

Abstract
This video and article contribution gives a comprehensive description of microinjection and electroporation of mouse testis in vivo. This particular transfection technique for testicular mouse cells allows the study of unique processes in spermatogenesis.
The following protocol focuses on transfection of testicular mouse cells with plasmid constructs. Specifically, we used the reporter vector pEGFP-C1, which expresses enhanced green fluorescent protein (eGFP) and also the pDsRed2-N1 vector expressing red fluorescent protein (DsRed2). Both encoded reporter genes were under the control of the human cytomegalovirus immediate-early promoter (CMV).
For performing gene transfer into mouse testes, the reporter plasmid constructs are injected into testes of living mice. To that end, the testis of an anaesthetized animal is exposed and the site of microinjection is prepared. Our preferred place of injection is the efferent duct, with the ultimately connected rete testis as the anatomical transport route of the spermatozoa between the testis and the epididymis. In this way, the filling of the seminiferous tubules after microinjection is excellently managed and controlled due to the use of stained DNA solutions. After observing a sufficient filling of the testis by its colored tubule structure, the organ is electroporated. This enables the transfer of the DNA solution into the testicular cells. Following 3 days of incubation, the testis is removed and investigated under the microscope for green or red fluorescence, illustrating transfection success.
Generally, this protocol can be employed for delivering DNA- or RNA- constructs into living mouse testis in order to (over)express or knock down genes, facilitating in vivo gene function analysis. Furthermore, it is suitable for studying reporter constructs or putative gene regulatory elements. Thus, the main advantages of the electroporation technique are fast performance in combination with low effort as well as the moderate technical equipment and skills required compared to alternative techniques.
Keywords: Molecular Biology, Issue 90, electroporation, transfection, microinjection, testis, sperm, spermatogenesis, reproduction
Introduction
Mammalian spermatogenesis is considered to be a sophisticated process of self-renewing stem cells successively undergoing mitosis, meiosis and differentiation in order to develop into mature haploid spermatozoa. These morphological changes are orchestrated by different cell types and despite profound attempts, it is still impossible to mimic these processes in cell culture1,2. Hence, research on spermatogenesis up to now relies on living organisms as in vivo models. In general, gene function studies are usually based on transgenic animals. However, generating and sustaining this kind of animal model is time-consuming, cost-intensive and quite elaborate. This is attributed to the required long breeding process for generating and maintaining the transgene over the generations. Additionally, the genetic manipulation of the entire organism by the transgenic or knockout approach is prone to cause physiological impairments when targeting genes with essential functions in multiple regions, e.g., outside the testis or systemically.
Further, some transient transfection methods are associated with some crucial disadvantages. For example, typical drawbacks of virus-mediated gene transfer are the possible provocation of immunoreactions and additional safety regulations, whereas lipofection3 and microparticle bombardment4 might damage the tissue and are limited to a certain cell depth for sufficient transfection efficiency.
In contrast, electroporation (EP) as another common way of transient transfection, seems to constitute a promising technique for enabling in vivo transfection and consistent in vivo gene analysis. In general, EP is referred to as a dynamic phenomenon that depends on local transmembrane voltage with consequently mediated pores within nanoseconds. These gaps can be maintained for milliseconds, sufficient to grant access to DNA, RNA or small molecules5. When the applied voltage is too high, the usually transient character of EP is counteracted due to heat production and induction of too comprehensive permeabilization with consequent irreversible damage of the cell5.
Here, we show that electroporation is an effective and economical transfection system which is capable of being utilized for genetic testis transformation in order to elucidate testicular gene characteristics in vivo. This article addresses plasmid preparation, microinjection via the efferent duct and the subsequent electroporation of mouse testis. This procedure can be the means of choice to achieve fast, specific and efficient transfection of seminiferous tubules of mouse testis in vivo in order to investigate processes of spermatogenesis.
Protocol
All performed animal experiments have been approved by the local ethics committee (Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei, Mecklenburg-Vorpommern, Germany).
1. Plasmid Preparation
For plasmid preparation, use plasmid purification kits (see Materials table) or similar methods with endotoxin removal buffer so that immune reactions of the animal can be avoided. Follow the instructions of the manual. Employ ddH2O to dilute the plasmid solution.
Eliminate debris by spinning the DNA solution at maximum speed (20,000 x g). Then collect the supernatant.
Determine plasmid concentration with a spectrophotometer (see materials).
Adjust plasmid concentration with ddH2O to 1-3 µg(DNA)/µl. Note: Lower concentrations will reduce transfection efficiency, while higher concentrations might cause a too viscous injection solution. Besides, the transfection efficiency depends on the size of the plasmid and has to be tested individually.
Prior to injection, prepare a solution mix with for example 40 µl plasmid together with 5 µl PBS (10x) and 5 µl Fast Green (0.5%). Fast Green is needed for tracking the injection process. Preferably, use thin wall PCR-tubes or Parafilm. Note: Depending of the size of the testis, a volume of 20-50 µl will be needed for each testis.
2. Preparation of Microinjection Pipette
Use borosilicate capillaries (see Materials table) for preparing the microinjection pipettes.
Pull glass capillaries with a vertical capillary puller (see Materials table).
Break the capillary tip with forceps by softly banding. The tip is usually 50-80 µm in diameter and about 1 cm long. Try to avoid tips that are too long as these are not stiff enough to penetrate the tissue.
At last, sharpen the tip in a 30° to 45° angle by using a micropipette beveler (see Materials table).
Load the microinjection pipette with the injection solution mix (see Plasmid Preparation). Apply the solution into the back of the glass capillary with a small syringe. Note: Try to avoid air bubbles while loading, otherwise these will be injected into the seminiferous tubules along with the plasmid solution and hence will reduce conductivity as well as possibly cause tissue damage.
3. Anesthesia and Surgery
Perform the operation under aseptic conditions by using sterile material (i.e., syringe, needle, surgical instruments, etc.). This will reduce infection of the animal and ensure good survival rates.
Use male mice for electroporation at an age of 6-8 weeks.
To initialize postsurgical analgesic treatment, provide the mouse with analgesics added drinking water on the day prior to surgery. This assures a preemptive analgesia in case of highly probable postsurgical hypodipsia (diminished water intake). To this end, expose drinking water with for example a pediatric ibuprofen suspension (20 mg/ml). The medicated drinking water should also be supplied on day one and two of recovery in identical concentration. This means a daily dose of 7.5 mg/kg, valid for 5 ml/d drinking water and body weight of 25g for 8 weeks aged C57BL/6J. This analgesic regimen guarantees a fundamental pain relief and accelerated recovery along with high welfare26.
For preparing the anesthesia working solution on the day of surgery, mix 10% Ketamin and 2% Xylazin in a 1:1 ratio (see Materials table).
To anesthetize the animal, apply 0.25 ml/100 mg body weight of anesthetic solution subcutaneously (Ketamin = 0.125 g/kg; Xylazin = 0.025 g/kg). Use an injection site between pelvic and limb in order to prevent organ damage and injury of the mice. Usually, 10 min are needed until the mouse is deeply anesthetized.
Cover the mouse eyes with vet ointment to prevent dryness during anesthesia.
Test deep anesthetic arrest, which is noticeable by a total lack of response. To this end, just pinch the toe of the animal.
To start the surgery, remove abdominal hair with an electric shaver or similar and disinfect the operation area with sterilium or similar.
Make a ventral incision directly above the preputial glands in the center of the abdominal area. At that place, first pull the skin away and conduct a small transversal cutaneous cut of about 8-14 mm. Then, continue along the abdominal muscular layer. Note: The lesion should be as small as possible to reduce harm to the animal.
Pull up the abdominal fat pads carefully to expose the attached testis and place it on a prior prepared waterproof disposable paper drape just beside the incision site (Figure 2A).
Use binoculars to find the efferent duct as the injection target. The ductus efferent is identified as the fine vessel junction between the testis and the epididymis. It is located adjacent to the prominent testicular artery. The ductus runs almost at an angle of 45° to the artery and visibly enters the caput epididymidis. Note: Depending on the mouse’s age, the ductus is usually buried in fatty tissue.
Use fine forceps to clear the efferent duct of this fatty tissue. After that, place the released ductus on a sterile paper strip to ensure the clear visibility of the duct without impairing surroundings.
4. Microinjection and DNA Application
Connect the microinjection pipette, which is loaded with the plasmid solution to the micromanipulator/injector unit.
Place the microinjection needle parallel to the efferent duct with the tip pointing towards the rete testis (Figure 2B).
Use fine tweezers and strip the duct over the microinjection pipette. Make sure that the capillary is kept parallel to the duct while pulling it over. Note: This procedure is more convenient than penetrating the vessel by moving the needle with the micromanipulator.
Direct the needle carefully towards the testis and stop just as it penetrates the rete testis directly beneath the tunica albuginea (Figure 2C). Note: In the case of overreaching the rete testis, the plasmid solution will leak into the interstitial space. This commonly leads to failure of transfecting the seminiferous tubules.
For injection of the plasmid solution utilize the microinjector with following settings: pi: 100 hPa, ti: 0.2 sec, and pc: 0 hPa (Figure 2D).
Monitor the entire injection process by observing the testis filling status with the help of the green color. Take care that the testis is only filled up to 2/3 of its volume with the plasmid solution (Figure 2E). Note: If the injection volume is exceeded, the testis tissue could be harmed.
5. Electroporation of Testis
To enable effective electroporation, soak the tweezer electrodes in PBS (1x). This ensures adequate conductance.
Smoothly squeeze the testis between the wet electrodes (Figure 2F). Measure the electrical resistance with the electroporator.
Apply current to the testis. Perform eight square pulses of 40 V at 4 different testis sites with a constant duration of 50 msec pulse time and 950 msec interval time.
6. Wound Closure and Post-surgery
When electroporation is finished, place the testis back in the original location.
Sew the inner muscular layer with surgical suture (see materials).
Close the skin by employing suture clips (see Materials table). Pull the skin up with tweezers. Make sure to exclude the muscular layer. Place the clips, each at a distance of not more than 5 mm. Note: Because surgical suture can be swallowed by the mouse, suture clips are favored for closing the skin lesion.
Allow the mouse to recover from anesthesia on sterile paper towels placed in a sterile empty mouse cage, which is tempered on a 37 °C warm pad. The pad should ensure the warming of one half of the cage creating a heat gradient. For the animal this makes possible to search for the individual, most comfortable area due to the temperature variety in the cage. In addition, cover the mouse with a sheet of sterile paper towel so that stress can be further reduced. Note: Since the mouse’s body surface is uncommonly high in relation to body mass, when compared with larger animals, the thermal support is of particular importance for successful recovery of rodents.
When the operated animal has fully awakened, transfer it for further recovery to a completely equipped new mouse cage. Do not put the animal back to its old cage or to a group of mice. Ensure that the cage is as clean and sterile as possible to prevent wound infection. Monitor the entire recovery process. To transfer the mouse back to the animal care facility, wait until it has fully recovered and appears to behave normally. Note: Additional per- and post-surgical strategies are discussed by Pritchett-Corning KR et al6.
Representative Results
The experimental setting for performing microinjection and electroporation of mouse testis in vivo as it is used according to the protocol is illustrated in Figure 1. Even though it is possible to acquire industrially manufactured micropipettes, we preferred to generate our own pipettes by pulling (Figure 1A) and beveling (Figure 1B) glass capillaries so that they fitted our needs. The equipment for microinjection and electroporation is illustrated in Figure 1C. For reasons of transfection efficiency, it is of importance to utilize a square wave electroporator.
Figure 2 displays the key steps of the mouse surgery with special focus on the preparation of the efferent duct. In this context the testis is exposed (Figure 2A) and the efferent duct is identified by binocular observation and released from fatty tissue (Figure 2B). Subsequently, the efferent duct is punctured with a microinjection pipette and the plasmid solution is administered (Figures 2C-E). Finally, the loaded testis is squeezed between two electrodes and transfected by applied square wave pulses (Figure 2F).
This in vivo transfection approach allows the use of different plasmids encoding various reporter genes. Here we used the reporter vector pEGFP-C1 (Figure 3A), which expresses enhanced green fluorescent protein (eGFP) and also the pDsRed2-N1 (Figure 3C) vector enabling transactivation of red fluorescent protein (DsRed2). In our case, both reporter genes were under control of the human cytomegalovirus immediate-early promoter (CMV).
In order to evaluate transfection success, the microinjected and electroporated testes were harvested 3 days after the entire procedure and observed under fluorescence microscopy (Figure 3). For detailed investigation, testes were formaldehyde-fixed, embedded in paraffin, sliced (5 µm) as well as counterstained with To-Pro-3, resulting in blue colored nuclei. Eventually, the sections were examined by fluorescence microscopy. Two examples of possible transfection results can be seen in Figure 4. The expression of the reporter gene EGFP is indicated by green fluorescence, the transactivation of DsRed2 is designated by red emission.
 Figure 1. Equipment for in vivo microinjection and electroporation of mouse testis. Glass capillaries are formed using micropipette puller (A) while the tips are sharpened utilizing the micropipette beveler (B). The unit for both techniques microinjection and electroporation is illustrated in (C). Instead of employing a commercial micromanipulator we used a XYZ-cross table of a microscope and attached a unit to hold the micropipette. Please click here to view a larger version of this figure.
Figure 1. Equipment for in vivo microinjection and electroporation of mouse testis. Glass capillaries are formed using micropipette puller (A) while the tips are sharpened utilizing the micropipette beveler (B). The unit for both techniques microinjection and electroporation is illustrated in (C). Instead of employing a commercial micromanipulator we used a XYZ-cross table of a microscope and attached a unit to hold the micropipette. Please click here to view a larger version of this figure.
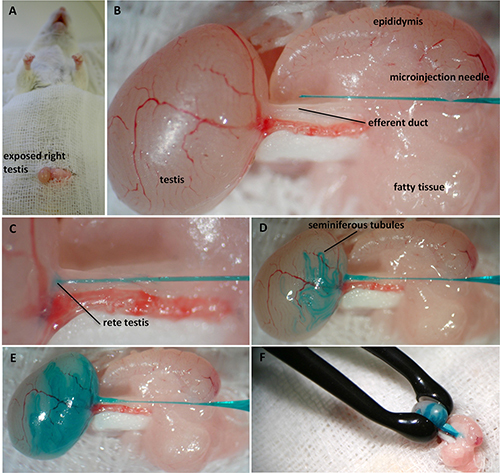 Figure 2. Illustration of the in vivo testis microinjection and electroporation proceeding in mouse including prior surgical intervention. Access to the testis is granted via opening of the mouse’s abdomen which has been anesthetized before. The cut is made just above the preputial glands. After revealing the testis, it is released from abdominal fat pads (A). Under binocular observation, the efferent duct is identified and fat-freed. The microinjection pipette is positioned beside the ductus while the sharp end of the needle is pointing towards the efferent duct (B). The glass capillary is inserted into the ductus and is moved towards the testis to be positioned directly within the rete testis (C). The procedure of DNA application and filling of the seminiferous tubules is monitored with the help of Fast Green (D). Only 2/3 of the testis gets filled in order to prevent harm of the tissue (E). For the final electroporation step, the testis is squeezed between tweezer-type electrodes to apply electric square pulses (F). Please click here to view a larger version of this figure.
Figure 2. Illustration of the in vivo testis microinjection and electroporation proceeding in mouse including prior surgical intervention. Access to the testis is granted via opening of the mouse’s abdomen which has been anesthetized before. The cut is made just above the preputial glands. After revealing the testis, it is released from abdominal fat pads (A). Under binocular observation, the efferent duct is identified and fat-freed. The microinjection pipette is positioned beside the ductus while the sharp end of the needle is pointing towards the efferent duct (B). The glass capillary is inserted into the ductus and is moved towards the testis to be positioned directly within the rete testis (C). The procedure of DNA application and filling of the seminiferous tubules is monitored with the help of Fast Green (D). Only 2/3 of the testis gets filled in order to prevent harm of the tissue (E). For the final electroporation step, the testis is squeezed between tweezer-type electrodes to apply electric square pulses (F). Please click here to view a larger version of this figure.
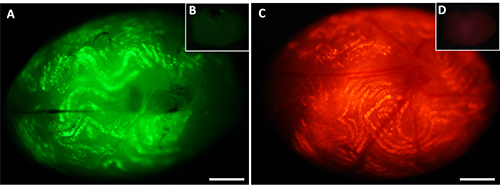 Figure 3. Whole mount testis 3 days after in vivo electroporation. Under fluorescence microscopy, transfected testis of C57BL/6 mouse show expression of enhanced green fluorescent protein - eGFP (A) and red fluorescent protein - DsRed2 (C) both encoded on pEGFP-C1 and pDsRed2-N1 plasmid, respectively. The expression of both reporter genes was under control of the ubiquitously active CMV-promotor element. The effective transfection of the targeted seminiferous tubules is demonstrated by fluorescence intensities. Panels (B) and (D) illustrate auto-fluorescence of untransfected control testis. Scale bar = 1 mm. Please click here to view a larger version of this figure.
Figure 3. Whole mount testis 3 days after in vivo electroporation. Under fluorescence microscopy, transfected testis of C57BL/6 mouse show expression of enhanced green fluorescent protein - eGFP (A) and red fluorescent protein - DsRed2 (C) both encoded on pEGFP-C1 and pDsRed2-N1 plasmid, respectively. The expression of both reporter genes was under control of the ubiquitously active CMV-promotor element. The effective transfection of the targeted seminiferous tubules is demonstrated by fluorescence intensities. Panels (B) and (D) illustrate auto-fluorescence of untransfected control testis. Scale bar = 1 mm. Please click here to view a larger version of this figure.
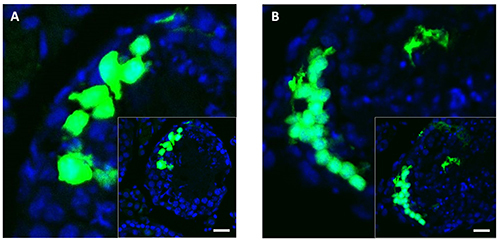 Figure 4. Sections of mouse testis 3 days after unilateral in vivo electroporation with pEGFP-C1 plasmid. Transfection outcome of fixed mouse testis sections (5 µm) are shown 3 days after unilateral electroporation with pEGFP-C1 plasmid. Successfully transfected testicular tubular cellsare indicated by green fluorescence. TO-PRO-3 counterstained nuclei are detectable by blue emission (A and B). The round structures are typical for the organization of cells within the seminiferous tubules. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Figure 4. Sections of mouse testis 3 days after unilateral in vivo electroporation with pEGFP-C1 plasmid. Transfection outcome of fixed mouse testis sections (5 µm) are shown 3 days after unilateral electroporation with pEGFP-C1 plasmid. Successfully transfected testicular tubular cellsare indicated by green fluorescence. TO-PRO-3 counterstained nuclei are detectable by blue emission (A and B). The round structures are typical for the organization of cells within the seminiferous tubules. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Discussion
Research in the field of reproductive biology, particularly in the area of male fertility and spermatogenesis inevitably relies on living organisms. In order to examine testicular function, no adequate cell culture/in vitro system has been established capable of reflecting all the crucial morphological changes from a diploid spermatogonium to a haploid mature spermatozoon1,2. Thus, the generation of genetically modified animals is often a necessary and as such a valuable tool in male reproductive biology. To this end, a considerable number of transgenic, knock-out and knock-in mice have been created which delivered insights into male fertility. Nevertheless, the generation of transgenic mice is usually done by injecting gene constructs into the pronucleus of fertilized eggs. However, this technique is time-consuming and is supposed to be done by specialized laboratories. The procedure to generate knock-out or knock-in animals is even more sophisticated.
Another way of studying gene function of living model organisms is by transient transfection. However, the common transduction with viral vectors may cause immunogenic or other unspecific reactions and requires special safety regulations. Lipofection3 and microparticle bombardment4 approaches are unfortunately restrained to a specific cell depth and even might trigger adverse effects on the tissue.
Nevertheless, by the means of directly applied electrical square pulses on the targeted tissue, so called electroporation, it is actually possible to transfect living tissue, e.g., herein mouse testis. Moreover, this method only requires low costs, low equipment and properly skilled lab personal.
A further noteworthy benefit associated with in vivo transfection methods in comparison to persistently gene-altered animal models is the possibility of co-transfection. While transgenic or knock-mice usually carry only one gene construct, in vivo transfection enables comprehensive, simultaneous analyses of different gene constructs within the same cell. This can be done by the use of various reporter genes, e.g., GFP versus YFP or Gaussia- versus Renilla-Luciferase, so that a comparative investigation of wild type and mutant gene effects is possible.
Furthermore, by the application of cell type specific promoters, the expression of gene constructs can be exquisitely directed onto specific cells within the testis. For example, this could allow a testicular gene expression just in Sertoli cells or spermatocytes.
The protocol presented in this article covers two important methods in the field of male reproductive biology. The first one is the microinjection into seminiferous tubules with the preparation of the injection capillary. This technique has been commonly implicated in germ/stem cell transplantation either from transgenic animals to the recipient7 or in the context of xenografting8. The second method is the electroporation transfection capable of inducing transient pores in the plasma membrane as well as a directed bulk flow of charged molecules like plasmids, ensuring entrance into certain cells or rather tissues. This specific direction of macromolecules by electroporation is frequently appreciated in unilaterally neural cell transfection often done in utero9,10 or in developing retina11.
The preceding detailed method protocol includes as the first crucial steps the anesthesia of male mice, the exposition of testis via abdominal lesion, the preparation of the efferent duct as well as the microinjection into the ductus. As these actions are quite demanding, it is strongly advised to first practice on dead animals before performing operations on living mice. Moreover, instructions are provided on how to inject and fill the seminiferous tubules with plasmid-DNA and how to conduct the electroporation of the testis appropriately.
Because of the application of electricity during EP, heat production might trigger tissue damage especially at high voltage. This increase in transfection efficiency12, 13, 15 by elevated voltage is accompanied by the adverse effect of testicular shrinking12-14. The most favorable voltage range seems to be between 30-50 V, guaranteeing small effects upon testicular integrity, a normal sperm quality16, a normal mating behavior17, the maintenance of offspring production ability16-20 as well as a sufficient transfection efficacy.
Regarding gene expression intensity of transferred gene constructs, Yomogida et al.21 have also revealed a markedly reduced expression of linearized vectors 3 days after EP implementation whereas circular plasmid-DNA obviously still sustains expression after 35 days with the highest one in Sertoli cells. This suggests that the best results are obtained when using plasmid constructs for electroporation as done here. In our approach, we investigated transfection efficiency 3 days after electroporation due to the observation that this time period is sufficient for adequate wound healing, reparation of cell integrity as well as the desired proper expression of the reporter proteins eGFP and DsRed2. Thus, an incubation time of 3 days following the surgical intervention and the entire transfection procedure seems to be necessary to guarantee appropriately restored cell functions including gene expression so that a fluorescence signal as intensive as possible can be detected ensuring a reliable transfection result. Concerning transfection efficiency, Yomogida et al.21 has revealed that the somatic Sertoli cells are apparently more responsive to EP-transfection than germ cells. We recommend co-transfection of testis with target and control plasmids, each coding for a different reporter gene.
The presented electroporation approach is applicable to a broad spectrum of possible issues referring to male germ cell and fertility processes. For instance, it can be utilized for analyzing regulatory elements of spermatogenesis-specific genes17,22. The method is also suitable for loss-of-function studies with small interfering RNA23 and gain-of-function studies24. Additionally, there are promising chances to even accomplish EP for therapeutic treatment and generation of transgenic offspring as research groups have already shown18,20,25.
Hence, the illustrated novel methodological approach of mouse testis in vivo electroporation in conjunction with efferent duct microinjection of plasmid constructs will hopefully rise to a valuable technique for successfully unraveling the nature of spermatogenesis.
Disclosures
Marten Michaelis, Alexander Sobczak, and Joachim M. Weitzel employed at the Institute of Reproductive Biology, Leibniz Institute for Farm Animal Biology (FBN), Dummerstorf, Germany, declare that they have no competing financial interests.
Acknowledgments
We thank Birgit Westernstroeer of the Centre of Reproductive Medicine and Andrology at the University of Muenster for teaching the testicular microinjection. Besides, we are grateful to Ursula Antkewitz and Petra Reckling for technical assistance. We thank the German Research Foundation (DFG) for supporting this work (WE2458/10-1).
References
- Reuter K, Schlatt S, Ehmcke J, Wistuba J. Fact or fiction: In vitro spermatogenesis. Spermatogenesis. 2012;2:245–252. doi: 10.4161/spmg.21983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter D, Anand-Ivell R, Danner S, Ivell R. Models of in vitro spermatogenesis. Spermatogenesis. 2012;2:32–43. doi: 10.4161/spmg.19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizzi A, et al. fluorescent protein as indicator of nonviral transient transfection efficiency in endometrial and testicular biopsies. Microsc. Res. Tech. 2010;73:229–233. doi: 10.1002/jemt.20779. [DOI] [PubMed] [Google Scholar]
- Williams RS, et al. Introduction of foreign genes into tissues of living mice by DNA-coated microprojectiles. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2726–2730. doi: 10.1073/pnas.88.7.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockmann RA, de Groot BL, Kakorin S, Neumann E, Grubmuller H, Kinetics statistics, and energetics of lipid membrane electroporation studied by molecular dynamics simulations. Biophys. J. 2008;95:1837–1850. doi: 10.1529/biophysj.108.129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchett-Corning KR, Luo Y, Mulder GB, White WJ. Principles of rodent surgery for the new surgeon. J. Vis. Exp. 2011. [DOI] [PMC free article] [PubMed]
- Brinster RL, Avarbock MR. Germline transmission of donor haplotype following spermatogonial transplantation. Proc. Natl. Acad. Sci. U.S.A. 1994;91:11303–11307. doi: 10.1073/pnas.91.24.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatt S, Von SV, Schepers AG. Male germ cell transplantation: an experimental approach with a clinical perspective. Br. Med. Bull. 2000;56:824–836. doi: 10.1258/0007142001903409. [DOI] [PubMed] [Google Scholar]
- Walantus W, Castaneda D, Elias L, Kriegstein A. In utero intraventricular injection and electroporation of E15 mouse embryos. J. Vis. Exp. 2007. [DOI] [PMC free article] [PubMed]
- Matsui A, Yoshida AC, Kubota M, Ogawa M, Shimogori T. Mouse in utero electroporation: controlled spatiotemporal gene transfection. J. Vis. Exp. 2011. [DOI] [PMC free article] [PubMed]
- Blackshaw S. In vivo electroporation of developing mouse retina. J. Vis. Exp. 2011. [DOI] [PMC free article] [PubMed]
- Yomogida K, Yagura Y, Nishimune Y. Electroporated transgene-rescued spermatogenesis in infertile mutant mice with a sertoli cell defect. Biol. Reprod. 2002;67:712–717. doi: 10.1095/biolreprod.101.001743. [DOI] [PubMed] [Google Scholar]
- Ryoki S, Park H, Ohmori Y, Shoji-Tanaka A, Muramatsu T. An integrase facilitates long-lasting foreign gene expression in vivo in mouse spermatogenic cells. J. Biosci. Bioeng. 2001;91:363–367. doi: 10.1263/jbb.91.363. [DOI] [PubMed] [Google Scholar]
- Umemoto Y, et al. Gene transfer to mouse testes by electroporation and its influence on spermatogenesis. J. Androl. 2005;26:264–271. doi: 10.1002/j.1939-4640.2005.tb01094.x. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Shibata O, Ryoki S, Ohmori Y, Okumura J. Foreign gene expression in the mouse testis by localized in vivo gene transfer. Biochem. Biophys. Res. Commun. 1997;233:45–49. doi: 10.1006/bbrc.1997.6361. [DOI] [PubMed] [Google Scholar]
- Hibbitt O, et al. In vivo gene transfer by electroporation allows expression of a fluorescent transgene in hamster testis and epididymal sperm and has no adverse effects upon testicular integrity or sperm quality. Biol. Reprod. 2006;74:95–101. doi: 10.1095/biolreprod.105.042267. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Yagi T, Ozaki T, Imoto K. In vivo gene transfer to mouse spermatogenic cells using green fluorescent protein as a. J. Exp. Zool. 2000;286:212–218. doi: 10.1002/(sici)1097-010x(20000201)286:2<212::aid-jez13>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Dhup S, Majumdar SS. Transgenesis via permanent integration of genes in repopulating spermatogonial cells in vivo. Nat. Methods. 2008;5:601–603. doi: 10.1038/nmeth.1225. [DOI] [PubMed] [Google Scholar]
- Huang Z, et al. In vivo transfection of testicular germ cells and transgenesis by using the mitochondrially localized jellyfish fluorescent protein gene. FEBS Lett. 2000;487:248–251. doi: 10.1016/s0014-5793(00)02271-7. [DOI] [PubMed] [Google Scholar]
- Majumdar SS, et al. A method for rapid generation of transgenic animals to evaluate testis genes during sexual maturation. J. Reprod. Immunol. 2009;83:36–39. doi: 10.1016/j.jri.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Yomogida K, Yagura Y, Tadokoro Y, Nishimune Y. Dramatic expansion of germinal stem cells by ectopically expressed human glial cell line-derived neurotrophic factor in mouse Sertoli cells. Biol. Reprod. 2003;69:1303–1307. doi: 10.1095/biolreprod.103.015958. [DOI] [PubMed] [Google Scholar]
- Ike A, et al. Transient expression analysis of the mouse ornithine decarboxylase antizyme haploid-specific promoter using in vivo electroporation. FEBS Lett. 2004;559:159–164. doi: 10.1016/S0014-5793(04)00065-1. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gonzalez E, Lopez-Casas PP, Del MJ. Gene silencing by RNAi in mouse Sertoli cells. Reprod. Biol. Endocrinol. 2008;6:29. doi: 10.1186/1477-7827-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Kung A, Goldberg E. Regulation of murine lactate dehydrogenase C (Ldhc) gene expression. Biol. Reprod. 2008;78:455–461. doi: 10.1095/biolreprod.107.064964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yomgogida K. Mammalian testis: a target of in vivo electroporation. Dev. Growth Differ. 2008;50:513–515. doi: 10.1111/j.1440-169X.2008.01042.x. [DOI] [PubMed] [Google Scholar]
- Hayes KE, et al. An Evaluation of Analgesic Regimens for Abdominal Surgery in Mice. J Am Assoc Lab Anim Sci. 2000;6:18–23. [PubMed] [Google Scholar]