

An approved anticancer drug selectively targets the first step in the molecular cascade resulting in Alzheimer’s disease.
Keywords: chemical kinetics, molecular mechanisms, protein misfolding, neurodegeneration
Abstract
The conversion of the β-amyloid (Aβ) peptide into pathogenic aggregates is linked to the onset and progression of Alzheimer’s disease. Although this observation has prompted an extensive search for therapeutic agents to modulate the concentration of Aβ or inhibit its aggregation, all clinical trials with these objectives have so far failed, at least in part because of a lack of understanding of the molecular mechanisms underlying the process of aggregation and its inhibition. To address this problem, we describe a chemical kinetics approach for rational drug discovery, in which the effects of small molecules on the rates of specific microscopic steps in the self-assembly of Aβ42, the most aggregation-prone variant of Aβ, are analyzed quantitatively. By applying this approach, we report that bexarotene, an anticancer drug approved by the U.S. Food and Drug Administration, selectively targets the primary nucleation step in Aβ42 aggregation, delays the formation of toxic species in neuroblastoma cells, and completely suppresses Aβ42 deposition and its consequences in a Caenorhabditis elegans model of Aβ42-mediated toxicity. These results suggest that the prevention of the primary nucleation of Aβ42 by compounds such as bexarotene could potentially reduce the risk of onset of Alzheimer’s disease and, more generally, that our strategy provides a general framework for the rational identification of a range of candidate drugs directed against neurodegenerative disorders.
INTRODUCTION
The incidence of Alzheimer’s disease (AD) is increasing rapidly as the global population ages. It is estimated that 44 million people currently suffer from AD and that this number will exceed 135 million by 2050 (1). AD is one of more than 40 related disorders (2) characterized by the misfolding of soluble proteins and their subsequent conversion into amyloid fibrils (3–9).
A key molecular pathway that underlies AD involves the aggregation of Aβ42, the 42-residue form of the β-amyloid (Aβ) peptide, which is a fragment produced by the proteolytic cleavage of the amyloid precursor protein (3–5, 8, 9). Aβ42 is an intrinsically disordered peptide (10) that self-assembles into fibrillar aggregates observed in the brains of AD patients (5, 9). Inhibiting the self-assembly of Aβ42 has, therefore, emerged as a major potential therapeutic strategy against AD (11–20), although no small molecule designed to achieve this effect has yet shown clinical efficacy (21). Such clinical failures are caused at least in part by the incomplete knowledge of the molecular mechanisms underlying the generation of toxic species and of the processes by which small molecules are able to interfere with the aggregation pathway of Aβ42. In addition, it is increasingly evident that prefibrillar oligomeric species, rather than mature amyloid fibrils and plaques, represent the main pathogenic agents in AD and other neurodegenerative conditions (3, 22–27). Accordingly, effective therapeutic strategies are unlikely to consist of a nonspecific suppression of Aβ42 fibril formation but involve the targeting of specific species in a controlled intervention at precise microscopic steps during the complex aggregation process of Aβ42 (28).
A therapeutic strategy of this type can now be proposed by exploiting recent major advances in our understanding of the molecular processes underlying amyloid formation. These advances are the result of the innovative application of chemical kinetics to the study of protein aggregation (29, 30). The availability of highly reproducible data obtained from kinetic measurements based on thioflavin T (ThT) fluorescence (31) has recently allowed us to define the Aβ42 aggregation mechanism in terms of its underlying molecular events (30). We observed that once a small but critical concentration of Aβ42 aggregates has been generated through primary nucleation of monomers, surface-catalyzed secondary nucleation becomes the dominant process, whereby the surfaces of the fibrils once formed serve as catalytic sites for the generation of toxic oligomeric species (30). These oligomers can then grow and convert into additional fibrils, thus further promoting the formation of additional toxic species in a highly effective catalytic cycle (30).
These results can now be taken further as they offer the possibility of investigating the mechanism of inhibition of the Aβ42 aggregation process by therapeutic molecules (28, 29, 32). Approaches using chemical kinetics, which do not require prior knowledge of the elusive structures of the toxic species and are not limited by the need for very tight binding of small molecules to the aggregation-prone proteins, provide highly sensitive methods for the quantitative detection of the effects of potential therapeutic molecules on the aggregation process. By adopting this strategy, we show here that bexarotene, which is an anticancer drug approved by the U.S. Food and Drug Administration (FDA), selectively targets the primary nucleation step in the self-assembly of Aβ, delays the formation of toxic species in neuroblastoma cells, and completely suppresses Aβ aggregation and its consequence in a Caenorhabditis elegans model of Aβ-mediated toxicity.
RESULTS
Chemical kinetics-based therapeutic strategies allow to combat Aβ aggregation
The drug discovery strategy that we describe in this work to target the aggregation of Aβ42 consists of four main steps (Fig. 1). First, a fragment-based approach is applied to identify small molecules that could interfere with Aβ aggregation (Fig. 1, step 1). Fragment-based drug design approaches are based on the screening of a limited number of small molecules to identify fragments with low binding affinities, typically with KD values in the high micromolar to millimolar range (33, 34). When combined together, some of these fragments result in molecules containing multiple favorable interactions that bind more tightly than the initial fragments to the target of interest. To implement this strategy, we generated a set of 164 fragments derived from 88 compounds reported in the literature to interact with Aβ. These fragments were then screened for chemically related compounds in four small-molecule databases (ChEMBL, PubChem, ZINC, and DrugBank), resulting in the identification of 16,850 molecules. This fragment-based library contains 386 FDA-approved drugs, which have the potential to be effective candidates for preclinical lead development. Indeed, such an approach offers exciting opportunities to repurpose existing licensed therapeutic compounds for use in AD with the benefit of providing a more rapid route to the clinic than through novel drug discovery approaches (35, 36).
Fig. 1. Schematic illustration of the drug discovery strategy described in this work.
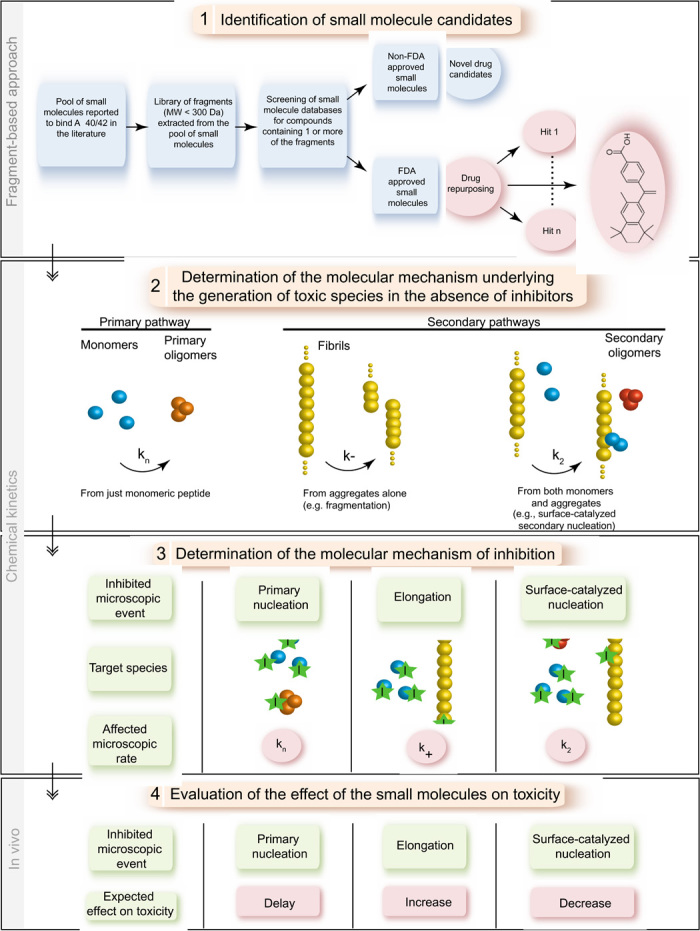
The strategy consists of four steps: (1) A fragment-based approach that allows the identification of small molecules that interact with the aggregation-prone system, here Aβ42, including FDA-approved molecules for drug repurposing. (2) An in vitro kinetic analysis that identifies the specific molecular steps in the Aβ42 aggregation mechanism responsible for the generation of toxic species. (3) A further kinetic analysis to determine the mechanism of inhibition associated with the molecules identified in step 1. (4) An evaluation of the effects of these molecules on the formation of toxic species in vivo. In particular, the inhibition of primary nucleation is predicted to delay the aggregation without affecting the total number of oligomers generated by the aggregation process, whereas inhibiting elongation or secondary nucleation is predicted either to increase or to decrease the number of toxic oligomers, respectively (see text).
To test this strategy, we selected two compounds from this library with different chemical scaffolds, bexarotene and tramiprosate (Fig. 1, step 1). Tramiprosate, which has been unsuccessful in phase 3 AD clinical trials (identifier: NCT0088673), was reported in preclinical development to bind soluble Aβ species, to maintain them in a nonfibrillar form, to decrease Aβ-induced neurotoxicity, and to reduce amyloid plaques and cerebral levels of Aβ in mice (37). By contrast, bexarotene is still in phase 2 AD clinical trials (identifier: NCT01782742). This small molecule is a retinoid X receptor agonist approved by the FDA for the treatment of cutaneous T cell lymphoma and has shown an ability to restore cognitive function to some degree in AD (38–44). Although this activity has been reported to be associated with an enhancement of apolipoprotein E levels, and thus of Aβ42 clearance, conflicting reports have been published and the detailed mechanism of action remains uncertain (38–44). Indeed, this molecule has also been reported to compete with cholesterol for binding to Aβ42, thus inhibiting its cholesterol-induced oligomerization (45).
The next step in our general strategy is to apply chemical kinetics to elucidate the mechanism of aggregation of the protein of interest (Fig. 1, step 2). In the specific case of Aβ42, as mentioned in the Introduction, we have recently provided a comprehensive description of the mechanism (32). The kinetic analysis is then repeated in the presence of selected compounds to identify the microscopic events that are specifically inhibited and hence to define the species with which the compound interacts (Fig. 1, step 3). Indeed, a potential drug candidate could bind to monomers, oligomers, or fibril surface or ends, and therefore different microscopic steps could be affected. Sequestration of monomers could result in a decrease in the rates of all microscopic events shown in Fig. 1 (that is, kn for primary nucleation, k2 for secondary nucleation, and k+ for elongation), whereas sequestering oligomers is expected to affect kn and k2. On the other hand, targeting fibrils could decrease either k+ or k2 depending on whether inhibitors bind to fibril ends or surfaces, respectively (28).
Finally, we evaluate the observed effect of the molecules on the formation of toxic species in vivo (Fig. 1, step 4). The inhibition of specific microscopic steps in Aβ42 aggregation is expected to have different effects on the generation of toxic oligomers (28). More specifically, inhibiting primary nucleation will strongly delay the aggregation reaction and should not affect the total load of toxic oligomers generated during the reaction. However, an increase or a decrease in the number of toxic oligomers is anticipated to be the result of the suppression of either elongation or secondary nucleation, respectively (28). These two latter processes represent interchangeable pathways as inhibiting elongation is expected to redirect the aggregation reaction toward secondary nucleation, which will consequently increase the number of toxic oligomers, and vice versa (28).
Bexarotene, but not tramiprosate, delays Aβ42 fibril formation
To investigate the effects of the two small molecules selected for this study on the individual microscopic steps underlying the aggregation process of Aβ42, we carried out a global analysis of the aggregation profiles acquired at different concentrations of both Aβ42 and each of the two compounds in turn (Fig. 2). We monitored Aβ42 fibril formation in vitro in the absence and in the presence of tramiprosate and of bexarotene using a highly reproducible ThT-based protocol described previously (31). Unlike earlier reports (37), we observed no effects of tramiprosate on Aβ42 aggregation even when present in 20-fold excess (Fig. 2A and fig. S1A). By contrast, we observed a progressive delay in Aβ42 aggregation with increasing concentrations of bexarotene (Fig. 2, B and C, and fig. S1B). The scaling of the half-times in such reaction profiles as a function of the total protein concentration follows a power law whose exponent contains important information about the microscopic events underlying the macroscopic aggregation behavior (32). In the presence of bexarotene, although the lag times of the aggregation reaction increase as the concentration is increased, the scaling exponent remains unaffected. This result suggests that, under these conditions, bexarotene delays the aggregation reaction by inhibiting primary nucleation but does not affect the relative contributions of primary and secondary nucleation to the overall aggregation reaction.
Fig. 2. Bexarotene, but not tramiprosate, delays the formation of Aβ42 fibril formation.

(A) Kinetic profiles of Aβ42 aggregation under quiescent conditions at a concentration of 3 μM (open circles) and 4 μM (open squares) in the absence or in the presence of a fourfold excess of tramiprosate. (B) Kinetic profiles of Aβ42 aggregation under quiescent conditions at a concentration of 3 μM (open circles) and 4 μM (open squares) in the absence or in the presence of a fourfold excess of bexarotene. (C) Average half-time of the aggregation reaction as a function of the initial monomer concentration in the absence or in the presence of fourfold excess of bexarotene.
These experiments show that the effect of bexarotene on Aβ42 aggregation is substantial. The addition of a fourfold excess of bexarotene to a 2 μM sample of Aβ42 increased the time to half-completion of the aggregation reaction compared to that of Aβ42 alone by a factor of 2 (that is, 2.30 ± 0.02 hours for Aβ42 alone and 4.50 ± 0.06 hours for Aβ42 in the presence of bexarotene) (Fig. 3A). To further probe this effect, and to exclude any possible interference of bexarotene on ThT binding to Aβ42 fibrils and hence on the resulting fluorescence, we monitored fibril formation under conditions where ThT was not present by removing aliquots of solution at a series of different time points and measuring the extent of fibril formation using atomic force microscopy (AFM) and immunochemistry (Fig. 3, B to D). Fractions were collected throughout the entire reaction to monitor the events during the three phases of Aβ42 aggregation (that is, lag, growth, and saturation) as completely as possible. AFM images show that after 2.1 hours, fibrillar structures are visible in the absence of bexarotene but not in its presence, thus providing independent evidence for a bexarotene-induced delay of fibril formation (Fig. 3B). These results were extended (Fig. 3C) by probing the quantities of Aβ42 at nine different time points during the aggregation using either sequence-sensitive W0-2 or fibril-sensitive OC primary antibodies (see Materials and Methods). W0-2 antibodies recognize the sequence spanning residues 4 to 10 (that is, the N terminus) that is not likely to be involved in the hydrophobic core of any aggregated species and thus bind to all types of Aβ species. W0-2 antibodies indicated the presence of similar quantities of Aβ42 at the different time points during its aggregation reaction (Fig. 3C, upper panel), whereas fibril-specific OC antibodies that recognize only fibrillar species (Fig. 3C, lower panel) showed, in agreement with the kinetic analysis and the AFM images, a delay of 2 hours in the time required for half-completion of the aggregation process.
Fig. 3. Bexarotene delays Aβ42 fibril formation in a label-free environment.

(A) Kinetic profiles of the aggregation of 2 μM Aβ42 under quiescent conditions in the absence and in the presence of a fourfold excess of bexarotene; the table below the graph shows the equivalent of the different time points in hours (represented in black solid lines in the graph) at which aliquots of Aβ42 were removed from a solution of 2 μM peptide undergoing aggregation. (B) AFM images of Aβ42 species in the absence and in the presence of a fourfold excess of bexarotene. Images were acquired with tapping mode in air on aliquots of the Aβ42 solutions that were removed from the aggregation reaction at the 1.2- and 2.1-hour time points. Fibrillar structures can be observed after 2.1 hours only in the absence of bexarotene. (C) Time course of the formation of 2 μM Aβ42 fibrils as assessed by antibody binding. The quantity of Aβ42 that was detected by the sequence-specific W0-2 antibody (upper panel) remained unchanged during the complete time course of the reaction on the total quantity of Aβ42 (in solution or as aggregates). The fibril-specific OC antibody (lower panel), however, probes only fibrillar structures that can be seen to have formed earlier in the absence of bexarotene than in its presence. The extent of the observed delay (highlighted in red) is in complete accord with the aggregation profiles shown in (A). (D) Calibration of the dependence of Aβ42 fibril mass concentration to the dot-blot intensity of the fibril-specific OC antibody. (Top) Kinetic profile of 4 μM Aβ42 by means of ThT fluorescence. AFM image of typical mature Aβ42 fibrils acquired with tapping mode in air, formed at pH 8. (Bottom) Dot-blot intensities obtained from binding of the fibril-specific OC antibody to a serial dilution of Aβ42 fibrils that were collected after 3 hours of incubation of a fresh 4 μM Aβ42 monomer. Fibril concentrations were in the range of 4 to 0.1 μM.
We further probed the quantity of Aβ42 converted into fibrils by comparing the intensities of the dots to a dot-blot assay that was performed on a range of concentrations between 4 and 0.1 μM Aβ42 fibrils using the OC fibril antibodies (Fig. 3D). Fibrils were collected after incubation for 3 hours of a freshly prepared 4 μM solution of monomeric Aβ42 (Fig. 3D, lower panel), and then the solution was diluted to yield 12 samples with concentrations ranging from 4 to 0.1 μM (Fig. 3D, lower panel). Analysis of the dot-blot data indicated that the quantities of fibrils formed at the reaction half-times of the 2 μM Aβ42 sample in the absence and in the presence of bexarotene (Fig. 3C, lower panel, time points 6 and 7) were indeed closely similar to those formed during the aggregation reaction of an Aβ42 sample of 1 μM concentration, consistent with no interference from ThT on the aggregation reaction (Fig. 3D, lower panel).
Bexarotene specifically inhibits the primary nucleation of Aβ42 aggregation
We then carried out a quantitative analysis of the effects of bexarotene by matching the aggregation profiles on the basis of the rate laws derived from a master equation that relates the macroscopic time evolution of the quantity of fibrils to the rate constants of the different microscopic events (28, 29). In this approach, the aggregation profiles in the presence of the inhibitor are described by introducing into the rate laws suitable perturbations to each of the microscopic rate constants evaluated in the absence of the inhibitor. The modifications of the rate constants required to describe the aggregation profiles in the presence of different inhibitor concentrations are then indicative of the specific process affected by the presence of the compound. The aggregation profiles of 5 μM Aβ42 in the presence of concentrations of bexarotene in the range of 5 to 25 μM show that the experimental data are extremely well described when the primary nucleation rate constant, kn, is specifically decreased. By contrast, the experimental data are not consistent with predictions made by altering the rate constants of secondary nucleation or of elongation, k2 and k+, respectively (Fig. 4, A to C). The data therefore reveal that, under these conditions, bexarotene specifically modifies the primary nucleation pathway (that is, knk+) with no (or very little) detectable effect on secondary pathways in the aggregation reaction (that is, k2k+) (Fig. 4D). Similar results were obtained at different Aβ42 concentrations (fig. S2).
Fig. 4. Bexarotene selectively targets the primary nucleation of Aβ42.

(A to C) Kinetic profiles of the aggregation reaction of 5 μM Aβ42 in the absence or in the presence of a 1:1 or 5:1 concentration ratio of bexarotene to Aβ42 (represented by different colors). The solid lines show predictions for the resulting reaction profiles when secondary nucleation (A), fibril elongation (B), or primary nucleation (C) is inhibited by bexarotene. Only the prediction for the case where primary nucleation alone is inhibited closely fits the experimental data. (D) Evolution of the apparent reaction rate constants with increasing concentration ratios of bexarotene (kn is the rate of primary nucleation, k+ is the rate of elongation, and k2 is the rate of secondary nucleation; K represents in each case either knk+ or k2k+). Note the significant decrease in primary pathways, knk+, when compared to secondary pathways, k2k+, as the concentration of bexarotene is increased. (E) Kinetic profiles of 2 μM Aβ42 without (blue) and with the addition of 10% of preformed seed fibrils in the absence or in the presence of a 2-, 5-, 7-, and 10-fold excess of bexarotene (represented by different colors). Note the rapid increase in theslope of the aggregation reaction in the presence of preformed seed fibrils compared to that of the reaction without the addition of preformed fibrils. (F) Simulations showing identical curves for the aggregation profile of a 2 μM Aβ42 sample in the presence of 5% of preformed fibril seeds where primary nucleation events either contribute (gray) or are negligible (brown). (G) Effect of 0.5- and 5-fold excess of bexarotene on the aggregation kinetics of a 2 μM Aβ42 sample in the presence of 5% of preformed fibril seeds. (H) Effect of 0.5- and 5-fold excess of bexarotene on the rates of surface-catalyzed secondary nucleation (k2) as obtained from the aggregation kinetics in (G).
To further strengthen these conclusions, we carried out an additional series of measurements of the aggregation kinetics of Aβ42 under conditions where the primary nucleation step was bypassed by the introduction of preformed fibrils (that is, seeds) to the reaction mixture. In such a situation, the contribution of primary nucleation to the reaction kinetics is negligible, as the conversion of soluble peptide into mature fibrils is greatly accelerated by secondary nucleation and elongation reactions promoted by the seeds (22). In the presence of 10% fibril seeds, where elongation of the preformed fibril seeds is the dominant mechanism, no effect was observed on the aggregation kinetics of 2 μM Aβ42 even at a 10-fold excess of bexarotene (Fig. 4E), whereas the corresponding aggregation process under unseeded conditions was completely inhibited for 15 hours (fig. S2A). Furthermore, we also measured the aggregation kinetics of a 2 μM sample of Aβ42 in the presence of 5% of preformed fibril seeds. Under these conditions, primary nucleation is completely bypassed, whereas both surface-catalyzed secondary nucleation and elongation significantly contribute to the overall kinetics (Fig. 4F). These experiments show a concentration-dependent bexarotene-induced delay that corresponds to a decrease in the rate of surface-catalyzed secondary nucleation (Fig. 4, G and H). These findings show that, although the elongation of fibrils is essentially unaffected by bexarotene, this compound has a large and highly selective effect on the nucleation of Aβ42, with the effect being more selective toward primary rather than secondary nucleation.
The observation that bexarotene inhibits Aβ42 aggregation by specifically perturbing both primary and secondary nucleation could, in principle, result from the interaction of bexarotene with Aβ42 monomers, with primary and secondary oligomers, or indeed with both monomers and oligomers (Fig. 4F). However, binding to monomers would affect the rates of all the microscopic steps in the overall reaction under the conditions studied here (28), enabling us to conclude that bexarotene specifically interacts primarily with Aβ42 oligomers. This conclusion is further supported by nuclear magnetic resonance (NMR) spectroscopy measurements, where no significant perturbations of the chemical shifts could be observed in the heteronuclear single-quantum correlation (HSQC) spectra of 25 μM 15N-labeled monomeric Aβ42 before and after the addition of a fivefold excess of bexarotene, indicating that binding to the monomeric form of Aβ42 is likely to be negligible (figs. S3 and S4). Furthermore, these findings provide novel insights into the structural features of primary and secondary oligomers of Aβ42, suggesting that primary and secondary nuclei may have similar structural features, and hence can interact with bexarotene in a similar manner, and that the same residues of Aβ42 may be involved in both primary and secondary nucleation events.
Targeting primary nucleation delays the formation of toxic species of Aβ42
We next explored whether the delay in Aβ42 fibril formation resulting from inhibiting primary nucleation by bexarotene could be associated with a delay in the formation of neurotoxic species, as the perturbation of different microscopic steps in Aβ42 aggregation has different effects on the rate of generation of toxic oligomers (28). Thus, for example, decreasing the primary nucleation rate is expected to delay the overall aggregation reaction but not to affect the total number of toxic oligomers generated during the reaction (28). An increase or decrease in the number of toxic oligomers is, however, likely to result from the suppression of elongation or secondary nucleation, respectively (28).
To examine this issue, we performed numerical simulations of the total rate of formation of oligomers, from both primary and secondary processes in the aggregation reaction of a 0.5 μM solution of Aβ42 in the absence and presence of a 20-fold excess of bexarotene (see Materials and Methods). Because bexarotene preferentially targets primary nucleation, it is expected to delay the entire aggregation process, that is, to delay the generation of toxic species without necessarily decreasing the amount of the toxic species. On the basis of our previous findings, primary nucleation is a very rapid process that is directly bypassed by secondary nucleation once a small but critical concentration of seeds has been formed (22). We have therefore used the lowest concentration of Aβ42 at which aggregation can be observed (31) and monitored its aggregation in the presence of high concentrations of bexarotene to inhibit the primary nucleation step as strongly as possible. The simulations show that by delaying the primary nucleation of Aβ42 in the presence of bexarotene, a delay in the formation of Aβ42 oligomers occurs but without decreasing the total amount of oligomers formed over the time course of the aggregation process (Fig. 5, A and B). Indeed, the formation of oligomers follows a parabolic evolution in that the number of oligomers formed during the aggregation of Aβ42 alone starts to decrease at the point where the number in the presence of bexarotene started to increase.
Fig. 5. Bexarotene delays the formation of Aβ42 toxic species in neuroblastoma cells.

(A and B) Numerical simulations of the reaction profiles (A) and nucleation rates (B) for a solution of 0.5 μM Aβ42 in the absence and presence of a 20-fold excess of bexarotene. Blue lines correspond to a control aggregation reaction in the absence of bexarotene, with reaction rate constants k2 = 1 × 106 M−2 s−1, k+ = 3 × 106 M−1 s−1, and kn = 1 × 104 M−1 s−1. Dotted red lines show the behavior in the presence of bexarotene, where the nucleation rate constant, kn, has been decreased to 1 × 102 1 M−1 s−1. A delay in the evolution of the total nucleation rate (that is, of both primary and secondary nucleation) is observed. (C to F) Levels of activated caspase-3 as an indicator of the cytotoxic effects of Aβ42 species on a human neuroblastoma cell line (SH-SY5Y); the fluorescence values have been normalized (see Materials and Methods). Aliquots of 0.5 μM Aβ42, in the absence and in the presence of 10 μM bexarotene, were removed from the aggregation reaction at 0 hour (C), 0.3 hour (D), 4 hours (E), and 7 hours (F). A.U., arbitrary units. Percentage differences between the fluorescence values of Aβ42 in the absence or in the presence of bexarotene (gray circles). These results show that detectable quantities of toxic Aβ42 species are formed in the presence of bexarotene only after 7 hours of incubation at 37°C, in agreement with a bexarotene delay of the formation of Aβ42 toxic species.
To verify these predictions, we measured the cytotoxicity in human neuroblastoma cells (SH-SY5Y) resulting from the aggregation of 0.5 μM Aβ42 in the absence and presence of a 20-fold excess (that is, 10 μM) of bexarotene by evaluating the levels of caspase-3, an early marker of apoptosis (see Materials and Methods). Indeed, caspase-9 is an initiator enzyme that mediates apoptotic pathways after mitochondrial damage and activates effector caspases, such as caspase-3, by cleaving their inactive proforms and initiating the apoptotic cascade. Staurosporine, which is a broad-spectrum kinase inhibitor known to activate the apoptosis pathway, was used as a positive control (see Materials and Methods). Aliquots of Aβ42 solutions were removed at different time points during the lag phase of the aggregation reaction in the absence and presence of bexarotene and incubated with SH-SY5Y neuroblastoma cells. In its monomeric form (before incubation, Fig. 5C), Aβ42 showed no detectable toxicity irrespective of the presence of bexarotene. In the absence of bexarotene, the species formed after incubation for 0.3 hours (Fig. 5D) and 4 hours (Fig. 5E) exhibited a level of toxicity 65 ± 5% higher than that in the presence of bexarotene. However, for the samples incubated for 7 hours, the level of toxicity generated in the presence of bexarotene increased to a value similar to that observed in its absence whereas the toxicity of Aβ alone decreased. This further supports a bexarotene-induced delay of the formation of Aβ42 toxic species in cell (Fig. 5F).
Bexarotene rescues Aβ42-mediated dysfunction in C. elegans models
We further evaluated the effects of bexarotene on the formation of toxic Aβ42 species in a C. elegans model of Aβ42-mediated dysfunction, denoted GMC101 (termed the Aβ worm model) (46), and compared the resulting effects of the addition of bexarotene to those observed in a control worm model, CL2122, which is free of Aβ42 (see Materials and Methods). In the GMC101 model, Aβ42 is expressed in body wall muscle cells where it forms aggregates and results in severe age-progressive paralysis (46). We exposed the worms to increasing concentrations of bexarotene ranging from 5 to 10 μM and measured the frequency of body bends, which is indicative of the state of the muscle cells, and also the quantity of Aβ42 aggregates within the body of the worms (Fig. 6) (see Materials and Methods). In the absence of bexarotene, the frequency of the body bends of the Aβ worm model decreased significantly after just 2 days compared to the situation in the control worm model where a similar decrease in the motility was observed only after 9 days (Fig. 6B). In agreement with such a difference, the levels of aggregates monitored using the fluorescence intensity of the amyloid-specific dye NIAD-4 (47) differ significantly between the two models, with essentially no aggregates being detected on the same day the worms reached adulthood in the Aβ worm model in the presence of bexarotene (Fig. 6, C and D).
Fig. 6. Bexarotene restores the motility of C. elegans models of Aβ42-mediated toxicity by preventing Aβ42 aggregation.

(A) Experimental procedure for the measurement of the effects of bexarotene on the frequency of body bends and on the quantity of aggregates in C. elegans GMC101 (that is, the Aβ worm model) and CL2122 (that is, the control worm model) models. Bexarotene was given to the worms at larval stages L1 and L4. (B) Measurements of the effect of increasing concentrations of bexarotene ranging from 5 to 10 μM on the frequency of body bends in the Aβ worm model. Normalized values with respect to day 0 are shown. The experimental data are shown for a single experiment but are representative in each case of three independent experiments. Complete recovery of the motility of the Aβ worm model can be observed at 10 μM bexarotene; the inset shows the dose dependence of the effects of bexarotene on Aβ worms at day 3 of adulthood. (C) In vivo imaging of aggregates stained using the amyloid-specific dye NIAD-4 in the absence and in the presence of 10 μM bexarotene; images from days 6 and 9 only are shown for clarity. (D) Time course of the reaction of amyloid aggregates formed in the Aβ worms in the absence and in the presence of 1 μM bexarotene. Quantification of fluorescence intensity was performed using ImageJ software (see Materials and Methods). In all panels, error bars represent the SEM. (E) Insoluble fraction of the protein extracts from C. elegans in the presence and in the absence of bexarotene with immunodetection of Aβ and α-tubulin (see Materials and Methods).
We then explored the effect of increasing concentrations of bexarotene added at the larval stages of the C. elegans life cycle (Fig. 6A) and observed a concentration-dependent maintenance of the motility of the Aβ worm model. Indeed, the motility of the Aβ worms was maintained completely upon addition of 10 μM bexarotene in two bursts at the L1 and L4 larval stages, that is, reached the level observed in the control worms (Fig. 4, A and B). In addition, imaging studies (see Materials and Methods) showed that the addition of 10 μM bexarotene to the Aβ worms suppressed the formation of Aβ42 fibrils for 9 days of adulthood (Fig. 6D). The level of Aβ42 expression in the Aβ worms in the absence and in the presence of bexarotene was found to be closely similar (Fig. 6E). This result indicates that maintaining the level of motility in the Aβ worms could be achieved by preventing the aggregation of Aβ42 by bexarotene. In accord with this conclusion, the addition of bexarotene did not show any effects in the control worm model (fig. S5A). The normal motility observed for the Aβ worms in the presence of bexarotene could be consistent with two distinct scenarios. One possibility could be a strong delay in primary nucleation, which would substantially delay the aggregation of Aβ42 and hence maintain the motility of the Aβ worms to values similar to those of the control worms. Alternatively, these results could arise from a combination of effects on primary and secondary nucleation. This latter possibility is particularly relevant because although bexarotene preferentially inhibits primary nucleation, it could also affect secondary nucleation when present in excess. Indeed, our experiments using 5% seeds show that bexarotene is capable of acting on surface-catalyzed secondary nucleation as well as on primary nucleation (Fig. 4, F to H). Furthermore, we also observed that adding bexarotene only at day 2 did not show any detectable effect on the motility of the Aβ worms (fig. S5, B and C), suggesting that the finding from the in vitro studies that bexarotene specifically affects nucleation events in Aβ42 aggregation is also the case in vivo.
DISCUSSION
We have described a drug discovery strategy that aims at targeting Aβ42 aggregation with small molecules. This strategy involves the identification of compounds from an Aβ-specific fragment-based library and the subsequent application of chemical kinetics for gaining a fundamental understanding of the molecular mechanism of inhibition (Fig. 1). We have found that although tramiprosate has no effect on Aβ42 aggregation, bexarotene interferes selectively with the earliest stage of Aβ42 aggregation by reducing the rate of primary nucleation. We have also observed that this effect significantly inhibits the rate of the overall aggregation reaction not only in vitro but also in cells and in vivo, as bexarotene was found to suppress the formation of toxic species both in cells and in C. elegans. Furthermore, we have shown that a sufficient concentration of bexarotene given at the earliest stages of the C. elegans life cycle can maintain the motility of the worms expressing Aβ42 at a level comparable to those in which Aβ42 is not expressed, thus further supporting the preventive effect of this molecule (48–50).
We anticipate that the strategy that we have described in this paper will enable the identification of further compounds capable of inhibiting Aβ42 aggregation and the definition of the specific microscopic steps affected by each compound (that is, primary or secondary nucleation or elongation). These results should enable a range of compounds to be identified to control and modulate the onset of aggregation and the rate of its progression first in vitro and then in vivo. In addition, the results described in the present work indicate that bexarotene and other inhibitors of primary nucleation have the potential to be efficient means of delaying aggregation by reducing the probability that primary nuclei are formed and proliferate, such that our natural protection mechanisms could remain effective to more advanced ages. Indeed, we draw an analogy between the strategy presented here and that of using statins, which reduce the level of cholesterol and thus the risk of heart conditions, and suggest that such molecules could effectively act as “neurostatins.” In addition to the use of molecules such as bexarotene for preventive purposes, it is possible to speculate that targeting secondary nucleation, which is largely responsible for the production of toxic species (30), would form the basis to developing treatments to reduce the rate of progression of AD.
CONCLUSIONS
We have described in this study the development and application of a comprehensive strategy for the identification of potent anti-AD drugs. This strategy is based on understanding the molecular details of the inhibition processes, creating opportunities to identify effective therapeutic agents in a rational manner that could eventually lead to successful drugs to combat AD. Finally, given the generality of the phenomenon of protein aggregation, the present strategy offers a framework for the rational design and evaluation of novel therapeutics to target other forms of neurodegenerative disorders.
MATERIALS AND METHODS
Preparation of Aβ peptides
The recombinant Aβ (M1-42) peptide (MDAEFRHDSGY EVHHQKLVFF AEDVGSNKGA IIGLMVGGVV IA), here called Aβ42, was expressed in the Escherichia coli BL21 Gold (DE3) strain (Stratagene) and purified as described previously with slight modifications (51). Briefly, the purification procedure involved sonication of E. coli cells, dissolution of inclusion bodies in 8 M urea, ion exchange in batch mode on diethylaminoethyl cellulose resin, and lyophilization. The lyophilized fractions were further purified using a Superdex 75 HR 26/60 column (GE Healthcare), and eluates were analyzed using SDS–polyacrylamide gel electrophoresis for the presence of the desired protein product. The fractions containing the recombinant protein were combined, frozen using liquid nitrogen, and lyophilized again.
Isotopically labeled 15N-Aβ42 was prepared by growing transformed E. coli BL21 Gold (DE3) strain (Stratagene) in 1-liter flasks at 37°C with 500 ml of minimal M9 batch medium. Briefly, a 20-ml preculture grown overnight to saturation in LB medium containing ampicilin (100 μg/liter) was diluted 1:25 in 2YT medium supplemented with ampicilin (100 μg/liter) and grown at 37°C. When the optical density at 600 nm reached 0.5, the cells were harvested, washed in minimal medium, and inoculated in one-quarter of the initial culture volume of minimal M9 medium containing ampicilin (100 μg/liter) and supplemented with 15NH4Cl (2 g/liter) (Cambridge Isotope Laboratories). Cells were grown at 37°C for 2 hours before induction. Isopropyl-β-d-thiogalactopyranoside was then added to a final concentration of 1 mM, and cells were grown at 37°C overnight. Purification of 15N-Aβ42 was performed as described above. Chemicals, including bexarotene and tramiprosate, were obtained from Sigma-Aldrich and were of the highest purity available.
Preparation of samples for kinetic experiments
Solutions of monomeric peptides were prepared by dissolving the lyophilized Aβ42 peptide in 6 M GuHCl. Monomeric forms were purified from potential oligomeric species and salt using a Superdex 75 10/300 GL column (GE Healthcare) at a flow rate of 0.5 ml/min and were eluted in 20 mM sodium phosphate buffer (pH 8) supplemented with 200 μM EDTA and 0.02% NaN3. The center of the peak was collected, and the peptide concentration was determined from the absorbance of the integrated peak area using ε280 = 1400 liter mol−1 cm−1. The obtained monomer was diluted with buffer to the desired concentration and supplemented with 20 μM ThT from a 1 mM stock. All samples were prepared in low-binding Eppendorf tubes on ice using careful pipetting to avoid introduction of air bubbles. Each sample was then pipetted into multiple wells of a 96-well half-area, low-binding polyethylene glycol coating plate (Corning 3881) with a clear bottom, at 80 μl per well.
For the seeded experiments, preformed fibrils were prepared just before the experiment. Kinetic experiments were set up just as above for a 4 μM Aβ42 sample in 20 mM sodium phosphate buffer (pH 8) with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. The ThT fluorescence was monitored for 3 hours to verify the formation of fibrils. Samples were then collected from the wells into low-binding tubes and sonicated for 2 min in a sonicator bath at room temperature. Under the conditions used here (4 μM Aβ42), the monomer concentration is negligible at equilibrium (31). The final concentration of fibrils, in monomer equivalents, was considered equal to the initial concentration of the monomer. Fibrils were then added to the freshly prepared monomer to reach either 5 or 10% final concentration of fibrils.
In all cases, bexarotene was first solubilized in 100% dimethyl sulfoxide (DMSO) to a concentration of 5 mM and then diluted in the peptide solution to reach a final DMSO concentration of maximum 1%. We verified that the addition of 1% DMSO in the reaction mixture has no effect on Aβ42 aggregation (fig. S6).
Kinetic assays
Assays were initiated by placing the 96-well plate at 37°C under quiescent conditions in a plate reader (Fluostar Omega, Fluostar Optima, or Fluostar Galaxy; BMG Labtech). The ThT fluorescence was measured through the bottom of the plate with a 440-nm excitation filter and a 480-nm emission filter. The ThT fluorescence was followed for three repeats of each sample.
Theoretical analysis
The time evolution of the total fibril mass concentration, M(t), is described by the following integrated rate law (30, 32)
| (1) |
where the kinetic parameters B±, C±, k, k∞, and are functions of the two combinations of the microscopic rate constants k+k2 and knk2, where kn, k+, and k2 are the primary nucleation, elongation, and secondary nucleation rate constants, respectively.
Bexarotene can interfere with the aggregation process by inhibiting one or more of the individual microscopic reactions. We can identify the microscopic events that are inhibited by the chemical compound by applying the above equation to describe the macroscopic aggregation profiles shown in Fig. 4 and fig. S2 and comparing the set of microscopic rate constants k+k2 and knk2 required to describe the time evolution of the fibril formation in the absence and presence of bexarotene. As shown in Fig. 4 and fig. S2, in unseeded aggregation reactions, the presence of the chemical compound mainly perturbs the primary nucleation rate.
The numerical simulations reported in Fig. 5 (A and B) show the reaction profiles in the presence and absence of bexarotene simulated according to Eq. 1. The time evolution of the nucleation rate, r(t), was simulated according to the equation
| (2) |
Model simulations in Fig. 4F were performed by fixing the k+ and k2 parameters for a 2 μM Aβ42 solution seeded with 5% of preformed seed fibrils. The kn parameter was then decreased to very negligible values (~0). This showed insignificant perturbation to the reaction profile when the primary nucleation process does not occur.
AFM imaging
Aβ42 samples were removed from the aggregation reaction and were directly deposited onto freshly cleaved mica surfaces and allowed to dry for 30 min. Samples were then washed with Milli-Q water and then dried with nitrogen. AFM images were acquired using a VEECO Dimension 3100 atomic force microscope (Bruker) and JPK Nanowizard software. The instrument was operated in tapping mode in air using n-type silicon cantilevers with resonant frequencies between 65 and 130 kHz.
Dot-blot assay
Blotting was performed using either Aβ42 fibril-specific antibody (OC, Millipore) or Aβ42 sequence-specific antibody (W0-2, Millipore). During the time course of the aggregation of 2 μM Aβ42 in the absence and in the presence of fourfold excess of bexarotene (Fig. 3), 4- and 2-μl Aβ42 aliquots were removed from the mixture at different time points for blotting with either OC or W0-2, respectively. Aβ42 aliquots were spotted onto a nitrocellulose membrane (0.2 μm; Whatman), and then the membranes were dried and then blocked with Blocking One (Nacalai Tesque) before immunodetection. OC and W0-2 were used according to the manufacturer’s instructions. Alexa Fluor 488–conjugated secondary antibodies (Life Technologies) were subsequently added, and fluorescence detection was performed using Typhoon Trio Imager (GE Healthcare). DMSO-solubilized bexarotene was added to Aβ42 with a final concentration of 1% DMSO. For the calibration curve (Fig. 3D), a series of dilutions between 4 and 0.1 μM were performed on freshly prepared Aβ42 fibrils and 4-μl aliquots were deposited onto the same nitrocellulose membrane (0.2 μm; Whatman) as the one used for OC fibril-specific antibody. An oligomer-specific A11 antibody was also used to detect the evolution of the formation of Aβ42 oligomers in the absence and in the presence of bexarotene. However, under these conditions, we could not detect oligomers because of their very low concentration during Aβ42 aggregation even when depositing on the membrane volumes as high as 20 μl.
NMR experiments
For NMR analyses, 15N-Aβ42 was purified as described above except that the buffer of the gel filtration was 50 mM ammonium acetate (pH 8.5). Lyophilized powder of 15N-Aβ42 was dissolved at an approximate concentration of 1 mM in 0.2% (v/v) ammonium solution and then collected and stored in aliquots at −80°C until use. NMR samples were prepared by dissolving the lyophilized powder in 20 mM potassium phosphate buffer (pH 7.4) at a concentration of 25 μM containing 10% (v/v) 2H2O in the presence and absence of a fivefold excess of bexarotene in 2.5% DMSO. The pH of the mixture was checked for possible pH variations that could occur from the addition of bexarotene and was found to be unchanged. NMR spectral measurements were made on a Bruker AVANCE-500 spectrometer equipped with a cryogenic probe. The probe temperature was set to 278 K. 1H-15N HSQC spectra were recorded at a 1H observation frequency of 500 MHz with 128 (t1) × 1024 (t2) complex points and 32 scans per t1 increment. The spectral width was 1216 Hz for the 15N dimension and 6010 Hz for the 1H dimension. Chemical shift perturbation (CSP) and intensity changes were monitored using 1H-15N HSQC spectra at increasing molar ratios of bexarotene to monomeric Aβ42. A reference sample of DMSO without compound was also titrated into Aβ42 samples. CSP and intensity ratio due to the addition of DMSO was subtracted from those observed during titrations with compound. CSP was calculated as Δδ = ((ΔδN/5)2 + (ΔδH)2)1/2. NMR spectra were processed by TopSpin 2.1 (Bruker). Resonance assignment and intensity calculations were performed using the program Sparky (www.cgl.ucsf.edu/home/sparky/). The sample preparation protocol and the low NMR probe temperature (5°C) were chosen to ensure that the Aβ42 peptide remained monomeric during the entire data acquisition. The HSQC spectra are typical of intrinsically disordered peptides with a very low dispersion of resonant frequencies (figs. S3 and S4).
Caspase-3 cytotoxicity assay
Aβ42 oligomers are known to be toxic to cells leading to apoptosis and to cellular death. Cytotoxicity assays were performed by measuring the activity of caspase-3, an earlier marker of apoptosis, in a neuroblastoma cell line when incubated with Aβ42 either in the presence or in the absence of bexarotene (52). SH-SY5Y human neuroblastoma cells were cultured under standard conditions, at 37°C in a humidified incubator with 5% CO2. Cells were seeded at a density of 25,000 per well in a white-walled, clear-bottomed 96-well plate and cultured for 24 hours in minimum essential medium–F12 (1:1)/10% fetal bovine serum (FBS) (Gibco). The culture medium was then replaced with prewarmed phenol red–free Dulbecco’s modified Eagle’s medium without FBS into which the peptide samples or NaPO4 buffer were diluted 1:1. Aβ samples at roughly 0.5 μM, either alone or in the presence of 20-fold excess of bexarotene in 1% DMSO final concentration, were removed from an ongoing aggregation reaction at different time points (0, 0.3, 4, and 7 hours) and were followed in parallel by means of ThT fluorescence. The aggregation reaction was set up as above except for the buffer from which NaN3 was removed because of its toxicity. At 10 μM, DMSO-solubilized bexarotene did not show any toxicity and thus was comparable to the control values (fig. S7). All the specified time points correspond to Aβ42 in the lag phase to monitor inhibition at the early stage of the aggregation. We used one separate plate for each time point with 25 replicates of Aβ either alone or in the presence of bexarotene. The cells were cultured in the presence of the peptides, buffer, and media for an additional 24 hours before the cytotoxic effect was measured. Cell medium diluted with the same buffer that was used in these experiments was included in some wells to evaluate the intrinsic fluorescence of the media and bexarotene.
As a negative control, we added to the cells the cell media diluted with the buffer that was used to prepare bexarotene (phosphate buffer with 1% DMSO). For the positive control, staurosporine (1 μM) was added to the cells for 2 hours. Caspase-3/7 activity was measured using the Apo-ONE Homogeneous Caspase-3/7 Assay (Promega). The fluorogenic caspase-3/7 substrate rhodamine 110 bis-(N-CBZ-l-aspartyl-l-glutamyl-l-valyl-aspartic acid amide) was diluted 1:100 in the lysis buffer provided and added to the cell medium at a 1:1 ratio. The fluorescence was measured at an excitation of 480 nm and an emission of 520 nm in an Optima Fluostar plate reader. In each of the used plates, the toxicity was assessed according to a negative and a positive control.
For comparative purposes between different plates, we normalized the fluorescence values using a parabolic equation (y = axb), which corresponds to the evolution of the oligomers as a function of time. Negative and positive controls were set to 1 and 10, respectively. According to these calculations, the toxicity of Aβ42 in the absence and in the presence of bexarotene has normalized values between 1 and 10 and hence could be compared between different plates. Statistical analyses were performed using GraphPad Prism 6.0. Results are expressed as means ± SEM. Statistical analyses were performed using Student t test or one-way analysis of variance followed by Tukey’s post hoc test.
C. elegans experiments
Standard conditions were used for C. elegans propagation at 20°C (53). Briefly, animals were synchronized by hypochlorite bleaching, hatched overnight in M9 buffer, and subsequently cultured on nematode growth medium (NGM; USP agar) plates seeded with the E. coli strain OP50. Saturated cultures of OP50 were grown by inoculating 50 ml of LB medium with OP50 and incubating the culture for 16 hours at 37°C. NGM plates were seeded with bacteria by adding 350 μl of saturated OP50 to each plate and leaving the plates at room temperature for 2 to 3 days. On day 3 after synchronization, animals were placed on NGM plates containing 5-fluoro-2′-deoxyuridine (75 mM, unless stated otherwise) to inhibit growth of offspring.
The following strains were used: dvIs100[unc-54p::A-beta-1-42::unc-54 3′UTR + mtl-2p::GFP] (GMC101). mtl-2p::GFP produces constitutive expression of GFP in intestinal cells. unc-54p::A-beta-1-42 expresses full-length human Aβ42 peptide in body wall muscle cells that aggregates in vivo. Shifting L4 or young adult animals from 20° to 25°C causes paralysis (46). dvIs15[pPD30.38 (unc-54 vector) + mtl-2::GFP (pCL26)] (CL2122) is the control strain for GMC101 (46). C. elegans var. Bristol (N2) was isolated from the mushroom compost near Bristol, England. Generation time is about 3 days and mean brood size is about 350 progeny (53).
Bexarotene stock (5 mM in 100% DMSO) was used at an appropriate concentration to seed 9-cm NGM plates. Plates were placed in a laminar flow hood at room temperature (22°C) for 1 hour to dry. C. elegans cultures were transferred onto media with compound as L1 larvae for 64 to 72 hours at 20°C. Cultures were then transferred to freshly seeded 9-cm plates as young adults and incubated at 23° or 25°C for the whole experiment. Experiments were carried out at different bexarotene concentrations ranging from 5 to 10 μM in 1% DMSO. As controls, plates seeded only with 1% DMSO were used.
C. elegans motility assay
All populations were cultured at 20°C and developmentally synchronized from a 4-hour egg lay. At 64 to 72 hours after egg lay (time 0), individuals were shifted to 23° or 25°C, and body movement was assessed over time as indicated. At different ages, animals were washed out of the plates with M9 and spread over an unseeded 6-cm plate, after which their movements were recorded at 30 frames per second using a homemade microscopic setup, at ×0.75 magnification, for 30 s or 1 min. Two hundred animals were counted per experiment unless stated otherwise. All experiments were carried out in triplicate and the data from one representative experiment are shown. Statistical analyses were performed using the GraphPad Prism software (GraphPad Software).
Videos were elaborated using ImageJ (National Institutes of Health, Bethesda, MA), and body bends were quantified using the wrMTrck plug-in (www.phage.dk/plugins/wrmtrck.html), developed by Jesper Søndergaard Pedersen.
Staining and microscopy in living C. elegans
Live transgenic animals were incubated with NIAD-4 over a range of concentrations and times, and it was empirically determined that incubation of living animals with 1 μM NIAD-4 (0.1% DMSO in M9 buffer) for 4 hours at room temperature gave robust and reproducible staining. After staining, animals were allowed to recover on NGM plates for about 24 hours to allow destaining via normal metabolism. Stained animals were mounted on 2% agarose pads containing 40 mM NaN3 as anesthetic on glass microscope slides for imaging. Images were captured with a Zeiss Axio Observer D1 fluorescence microscope (Carl Zeiss Microscopy GmbH) with a 20× objective and a 49004 ET-CY3/TRITC filter (Chroma Technology Corp). Fluorescence intensity was calculated using ImageJ software (National Institutes of Health) and then normalized as the corrected total cell fluorescence. Only the head region was considered because of the high background signal in the guts. All experiments were carried out in triplicate, and the data from one representative experiment are shown. Statistical significance was determined using t tests.
Western blot analysis
For comparison of the Aβ42 expression level in the absence and in the presence of bexarotene, ~3000 adults were collected in S-basal (53) in triplicate and then frozen in liquid N2. Samples were then extracted in radioimmunoprecipitation assay buffer [50 mM tris (pH 8), 150 mM NaCl, 5 mM EDTA, 0.5% SDS, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, Roche Complete inhibitors 1×; Roche Holding AG], disrupted via sonication, and then centrifuged at 16,000 rpm for 10 min at 4°C. Pellets and supernatants were then separated. Pellets were solubilized using urea/SDS buffer (8 M urea, 2% SDS, 50 mM tris). Samples at the appropriate concentration were added to NuPAGE LDS Sample Buffer (1×) and NuPAGE Sample Reducing Agent (1×) (Life Technologies) and heated at 70°C for 10 min. Material was resolved via NuPAGE Novex 4 to 12% Bis-Tris Protein Gels (Life Technologies), transferred to nitrocellulose membranes using an iBlot Dry Blotting System (Life Technologies), and blocked overnight at 4°C in 5% bovine serum albumin. Membranes were then probed at room temperature with MABN10 anti-Aβ antibody, clone W0-2 (Millipore) diluted to 1:500. Blots were reprobed with anti–α-tubulin, clone B-5-1-2 (Sigma-Aldrich) diluted to 1:5000, to standardize total protein loading. Alexa 488–conjugated secondary antibody was used for detection.
Supplementary Material
Acknowledgments
We thank C. D. Link for his valuable advice in setting up the NIAD-4 staining protocol. Funding: This work was supported by the Centre for Misfolding Diseases, University of Cambridge. Author contributions: J.H., M.P., P.J., S.C., S.L., E.A.A.N., T.P.J.K., C.M.D., and M.V. designed the research. J.H., P.A., M.P., A.R.C., M.Y.-U., P.J., S.C., and M.B.D.M. performed research. J.H., P.A., M.P., A.R.C., M.Y.-U., P.J., S.C., M.B.D.M., S.I.A.C., S.L., E.A.A.N., T.P.J.K., C.M.D., and M.V. analyzed the data. J.H., P.A., M.P., S.L., E.A.A.N., T.P.J.K., C.M.D., and M.V. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper itself and the Supplementary Materials or available upon request from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/2/e1501244/DC1
Fig. S1. Comparison of the effects of bexarotene and tramiprosate on Aβ42 aggregation.
Fig. S2. Quantitative evaluation of the effect of bexarotene on the rate constants of Aβ42 aggregation.
Fig. S3. HSQC spectra of 15N-Aβ42 in the absence and in the presence of fivefold excess of bexarotene.
Fig. S4. Interaction between 15N-Aβ42 and bexarotene.
Fig. S5. Effect of bexarotene on Aβ42 aggregation in C. elegans.
Fig. S6. Effect of DMSO on Aβ42 aggregation.
Fig. S7. Toxicity induced by bexarotene in SH-SY5Y human neuroblastoma cells.
REFERENCES AND NOTES
- 1.Alzheimer’s Association, 2012 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 8, 131–168 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Chiti F., Dobson C. M., Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Dobson C. M., Protein folding and misfolding. Nature 426, 884–890 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Selkoe D. J., Folding proteins in fatal ways. Nature 426, 900–904 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Haass C., Selkoe D. J., Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Balch W. E., Morimoto R. I., Dillin A., Kelly J. W., Adapting proteostasis for disease intervention. Science 319, 916–919 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Hartl F. U., Bracher A., Hayer-Hartl M., Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Eisenberg D., Jucker M., The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knowles T. P. J., Vendruscolo M., Dobson C. M., The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Habchi J., Tompa P., Longhi S., Uversky V. N., Introducing protein intrinsic disorder. Chem. Rev. 114, 6561–6588 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Bieschke J., Natural compounds may open new routes to treatment of amyloid diseases. Neurotherapeutics 10, 429–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J., Armstrong A. H., Koehler A. N., Hecht M. H., Small molecule microarrays enable the discovery of compounds that bind the Alzheimer’s Aβ peptide and reduce its cytotoxicity. J. Am. Chem. Soc. 132, 17015–17022 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito M., Johansson J., Strömberg R., Nilsson L., Effects of ligands on unfolding of the amyloid β-peptide central helix: Mechanistic insights from molecular dynamics simulations. PLOS One 7, e30510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroth H., Ansaloni A., Varisco Y., Jan A., Sreenivasachary N., Rezaei-Ghaleh N., Giriens V., Lohmann S., López-Deber M. P., Adolfsson O., Pihlgren M., Paganetti P., Froestl W., Nagel-Steger L., Willbold D., Schrader T., Zweckstetter M., Pfeifer A., Lashuel H. A., Muhs A., Discovery and structure activity relationship of small molecule inhibitors of toxic β-amyloid-42 fibril formation. J. Biol. Chem. 287, 34786–34800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Necula M., Kayed R., Milton S., Glabe C. G., Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Nie Q., Du X., Geng M., Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 32, 545–551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porat Y., Abramowitz A., Gazit E., Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 67, 27–37 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Sinha S., Lopes D. H. J., Du Z., Pang E. S., Shanmugam A., Lomakin A., Talbiersky P., Tennstaedt A., McDaniel K., Bakshi R., Kuo P.-Y., Ehrmann M., Benedek G. B., Loo J. A., Klärner F.-G., Schrader T., Wang C., Bitan G., Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 133, 16958–16969 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abelein A., Lang L., Lendel C., Gräslund A., Danielsson J., Transient small molecule interactions kinetically modulate amyloid β peptide self-assembly. FEBS Lett. 586, 3991–3995 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Lansbury P. T., Lashuel H. A., A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Cummings J. L., Morstorf T., Zhong K., Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 6, 37 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arosio P., Cukalevski R., Frohm B., Knowles T. P. J., Linse S., Quantification of the concentration of Aβ42 propagons during the lag phase by an amyloid chain reaction assay. J. Am. Chem. Soc. 136, 219–225 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Butterfield S. M., Lashuel H. A., Amyloidogenic protein–membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. 49, 5628–5654 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C. M., Cecchi C., Chiti F., A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J., Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh D. M., Selkoe D. J., Aβ oligomers—A decade of discovery. J. Neurochem. 101, 1172–1184 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Mannini B., Mulvihill E., Sgromo C., Cascella R., Khodarahmi R., Ramazzotti M., Dobson C. M., Cecchi C., Chiti F., Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 9, 2309–2317 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Arosio P., Vendruscolo M., Dobson C. M., Knowles T. P. J., Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Knowles T. P. J., Waudby C. A., Devlin G. L., Cohen S. I. A., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., Dobson C. M., An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J., Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hellstrand E., Boland B., Walsh D. M., Linse S., Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 1, 13–18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen S. I. A., Vendruscolo M., Dobson C. M., Knowles T. P. J., From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 421, 160–171 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Hajduk P. J., Galloway W. R. J. D., Spring D. R., Drug discovery: A question of library design. Nature 470, 42–43 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Rees D. C., Congreve M., Murray C. W., Carr R., Fragment-based lead discovery. Nat. Rev. Drug Discov. 3, 660–672 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Appleby B. S., Cummings J. L., Discovering new treatments for Alzheimer’s disease by repurposing approved medications. Curr. Top. Med. Chem. 13, 2306–2327 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Corbett A., Williams G., Ballard C., Drug repositioning: An opportunity to develop novel treatments for Alzheimer’s disease. Pharmaceuticals 6, 1304–1321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caltagirone C., Ferrannini L., Marchionni N., Nappi G., Scapagnini G., Trabucchi M., The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: A review. Aging Clin. Exp. Res. 24, 580–587 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Bomben V., Holth J., Reed J., Cramer P., Landreth G., Noebels J., Bexarotene reduces network excitability in models of Alzheimer’s disease and epilepsy. Neurobiol. Aging 35, 2091–2095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cramer P. E., Cirrito J. R., Wesson D. W., Lee C. Y. D., Karlo J. C., Zinn A. E., Casali B. T., Restivo J. L., Goebel W. D., James M. J., Brunden K. R., Wilson D. A., Landreth G. E., ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fitz N. F., Cronican A. A., Lefterov I., Koldamova R., Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-c (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price A. R., Xu G., Siemienski Z. B., Smithson L. A., Borchelt D. R., Golde T. E., Felsenstein K. M., Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-d (2013). [DOI] [PubMed] [Google Scholar]
- 42.Tesseur I., Lo A. C., Roberfroid A., Dietvorst S., Van Broeck B., Borgers M., Gijsen H., Moechars D., Mercken M., Kemp J., D’Hooge R., De Strooper B., Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-e (2013). [DOI] [PubMed] [Google Scholar]
- 43.Veeraraghavalu K., Zhang C., Miller S., Hefendehl J. K., Rajapaksha T. W., Ulrich J., Jucker M., Holtzman D. M., Tanzi R. E., Vassar R., Sisodia S. S., , Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-f (2013). [DOI] [PubMed] [Google Scholar]
- 44.LaClair K. D., Manaye K. F., Lee D. L., Allard J. S., Savonenko A. V., Troncoso J. C., Wong P. C., Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol. Neurodegener. 8, 18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fantini J., Di Scala C., Yahi N., Troadec J.-D., Sadelli K., Chahinian H., Garmy N., Bexarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer’s β-amyloid peptides. ACS Chem. Neurosci. 5, 216–224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McColl G., Roberts B. R., Pukala T. L., Kenche V. B., Roberts C. M., Link C. D., Ryan T. M., Masters C. L., Barnham K. J., Bush A. I., Cherny R. A., Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 7, 57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olzscha H., Schermann S. M., Woerner A. C., Pinkert S., Hecht M. H., Tartaglia G. G., Vendruscolo M., Hayer-Hartl M., Hartl F. U., Vabulas R. M., Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Cohen S. I. A., Arosio P., Presto J., Kurudenkandy F. R., Biverstål H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P. J., Linse S., A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sperling R. A., Aisen P. S., Beckett L. A., Bennett D. A., Craft S., Fagan A. M., Iwatsubo T., Jack C. R. Jr, Kaye J., Montine T. J., Park D. C., Reiman E. M., Rowe C. C., Siemers E., Stern Y., Yaffe K., Carrillo M. C., Thies B., Morrison-Bogorad M., Wagster M. V., Phelps C. H., Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 7, 280–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selkoe D. J., Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 17, 1060–1065 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Walsh D. M., Thulin E., Minogue A. M., Gustavsson N., Pang E., Teplow D. B., Linse S., A facile method for expression and purification of the Alzheimer’s disease-associated amyloid β-peptide. FEBS J. 276, 1266–1281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roychaudhuri R., Yang M., Hoshi M. M., Teplow D. B., Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 284, 4749–4753 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brenner S., The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/2/e1501244/DC1
Fig. S1. Comparison of the effects of bexarotene and tramiprosate on Aβ42 aggregation.
Fig. S2. Quantitative evaluation of the effect of bexarotene on the rate constants of Aβ42 aggregation.
Fig. S3. HSQC spectra of 15N-Aβ42 in the absence and in the presence of fivefold excess of bexarotene.
Fig. S4. Interaction between 15N-Aβ42 and bexarotene.
Fig. S5. Effect of bexarotene on Aβ42 aggregation in C. elegans.
Fig. S6. Effect of DMSO on Aβ42 aggregation.
Fig. S7. Toxicity induced by bexarotene in SH-SY5Y human neuroblastoma cells.