103,598). The region was predicted to contain a recombination track. The three lower frames show depth of coverage graphs for the mapping of the sequenced reads against the reference for PEXP, PEX1, and ATCC 24692 strains, in this order. SNPs are indicated with colors. Note that the reference strain, as expected, has no SNPs, and that PEX1 shows a region of possible recombination (highlighted with a red box) in which the SNP pattern is similar to that in ATCC 24692 whereas the surrounding regions have much fewer and distinct SNPs. (B) Schematic representation of the selected region, in which the two SNP patterns in (A) are indicated as different colors. The red square shows a region in which the strain PEX1 has a SNP pattern (green) which is different from the overall pattern of the rest of the region and that in PEXP (yellow), and similar to the one found in ATCC 24692.
Consistent with sexual crossing as the source of recombination between lineages we found reproduction idiomorphs of alternative mating types (MAT genes) among the sequenced P. expansum strains. Indeed, we found that the strains NRRL 62431 and R19 of P. expansum carry the MAT1-1 idiomorph as was previously reported for the PEXP, PEX1, PEX2 strains (Ballester et al. 2014). However, the ATCC 24692 and T01 strains had the MAT1-2 idiomorph (supplementary fig. S1, Supplementary Material online), suggesting the possibility of sexual reproduction in P. expansum as an heterothallic species. The upstream and downstream flanking regions of the MAT genes included, respectively, the genes APN2 and SLA2, which is a structural organization similar to the mating-type loci in other ascomycetes that have sexual reproduction, like P. roqueforti and P. chrysogenum (Ropars et al. 2012; Böhm et al. 2015). These results indicate that P. expansum is an heterothallic species and that mating among diverged strains may have contributed to the observed patterns of genomic variation. To the best of our knowledge this is the first indication of heterothallism in P. expansum.
Concluding Remarks
The results reported here from the analysis of the genomes of P. expansum and P. digitatum strains sampled worldwide unveil contrasting patterns of genomic variation. On the one hand P. expansum has a large genetic diversity. Consistently, pairwise comparisons of strains show a relatively high sequence divergence, which does not correlate with the geographical distance between the compared isolates. This indicates that sampled isolates diverged a long time ago but that the geographical structure of the populations may have been broken by migrations. In addition we find compelling evidence for recombination between diverged P. expansum lineages. In this regard the demonstration of the presence of two alternative mating type loci in P. expansum support heterothallism and sexual recombination in this species, thus providing a mechanistic basis for the observed introgressions between diverged lineages. On the other hand P. digitatum shows a remarkably low genetic diversity and only one mating type locus was present among the analyzed strains. In addition we detected a relative excess of nonsynonymous SNPs as compared with P. expansum. Taken together these results suggest a very recent clonal expansion for P. digitatum.
How can these two contrasting patterns be explained in the light of their association with species of agricultural interests?. One of the main differences between these two species is their contrasting host specificity. As explained above, P. expansum is able to infect a wide range of hosts, whereas P. digitatum is specific to citrus fruits. Although any consideration must remain highly speculative at this point, we hypothesize that differences in host specificity coupled with present and past agricultural practices in the respective hosts have shaped the two species differently. Of note, although most fruits were first domesticated several millennia ago, the industrial expansion of key varieties and the start of significant global commerce can be traced back in most cases to the last century. Hence, it is reasonable to consider that expansion and industrialization of agriculture have favored the movement of strains of both species around the world in recent times, which is compatible with the observed absence of a clear geographical structure of the genetic diversity. However, we argue that, being P. digitatum a highly host-specific species, its original distribution was narrow, perhaps restricted to certain geographical areas, and thus the expansion affected a single population that thereby colonized new areas. The fact that only one mating type is found in sequenced P. digitatum strains may be related to this clonal expansion, although the number of analyzed strains is still too low to reach any conclusion. In contrast, the more versatile P. expansum may have had a wider, perhaps global, geographical distribution preceding the start of global commerce. Thus when commercial expansion of several of its host species favored migration, this resulted in favoring genetic admixture between the newly encountered populations, particularly considering that the species is able to reproduce sexually. Such scenario could explain how a similar process (global migration) can result in totally different population genomics patterns depending on the preceding diversity and geographical distribution of the species at hand. Thus, whereas global commerce may favor fast clonal expansion of niche specialists it will trigger population admixture in generalists. Naturally, further research is needed to better understand the present and past population dynamics in these and other important plant pathogens.
Supplementary Material
Supplementary figure S1 and table S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported in part by Grants from the Spanish Ministry of Economy and Competitiveness: “Centro de Excelencia Severo Ochoa 2013–2017” SEV-2012-0208, and BIO2012-37161 cofounded by European Regional Development Fund (ERDF); from the European Union and ERC Seventh Framework Programme (FP7/2007-2013) under grant agreement ERC-2012-StG-310325, and grant from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No H2020-MSCA-ITN-2014-642095; and Peruvian Ministry of Education: “Beca Presidente de la República” (2013-III).
Literature Cited
- Almeida JMGCF, Cissé OH, Fonseca Á, Pagni M, Hauser PM. 2015. Comparative genomics suggests primary homothallism of Pneumocystis species. MBio 6(1):e02250–e02214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen MR, et al. 2011. Comparative genomics of citric-acid-producing Aspergillus Niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res. 21(6):885–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, McVean G. 2007. Recombination rate estimation in the presence of hotspots. Genome Res. 17(8):1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester A-R, et al. 2014. Genome, transcriptome, and functional analyses of Penicillium expansum provide new insights into secondary metabolism and pathogenicity. Mol Plant-Microbe Interact. 28(6):635–647. [DOI] [PubMed] [Google Scholar]
- Billiard S, López-Villavicencio M, Hood ME, Giraud T. 2012. Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J Evol Biol. 25(6):1020–1038. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27(4):578–579. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol. 13(6):R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm J, Dahlmann TA, Gümüşer H, Kück U. 2015. A MAT1-2 wild-type strain from Penicillium chrysogenum: functional mating-type locus characterization, genome sequencing and mating with an industrial penicillin-producing strain. Mol Microbiol. 95(5):859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buron-Moles G, et al. 2015. Characterizing the proteome and oxi-proteome of apple in response to a host (Penicillium expansum) and a non-host (Penicillium digitatum) pathogen. J Proteomics. 114:136–151. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, et al. 2008. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18(1):188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Li H, Tian S, Qin G. 2012. Boron improves the biocontrol activity of Cryptococcus laurentii against Penicillium expansum in jujube fruit. Postharvest Biol Technol. 68:16–21. [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25(15):1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PS. 2008. Evolutionary biology: genomic clues to original sex in fungi. Curr Biol. 18(5):R207–R209. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova ND, et al. 2008. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 4(4):e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler TJ, DeSimone SM, Mitton MF, Kurjan J, Raper CA. 1999. Multiple sex pheromones and receptors of a mushroom-producing fungus elicit mating in yeast. Mol Biol Cell. 10(8):2559–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldón T. 2008. Comparative genomics-based prediction of protein function. Methods Mol Biol. 439: 387–401. [DOI] [PubMed] [Google Scholar]
- Geer LY, et al. 2010. The NCBI biosystems database. Nucleic Acids Res. 38(Database issue):D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, et al. 2012. The evolutionary imprint of domestication on genome variation and function of the filamentous fungus Aspergillus oryzae. Curr Biol. 22(15):1403–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52(5):696–704. [DOI] [PubMed] [Google Scholar]
- Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. 2015. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn BW, Ramirez-Prado JH, Carbone I. 2009. Sexual reproduction and recombination in the aflatoxin-producing fungus Aspergillus parasiticus. Fungal Genet Biol. 46(2):169–175. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. 2011. PhylomeDB v3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 39(Database issue):D556–D560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha DK, Sharma GD, Mishra RR. 1992. Ecology of soil microflora and mycorrhizal symbionts in degraded forests at two altitudes. Biol Fertil Soils. 12(4):272–278. [Google Scholar]
- Judet-Correia D, et al. 2010. Validation of a predictive model for the growth of Botrytis cinerea and Penicillium expansum on grape berries. Int J Food Microbiol. 142(1–2):106–113. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-I, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T, Sonnhammer ELL. 2005. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, et al. 2015. Genomic characterization reveals insights into patulin biosynthesis and pathogenicity in Penicillium species. Mol Plant-Microbe Interact 28(6):635–647. [DOI] [PubMed] [Google Scholar]
- Li H. 2012. Wgsim—read simulator for next generation sequencing. Bioinformatics 25:1754–1760.19451168 [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, et al. 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451(7181):994–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Houben M, et al. 2012. Genome sequence of the necrotrophic fungus Penicillium digitatum, the main postharvest pathogen of citrus. BMC Genomics 13:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16(6):562–563. [DOI] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales H, Marín S, Rovira A, Ramos AJ, Sanchis V. 2007. Patulin accumulation in apples by Penicillium expansum during postharvest stages. Lett Appl Microbiol. 44(1):30–35. [DOI] [PubMed] [Google Scholar]
- Nei M. 1973. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A. 70(12):3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Feretzaki M, Sun S, Wang X, Heitman J. 2011. Sex in fungi. Annu Rev Genet. 45:405–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg H, et al. 2014. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 42(Database issue):D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 302(1):205–217. [DOI] [PubMed] [Google Scholar]
- Paster N, Huppert D, Barkai-Golan R. 2009. Production of patulin by different strains of Penicillium expansum in pear and apple cultivars stored at different temperatures and modified atmospheres. Food Addit Contam. 12(1):51–58. [DOI] [PubMed] [Google Scholar]
- Peng Q, et al. 2014. Genomic characteristics and comparative genomics analysis of Penicillium chrysogenum KF-25. BMC Genomics 15:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropars J, et al. 2012. Sex in cheese: evidence for sexuality in the fungus Penicillium roqueforti. PLoS One 7(11):e49665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster E, Dunn-Coleman N, Frisvad JC, Van Dijck PWM. 2002. On the safety of Aspergillus niger—a review. Appl Microbiol Biotechnol. 59(4–5):426–435. [DOI] [PubMed] [Google Scholar]
- Simpson JT, et al. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19(6):1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonjak S, Frisvad JC, Gunde-Cimerman N. 2006. Penicillium mycobiota in Arctic subglacial ice. Microb Ecol. 52(2):207–216. [DOI] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32 (Web Server issue):W309–W312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, et al. 2013. Genomewide investigation into DNA elements and ABC transporters involved in imazalil resistance in Penicillium digitatum. FEMS Microbiol Lett. 348:11–18. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Hann-Soden C, Branco S, Sylvain I, Ellison CE. 2015. Clonal reproduction in fungi. Proc Natl Acad Sci U S A. 112(29):8901–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira J, Amorim A, Araujo R. 2015. Recombination detection in Aspergillus fumigatus through single nucleotide polymorphisms typing. Environ Microbiol Rep. doi: 10.1111/1758-2229.12321. [DOI] [PubMed] [Google Scholar]
- Travis JMJ, et al. 2007. Deleterious mutations can surf to high densities on the wave front of an expanding population. Mol Biol Evol. 24(10):2334–2343. [DOI] [PubMed] [Google Scholar]
- Umemura M, et al. 2012. Comparative genome analysis between Aspergillus oryzae strains reveals close relationship between sites of mutation localization and regions of highly divergent genes among Aspergillus species. DNA Res. 19(5):375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F-Q, et al. 2014. Genome sequencing of high-penicillin producing industrial strain of Penicillium chrysogenum. BMC Genomics 15(Suppl 1):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. 2014. Genome sequencing and analysis of the paclitaxel-producing endophytic fungus Penicillium aurantiogriseum NRRL 62431. BMC Genomics 15(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Cao S, Cai Y, Zheng Y. 2011. Combination of salicylic acid and ultrasound to control postharvest blue mold caused by Penicillium expansum in peach fruit. Innov Food Sci Emerg Technol. 12(3):310–314. [Google Scholar]
- Yin C, Wang B, He P, Lin Y, Pan L. 2014. Genomic analysis of the aconidial and high-performance protein producer, industrially relevant Aspergillus niger SH2 strain. Gene 541(2):107–114. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. 2014. Draft genome sequence of Penicillium expansum strain R19, which causes postharvest decay of apple fruit. Genome Announc. 2(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.