

Abstract
Transient global ischemia is a neuronal insult that induces delayed cell death. A hallmark event in the early post-ischemic period is enhanced permeability of mitochondrial membranes. The precise mechanisms by which mitochondrial function is disrupted are, as yet, unclear. Here we show that global ischemia promotes alterations in mitochondrial membrane contact points, a rise in intramitochondrial Zn2+, and activation of large, multi-conductance channels in mitochondrial outer membranes by 1 h after insult. Mitochondrial channel activity was associated with enhanced protease activity and proteolytic cleavage of BCL-xL to generate its pro-death counterpart, ΔN-BCL-xL. The findings implicate ΔN-BCL-xL in large, multi-conductance channel activity. Consistent with this, large channel activity was mimicked by introduction of recombinant ΔN-BCL-xL to control mitochondria and blocked by introduction of a functional BCL-xL antibody to post-ischemic mitochondria via the patch pipette. Channel activity was also inhibited by nicotinamide adenine dinucleotide, indicative of a role for the voltage-dependent anion channel (VDAC) of the outer mitochondrial membrane. In vivo administration of the membrane-impermeant Zn2+ chelator CaEDTA before ischemia or in vitro application of the membrane-permeant Zn2+ chelator tetrakis-(2-pyridylmethyl) ethylenediamine attenuated channel activity, suggesting a requirement for Zn2+. These findings reveal a novel mechanism by which ischemic insults disrupt the functional integrity of the outer mitochondrial membrane and implicate ΔN-BCL-xL and VDAC in the large, Zn2+-dependent mitochondrial channels observed in post-ischemic hippocampal mitochondria.
Keywords: global ischemia, zinc, mitochondria, BCL-xL, ion channels, programmed cell death
Introduction
Transient global ischemia induces delayed death of hippocampal CA1 pyramidal neurons (Lee et al., 1999; Lipton, 1999; Banasiak et al., 2000; Zukin et al., 2004). During ischemia, mitochondria sense oxygen levels and decrease ATP production (Sugawara et al., 1999; Wang et al., 2003). The effects of energy depletion on neurons (Howard et al., 1998; Tian and Baker, 2000; Fleidervish et al., 2001; Bolay et al., 2002) include neuronal membrane depolarization, synaptic glutamate release, a rise in cytosolic Ca2+, and swelling of cells (Choi, 1994; Sattler and Tymianski, 2000; Nishizawa, 2001; Zukin et al., 2004). Just after reperfusion, ATP levels and membrane polarization are restored (Zukin et al., 2004). Although histologically detectable neuronal death does not occur until 48 h, a number of cell death cascades (Alkayed et al., 2001; Northington et al., 2001), including the release of cytochrome c from mitochondria (Ouyang et al., 1999; Sugawara et al., 1999), are activated by 1–3 h after insult.
BCL-2 family members are critical to the response to cell death stimuli (Kroemer and Reed, 2000; Hengartner, 2000). BCL-xL is abundantly expressed in adult neurons (Boise et al., 1993; Krajewski et al., 1994; González-Garcia et al., 1995) and localizes to outer mitochondrial membranes (Kaufmann et al., 2003). It protects cells by its metabolic effects and/or by blocking activation of pro-death BCL-2 family members (Vander Heiden et al., 2000; Cheng et al., 2001; Basañez et al., 2002). Injurious stimuli promote the N-terminal proteolytic cleavage of BCL-xL to form the killer protein ΔN-BCL-xL (Cheng et al., 1997), which induces large conductance channels in mitochondrial outer membranes (Jonas et al., 2004, 2005) and cytochrome c release (Clem et al., 1998; Fujita et al., 1998).
Zn2+ is normally abundant in hippocampus (Frederickson et al., 1982; Assaf and Chung, 1984; Frederickson and Danscher, 1990; Tonder et al., 1990; Danscher et al., 1997; Li et al., 2001) (but see Kay, 2003) and at high concentrations is a mediator of neuronal death (Koh et al., 1996; Choi and Koh, 1998; Weiss and Sensi, 2000; Bossy-Wetzel et al., 2004; Sensi and Jeng, 2004; Zukin et al., 2004). Global ischemia elicits a delayed rise in Zn2+ in CA1 neurons before death (Koh et al., 1996; Choi and Koh, 1998; Calderone et al., 2004); evidence suggests that Zn2+ enters through GluR2 (glutamate receptor subunit 2)-lacking AMPA receptors (Sensi et al., 1999; Yin et al., 2002) and is released from intracellular stores (Aizenman et al., 2000; Lee et al., 2000; Sensi et al., 2003a; Bossy-Wetzel et al., 2004). The rise in cytosolic Zn2+ is accompanied by a rise in mitochondrial Zn2+ (Sensi et al., 2000, 2003a; Jiang et al., 2001; Bossy-Wetzel et al., 2004), which disrupts mitochondrial function (Nicholls and Budd, 2000; Kim et al., 2003). After ischemia, Zn2+ is required for release of cytochrome c and Smac/DIABLO (Calderone et al., 2004).
The mechanisms underlying Zn2+-induced neuronal death are complex (Sensi and Jeng, 2004). Here we show that transient global ischemia induces the formation of large multi-conductance channels in the mitochondrial outer membrane by 1 h after insult. The large channels are inhibited by nicotinamide adenine dinucleotide (NADH), an inhibitor of the mitochondrial voltage-dependent anion channel (VDAC), and by Zn2+ chelation, suggesting a requirement for intra-mitochondrial Zn2+. Channel activity correlates with appearance of ΔN-BCL-xL in mitochondria, is mimicked by introduction of recombinant ΔN-BCL-xL to the outer membrane of control mitochondria with a patch pipette, and is inhibited with a functional BCL-xL antibody. These findings are consistent with a role for intramitochondrial Zn2+ in the induction of outer membrane channel activity during global ischemia.
Materials and Methods
Global ischemia.
Male Sprague Dawley rats (150–200 g; Charles River Laboratories, Wilmington, MA) were maintained in a temperature- and light-controlled environment and were treated in accordance with the principles and procedures of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine. Animals were subjected to global ischemia by four-vessel occlusion (10 min), followed by reperfusion, as described previously (Calderone et al., 2003). Briefly, on the day before surgery, rats were anesthetized with halothane (2% was decreased to 1% after the induction of anesthesia) in a mixture of N2/O2 (70:30) delivered by mask attached to a Vapomatic anesthetic vaporizer (CWE, Ardmore, PA). The vertebral arteries were permanently occluded by electrocauterization, the common carotid arteries were exposed through a ventral midline neck incision and isolated with 3-0 silk ligatures, and the wound was closed. Rats were fasted overnight and the next day were again anesthetized, the wound was reopened, and the carotid arteries were occluded with aneurism clips (10 min). After removal of clips, the arteries were visually inspected to ensure adequate reflow. Anesthesia was discontinued immediately after initiation of occlusion. Body temperature was maintained at 37.5 ± 0.5°C with a rectal thermistor and heat lamp until recovery from anesthesia. Animals that failed to show complete loss of the righting reflex and dilation of the pupils from 2 min after occlusion was initiated until the end of occlusion, and the rare animals that subsequently exhibited obvious behavioral manifestations (abnormal vocalization when handled, generalized convulsions, hypoactivity) were excluded from the study. Sham animals were subjected to the identical procedure, except that carotid arteries were not occluded.
Isolation of mitochondria and synaptoneurosomes.
Mitochondria and synaptoneurosomes were isolated from control and experimental rat brains and purified by centrifugation over a discontinuous Ficoll gradient (Sullivan et al., 2004). In brief, rats (n = 3–6 per group; tissue from three to six animals pooled for each assay) were killed by decapitation 1 h after ischemia, and brains or hippocampi were dissected on ice and transferred to isolation buffer (250 mm sucrose, 20 mm HEPES, pH 7.2, 1 mm EDTA, and 0.5% BSA). Tissue was minced, homogenized (two times with a homogenizer; Wheaton, Millville, NJ), and centrifuged at 1300 × g to pellet nuclear material. The supernatant was centrifuged at high speed (13,000 × g for 10 min at 4°C); the pellet containing the mitochondria and synaptosomes was resuspended in isolation buffer and layered onto a Ficoll gradient ranging from 7.5 to 10%. After 30 min ultracentrifugation, at 32,500 rpm, 4°C, the mitochondrial and synaptosomal layers were removed and washed in isolation buffer by centrifugation at 13,000 × g. Mitochondria and synaptosomal fractions were frozen on dry ice and stored at − 80°C until use. Previously frozen mitochondria were used for electrophysiology, electron micrographs, and immunoblots. Synaptosomal protein concentrations were determined using the Pierce (Rockford, IL) BCA kit, and equivalent amounts of synaptosomal protein were used in all experiments. For all variables examined, mean values from control tissue were obtained and expressed as a percentage of mean control values, and the data were analyzed using a two-tailed t test.
Electron microscopy.
Mitochondrial pellets were fixed in 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, postfixed in the same buffer containing 1% osmium tetroxide, stained with 2% uranyl acetate, dehydrated in a graded series of ethanol, and embedded in Embed 812 resin (Electron Microscopy Sciences, Fort Washington, PA). Ultrathin sections (60 nm) were cut on a Reichert Ultracut E ultramicrotome, collected on Formvar and carbon-coated grids, stained with uranyl acetate and lead citrate, and observed in a Tecnai (Hillsboro, OR) 12 electron microscope. Measurements of density were acquired from different areas of the micrographs using Adobe Photoshop software (Adobe Systems, San Jose, CA), as described in the legend of Figure 1. Contact point density measurements were made on photomicrographs of high-power images of mitochondria that had intact outer membranes. Contact points were strictly defined as areas of increased density between the inner and outer mitochondrial membranes found at points at which the membranes curved in toward each other. Contact point length was defined as the measurement in micrometers of the lateral extent of the increase in density from one end of the dense area to the other. If the end of a dense area could not be detected, the length measurement was omitted. Each contact point density measurement was corrected for background density by subtracting a density measurement of an area of the background free of organelles and adjacent to the mitochondrion. The investigator was blind to the treatment conditions during the measurements.
Figure 1.
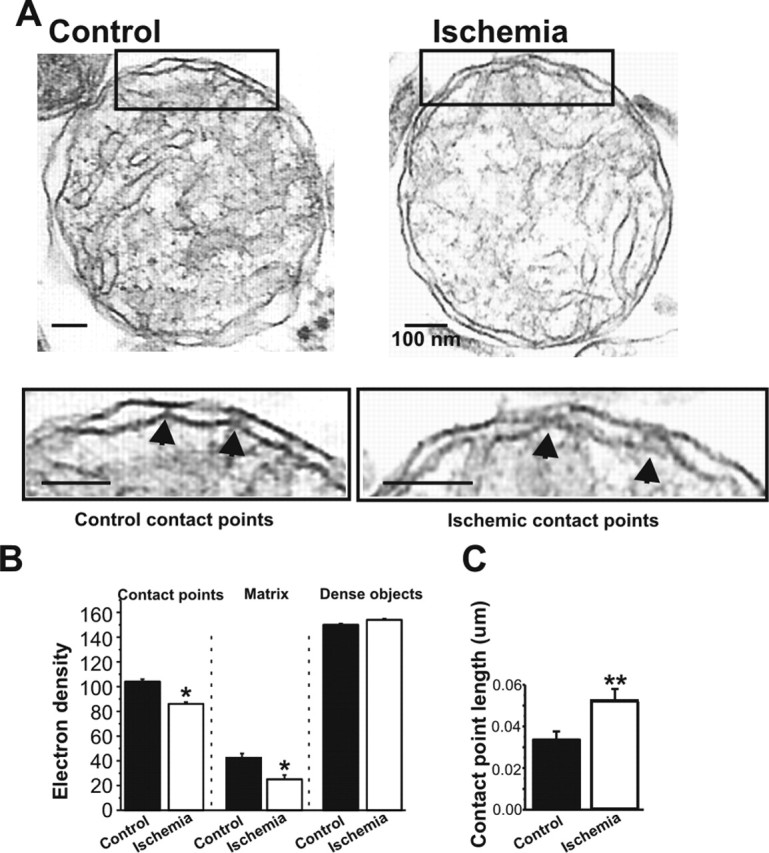
Mitochondrial morphology is altered in post-ischemic mitochondria. A, Electron micrographs of mitochondria isolated from the hippocampus of post-ischemic and control rats at 50 min after reperfusion. Insets show areas of contact between outer and inner membranes. B, The density of contact points is reduced in micrographs of post-ischemic mitochondria compared with control mitochondria (n = 6 control mitochondria, 48 contact points; n = 7 ischemic mitochondria, 65 contact points; ∗p < 0.02; mitochondria were pooled from 5 ischemic rat brains and 5 control rat brains from animals that had been treated during 1 experiment). Filled arrows in the insets of A show representative contact points used for the morphological analysis. Matrix density was lower in ischemic mitochondria compared with controls (20 measurements were made in each of three mitochondria in each of 4 micrographs of control pellets for a total of 240 control measurements; 20 measurements were made in each of 3 mitochondria in each of 6 micrographs of ischemic pellets for a total of 360 ischemic measurements; ∗p < 0.03). The density of the densest nonmitochondrial organelles in each micrograph was not different for control versus ischemic micrographs (n = 30 control measurements from 4 micrographs; n = 45 ischemic measurements from 6 micrographs; same experimental day as for contact point and matrix density measurements). C, The length of contact points in micrometers is increased in ischemic mitochondria over controls (n = 6 control mitochondria, 38 total contact points; n = 8 ischemic mitochondria, 33 total contact points; ∗∗p < 0.009; same experimental day as above). Scale bars, 100 nm.
Electrophysiology.
For patch-clamp recordings, the mitochondria were placed in the internal patch solution (containing 150 mm KCl and 20 mm HEPES, pH 7.2). Patch-clamp pipettes (80–100 MΩ) were filled with intracellular solution that contained 150 mm KCl and 20 mm HEPES, pH 7.2. The membrane potential was maintained at voltages ranging from −100 to +100 mV for periods of 10 s. Data were collected at 20 kHz and filtered at 500–1000 Hz. Zn2+ chelators [EDTA and tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN)] were obtained from Sigma (St. Louis, MO). In recordings with recombinant ΔN-BCL-xL (8 μg/ml), the protein was added to the internal patch pipette solution of PBS, and the bath perfusate was changed to PBS. The anti-BCL-xL antibody used in recordings of ischemic mitochondria was a polyclonal antibody obtained from A.G. Scientific (San Diego, CA).
Western blot analysis.
For detection of the proteolytic fragment of BCL-xL, protein samples (20 μg) isolated from control and experimental hippocampal mitochondria were subjected to gel electrophoresis on a 12% SDS gel and probed with rabbit anti-chicken BCL-xL polyclonal antibody (1:1000 dilution; gift from Craig Thompson, University of Pennsylvania, Philadelphia, PA) or a polyclonal antibody against BCL-xL (A.G. Scientific). As a positive control, 10 ng of recombinant human BCL-xL C-terminal truncation (lacking the last 76 amino acids) was also subjected to gel electrophoresis (data not shown). All blots were routinely reprobed with an antibody to the mitochondrial marker cyclooxygenase IV (COX IV) to monitor the quality and purity of mitochondrial preparations and to provide a loading control.
Caspase activity assay.
Caspase assays were performed in samples of synaptoneurosomes using the FAM-DEVD-FMK (carboxyfluorescein analog of benzyloxycarbonylaspartylglutamylvalylaspartic acid, a peptide inhibitor of caspase-3) caspase detection kit according to the instructions of the manufacturer (APO LOGIX; Bachem, Bubendorf, Switzerland) (Tanaka et al., 2004). FAM-DEVD-FMK is a carboxy-fluorescein analog of zDEVD-FMK (z-Asp-Glu-Val-fluoromethyl ketone), a broad-spectrum cysteine protease inhibitor that enters cells and irreversibly binds activated caspases. FAM-DEVD-FMK exhibits higher affinity for caspase-3 than for caspase-8 or caspase-7 and exhibits much lower affinity for the calpains than for caspases; thus, at 5 μm, FAM-DEVD is a relatively selective inhibitor of caspase-3 (Pozarowski et al., 2003). Moreover, FAM-DEVD-FMK labeling of CA1 neurons correlates well with caspase-3 activation, as assessed by Western blot analysis (Tanaka et al., 2004). The synaptosomal fraction was isolated from control and experimental brains as described above. Synaptoneurosomal protein concentration was determined using the Pierce BCA kit, and equivalent amounts of synaptosomal protein were used in all experiments. Samples were incubated with 5 μm FAM-DEVD-FMK (1 h, 37°C) and washed three times. For quantitation of protease activity, cells were sorted by flow cytometric analysis using a fluorescence-activated cell sorter. Statistical analysis was by ANOVA, followed by a Newman–Keuls test.
Imaging.
Mitochondrial studies were performed using the fluorescent Zn2+ indicator dye RhodZin-3 (Invitrogen, Carlsbad, CA). RhodZin-3 has a nanomolar affinity for Zn2+ (Kd of ∼65 nm) and exhibits no sensitivity to physiological concentrations of Ca2+. Among other ions, RhodZin-3 shows only modest sensitivity to Fe3+ (Kd of ∼5μm), very weak sensitivity to Hg2+ and Cd2+, and no response to 500 μm Pb2+, Ni2+, Mn2+, Mg2+, or Gd3+ or 200 mm Na+. Fe2+ and Cu2+ completely quench the probe, as expected with paramagnetic metals (Sensi et al., 2003b).
In neurons, RhodZin-3 localizes to mitochondria and detects changes of intramitochondrial-free Zn2+ (Sensi et al., 2003a,b). Mitochondria from control and experimental brains were loaded with RhodZin-3 AM (100 nm, 30 min at room temperature) and then incubated (30 min at room temperature) to allow for de-esterification. Fluorescence imaging was performed on an inverted microscope (Zeiss, Oberkochen, Germany) with a mercury lamp, a 63× epifluorescence oil-immersion objective, and a green fluorescence cube (excitation, 450–490 nm; dichroic, 510 nm; emission, 530 nm). Images were acquired with a Zeiss Axiocam MRM digital camera and analyzed with Zeiss Axiovision software and Adobe Photoshop. Six boxes of equal area were selected from each micrograph by an investigator blinded to the treatments. The average fluorescence intensity per box was calculated after subtraction of background fluorescence intensity, assessed by densitometry of a box of equivalent area from a mitochondria-free region of the dish.
Results
Ischemia alters the density of the contact points between the inner and outer mitochondrial membranes
Transient global ischemia is a neurological insult in which the entire brain is transiently deprived of oxygen and undergoes ATP depletion. We first examined the early morphological changes of mitochondria isolated from the hippocampi of rats subjected to sham surgery (control) or transient global ischemia (ischemia) at 50 min after reperfusion. Electron micrographs of isolated mitochondria also revealed the presence of synaptosomes and lysosomes. Neither the percentage of completely intact double membranes (45.9 ± 5.2% for control mitochondria, n = 178; 48.3 ± 2.3% for ischemic mitochondria, n = 145) nor the average diameter of intact mitochondria (0.89 ± 0.07 μm for control mitochondria, n = 9; 0.85 ± 0.08 μm for ischemic mitochondria, n = 13) differed significantly in samples from ischemic versus control brains. These findings indicate little or no swelling, permeability transition, or generalized changes in solute transport in mitochondria in the first hour after ischemia (Fig. 1A) (Hunter et al., 1976; Gottlieb et al., 2002) and suggest little or no difference in physical injury to the mitochondria during the isolation procedure between the control and ischemic groups. On closer inspection, however, subtle differences could be observed between ischemic and control mitochondria. For example, control mitochondria exhibited bright areas in the intermembrane space separated by electron-dense points of contact connecting the outer and inner membranes (Fig. 1A,B and insets in A) and a slightly more electron-dense matrix than did ischemic mitochondria (Fig. 1A,B). Post-ischemic mitochondria exhibited a reduction in the electron density of the contact points (Fig. 1A,B and insets in A) relative to that of control mitochondria. In contrast, the density of other dense organelles in the same micrographs did not differ significantly between the two groups (Fig. 1B). Consistent with a change in structure of the contact points, the length of the contact points within the intermembrane space was increased in post-ischemic mitochondria versus controls (Fig. 1C). These findings suggest that global ischemia induces subtle alterations in the architecture of the matrix and intermembrane space of hippocampal mitochondria at early times after ischemia. These changes may precede more marked changes such as dilated and distorted mitochondria (and other organelles) at later times (4–60 d) after global ischemia (Colbourne et al., 1999).
Large conductance channel activity occurs in mitochondria from ischemic brain
Contact points are thought to represent the site of connection between ion channels present on the outer membrane and those of the inner membrane (Frey and Mannella, 2000) and are the presumed site of the permeability transition pore (Halestrap, 2005). We therefore examined whether global ischemia induces alterations in the permeability of mitochondrial membranes in the period after insult. Toward this end, we undertook patch-clamp recordings of mitochondria isolated from whole brains of control and ischemic rats at 50 min after reperfusion. Control mitochondria exhibited occasional channel activity (Fig. 2A,B). Discrete channel openings to a variety of amplitudes were recorded (Figs. 2A,B) (see Figs. 4A, 5A). Conductances were classified as small (<180 pS), intermediate (180–760 pS), and large (>760 pS) (Jonas et al., 2003, 2004). This classification has been useful in comparing the outer membrane activity of hypoxic and control mitochondria within living squid presynaptic endings (Jonas et al., 2005). To examine the frequency of occurrence of different conductance levels, amplitudes were converted to conductances by assuming a linear relationship between current and evoked voltage in patches (Fig. 2A). Bar graphs of the frequency of occurrence of different conductance levels in controls revealed that the closed state of the channel predominated over the open state.
Figure 2.
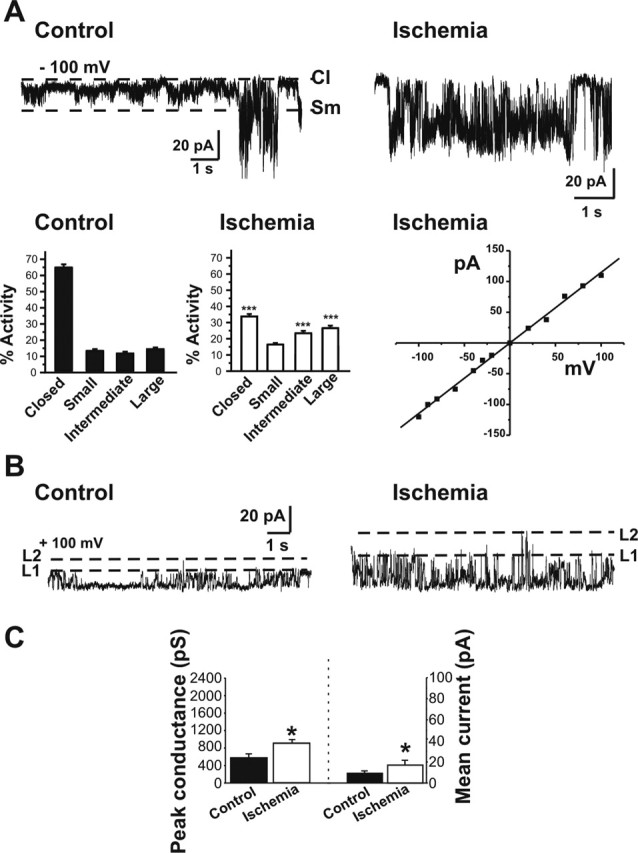
Post-ischemic mitochondria exhibit an increase in large conductance channels. Recordings were made from organelle-attached patches on the membranes of isolated post-ischemic and control whole-brain mitochondria. A, Sample recordings at −100 mV in control (left) and post-ischemic (right) whole-brain mitochondria. Bar graphs show histograms of channel activity for recordings like those illustrated in A. Channel activity was defined as closed channels; small activity, <180 pS; intermediate activity, 180–760 pS; and large activity, >760 pS. Whole-brain mitochondria from post-ischemic animals contain more frequent large and intermediate conductance activity and less frequent closed times (n = 29 ischemic mitochondrial recordings; n = 26 controls; the isolations were from 2 different experiments; in 1 experiment, isolated mitochondria were pooled from 3 ischemic rat brains and 3 control rat brains; in the other experiment, mitochondria were pooled from 6 ischemic rat brains and 6 control rat brains; ∗∗∗p < 0.0001). The current–voltage relationship for a representative large-conductance channel recorded from post-ischemic mitochondria shows the absence of voltage dependence. B, Traces at +100 mV demonstrate the larger conductance and increased frequency of occurrence of discrete single channel openings in patches from post-ischemic mitochondria than in patches from controls. First and second levels (L1, L2) of amplitudes of opening of the multi-conductance single channels are indicated by dotted lines. C, Peak conductance is significantly larger in post-ischemic mitochondria than in controls (n = 36 different mitochondrial recordings; in each group, ∗p < 0.02). The mean current conducted through patches from post-ischemic mitochondria (n = 22 different recordings) is greater than that through control patches (n = 11; ∗p < 0.01). Multiplying the frequency of occurrence of a single channel event by the conductance level of that event gives the mean current conducted through a patch.
Figure 4.
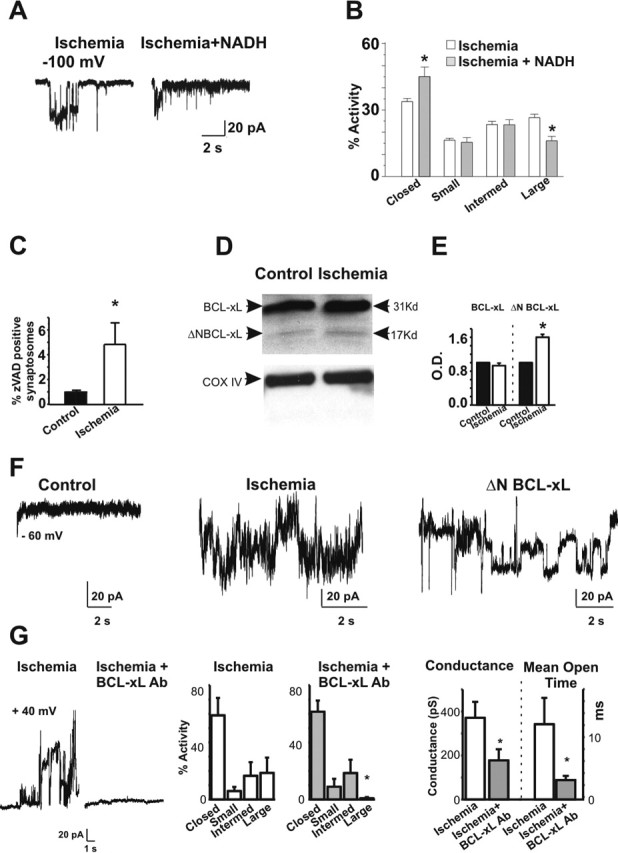
Large conductance channels are inhibited by NADH and an antibody against BCL-xL. A, Application of NADH attenuates large conductance openings of post-ischemic mitochondria from whole brain. Examples of 10 s periods of recording at the indicated voltage made from a post-ischemic mitochondrion (left) and a post-ischemic mitochondrion in the presence of 2 mm NADH in the bath and the pipette (right). B, Histograms combining all experiments show the percentage activity of different levels of conductance in the post-ischemic patches with and without NADH. Large conductances are reduced in the presence of NADH. The conductances based on peaks in amplitude histograms are as follows: closed channels, small activity at <180 pS, intermediate activity between 180 and 760 pS, and large activity at >760 pS. C, Protease activation in nerve terminals occurs early after a brief ischemic insult. A higher percentage of synaptosomes from post-ischemic whole brain or from post-ischemic hippocampus contain activated proteases compared with controls (n = 12 control samples; n = 11 post-ischemic samples from three different experiments; ∗p < 0.03; in each experiment, between 3 and 6 rat brains were used for ischemic and control conditions). The amount of fluorescent zVAD binding represents activated proteases. D, Western blots show that BCL-xL is present at high levels in mitochondria isolated from hippocampus in both post-ischemic mitochondria and controls. Protein yields are not significantly different between ischemic and control (5.4 μg/μl ischemic vs 5.46 μg/μl control), yet an N-terminal proteolytic cleavage fragment of BCL-xL is present at higher levels in post-ischemic hippocampal mitochondria than in controls. Immunoblots against COX IV are provided as an additional control for equivalence of protein samples. E, Densitometric analysis of immunoblots of BCL-xL and ΔN-BCL-xL (n = 11 controls; n = 13 post-ischemic; ∗p < 0.05). Densities of BCL-xL or ΔN-BCL-xL were normalized to values in controls. F, Recordings of isolated hippocampal mitochondria from control (left), ischemic (middle), and a control mitochondrion exposed to ΔN-BCL-xL in the patch pipette (right). Large and multiple conductances were recorded from ischemic mitochondria and from control mitochondria exposed to ΔN-BCL-xL (n = 4 recordings from control mitochondria in the presence of ΔN-BCL-xL). G, A polyclonal antibody against BCL-xL blocks large conductance activity in the outer membrane of post-ischemic mitochondria (antibody 1:1000, n = 4; or 1:500, n = 2). The left panel shows a recording without antibody, and the right panel shows a recording with antibody. Histograms combining all experiments show the frequency of channel activity in mitochondria from ischemic brain (n = 40 different mitochondria; left) and mitochondria from ischemic brain exposed to anti-BCL-xL antibody (n = 6 different mitochondria, right; ∗p < 0.0001). Closed, Absence of activity; Small, activity <180 pS; Intermed, activity between 180 and 760 pS; Large, activity >760 pS. Ischemic mitochondrial recordings in this experiment did not differ significantly from those in Figure 2; therefore, the data from the two sets of experiments were pooled. Histograms at right show the conductance level and mean channel open time of activity recorded from post-ischemic mitochondria in the presence or absence of antibody. All recordings that contained discrete single channel openings were analyzed (n = 6 ischemic mitochondrial traces in the absence of antibody; n = 7 ischemic mitochondrial traces in the presence of antibody. ∗p < 0.05).
Figure 5.
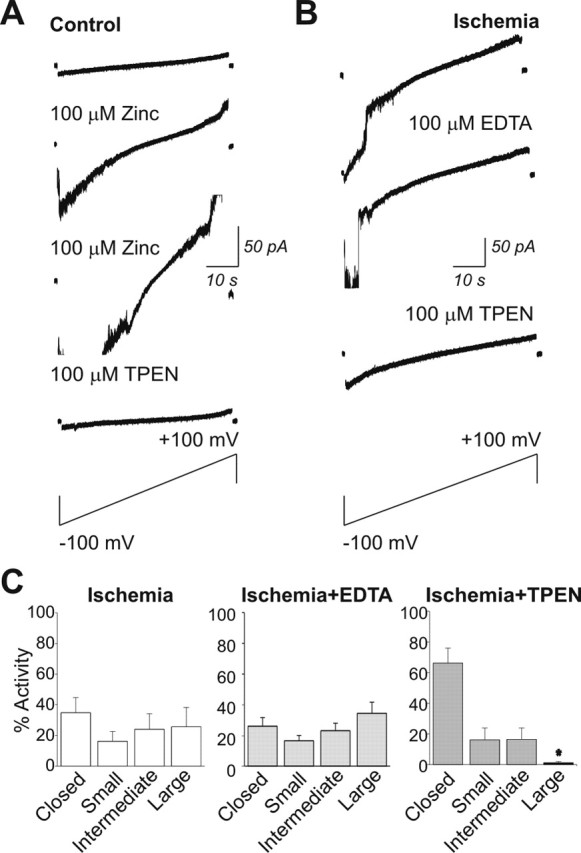
Zn2+ is critical to large channel activity in the outer membrane of whole-brain mitochondria. A, Example of a patch-clamp recording from a control mitochondrion isolated from whole brain. Bath application of Zn2+ (100 μm) enhances the conductance of channels in the patch. A second application of Zn2+ further increases the conductance of channel activity in the patch. Subsequent application of the selective, membrane-permeant Zn2+ chelator TPEN reduces channel conductance in the patch. Traces shown are current responses elicited by a voltage ramp from −100 to +100 mV. B, Large conductance channel activity of post-ischemic mitochondria is attenuated by application of the membrane-permeant Zn2+ chelator TPEN. Shown are patch-clamp traces recorded during a voltage ramp from −100 to +100 mV. The membrane-permeant Zn2+ chelator TPEN, but not the membrane-impermeant Zn2+ chelator EDTA, inhibits large conductance activity in the post-ischemic mitochondrial patch. C, Histograms of activity of different size conductances recorded from post-ischemic whole-brain mitochondria before and after application of TPEN or EDTA. TPEN, but not EDTA, decreases the large conductance activity (n = 6 control and with TPEN, ∗p < 0.05; n = 3 EDTA).
Global ischemia induced large channels with multiple levels of conductance in mitochondrial membranes (Fig. 2A,B) (see Figs. 4A, 5B). Compared with control recordings, this activity had a higher frequency of openings in the intermediate (180–760 pS) and large (>760 pS) conductance range (Fig. 2A,B) (see Figs. 4A, 5B). Moreover, the maximal unitary current for ischemic mitochondria was significantly greater than that of control mitochondria (Fig. 2C). It is possible that the different conductances observed within a trace result from multiple conductances of one channel, because the opening to a single conductance level often occurred for long periods of time, followed by a transition to a different conductance level. The current–voltage relationships for the large conductance channels were measured by rapidly stepping the holding potential across a range of voltages during a series of openings at one conductance level. The large conductance channels exhibited a linear current–voltage relationship, with a reversal potential close to 0 mV (Fig. 2A), and opened at all voltages between −100 and +100 mV (Fig. 2A). The large conductance channel activity resembles the channel activity triggered in mitochondria within the giant presynaptic terminal of the adult squid stellate ganglion by hypoxia (Jonas et al., 2005) or by introduction of recombinant proapoptotic ΔN BCL-xL via a patch pipette directly to mitochondria (Jonas et al., 2004).
Intermediate conductance channel activity is altered in post-ischemic mitochondria
The channel activity found in recordings of ischemic mitochondria from whole brain was enriched in activity in the intermediate conductance range. Although this activity may represent subconductance states of proapoptotic channels such as BAX or ΔN-BCL-xL it is also possible that full-length BCL-xL, which forms an intermediate conductance channel when added exogenously to mitochondrial membranes (Jonas et al., 2003), also becomes activated or translocates to mitochondria during ischemia (Alkayed et al., 2001), possibly contributing to the recorded channel activity. Because ischemia increased the frequency of intermediate channel openings in whole-brain mitochondria (Fig. 2A), we further characterized these channels. We selected single traces from recordings of different mitochondrial patches that had high-resistance seals; a sufficiently low frequency of events that a closed baseline could be detected and consistent unitary openings. The amplitude and frequency of each level of single channel conductance in these traces were compared. Because more traces in control recordings contained no channel activity, we were more likely to detect openings in ischemic mitochondria compared with controls. In fact, single channel traces from 22 mitochondria from the brains of ischemic animals were compared with 12 from the control group. The lowest amplitude of discrete unitary events found in each trace was called level 1, and the second lowest, level 2 (Fig. 2B). Level 1, the smallest discrete conductance level detected in these traces, was significantly larger in ischemic mitochondria compared with control mitochondria (386 ± 50 pS in ischemic mitochondria vs 249.6 ± 41 in controls; p < 0.02), although there were not significantly more frequent openings at this level. Level 2 was significantly more frequently open in ischemic mitochondria compared with controls (0.15 ± 0.03 in ischemic vs 0.097 ± 0.05 in controls; p < 0.03), although the amplitude was not significantly different from that of controls (618 ± 6.3 in ischemic vs 553.7 ± 12.6 in controls). In summary, the overall current conducted through intermediate channels in the outer membranes of post-ischemic mitochondria was significantly greater than that of controls (Fig. 2C).
Large conductance outer mitochondrial membrane channel activity is enhanced in hippocampus
Transient global ischemia induces delayed, selective death, primarily of hippocampal pyramidal neurons (Kirino, 1982; Pulsinelli et al., 1982; Koh et al., 1996; Tanaka et al., 2002). We next examined channel activity in mitochondria isolated from the hippocampus of control and post-ischemic animals at 50 min after reperfusion. Mitochondria from the hippocampus of post-ischemic animals exhibited a significant increase in frequency of large channel openings relative to those of control mitochondria (Fig. 3A) that was markedly enhanced over the effect in whole-brain mitochondria. Specifically, in hippocampus, the frequency of occurrence of large conductance activity was six times that of controls, whereas the frequency of large conductance activity was only twice that of controls in ischemic mitochondria isolated from whole brain. The finding was consistent with the known predisposition of hippocampal neurons to undergo delayed death associated with the release of the large protein cytochrome c from mitochondria. In addition, the maximal unitary current recorded from mitochondria from post-ischemic hippocampus (Fig. 3B) was twice as large as the maximal unitary current recorded from whole-brain mitochondria, implying that the largest channels were larger in ischemic hippocampus than in ischemic whole brain and could be large enough to release proapoptotic factors (Hille, 2001; Dejean et al., 2005). Nevertheless, despite the finding that the mean current conducted through level 1 channels was significantly higher than that of controls (Fig. 3B), there was not a significant difference in the overall frequency of opening of intermediate conductance channels in recordings of ischemic hippocampal mitochondria compared with controls, suggesting that a change in frequency of intermediate conductance openings serves a less important function in hippocampus than in other parts of the brain after ischemic injury.
Figure 3.
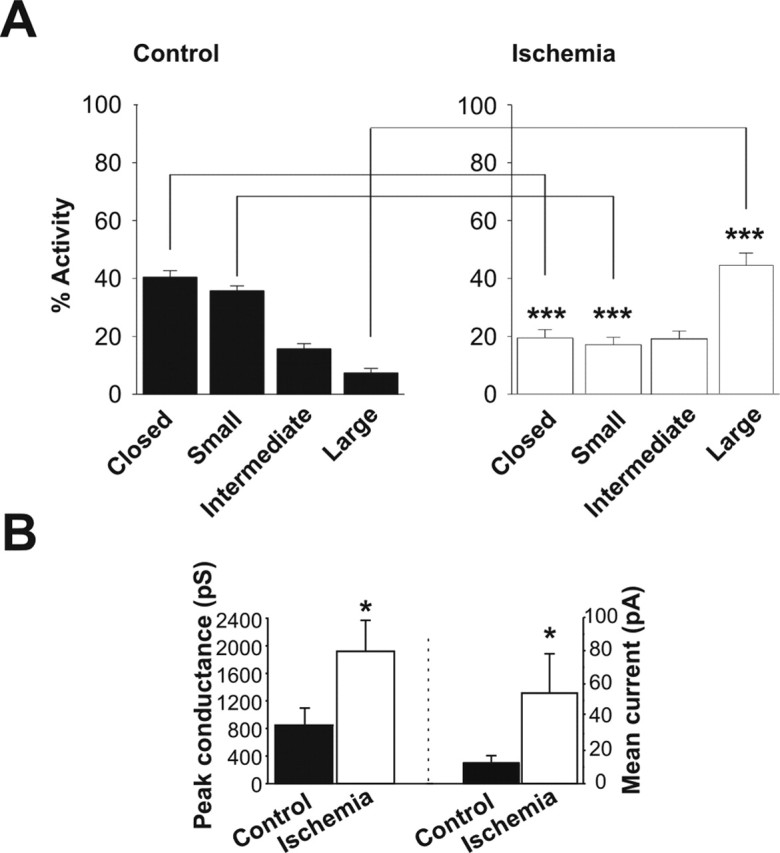
Post-ischemic mitochondria contain more frequent and larger conductance channel activity than do control mitochondria. A, Distribution of activity level of different conductances in recordings of isolated hippocampal mitochondria. All-points histograms show the absence of activity (Closed), activity at <180 pS (Small), activity between 180 and 760 pS (Intermediate), and activity at >760 pS (Large). Hippocampal mitochondria contain more frequent large conductance activity and less frequent small conductance activity (∗∗∗p < 0.0001). B, Peak conductance is significantly larger in post-ischemic mitochondria from hippocampus than in controls from hippocampus (n = 9 different control hippocampal recordings; n = 4 different hippocampal ischemic recordings; ∗p < 0.05). The mean current conducted through patches containing discrete single channel openings is increased in ischemic patches from hippocampal mitochondria compared with controls (n = 6 hippocampal ischemic patches; n = 10 hippocampal control patches; ∗p < 0.05). The mean current conducted through a patch was calculated by multiplying the frequency of occurrence of a single channel event by its conductance level. The hippocampal mitochondria were isolated from 10 ischemic animals and eight sham-operated controls from two independent experiments.
The large channels are sensitive to NADH and are associated with pro-death, truncated BCL-xL
Because the gigaohm seals were formed directly on predominantly intact mitochondria, it is likely that the membrane contacted by the patch pipette is the outer mitochondrial membrane. The conductance of this membrane is reduced by millimolar concentrations of NADH (Lee et al., 1994). In lipid bilayers, NADH has also been shown to reduce the conductance of VDAC, a relatively nonselective channel that is believed to be the major conductance pathway across the outer membrane (Colombini et al., 1996). In addition, a pro-death proteolytic cleavage fragment of BCL-xL produces large conductance mitochondrial channel openings that are attenuated by NADH in both the squid presynaptic terminal and in isolated yeast mitochondria (Jonas et al., 2004). We therefore tested the effect of this agent in recordings of channel activity in post-ischemic mitochondria from whole brain. Application of NADH (2 mm) via the bath perfusate and the patch pipette significantly reduced the frequency of large conductance activity in recordings from post-ischemic mitochondria (Fig. 4A,B), suggesting a role for VDAC in the large conductance channel activity observed after ischemic insult.
Cell death stimuli such as global ischemia enhance permeability of the outer membranes of mitochondria (Green and Kroemer, 2004), enabling release of cytochrome c and other mitochondrial components and triggering activation of proteases such as calpain and caspases (Cheng et al., 1997; Clem et al., 1998; Li et al., 1998; Luo et al., 1998; Nakagawa and Yuan, 2000; Wood and Newcomb, 2000; Condorelli et al., 2001), which break down integral cellular components and thereby promote neuronal death. Hypoxia activates protease activity even more rapidly, before the release of cytochrome c (Jonas et al., 2004, 2005). This early protease activity is associated with N-terminal cleavage of BCL-xL to generate its pro-death counterpart ΔN-BCL-xL and with appearance of large conductance channels in the mitochondrial outer membrane. In keeping with this, global ischemia induced pronounced protease activity in synaptosomes isolated from whole brain or hippocampus, as assessed by zVAD-FMK [N-benzyloxycarbonyl-Val-Ala-Asp-(OMe) fluoromethyl ketone] fluorescence at 50 min after insult (Fig. 4C). Moreover, the protease activity coincided with a dramatic increase in ΔN-BCL-xL in the mitochondrial fraction, as assessed by Western blot analysis (Fig. 4D,E). These findings raise the enticing possibility that N-terminally truncated BCL-xL plays a critical role in the appearance of the large conductance activity in post-ischemic mitochondria.
To test this hypothesis directly, we performed two experiments. First, we introduced recombinant ΔN-BCL-xL via the patch pipette to hippocampal mitochondria isolated from sham-operated (control) rats (Fig. 4F). ΔN-BCL-xL produced large, multi-conductance activity of control mitochondrial membranes that closely resembled in single channel conductance and frequency of opening, the channel activity recorded from post-ischemic mitochondria. Second, we introduced a functional antibody against BCL-xL to mitochondrial patches and examined the impact on channel activity. We reasoned that, if the outer membrane channel does indeed contain the BCL-xL protein, then an antibody against BCL-xL could bind to the protein and produce changes in channel structure and/or activity (Dallas et al., 2005). We therefore applied a specific polyclonal anti-BCL-xL antibody via the patch pipette and examined its effects on channel activity recorded from mitochondria of ischemic and control brain. Inclusion of the antibody in the patch pipette abolished the large conductance channel activity typically observed in post-ischemic mitochondria (Fig. 4G). To evaluate the impact of the BCL-xL antibody on the intermediate conductance activity possibly induced by full-length BCL-xL (Jonas et al., 2003), we selected all traces from patches that contained small or intermediate conductance events and a sufficiently low frequency of events such that a closed baseline could be detected and unitary openings could be discerned. The anti-BCL-xL antibody significantly reduced the mean conductance level and the mean open time of these small and intermediate events (Fig. 4G), indicative of BCL-xL-containing channels. These experiments suggest a critical role for BCL-xL in the large and intermediate conductance channel activity observed in the outer membranes of post-ischemic mitochondria.
Large channels in post-ischemic mitochondria are blocked by the Zn2+ chelator TPEN
Permeability of the mitochondrial outer membrane is tightly controlled (Tedeschi and Kinnally, 1987; Tedeschi et al., 1989; Jonas et al., 1999; Pavlov et al., 2001). Thus, channel activity in the outer membrane may be modulated by effectors and/or second messengers acting at the intermembrane face of the outer membrane or within the cytosol. Alternatively, a channel spanning both inner and outer membranes could be modulated by messengers on the matrix side of the inner membrane. Introduction of Ca2+ or other divalent ions to the matrix side of patches excised from isolated inner membranes (“mitoplasts”) (Kinnally et al., 1989; Petronilli et al., 1989), and/or application of Zn2+ via the bath perfusion (Sensi et al., 2003a) activates a channel of ∼1 nS conductance in the inner membrane. This inner membrane channel is blocked by cyclosporin A (Szabo and Zoratti, 1991), indicative of a role for cyclophilin D, or by the membrane-permeant Zn2+ chelator TPEN, indicative of a role for Zn2+ in channel activity (Arslan et al., 1985; Sensi et al., 2003a).
Large conductance openings were rare in whole-brain mitochondria isolated from control animals under basal conditions (Fig. 5A, first trace). Application of Zn2+ via the bath perfusion induced large channel activity (Fig. 5A, second and third traces), consistent with the presence of Zn2+-sensitive channels recorded in the outer membrane. Application of the membrane-permeant Zn2+ chelator TPEN inhibited the Zn2+-induced channel activity recorded from control mitochondria (Fig. 5A, fourth trace) but had little or no effect on the occasional spontaneous large channel openings recorded from control mitochondria in the absence of exogenous Zn2+ (inhibition of large conductance events in 3 of 10 patches; data not illustrated).
In contrast, TPEN (20 or 100 μm) essentially abolished the large conductance activity recorded from the outer membranes of post-ischemic mitochondria (inhibition by TPEN in 9 of 10 patches) (Fig. 5B,C). In contrast, application of the nonpermeant Zn2+ chelator EDTA via the bath perfusate had little or no effect (Fig. 5B,C), indicative of a role for intramitochondrial Zn2+ in the modulation of large channel openings.
Ischemic mitochondria contain higher levels of chelatable zinc
We next examined the impact of global ischemia on the concentration of free, intramitochondrial Zn2+. To visualize Zn2+ in mitochondria from control and ischemic animals, we used the membrane-permeant Zn2+-specific fluorescent indicator RhodZin-3. This indicator is useful for assessing changes in intramitochondrial free Zn2+ and has been used to demonstrate the presence of Zn2+ in the mitochondria of cortical neurons in culture (Sensi et al., 2003a,b). In neurons, RhodZin-3 localizes to mitochondria and detects changes of intramitochondrial free Zn2+. To examine whether ischemia alters mitochondrial free Zn2+ at early times after insult, we loaded mitochondria isolated from control and post-ischemic hippocampus with RhodZin-3 and visualized Zn2+ fluorescence at 50 min after insult. Global ischemia induced a substantial increase in mitochondrial free Zn2+, as assessed by RhodZin-3 fluorescence (Fig. 6A,B). Addition of the selective Zn2+ chelator TPEN to isolated mitochondria from post-ischemic animals significantly decreased Zn2+ fluorescence (Fig. 6A,B), whereas it had little effect on controls, validating RhodZin-3 as a Zn2+ indicator. These findings suggest that ischemia elicits a rise in mitochondrial Zn2+ in hippocampus and that Zn2+ is critical to activation of large conductance channels recorded in the outer membrane. To test this prediction, we took advantage of the membrane-impermeant Zn2+ chelator CaEDTA. Previous studies suggest that CaEDTA administered before ischemia depletes not only extracellular Zn2+ but also intracellular Zn2+, which leeches out of CA1 neurons as extracellular Zn2+ is depleted (Frederickson et al., 2002; Calderone et al., 2004). Thus, unlike EDTA applied in vitro to isolated post-ischemic mitochondria (above), CaEDTA administered in vivo before ischemia would be expected to prevent the rise in intracellular (and presumably intramitochondrial) Zn2+. CaEDTA administered into the ventricles of rats 30 min before global ischemia prevented the induction of large channel activity in the outer membrane of hippocampal mitochondria (Fig. 6C). This finding is consistent with the possibility that global ischemia induces a rise in Zn2+ in hippocampal neurons (and mitochondria) and that the rise in Zn2+ is critical to activation of large conductance channel activity. Moreover, CaEDTA prevented the morphological alterations in intramembrane contact points, as assessed by electron microscopy. Administration of the membrane-impermeant Zn2+ chelator CaEDTA into the ventricles of rats 30 min before global ischemia mostly prevented the alterations in electron density of contact points between the inner and outer membranes in post-ischemic mitochondria (contact point density of mitochondria from CaEDTA-treated hippocampi, 115.9 ± 4.2, n = 8 mitochondrial micrographs; contact point density of mitochondria from CaEDTA plus ischemia-treated hippocampi, 116.7 ± 3.9, n = 8 mitochondrial micrographs; 24 measurements per group). The findings that CaEDTA not only prevents large channel activity but also prevents alterations in the morphology of contact points are consistent with a role for the contact points as sites of interaction for channels that span the two membranes.
Figure 6.
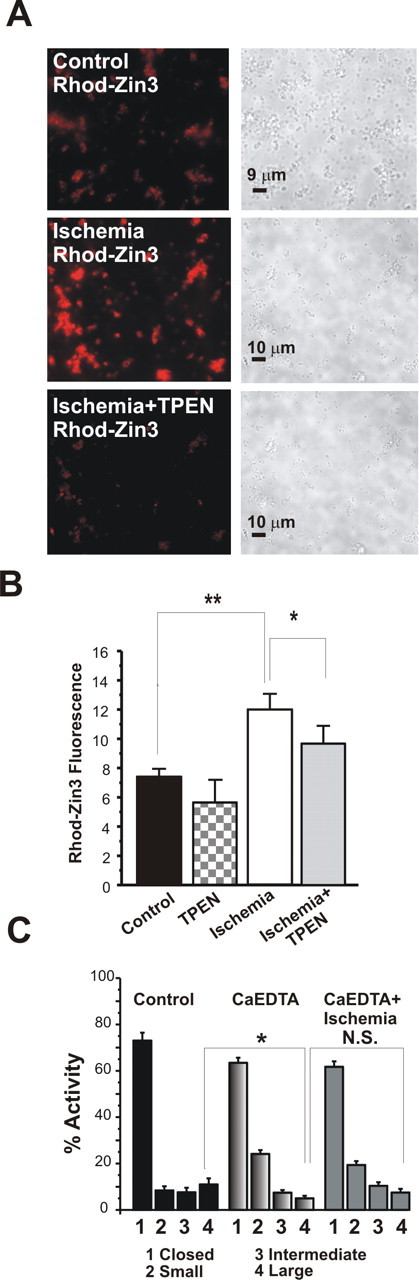
Zn2+ fluorescence is greater in post-ischemic versus control mitochondria. A, Examples of micrographs of mitochondria isolated from the hippocampus of control (top) and post-ischemic (middle) animals and treated with the Zn2+-specific fluorescent indicator dye RhodZin-3 (100 nm). Phase images are shown to the right of fluorescent images. Middle and bottom panels show a paired experiment in which micrographs of post-ischemic hippocampal mitochondria were obtained before and 10 min after application of TPEN (20 μm). B, Data from all images were obtained by determining the intensity of fluorescence within six boxes set over clusters of mitochondria on each dish. All measurements were background subtracted (n = 61 control measurements; n = 102 post-ischemic measurements; ∗∗p < 0.003). Data were obtained from three ischemic rat brains and three control rat brains on the same day. In paired experiments, TPEN significantly decreased RhodZin-3 fluorescence in post-ischemic mitochondria (n = 14 controls after TPEN; n = 48 ischemic measurements after TPEN; fluorescence significantly decreased after TPEN in ischemic mitochondria; ∗p < 0.03). C, Appearance of large mitochondrial membrane channels is inhibited by injection of rat ventricles with the Zn2+ chelator CaEDTA 30 min before the ischemic episode. Histograms combining all recordings show the percentage activity of different levels of conductance in control patches or in post-ischemic patches with and without CaEDTA. The conductances are as follows: closed channels; small activity at <180 pS; intermediate activity between 180 and 760 pS; and large activity at >760 pS. Two rats were treated in each group (control, CaEDTA, and ischemia plus Ca EDTA). n = 10 mitochondrial recordings in the control group and n = 16 mitochondrial recordings in each of CaEDTA or ischemia plus CaEDTA (∗p < 0.05).
Discussion
Transient forebrain or global ischemia is a neuronal insult that induces delayed cell death with many of the features of programmed cell death. A hallmark event in the early post-ischemic period is enhanced permeability and disruption of the functional integrity of the outer mitochondrial membrane. The precise mechanisms by which mitochondrial function is disrupted are, as yet, unclear. Here we show that global ischemia promotes alterations in mitochondrial morphology, a rise in intramitochondrial Zn2+, and activation of large, Zn2+-dependent multi-conductance channels in the mitochondrial outer membrane by 1 h after insult. The channel activity was associated with appearance of the proapoptotic N-terminal proteolytic cleavage fragment ΔN-BCL-xL and protease activity in post-ischemic mitochondria, consistent with a role for cleaved BCL-xL in channel formation. In keeping with this, channel activity was mimicked by introduction of recombinant ΔN-BCL-xL to control mitochondria and blocked by introduction of a functional BCL-xL antibody to post-ischemic mitochondria via the patch pipette. The large channels were also inhibited by NADH, indicative of an additional role for VDAC, a voltage-dependent anion channel present in the outer mitochondrial membrane. We further show that global ischemia induces a rise in intramitochondrial Zn2+ and that Zn2+ is critical to large channel opening. The Zn2+ requirement is striking in that channel activity induced by global ischemia in vivo was reversed by Zn2+ chelators given in vitro to mitochondria isolated from post-ischemic hippocampus or applied in vivo to intact animals before ischemia. Moreover, application of Zn2+ to isolated control mitochondria induced large channel activity reminiscent of post-ischemic mitochondria. Our study reveals a novel mechanism by which ischemic insults disrupt the functional integrity of the outer mitochondrial membrane and implicate the large, Zn2+-dependent mitochondrial channels in global ischemia-induced neuronal death.
Significance of altered mitochondrial morphology
Previous studies have delineated morphological alterations in mitochondria of hippocampal CA1 neurons many days after ischemia. At this late time, mitochondria are swollen, and fragments of degenerated mitochondria are present within extensive networks of lysosomes in the cytosol of damaged neurons (Colbourne et al., 1999). A novel finding of the present study is that global ischemia induces morphological changes in brain mitochondria in the early post-ischemic period as early as 1 h after insult. Mitochondria isolated from early post-ischemic hippocampus exhibit two hallmark morphological features. First, the matrix of post-ischemic mitochondria is less electron dense than that of control mitochondria. Second, the points of contact between the inner and outer mitochondrial membranes exhibit reduced electron density, as well as a lateral splaying of the area of contact between the two membranes. Contact points are likely to provide the conduit between the matrix and the cytoplasm involving an ion channel complex spanning both membranes (Frey and Mannella, 2000) and are the presumed site of the mitochondrial permeability transition pore (mPTP) (Halestrap, 2005). Our observation that a rise in intramitochondrial Zn2+ opens channels in the mitochondrial outer membrane is consistent with either activation of an outer membrane channel by binding of Zn2+ in the intermembrane space and/or activation of a channel that spans the two mitochondrial membranes. The opening of such a channel might be expected to produce changes visible in electron micrographs. Accordingly, the alterations in mitochondrial morphology coincide with the increase in large channel openings in the outer membrane. Moreover, the reduction in electron density and lateral length of the contact points are consistent with a change in channel open state and/or a change in position of members of the channel complex.
A possible explanation for the reduced electron density of the matrix observed in post-ischemic mitochondria is a change in the state of cellular metabolism (Lehninger, 1982). Under resting conditions, ADP is depleted and mitochondria assume the orthodox configuration in which the matrix and inner membrane swell to encompass the space bounded by the outer membrane (Hackenbrock, 1966). In response to exogenous ADP, mitochondria undergo metamorphosis to the condensed state in which the matrix and inner membrane are tightly folded and highly contorted. During the ischemic episode, although the ratio of ADP to ATP increases, respiration is arrested and, as a consequence, mitochondria fail to enter the condensed configuration (Lehninger, 1982). During reoxygenation, ATP is once again produced, but reactive oxygen species generated in response to oxidative stress impair function of the electron transport chain (Fiskum et al., 2004) and may impair the ability of the mitochondria to condense, hence resulting in matrices with reduced electron density.
Significance of large, multi-conductance channels in post-ischemic hippocampal mitochondria
Injurious stimuli such as global ischemia promote the translocation of BCL-2 family molecules from the cytosol to the mitochondria, and their cleavage to generate their pro-death counterparts (Wolter et al., 1997; Wei et al., 2001; Krajewska et al., 2004). An attractive scenario is that ischemia promotes cleavage of BCL-xL to generate ΔN-BCL-xL, which interacts with VDAC to form large channels in hippocampal mitochondria. Several findings support this concept. First, global ischemia promotes the appearance of cleaved ΔN-BCL-xL and large channel activity in hippocampal mitochondria by 1 h after insult. Second, the large channel activity can be mimicked in control mitochondria by introduction of ΔN-BCL-xL via the patch pipette. Third, the channel activity can be blocked by a functional antibody against BCL-xL. Fourth, the large channel activity was reduced by NADH, implicating a role for the endogenous mitochondrial channel VDAC. Our findings do not, however, rule out involvement of other proapoptotic BCL-2 family members such as BAX and BAK.
The channel activity observed in post-ischemic hippocampal mitochondria differs in several respects from that of whole-brain mitochondria. Whereas hippocampal mitochondria exhibit primarily large channel activity, whole-brain mitochondria exhibit prominent intermediate channel activity. The finding of intermediate-size channels in brain mitochondria and alteration of their biophysical properties by application of the BCL-xL antibody is consistent with a role for full-length, anti-apoptotic BCL-xL, which forms intermediate conductance channels when applied to control mitochondria inside living presynaptic terminals (Jonas et al., 2003) and is upregulated in response to global ischemia (Honkaniemi et al., 1996; Zhu et al., 1999). A prediction of the present study is that, in brain areas outside of hippocampus, BCL-xL protects neurons from apoptosis. In hippocampus, BCL-xL is cleaved to generate its pro-death counterpart ΔN-BCL-xL, which assembles with VDAC to form large channels in the outer membrane. The finding that BCL-xL abundance is unchanged in post-ischemic hippocampal mitochondria is consistent with the notion that, whereas ischemia promotes translocation of BCL-xL from the cytosol to the mitochondria, it also promotes cleavage of BCL-xL to generate ΔN-BCL-xL.
Although not directly addressed by the present study, several findings suggest that the appearance of large channel activity in post-ischemic hippocampal mitochondria is causally related to ischemia-induced neuronal death. First, in the present study as well as in previous studies, large multi-conductance channels are not observed with great frequency in normal mitochondrial recordings under physiological conditions (Jonas et al., 2004, 2005). Second, ischemia promotes large channel openings (Jonas et al., 2005) and neuronal death that is accompanied by cytochrome c release in the vulnerable CA1 region of the hippocampus (Sugawara et al., 1999; Calderone et al., 2004; Tanaka et al., 2004). Third, occurrence of large channel activity is enhanced in vulnerable hippocampal neurons. Fourth, neuroprotective agents such as CaEDTA prevent large channel activity, cytochrome c release, and neuronal death (Calderone et al., 2004). To definitively establish a causal relationship between channel activity in the outer membrane of post-ischemic mitochondria and ischemia-induced neuronal death would require the availability of specific blockers of the large conductance mitochondrial channels in sufficient quantity as to be administered in vivo to animals. To our knowledge, no such specific pharmacological agent is available commercially at the present time.
The role of Zn2+ in channel activity
The present study also shows that global ischemia induces a rise in intramitochondrial Zn2+ by 1 h after insults and that Zn2+ is critical to large channel opening. The membrane-permeant Zn2+ chelator TPEN applied to isolated mitochondria abolished large channel activity. Moreover, addition of Zn2+ to mitochondria from control brain induced large conductance activity. One possibility is that ischemia induces a conformational change in the channel, thereby exposing the Zn2+-binding site. In keeping with this, contact points between the inner and outer mitochondrial membranes, which are the presumptive site of membrane-spanning channels, exhibited a reduction in electron density. CaEDTA administered intracerebroventricularly before ischemia prevented not only large conductance channel activity but also the morphological alterations.
Injurious stimuli such as global ischemia elicit a rise in intracellular Zn2+ in hippocampal neurons destined to die (Koh et al., 1996; Choi and Koh, 1998). A mechanism by which Zn2+ induces neuronal death is by injury to the mitochondria (Jiang et al., 2001; Bossy-Wetzel et al., 2004; Calderone et al., 2004). As cytosolic Zn2+ rises, it promotes rapid dissipation of the mitochondrial membrane potential (Jiang et al., 2001). In isolated neural mitochondria, Zn2+ is taken up by mitochondria, inhibits respiration, and induces opening of the mPTP (Jiang et al., 2001). CaEDTA administered before ischemia preserves the functional integrity of the mitochondrial outer membrane, preventing release of cytochrome c and Smac/DIABLO (Calderone et al., 2004). In the present study, we show that CaEDTA administered before ischemia prevents the appearance of large channel activity in the mitochondrial outer membrane. The findings that CaEDTA prevents not only large channel activity (present study) but also the release of cytochrome c and Smac/DIABLO (Calderone et al., 2004) suggest a causal relationship between the large channel activity and cytochrome c release. An important unanswered question is how Zn2+ activates the large, outer membrane channel activity. An attractive scenario is that Zn2+ rises in the intermembrane space and directly activates large channels in the outer membrane. Alternatively, Zn2+ might rise in the matrix and activate the mPTP, resulting in release of a second messenger from the matrix to the intermembrane space and subsequent activation of large outer membrane channels. When activated early in the course of mitochondrial dysfunction, a flickering opening of the pore might not give rise to severe mitochondrial swelling (Green and Kroemer, 2004) but rather to the subtle alterations in matrix morphology observed in the present study. BCL-2 family proteins can interact with and modulate the activity of proteins in the inner membrane, such as mPTP and the adenine nucleotide transporter (Marzo et al., 1998; De Giorgi et al., 2002; Vyssokikh et al., 2002). Clearly, additional experiments are warranted to elucidate the mechanism by which Zn2+ activates large conductance channels in post-ischemic mitochondria.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants RO1NS45876 (E.A.J.) and RO1 NS46742 (R.S.Z.), American Heart Association Established Investigator Award (E.A.J.), NIH–National Institute on Aging Grant AG00919 (S.L.S.), and Progetti di Rilevante Interesse Nazionale 2004 grants (S.L.S.). We acknowledge Ilsa Schwartz and Carmen Mannella for discussions.
References
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ (2000). Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem 75:1878–1888. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD (2001). Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci 21:7543–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan P, Di Virgilio F, Beltrame M, Tsien RY, Pozzan T (1985). Cytosolic Ca2+ homeostasis in Ehrlich and Yoshida carcinomas. A new, membrane-permeant chelator of heavy metals reveals that these ascites tumor cell lines have normal cytosolic free Ca2+ J Biol Chem 260:2719–2727. [PubMed] [Google Scholar]
- Assaf SY, Chung SH (1984). Release of endogenous Zn2+ from brain tissue during activity. Nature 308:734–736. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Xia Y, Haddad GG (2000). Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol 62:215–249. [DOI] [PubMed] [Google Scholar]
- Basañez G, Sharpe JC, Galanis J, Brandt TA, Hardwick JM, Zimmerber J (2002). Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem 277:49360–49365. [DOI] [PubMed] [Google Scholar]
- Boise LH, González-Garcia M, Postema CE, Ding L, Lindstein T, Turka LA, Mao X, N̂uñez G, Thompson CB (1993). bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74:597–608. [DOI] [PubMed] [Google Scholar]
- Bolay H, Gursoy-Ozdemir Y, Sara Y, Onur R, Can A, Dalkara T (2002). Persistent defect in transmitter release and synapsin phosphorylation in cerebral cortex after transient moderate ischemic injury. Stroke 33:1369–1375. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA (2004). Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41:351–365. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS (2003). Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci 23:2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh K-M, Tanaka H, Bennett MVL, Zukin RS (2004). Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci 24:9903–9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001). BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8:705–711. [DOI] [PubMed] [Google Scholar]
- Cheng EHY, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM (1997). Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278:1966–1968. [DOI] [PubMed] [Google Scholar]
- Choi DW (1994). Calcium and excitotoxic neuronal injury. Ann NY Acad Sci 747:162–171. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY (1998). Zinc and brain injury. Annu Rev Neurosci 21:347–375. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EHY, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM (1998). Modulation of cell death by Bcl-xL through caspase interaction. Proc Natl Acad Sci USA 95:554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne F, Sutherland GR, Auer RN (1999). Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci 19:4200–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini M, Blachly-Dyson E, Forte M (1996). VDAC, a channel in the outer mitochondrial membrane. Ion Channels 4:169–202. [DOI] [PubMed] [Google Scholar]
- Condorelli F, Salomoni P, Cotteret S, Cesi V, Srinivasula SM, Elnemri ES, Calabretta B (2001). Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol Cell Biol 21:3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas M, Deuchars SA, Deuchars J (2005). Immunopharmacology: antibodies for specific modulation of proteins involved in neuronal function. J Neurosci Methods 146:133–148. [DOI] [PubMed] [Google Scholar]
- Danscher G, Jensen KB, Frederickson CJ, Kemp K, Andreasen A, Juhl S, Stoltenberg M, Ravid R (1997). Increased amount of zinc in the hippocampus and amygdala of Alzheimer's diseased brains: a proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J Neurosci Methods 76:53–59. [DOI] [PubMed] [Google Scholar]
- De Giorgi F, Lartigue L, Bauer MK, Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ, Ichas F (2002). The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. FASEB J 16:607–609. [DOI] [PubMed] [Google Scholar]
- Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B, Jonas EA, Kinnally KW (2005). Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Molec Biol Cell 16:2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, Kowaltowski A (2004). Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J Bioenerg Biomem 36:347–352. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Gebhardt C, Astman N, Gutnick MJ, Heinemann U (2001). Enhanced spontaneous transmitter release is the earliest consequence of neocortical hypoxia that can explain the disruption of normal circuit function. J Neurosci 21:4600–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Danscher G (1990). Zinc-containing neurons in hippocampus and related CNS structures. Prog Brain Res 83:71–84. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Manton WI, Frederickson MH, Howell GA, Mallory MA (1982). Stable-isotope dilution measurement of zinc and lead in rat hippocampus and spinal cord. Brain Res 246:338–341. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Suh SW, Koh JY, Cha YK, Thompson RB, LaBuda CJ, Balaji RV, Cuajungco MP (2002). Depletion of intracellular zinc from neurons by use of an extracellular chelator in vivo and in vitro. J Histochem Cytochem 50:1659–1662. [DOI] [PubMed] [Google Scholar]
- Frey TG, Mannella CA (2000). The internal structure of mitochondria. Trends Biochem Sci 25:319–324. [DOI] [PubMed] [Google Scholar]
- Fujita N, Nagahashi A, Nagashima K, Rokudai S, Tsuruo T (1998). Acceleration of apoptotic cell death after the cleavage of Bcl-xL protein by caspase-3-like proteases. Oncogene 17:1295–1304. [DOI] [PubMed] [Google Scholar]
- González-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB, Nunez G (1995). Bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA 92:4304–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, Armour SM, Thompson CB (2002). Mitochondrial respiratory control is lost during growth factor deprivation. Proc Natl Acad Sci USA 99:12801–12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G (2004). The pathophysiology of mitochondrial cell death. Science 305:626–629. [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR (1966). Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 30:269–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A (2005). A pore way to die. Nature 434:578–579. [DOI] [PubMed] [Google Scholar]
- Hille B (2001). In: Ion channel of excitable membranes Ed 2 pp. 351–352. Sunderland, MA: Sinauer.
- Hengartner MO (2000). The biochemistry of apoptosis. Nature 407:770–776. [DOI] [PubMed] [Google Scholar]
- Honkaniemi J, Massa SM, Breckinridge M, Sharp FR (1996). Global ischemia induces apoptosis-associated genes in hippocampus. Brain Res Mol Brain Res 42:79–88. [DOI] [PubMed] [Google Scholar]
- Howard EM, Gao TM, Pulsinelli WA, Xu ZC (1998). Electrophysiological changes of CA3 neurons and dentate granule cells following transient forebrain ischemia. Brain Res 798:109–118. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, Southard JH (1976). Relationship between configuration, function and permeability in calcium-treated mitochondria. J Biol Chem 251:5069–5077. [PubMed] [Google Scholar]
- Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH (2001). Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem 276:47524–47529. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Buchanan J, Kaczmarek LK (1999). Prolonged activation of mitochondrial conductances during synaptic transmission. Science 286:1347–1350. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hoit D, Hickman JA, Brandt TA, Polster BM, Fannjiang Y, McCarthy E, Montanez MK, Hardwick JM, Kaczmarek LK (2003). Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J Neurosci 23:8423–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, Ivanovska I, Basañez G, Kinnally KW, Zimmerberg J, Hardwick JM, Kaczmarek LK (2004). Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci USA 101:13590–13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Hardwick JM, Kaczmarek LK (2005). Exposure to hypoxia rapidly induces mitochondrial channel activity within a living synapse. J Biol Chem 280:4491–4497. [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C (2003). Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 160:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR (2003). Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J Neurosci 23:6847–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, He L, Lemasters JJ (2003). Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304:463–470. [DOI] [PubMed] [Google Scholar]
- Kinnally KW, Campo ML, Tedeschi H (1989). Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr 21:497–506. [DOI] [PubMed] [Google Scholar]
- Kirino T (1982). Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239:57–69. [DOI] [PubMed] [Google Scholar]
- Koh J-Y, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW (1996). The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272:1013–1016. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Rosenthal RE, Mikolajczyk J, Stennicke HR, Wiesenthal T, Mai J, Naito M, Salvesen GS, Reed JC, Fiskum G, Krajewski S (2004). Early processing of Bid and caspase-6, -8, -10, -14 in the canine brain during cardiac arrest and resuscitation. Exp Neurol 189:261–279. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Krajewska M, Shabaik A, Wang HG, Irie S, Fong L, Reed JC (1994). Immunohistochemical analysis of in vivo patterns of bcl-x expression. Cancer Res 54:5501–5507. [PubMed] [Google Scholar]
- Kroemer G, Reed JC (2000). Mitochondrial control of cell death. Nat Med 6:513–519. [DOI] [PubMed] [Google Scholar]
- Lee AC, Zizi M, Colombini M (1994). Beta-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. J Biol Chem 269:30974–30980. [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW (1999). The changing landscape of ischaemic brain injury mechanisms. Nature 399:[Suppl], A7–A14. [DOI] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Koh JY (2000). Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci 20:RC79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehninger AL (1982). In: Principles of biochemistry New York: Worth.
- Li H, Zhu H, Xu C, Yuan J (1998). Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501. [DOI] [PubMed] [Google Scholar]
- Li Y, Hough CJ, Suh SW, Sarvey JM, Frederickson CJ (2001). Rapid translocation of Zn2+ from presynaptic terminals into postsynaptic hippocampal neurons after physiological stimulation. J Neurophysiol 86:2597–2604. [DOI] [PubMed] [Google Scholar]
- Lipton P (1999). Ischemic cell death in brain neurons. Physiol Rev 79:1431–1568. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998). Bid, a Bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490. [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G (1998). Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281:2027–2031. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J (2000). Cross-talk between two cysteine protease families: activation of caspase-12 by calpain in apoptosis. J Cell Biol 150:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000). Mitochondria and neuronal survival. Physiol Rev 80:315–360. [DOI] [PubMed] [Google Scholar]
- Nishizawa Y (2001). Glutamate release and neuronal damage in ischemia. Life Sci 69:369–381. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Flock DL, Martin LJ (2001). Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci 21:1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Tan Y, Comb M, Liu CL, Martone ME, Siesjo BK, Hu BR (1999). Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome. c and activation of caspase-like proteases. J Cereb Blood Flow Metab 19:1126–1135. [DOI] [PubMed] [Google Scholar]
- Pavlov EV, Priault M, Pietkiewicz D, Cheng EHY, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW (2001). A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol 155:725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Szabo I, Zoratti M (1989). The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett 259:137–143. [DOI] [PubMed] [Google Scholar]
- Pozarowski P, Huang X, Halicka DH, Lee B, Johnson G, Darzynkiewicz Z (2003). Interactions of fluorochrome-labeled caspase inhibitors with apoptotic cells: a caution in data interpretation. Cytometry A 55:50–60. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F (1982). Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11:491–498. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M (2000). Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med 78:3–13. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Jeng JM (2004). Rethinking the excitotoxic ionic milieu: the emerging role of Zn2+ in ischemic neuronal injury. Curr Mol Med 4:87–111. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH (1999). Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA 96:2414–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Weiss JH (2000). AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur J Neurosci 12:3813–3818. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH (2003a). Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci USA 100:6157–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Weiss JH, Rothe A, Gee KR (2003b). A new mitochondrial fluorescent zinc sensor. Cell Calcium 34:281–284. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH (1999). Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19:RC39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, Scheff SW (2004). Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. J Comp Neurol 474:524–534. [DOI] [PubMed] [Google Scholar]
- Szabo I, Zoratti M (1991). The giant channel of the inner mitochondrial membrane is inhibited by cyclosporine A. J Biol Chem 266:3376–3379. [PubMed] [Google Scholar]
- Tanaka H, Calderone A, Jover T, Grooms SY, Yokota H, Zukin RS, Bennett MVL (2002). Ischemic preconditioning acts upstream of GluR2 down-regulation to afford neuroprotection in the hippocampal CA1. Proc Natl Acad Sci USA 99:2362–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MVL, Zukin RS (2004). Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci 24:2750–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi H, Kinnally KW (1987). Channels in the mitochondrial outer membrane: evidence from patch clamp studies. J Bioenerg Biomembr 19:321–327. [DOI] [PubMed] [Google Scholar]
- Tedeschi H, Kinnally KW, Mannella CA (1989). Properties of channels in the mitochondrial outer membrane. J Bioenerg Biomembr 21:451–459. [DOI] [PubMed] [Google Scholar]
- Tian GF, Baker AJ (2000). Glycolysis prevents anoxia-induced synaptic transmission damage in rat hippocampal slices. J Neurophysiol 83:1830–1839. [DOI] [PubMed] [Google Scholar]
- Tonder N, Johansen FF, Frederickson CJ, Zimmer J, Diemer NH (1990). Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci Lett 109:247–252. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB (2000). Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci USA 97:4666–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyssokikh MY, Zorova L, Zorov D, Heimlich G, Jurgensmeier JJ, Brdiczka D (2002). Bax releases cytochrome c preferentially from a complex between porin and adenine nucleotide translocator. Hexokinase activity suppresses this effect. Mol Biol Rep 29:93–96. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Xiao AY, Sheline C, Hyrc K, Yang A, Goldberg MP, Choi DW, Ping Yu S (2003). Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J Cell Sci 116:2099–2110. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL (2000). Ca2+-Zn2+ permeable AMPA or kainate receptors: Possible key factors in selective neurodegeneration. Trends Neurosci 23:365–371. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi X-G, Youle RJ (1997). Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Newcomb EW (2000). Cleavage of Bax enhances its cell death function. Exp Cell Res 256:375–382. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Sensi SL, Ogoshi F, Weiss JH (2002). Blockade of Ca2+-permeable AMPA/kainate channels decreases oxygen-glucose deprivation-induced Zn2+ accumulation and neuronal loss in hippocampal pyramidal neurons. J Neurosci 22:1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Prehn JH, Culmsee C, Krieglstein J (1999). The beta2-adrenoceptor agonist clenbuterol modulates Bcl-2, Bcl-xl and Bax protein expression following transient forebrain ischemia. Neuroscience 90:1255–1263. [DOI] [PubMed] [Google Scholar]
- Zukin RS, Jover T, Yokota H, Calderone A, Simionescu M, Lau C-Y (2004). Molecular and cellular mechanisms of ischemia-induced neuronal death. In: Stroke: pathophysiology, diagnosis, and management (Mohr JP, Choi DW, Grotta JC, Weir B, Wolf PA, eds) pp. 829–854. Philadelphia: Churchill Livingstone.