

Abstract
Normal valve structures consist of stratified layers of specialized extracellular matrix (ECM) interspersed with valve interstitial cells (VICs) and surrounded by a monolayer of valve endothelial cells (VECs). VECs play essential roles in establishing the valve structures during embryonic development, and are important for maintaining life-long valve integrity and function. In contrast to a continuous endothelium over the surface of healthy valve leaflets, VEC disruption is commonly observed in malfunctioning valves and is associated with pathological processes that promote valve disease and dysfunction. Despite the clinical relevance, focused studies determining the contribution of VECs to development and disease processes are limited. The isolation of VECs from animal models would allow for cell-specific experimentation. VECs have been isolated from large animal adult models but due to their small population size, fragileness, and lack of specific markers, no reports of VEC isolations in embryos or adult small animal models have been reported. Here we describe a novel method that allows for the direct isolation of VECs from mice at embryonic and adult stages. Utilizing the Tie2-GFP reporter model that labels all endothelial cells with Green Fluorescent Protein (GFP), we have been successful in isolating GFP-positive (and negative) cells from the semilunar and atrioventricular valve regions using fluorescence activated cell sorting (FACS). Isolated GFP-positive VECs are enriched for endothelial markers, including CD31 and von Willebrand Factor (vWF), and retain endothelial cell expression when cultured; while, GFP-negative cells exhibit molecular profiles and cell shapes consistent with VIC phenotypes. The ability to isolate embryonic and adult murine VECs allows for previously unattainable molecular and functional studies to be carried out on a specific valve cell population, which will greatly improve our understanding of valve development and disease mechanisms.
Keywords: Cellular Biology, Issue 90, Heart valve, Valve Endothelial Cells (VEC), Fluorescence Activated Cell Sorting (FACS), Mouse, Embryo, Adult, GFP.
Introduction
The mature valve is composed of three stratified layers of specialized extracellular matrix (ECM) interspersed with valve interstitial cells (VICs) and encapsulated by a single layer of heart valve endothelial cells (VECs)1. The role of the ECM is to provide all the necessary biomechanical properties to withstand constant changes in hemodynamic force during the cardiac cycle. Turnover of the valve ECM in the adult valve is tightly regulated by VICs that are largely quiescent and fibroblast-like in the absence of disease. In addition to the VIC population, heart valve endothelial cells (VECs) form an uninterrupted endothelium over the surface of the valve cusps2. While the importance of ECM and VICs for valve structure and function have been described by us and others, the role of VECs is less well known. However, this comparatively small cell population is critical for valve formation in the embryo and is described as dysfunctional in valve disease.
Heart valve formation in the embryo begins when a subset of endothelial cells within the atrioventricular canal and outflow tract regions undergo endothelial to mesenchymal transformation (EMT) and form swellings known as endocardial cushions1,3. The newly transformed mesenchymal cells within these structures later differentiate into VICs and form the mature valves. Once EMT is complete, endothelial cells surrounding the cushions, termed VECs, form an uninterrupted endothelial cell layer that protects the mature valve against injury. In addition, VECs sense the hemodynamic environment and have been shown to molecularly communicate with underlying VICs to regulate ECM homeostasis1,3. In diseased valves, the VEC monolayer is disrupted in association with abnormal changes in ECM organization and altered biomechanics4,5. In addition, studies in mouse models suggest that VEC dysfunction is the underlying cause of valve disease1,6-9. As VECs play an important role in valve development, maintenance, and disease, it is important that we fully define their temporal phenotypes in order to advance the field and understand mechanisms of disease.
Previous work by several labs has successfully isolated VECs from porcine and ovine models10-12. Due to the large size of these valves, isolations through swabbing and/or enzymatic digestion, followed by a number of different isolation methods including magnetic bead cell separation and single cell clonal expansion has been effective to generate pure populations11-13. However, these models can be restrictive due to the incomplete annotation of the pig and sheep genomes limiting the availability of molecular tools in addition to the high costs. Therefore experimentation of porcine and ovine VECs after isolation can be restrictive. Mouse models are preferable due to the many possibilities for genetic manipulation and molecular tools in the embryo and adult, but to date, no VEC isolations in small animal models have been reported. This is likely due to the difficulty of working with small tissue samples that include a minority cell population that currently lack unique identity of VEC-specific markers, thereby preventing antibody-based isolation methods.
In this article, we report a new method for direct isolation of murine VECs at embryonic and adult stages. This protocol takes advantage of Tie2-GFP mice, which express GFP in all endothelial cell types and have been extensively used to study endothelial cell populations14. However, the novelty of this current study is that these mice, for the first time have been utilized to isolate endothelial cells from the valves. By careful dissection of the valve tissue and a series of nine enzymatic digestions followed by FACS sorting, VECs can be isolated and used for various experimental techniques including RNA extraction and culture, directly following sorting.
Protocol
1. Preparation of Equipment and Solutions
Sterilize the dissection tools - fine tissue scissors to extract adult hearts and 2 fine forceps for dissection of the valvular region - by autoclaving in a covered instrument tray. Spray tools with 70% ethanol (EtOH) prior to dissection.
- Prepare all solutions immediately prior to the experiment and sterilize solutions by passing them through a sterile 0.2 μm filter. Keep solutions on ice until use.
- Sterile Dissociation Buffer (15 ml total/sample). Combine 1.2 ml collagenase IV, 300 μl 2.5% trypsin and 150 μl chick serum. Bring volume up to 15 ml with Hank’s Balanced Salt Solution (HBSS).
- Sterile Sorting Buffer (12 ml total). Combine 25 μl 0.5 M ethylenediaminetetraacetic acid (EDTA) and 12 μl DNase I (RNase-free). Bring volume up to 12 ml with HBSS.
- VEC Culture Media. Mix endothelial growth media according to the manufacturer’s instructions (see material spreadsheet) by adding the aliquoted components from the kit to 500 ml of EBM2 media.
- GFP Negative Culture Media (non-endothelial cell media). Mix 445 ml of Medium 199 1x with 5 ml of penicillin/streptomycin (pen/strep) (1% final concentration) and 50 ml FBS (10% final concentration).
2. Dissection of the Valvular Region from Adult mice and E14.5 embryos
All animal procedures were approved and performed in accordance with The Nationwide Children’s Hospital Research Institute IACUC guidelines.
Tie2-GFP mice were purchased from Jackson Laboratories (stock number 003658)14 and were maintained as homozygous genotypes on an FVB/N background, and therefore ensuring that all off-spring express GFP in Tie2-positive endothelial cells.
For FACS sorting, prepare one age matched negative sample that does not contain GFP such as C57/Bl6 to set GFP gate parameters for FACS sorting. NOTE: This protocol and representative results are based on using three, four-month-old adult mice or one litter at embryonic day (E) 14.5.
Adult Mice: Immediately following sacrifice by CO2 anoxia, use dissection scissors to gently open the chest cavity and expose the heart and lungs. Once exposed, grip the heart with forceps at the great arteries and pull away from the body. Remove lung tissue if intact, and place hearts in cold HBSS to rinse. Remove the ventricle and pull away the atria leaving the atrioventricular canal ‘ring’ and aortic regions intact. Make one incision down the side of atrioventricular canal to open up the ‘ring’ and expose the valvular structures. Remove the myocardium and proximal aorta regions by gently teasing away with forceps. Identify the valve leaflets as white, dense tissue over the pink myocardium. Gently detach the chordae tendinae from the atrioventricular valves and remove as much of the remaining myocardium as possible. Be cautious to not pull or scrape the valve leaflets during this process since VECs may be dislodged. Place trimmed valvular regions in a 1.5 ml eppendorf tube containing 1 ml HBSS and keep on ice. NOTE: Careful dissection is critical to eliminate non-valvular endothelial cells that could contaminate the sample.
- Embryos: Sacrifice female mice 14.0 days after copulation plug is observed (morning of copulation plug = 0.5 days).
- Immediately following sacrifice, dissect the uterus (containing embryos) and wash in cold HBSS. Dissect out individual embryos from the uterus and remove the embryonic yolk sac surrounding each embryo. Remove the hearts from each of the embryos and follow step 2.1. (NOTE: gently squeezing the embryo with forceps just below the upper appendages should help to expose the heart and make removal easier.)
3. Cell Dissociation and Preparation for FACS
Using sterile pipette tips, remove HBSS from the eppendorf tube containing the valvular regions.
Replace with 1 ml dissociation buffer and 4 μl DNase I.
Rotate the tubes at 37 °C for 7 min.
Pipette up and down 3 times and then let the sample settle for 15 sec. Collect the supernatant (containing the dissociated cells) into a 15 ml conical tube.
To stop the collagenase reaction, add 125 μl of horse serum to the collection tube. Keep collection tube on ice.
Repeat dissociation steps (3.2 - 3.5) nine times to collect nine fractions of supernatant.
Pass the fractioned collection through a 70 μm nylon filter into a new collection tube to ensure a single cell suspension by removing any debris and clumps.
Spin the suspension at 400 g for 5 min to pellet the cells.
Resuspend the cell pellet in 1 ml Sorting Buffer and keep on ice until FACS.
4. FACS Sorting GFP positive VECs
Set gates for forward and side scatter to exclude debris and capture single cells; exclude doublets by using doublet discrimination gating. Record the negative control sample and define gate settings in order to compare and accurately define the GFP-positive cell population in the Tie2-GFP sample.
Analyze GFP positive cells via FACS and refine gate settings.
Sort the Tie2-GFP sample and collect both GFP-positive and GFP-negative cells. If cells will be used for RNA extraction, sort directly into the Sorting Buffer. To culture cells after FACS, collect GFP positive cells in a sterile tube containing 1 ml VEC media with 1 ml FBS, and collect GFP negative cells in 1 ml of non-endothelial media with 1 ml FBS. Use this 50% media/50% serum combination to improve cell viability. Keep on ice until post FACS analysis.
5. Post FACS Analysis
- RNA Extraction:
- Immediately following FACS, centrifuge GFP positive and negative cells at 1,500 x g for 8 min.
- Carefully pipette the supernatant (sorting buffer) and discard.
- Resuspend the cell pellet (pellet is not visible for the sample sizes recommended here) in 200 μl Trizol.
- Freeze at -80 °C or begin standard phenol-chloroform extraction to isolate total mRNA as previously described by our lab15.
- Use 50-200 ng of RNA for cDNA synthesis using the Mastermix according to the manufacturer’s instructions.
- Subject cDNA to quantitative PCR amplification using IDT Primetime qPCR probes against mouse endothelial cell markers (CD31, von Willebrand Factor (vWF)), valve interstitial cell markers (α-smooth muscle actin (α-SMA), Periostin (Postn)), myocyte markers (Mhc6, Mhc7) and GAPDH.
- Culturing Murine VEC:
- After FACS sorting and cell collection (step 4.3), centrifuge cells at 300 x g for 5 min.
- Carefully pipette off the supernatant (sorting buffer + media) and discard.
- Resuspend the GFP-positive cell pellet in 1 ml of VEC media and the GFP-negative pellet in 1 ml non-endothelial media.
- Plate each sample in one well of a plastic chamber slide and grow until confluent. Culture for about 1 week for GFP negative cells to become confluent and more than 2 weeks for GFP positive cells to become confluent (from a sample of 3, adult mice). Change media every two days.
- Immunofluorescent Staining:
- Remove media from cells and fix in 4% paraformaldehyde (PFA) for 30 min at RT. Wash fixed cells three times for 5 min each in Phosphate Buffered Saline solution (PBS).
- Remove chamber wells from slide and block in 5% bovine serum albumin/1x PBS for 1 hr at RT.
- Incubate slides O/N at 4 °C with primary antibodies (CD31, 1:1,000 or α-smooth muscle actin (α-SMA), 1:100). Wash 3x for 5 min in PBS.
- Dilute the secondary antibody (Alexa-Fluor 568 Goat anti-Rat) 1:400 in PBS, add to slides, and incubate 1 hr at RT. Rinse slides 3x for 5 min in PBS.
- Mount in Vectashield-containing DAPI and incubate 1 hr at 4° C prior to viewing.
Representative Results
Tie2-GFP cells co-localize with endothelial cell markers in the embryonic and adult heart valves.
In order to confirm the specificity of Tie2-GFP expression in VECs from embryonic and adult mice, immunofluorescence was performed to determine co-localization with the endothelial cell marker, CD31 in tissue sections prepared from E14.5 and 3 month old adult Tie2-GFP mice using methods previously published by our lab16. As shown in Figure 1, VECs co-express GFP (Figure 1 A,B,E,F) and CD31 (Figure 1 C,D,E,F) at both adult (Figure 1 B,D,F) and embryonic (Figure 1 A,C,E) stages, therefore validating our model for subsequent VEC isolation.
FACS analysis identified distinct GFP-positive and GFP-negative cell populations in embryonic and adult heart valves isolated from Tie2-GFP mice.
Wild type C57/Bl6 mice were used to set GFP parameters and approximately 2% of single-gated cells from Tie2-GFP mice show a significant enrichment in GFP-positive cells by FACS analysis in both embryonic and adult samples (Figure 2). Based on co-localization studies shown in Figure 1, these cells are considered VECs and yield an average of 61,800 total cells (samples range from 39,000-77,000 cells/sample) in adult samples (n = 3), and 8,928 cells (8,015-11,000 cells/litter) from one litter of E14.5 embryos.
PCR analysis confirms enrichment of endothelial cell markers in GFP-positive cells and valve interstitial cell markers in GFP-negative cells isolated from Tie2-GFP mice.
Gene expression analysis of GFP positive and negative cell populations by qPCR following FACS show distinct molecular profiles. Compared to GFP negative cell populations isolated from Tie2-GFP embryos and adults, GFP positive cells are enriched for expression of endothelial cell markers CD31 and vWF, (Figure 3). Further, expression of myocyte markers (Myh6, Myh7) in these cell populations is very low, demonstrating minimal contamination of non-endothelial cells. In contrast, GFP negative cells isolated from adult Tie2-GFP mice and E14.5 embryos are enriched for valve interstitial cell (VIC) markers, α-sma and Periostin (POSTN) relative to GFP positive cells. Enrichment of these markers is higher in GFP negative cells isolated from E14.5 samples compared to adults due to the quiescent phenotype of adult VICs and therefore low expression of activated markers.
GFP-positive cells isolated from Tie2-GFP adult mice can be cultured in vitro.
The ability to culture GFP positive and GFP negative cells isolated from adult samples was examined based on cell morphology and immunohistochemical staining. Using protocols previously described by us17 we show in Figure 4 that GFP positive cells appear round in morphology (Figure 4A) and co-express GFP with the endothelial cell marker CD31 after two weeks in culture (Figure 4C). In contrast, non-endothelial cells are negative for GFP, display a mesenchymal-like morphology (Figure 4B) and express the VIC marker α-SMA after nine days (Figure 4D).
 Figure 1. Tie2-GFP-positive cells co-localize with CD31 in embryonic and adult heart valves. Immunofluorescence to show association of (Tie2-)GFP expression (A, B) with the endothelial cell marker, CD31 (C, D) in the septal leaflets of the mitral valve at E14.5 (A, C, E) and adult (B, D, F) stages. The valve region is highlighted in A, C, E. (E, F) Merged images. Please click here to view a larger version of this figure.
Figure 1. Tie2-GFP-positive cells co-localize with CD31 in embryonic and adult heart valves. Immunofluorescence to show association of (Tie2-)GFP expression (A, B) with the endothelial cell marker, CD31 (C, D) in the septal leaflets of the mitral valve at E14.5 (A, C, E) and adult (B, D, F) stages. The valve region is highlighted in A, C, E. (E, F) Merged images. Please click here to view a larger version of this figure.
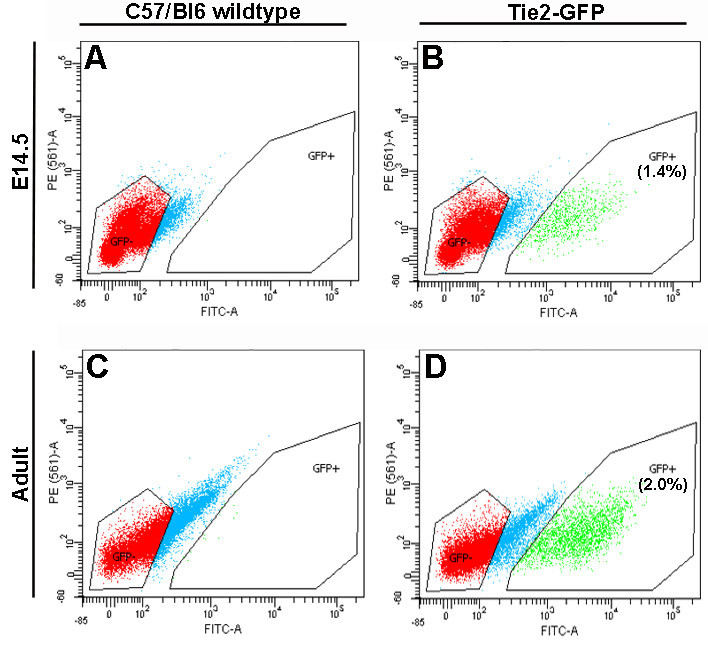 Figure 2. Tie2-GFP-positive VECs can be identified by FACS. Compared to age-matched C57/Bl6 controls (A, C), valves isolated from Tie2-GFP embryos (B) and adults (D) contain a distinct GFP-positive cell population, as indicated in green. GFP-negative events, shown in red were collected as a control. Numbers in (B) and (D) indicate the average % of GFP-positive cells from the total number of events sorted by FACS (n = 3). Please click here to view a larger version of this figure.
Figure 2. Tie2-GFP-positive VECs can be identified by FACS. Compared to age-matched C57/Bl6 controls (A, C), valves isolated from Tie2-GFP embryos (B) and adults (D) contain a distinct GFP-positive cell population, as indicated in green. GFP-negative events, shown in red were collected as a control. Numbers in (B) and (D) indicate the average % of GFP-positive cells from the total number of events sorted by FACS (n = 3). Please click here to view a larger version of this figure.
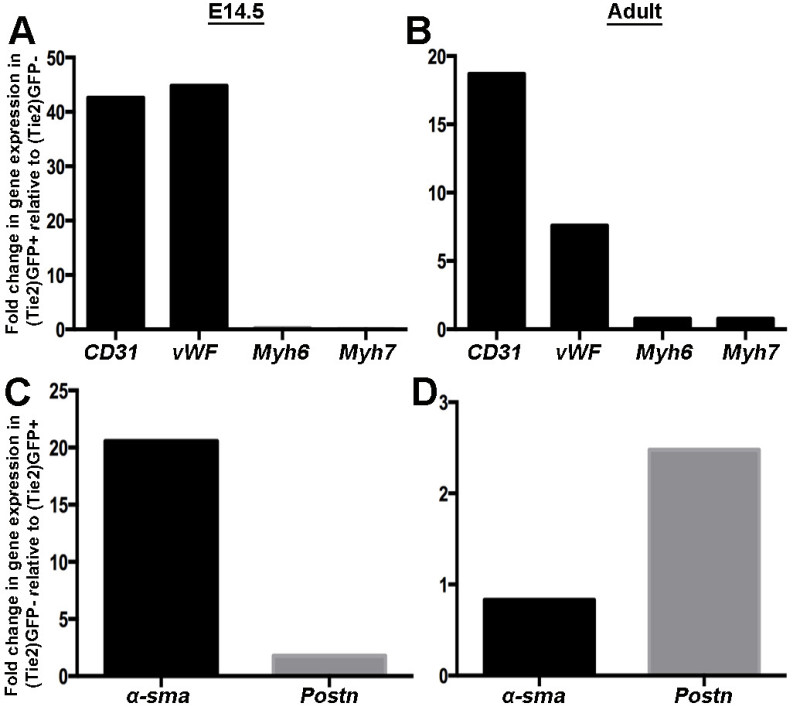 Figure 3. GFP-positive cells are enriched for endothelial cell markers whereas GFP-negative cells express genes associated with valve interstitial cells. Representative qPCR of endothelial cell markers (CD31, von Willebrand Factor (vWF)) and adult (Myh6) and fetal (Myh7) myocyte markers in GFP-positive cells isolated from valvular regions of E14.5 (A) and adult (B)
Tie2-GFP mice. Note enrichment of endothelial cell markers and very low levels of myocyte-associated genes. (C, D) Fold changes in expression of valve interstitial cell markers α-sma and Postn in isolated GFP-negative cells from E14.5 (C) and adult (D)
Tie2-GFP mice. Please click here to view a larger version of this figure.
Figure 3. GFP-positive cells are enriched for endothelial cell markers whereas GFP-negative cells express genes associated with valve interstitial cells. Representative qPCR of endothelial cell markers (CD31, von Willebrand Factor (vWF)) and adult (Myh6) and fetal (Myh7) myocyte markers in GFP-positive cells isolated from valvular regions of E14.5 (A) and adult (B)
Tie2-GFP mice. Note enrichment of endothelial cell markers and very low levels of myocyte-associated genes. (C, D) Fold changes in expression of valve interstitial cell markers α-sma and Postn in isolated GFP-negative cells from E14.5 (C) and adult (D)
Tie2-GFP mice. Please click here to view a larger version of this figure.
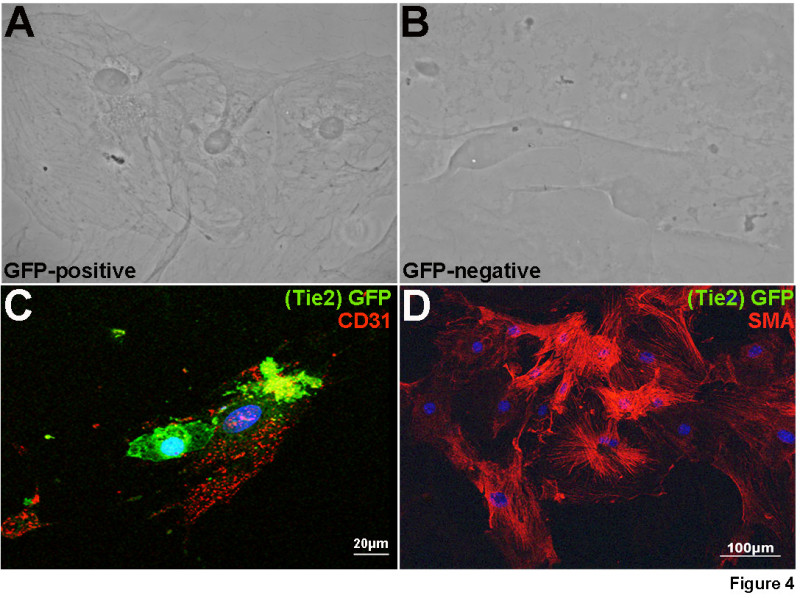 Figure 4. Tie2-GFP-positive cells maintain expression of endothelial cell markers in vitro. Following FACS, GFP positive (A, C) and negative (B, D) cell populations from adult mice were cultured until confluent and subject to phase contrast imaging (A, B) and fluorescent immunostaining (C, D). GFP-positive cells express CD31 (red) (C) after 10 days of culture. In contrast, GFP expression was not detected in the negative cell population, which stained positive for α-SMA, a marker of activated VICs (D).
Figure 4. Tie2-GFP-positive cells maintain expression of endothelial cell markers in vitro. Following FACS, GFP positive (A, C) and negative (B, D) cell populations from adult mice were cultured until confluent and subject to phase contrast imaging (A, B) and fluorescent immunostaining (C, D). GFP-positive cells express CD31 (red) (C) after 10 days of culture. In contrast, GFP expression was not detected in the negative cell population, which stained positive for α-SMA, a marker of activated VICs (D).
Discussion
Here we describe for the first time, a novel method for the isolation of embryonic and adult murine VECs from Tie2-GFP mice. While this mouse line has been extensively used for the isolation of endothelial cell populations, this is the first report showing selective isolation of VECs. Due to the fragility of the VEC population, we have developed a stringent protocol that allows for single cell isolation of GFP positive (and GFP negative) cells from heart valves of embryonic and adult mice. Compared to the original publication using whole embryos or organs from Tie2-GFP mice14, we have optimized collagenase and dissection steps based on the low abundance of this fragile endothelial cell population to isolate a select population of cells.
VEC isolations have previously only been reported in large animal models with limited genetic and biomolecular tools. These tools are well established in mice and therefore, the ability to isolate murine VECs allows for an expanded set of experimental designs to be used for heart valve research. Therefore, a significant advantage of this approach is that VECs can be isolated temporally from wild type mice, and models of valve disease and injury. A second advantage is that VECs can be isolated and analyzed almost immediately after dissection, preserving expression patterns that reflect the in vivo situation more accurately. Further, this isolation protocol is sufficient to eliminate cell culture for number expansion and therefore potential phenotypic changes induced by the in vitro environment are prevented.
Despite the novelties and experimental benefits of this protocol, we recognize that limitations still exist. First, the size of the VEC population within murine valves is very small and therefore, multiple timed embryonic litters are needed in order to generate sufficient RNA for gene expression analysis. While this can be overcome using multiple breeders, it could have an impact on the application of some post-isolation analysis tools. This limitation has been a challenge particularly for establishing confluent cultures of GFP-positive VECs in order to perform more thorough analyses of VEC phenotypes including molecular profiles and functional assays. Therefore we acknowledge that not all difficulties have been overcome by our approach but this is an area of interest that we are striving to overcome.
This approach of isolating VECs from valvular regions introduces the possibility of contamination from Tie-GFP-positive, non-valvular endothelial cells of the endocardium, or vascular structures within the ventricular myocardium. To date, molecular distinction of VECs from other cardiac endothelial cell populations have not been identified. However an enhancer region of the Nfatc1 gene has been identified and shown to specifically label VECs that do not undergo EMT, and no other endothelial cell population within the heart18. As a Cre model (Nfatc1enCre) is available, future studies could utilize the specificity of this line to minimize contamination risks. In addition to non-valvular Tie2-GFP-positive cells, there is always the possibility of contamination from myocytes at the point where the valve leaflets attach to the annular region of the septal and mural myocardial walls. In data not shown here, we initially began studies to isolate VECs from dissected valvular regions using antibody-conjugated beads coated with anti-GFP and anti-CD31. While this approach was successful for isolating the endothelial cells, we experienced significant cell clumping from adjacent, non-endothelial cell types and therefore VICs and myocardial cells contaminated our experimental sample. This has been avoided using FACS analysis as parameters have been set to isolate only single cell suspensions and PCR analysis to detect expression of myocyte-specific genes has controlled for this limitation (Figure 3). While our contamination can be deemed as minimal, it remains a potential experimental complexity which could be avoided in the future with the double selection of GFP and endothelial cell-specific surface markers.
Using this protocol, we have successfully isolated VECs from embryonic and adult mice and provided examples of how this approach can be used for RNA isolation and cell culture of GFP positive and GFP negative cell populations. However, this approach is not limited to these applications and can be used for a plethora of molecular, cellular and functional approaches. In addition, the Tie2-GFP background can be bred with genetic mouse models that will allow for comparative studies of VEC populations in health and disease. The development of this novel methodology will, for the first time, allow for focused studies examining the contribution of VECs in valve development and maintenance and could reveal previously unappreciated mechanisms of endothelial-dependent valve disease.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank The Ohio State University Comprehensive Cancer Center Analytical Cytometry Core facility, specifically Katrina Moore, for technical assistance with FACs analysis. In addition we recognize Dr. William Pu and his group for their scientific insights. This work was supported by NIH HL091878 (JL) and The Heart Center at Nationwide Children’s Hospital.
References
- Tao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: the role of endothelial cells. Current topics in developmental biology. 2012;100:203–232. doi: 10.1016/B978-0-12-387786-4.00006-3. [DOI] [PubMed] [Google Scholar]
- Lincoln J, Yutzey KE. Molecular and developmental mechanisms of congenital heart valve disease. Birth defects research. Part A, Clinical and molecular teratology. 2011;91:526–534. doi: 10.1002/bdra.20799. [DOI] [PubMed] [Google Scholar]
- Tao G, Levay AK, Gridley T, Lincoln J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Developmental biology. 2011;359:209–221. doi: 10.1016/j.ydbio.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg EJ, Mack PJ, Schoen FJ, Garcia-Cardena G, Kaazempur Mofrad MR. Hemodynamic environments from opposing sides of human aortic valve leaflets evoke distinct endothelial phenotypes in vitro. Cardiovasc Eng. 2010;10:5–11. doi: 10.1007/s10558-009-9089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton RB., Jr Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circulation research. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circulation research. 2013;113:186–197. doi: 10.1161/CIRCRESAHA.112.300154. [DOI] [PubMed] [Google Scholar]
- Bosse K, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. Journal of molecular and cellular cardiology. 2013;60:27–35. doi: 10.1016/j.yjmcc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. The Journal of clinical investigation. 2011;121:2876–2887. doi: 10.1172/JCI44555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann JJ, et al. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development. 2012;139:4449–4460. doi: 10.1242/dev.084871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie-Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:598–607. doi: 10.1161/ATVBAHA.110.216184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould RA, Butcher JT. Isolation of valvular endothelial cells. Journal of Visualized Experiments : JoVE. 2010. [DOI] [PMC free article] [PubMed]
- Butcher JT, Penrod AM, Garcia AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1429–1434. doi: 10.1161/01.ATV.0000130462.50769.5a. [DOI] [PubMed] [Google Scholar]
- Cheung WY, Young EW, Simmons CA. Techniques for isolating and purifying porcine aortic valve endothelial cells. The Journal of heart valve disease. 2008;17:674–681. [PubMed] [Google Scholar]
- Motoike T, et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Peacock JD, Lu Y, Koch M, Kadler KE, Lincoln J. Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Developmental dynamics : an official publication of the American Association of Anatomists. 2008;237:3051–3058. doi: 10.1002/dvdy.21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levay AK, et al. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circulation research. 2008;103:948–956. doi: 10.1161/CIRCRESAHA.108.177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Developmental biology. 2006;292:292–302. doi: 10.1016/j.ydbio.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Wu B, et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circulation research. 2011;109:183–192. doi: 10.1161/CIRCRESAHA.111.245035. [DOI] [PMC free article] [PubMed] [Google Scholar]