

Abstract
Importance
High placebo responses have been observed across a wide range of pathologies, severely impacting drug development.
Objective
Here we examined neurochemical mechanisms underlying the formation of placebo effects in patients with Major Depressive Disorder (MDD).
Participants
Thirty-five medication-free MDD patients.
Design and Intervention
We performed a single-blinded two-week cross-over randomized controlled trial of two identical oral placebos (described as having either “active” or “inactive” fast-acting antidepressant-like effects) followed by a 10-week open-label treatment with a selective serotonin reuptake inhibitor (SSRI) or in some cases, another agent as clinically indicated. The volunteers were studied with PET and the μ-opioid receptor (MOR)-selective radiotracer [11C]carfentanil after each 1-week “inactive” and “active” oral placebo treatment. In addition, 1 mL of isotonic saline was administered intravenously (i.v.) within sight of the volunteer during PET scanning every 4 min over 20 min only after the 1-week active placebo treatment, with instructions that the compound may be associated with the activation of brain systems involved in mood improvement. This challenge stimulus was utilized to test the individual capacity to acutely activate endogenous opioid neurotransmision under expectations of antidepressant effect.
Setting
A University Health System.
Main Outcomes and Measures
Changes in depressive symptoms in response to “active” placebo and antidepressant. Baseline and activation measures of MOR binding.
Results
Higher baseline MOR binding in the nucleus accumbens (NAc) was associated with better response to antidepressant treatment (r=0.48; p=0.02). Reductions in depressive symptoms after 1-week of “active” placebo treatment, compared to the “inactive”, were associated with increased placebo-induced μ-opioid neurotransmission in a network of regions implicated in emotion, stress regulation, and the pathophysiology of MDD, namely the subgenual anterior cingulate cortex, NAc, midline thalamus and amygdala (NAc: r=0.6, p<0.001). Placebo-induced endogenous opioid release in these regions was associated with better antidepressant treatment response, predicting 43% of the variance in symptom improvement at the end of the antidepressant trial.
Conclusions
These data demonstrate that placebo-induced activation of the μ-opioid system is implicated in the formation of placebo antidepressant effects in patients with MDD and also participate in antidepressant responses, conferring illness resiliency, during open administration.
Keywords: placebo effects, Major Depression, opioids, PET
Introduction
High rates of placebo responses are consistently reported across medical conditions, notably mood disorders, Parkinson Disease, and pain, but also schizophrenia, substance use disorders and surgeries1-5. Placebo response rates in antidepressant trials average 31-45% compared with ∼ 50% responses to antidepressants, and have increased over the last 30 years 4,5. The failure of antidepressant responses to separate from placebo has contributed to the reduction or discontinuation of research on new treatments for depression and other neuropsychiatric illnesses 6, hindering the development of novel neuropsychiatric treatments 7.
In conditions such as pain, where the neurobiological bases of placebo analgesic effects were first described 8, substantial headway has been made to identify their neural and molecular basis. Neural circuits involved in placebo analgesia 9-14 include the rostral anterior cingulate cortex (ACC), dorsolateral prefrontal cortex (DLPFC) and orbitofrontal cortex (OFC), insula (INS), nucleus accumbens (NAc), amygdala (AMY), midline thalamus (THA), and periaqueductal gray (PAG). Opioid and dopamine neurotransmission in these areas are known to modulate various elements of the analgesic placebo effect, including the representation of its subjective value, updates of expectations over time 15, the recall of pain and placebo experiences 16, and the changes in affective state and pain ratings 12,17,18. Furthermore, genetic variants have shown to modulate these neurotransmitter systems and placebo-associated symptom improvements 19-21.
In the only study examining the neural correlates of placebo effects in MDD 22, overlapping changes in metabolism were observed for placebo and SSRI arms of a RCT, albeit more extensively with the active agent. In addition, metabolic increases were noted in the ventral striatum and orbitofrontal regions at 1 week, regardless of treatment, regions implicated in reward expectation and monitoring, even in the absence of clinical effects 23. Here we investigate MOR-mediated neurotransmission as a potential candidate mechanism for the formation of placebo effects in MDD, given the MOR system's involvement in the regulation of emotion, stress and social rewards 24,25, and placebo analgesia 8,9,12,18,26. The study design incorporated a commonly used placebo lead-in phase with the administration of two identical placebos: one described as having fast-acting antidepressant effects (“active”) and one described as being a placebo with no antidepressant effects (“inactive”) (Fig.1). This was done to simulate common trial designs and to appropriately control for other statistical biases, such as the regression to the mean or response biases associated with study participation. In addition to evaluating the effects of sustained placebo pills, an intravenous placebo administration followed the 1-week active placebo, in order to investigate the effects of acute placebo administration on μ-opioid neurotransmission. Following each placebo intervention, patients underwent a 10-week open-label trial with a common SSRI treatment. We hypothesized that placebo-induced improvement in depressive symptoms would be associated with the capacity to activate endogenous MOR mediated neurotransmission in brain areas involved in stress and mood regulation 27-32 (i.e. subgenual –sg- ACC, NAc and AMY). In addition, we hypothesized that learning mechanisms involved during the administration of placebos will reinforce the response to common antidepressants, which might result in interactions between placebo and antidepressant effects.
Figure 1. Experimental Design.
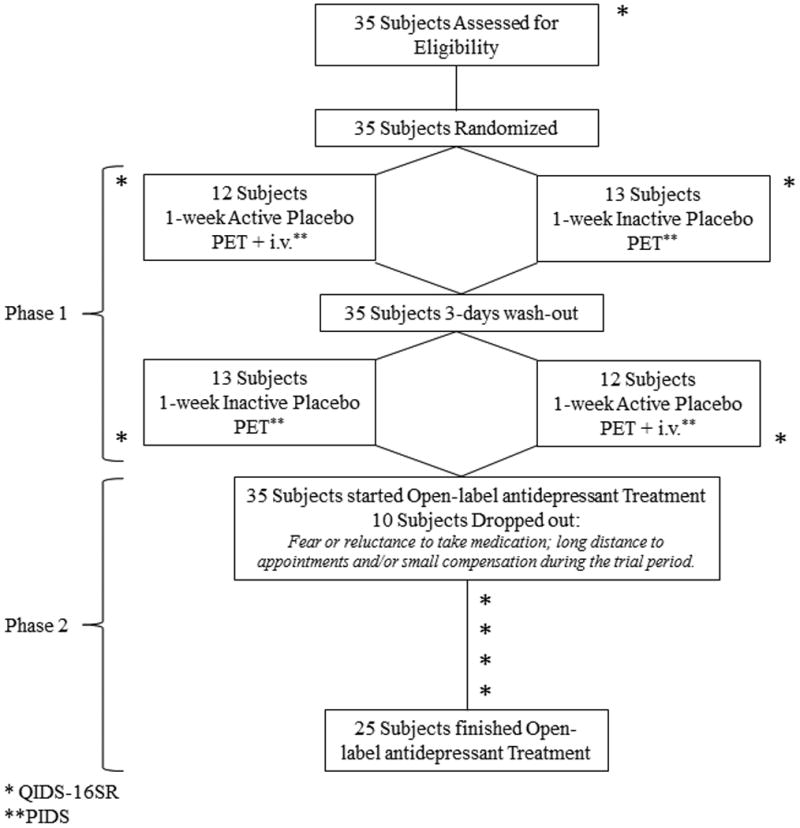
Abbreviations: PET: Positron Emission Tomography; i.v.: intravenous; QIDS-16SR: Quick Inventory of Depression Symptomatology; PIDS: Patient's Impression of Depression Severity.
Methods
1. Patients and Trial Design
Thirty five right-handed un-medicated participants with a DSM-V diagnosis of MDD (23 females; age range 19 to 59, mean ± S.D.: 35 ± 13), were recruited via advertisement (eMethods, Subjects). The study had two phases, a 2-week placebo single-blind RCT (starting 3-5 days after the screening interview) and a 10-week open-label flexible-dose antidepressant treatment (Fig. 1).
1.1. Placebo Phase
During the first phase, subjects were randomized to (1) 1-week “active” oral placebo treatment (2 pills/day), with expectations that it represented a fast-acting antidepressant agent, or (2) 1-week “inactive” oral placebo with disclosure that it was an inactive control. After a 3-day “washout” period without pills, participants were crossed over into the group to which they were not previously assigned. After each placebo week, participants underwent a PET scanning session (for data acquisition and statistical analysis see eMethods). As a challenge to induce endogenous opioid system activation and determine acute placebo effects, the PET session following the 1-week “active” oral placebo included the administration of an i.v. “active” placebo. This consisted 1mL of 0.9% isotonic saline introduced i.v. every 4 minutes during 20 minutes, starting at minute 42 and lasting for 15 seconds each time. Subjects were aware that the study drug was to be administered through a computer-generated human voice recording, followed by a second-by-second count of the infusion timing (15 seconds). No i.v. placebo followed the “inactive” placebo condition.
Depression symptoms were assessed using the Quick Inventory of Depressive Symptomatology (QIDS-SR16) 33 at pre- (baseline) and post- each placebo treatment. A single measure of sustained placebo response was created by subtracting the changes in QIDS-SR16 reductions from “active” and “inactive” placebo treatments [(QIDS-SR16 pre - post) “active” placebo – (QIDS-SR16 pre - post) “inactive” placebo]. Positive numbers then reflected reductions in depression symptoms as a result of oral placebo and this variable was used for correlational analysis and to dichotomize subjects into placebo responders (positive values, n = 14) and non-responders (0 or negative values, n = 21) (mean ± SD: 1.2 ± 5.4, range -10 to 16).
The i.v. placebo treatment was only administered during the scanning session that followed the active placebo. Patients' impression of severity (PIDS) ratings (“from 0 to 100 how depressed do you feel now?) were acquired every 4 minutes during the 2 PET scans, in the presence and absence of the i.v. placebo. Acute, i.v. placebo responses were assessed by the subtraction PIDSno i.v. –PIDSactive i.v.
1.2. Antidepressant Phase
Following the placebo phases and the two PET sessions, participants were invited to participate in an un-blind 10-week open-label trial with a commercially available SSRI, in most cases citalopram (starting at 20 mg/day and up to 40 mg/day in 77% of cases). An alternative agent was employed if clinically indicated (e.g. history of prior non-response to citalopram). Other treatments included sertraline (n=1), mirtazapine (n=1) fluoxetine (n=3) and bupropion (n=2). Participants were evaluated at weeks 0, 2, 4, 8 and 10 using the QIDS-16SR to evaluate symptom change.
Not-aggregated depression symptoms during the 10 week open-label treatment were assessed using linear mixed effects models 34,35 (Stata, version 1140). The longitudinal measurements of QIDS-16SR during the open trial were the repeated-measures outcomes. Baseline QIDS-16SR scores and week of the trial were included as covariates, which were also fit with random intercepts. Two continuous measures of placebo response were examined, sustained (oral) and acute (i.v.) placebo effects. By including a main effect and an interaction term with time for each predictor, the main effect is interpreted as the effect of the predictor on QIDS-16SR scores at the beginning of the open-label trial, and the interaction term is interpreted as the degree to which the trajectory of QIDS-16SR score over the 10-week trial varies by level of the predictor.
We also ran mixed effects models using a categorical variable that grouped participants as placebo responders or non-responders (as described above). A χ2 was used to evaluate the effect of placebo responsiveness group on remission rates. All statistical analyses were controlled by sex, order effects and QIDS-16SR pre-randomization scores.
Results
Placebo-Induced Changes in Depression Symptoms
Patient's characteristics are reported in eResults. As expected, no significant differences were observed between the 2 “pre” treatment QIDS scores (QIDS Baseline Inactive: 13.3 ± 4.9; QIDS Baseline Active: 14.2 ± 4.7; t=-1.2, p=0.24). The administration of 1-week of “active” placebo, compared to “inactive, was associated with significant reductions in depression symptoms [mean Δ ± SD: QIDS-SR16 pre - post “active” placebo: 1.75 ± 3.39; QIDS-SR16 pre – post “inactive” placebo: -0.15 ± 3.36, F=5, p=0.03)]. The i.v. acute placebo administration was also associated with a significant reduction in the average PIDS scores (PIDS active i.v. = 42 ± 26; PIDS no i.v. = 49 ± 22.4; F= 4.3, p= 0.04).
Oral placebo-induced improvement of depression symptoms (measured with ΔQIDS-SR16) were significantly correlated with the changes in PIDS scores after i.v. placebo administration (r= 0.35; p=0.04). Conversely, placebo-induced changes in QIDS-16SR and PIDS were not correlated with QIDS-16SR scores at baseline, patients' age, or initial expectations of recovery rated prior to the placebo treatments (for all, p>0.05). Females showed greater oral placebo-induced reductions in depression symptoms compared to males (mean ± SD: Females = 2.7 ± 5.3; Males =-1.6 ± 4.4; t=2.3; p=0.02), but not after the i.v. placebo administration. Finally, patients who received the “active” oral placebo first in order reported greater oral placebo-induced reductions in depressive symptoms that those who received the active oral placebo second (mean ± SD: First = 3.4 ± 5.7; Second = 0 ± 3.3; t=-2.1; p=0.04). Therefore, sex and order were introduced as a covariate in subsequent analyses, together with depression severity.
“Baseline” MOR Binding Potential (BPND) and Placebo-Induced Activation of MOR-Mediated Neurotransmission
We first evaluated the relationship between “baseline” MOR BPND (post-inactive placebo condition measures of MOR BPND), depression severity pre-randomization scores, acute and sustained placebo responses, and antidepressant responses, using BPND measures acquired 5-40 min post-tracer administration. While these post-inactive placebo condition measures of MOR BPND may not represent a true baseline, no significant differences in MOR BPND were observed between the active and the inactive condition during the early scan measures (5 to 40 minutes), when no i.v. placebo was administered, which confirms the stability of the BPND values in the absence of an acute challenge. A whole-brain voxel-by-voxel analysis showed a significant positive relationship between QIDS-16SR pre-randomization scores and “baseline” MOR BPND in the NAc (Fig. 2, Table 1, A). NAc MOR BPND was also significantly correlated with improvement in QIDS-16SR after 10 weeks of antidepressants (n=25; NAc: r=0.48; p=0.02). Instead, baseline μ-opioid receptor BPND was not associated with depression symptom improvements in response to 1-week oral or i.v. placebo administration, and therefore imaging analyses examining the effect of placebo on neurotransmitter release were not controlled for baseline BPND.
Figure 2. Voxel-by-voxel MOR availability at baseline is positively correlated with depression severity (pre-randomization QIDS-16SR, left) and with antidepressant treatment response (right).

Table 1. μ-opioid BPND at baseline and placebo-induced reductions in μ-opioid BPND: Correlations with depression improvement after placebo and antidepressant treatment.
| Region | Ha | x,y,z {mm}b | Cluster sizec | Zd | Effects of regional placebo-induced opioid release on antidepressant response | |||
|---|---|---|---|---|---|---|---|---|
| Estimate | SE | 95% C.I. | ||||||
| A. Baseline μ-opioid BPND correlation with QIDS-16SR at screening. | ||||||||
| NAc | L | -8 8 -8 | 200 | 3.76 | -0.71 | 0.18 | -1.05 | -0.36 |
| B. Placebo-induced decrease μ-opioid BPND. | ||||||||
| NAc | L | -8 2 -4 | 552 | 4.72 | -0.43 | 0.22 | -0.85 | -0.004 |
| C. Placebo-induced decrease μ-opioid BPND correlation with improvement of depression symptoms measure with the QIDS-16SR. | ||||||||
| THA | L | -4 -8 -4 | 4888 | 6.4 | -1.02 | 0.23 | -1.47 | -0.56 |
| NAc/sgACC | L | -14 10 -8 | 1064 | 4.49 | -0.8 | 0.2 | -1.18 | -0.41 |
| R | 8 8 -12 | 1136 | 4.43 | -0.68 | 0.23 | -1.14 | -0.22 | |
| AMY* | L | -36 -4 -18 | 352 | 3.84 | -0.38 | 0.28 | -0.93 | 0.17 |
| D. Placebo-induced decrease μ-opioid BPND correlation with improvement of depression symptoms measure with the PIDS. | ||||||||
| sgACC | L | -4 10 -6 | 256 | 4.19 | -0.61 | 0.2 | -1.01 | -0.22 |
| AMY | L | -22 -2 -24 | 272 | 4.19 | -0.39 | 0.2 | -0.79 | 0.01 |
| E. Placebo-induced decrease μ-opioid BPND correlation with improvement of depression symptoms after 10 weeks of antidepressant treatment. | ||||||||
| THA | R | 0 -8 -2 | 3584 | 5.41 | -0.98 | 0.23 | -1.44 | -0.52 |
| NAc/sgACC | R | 6 8 -8 | 1136 | 4.49 | -1.12 | 0.23 | -1.58 | -0.66 |
| AMY | L | -26 -10 -16 | 392 | 3.99 | -0.6 | 0.2 | -1 | -0.2 |
Footnote:
H: hemisphere.
Montreal Neurological Institute (MNI) coordinates of peak voxel.
Cluster size in mm3.
Two-sided voxel-level Z score at peak voxel and a p<0.001 uncorr. for AMY, sgACC and NAC and FWE-corr. for other regions. Abreviations: BPND: binding potential non-displaceble; NAc: nucleus accumbens; THA: thalamus; sgACC: subgenual anterior cingulate cortex; AMY: amygdala; PIDS: patients' impression of depression severity.
Second, we examined the main effect of i.v. placebo administration on μ-opioid system activation (reductions in BPND when compared to no i.v. placebo). Significant activation of μ-opioid neurotransmission after i.v. placebo was localized in the NAc (Table 1, B).
Third, we investigated the relationship between changes in MOR BPND in response to i.v. placebo and the sustained and acute placebo responses. Improvement in QIDS-16SR after the “active” oral placebo, compared to the “inactive”, was positively associated with placebo-induced opioid release in multiple brain areas, including the sgACC, NAc, AMY, and the midline THA, the latter peak extending to the hypothalamus, Fig. 3, Table 1, C). Reductions in PIDS during scanning after i.v. placebo administration were also associated with greater placebo MOR system activation in the sgACC and AMY (Table 1, D).
Figure 3. Voxel-by-voxel placebo-induced activation of MOR mediated neurotransmission is associated with placebo-induced improvement in depression symptoms.
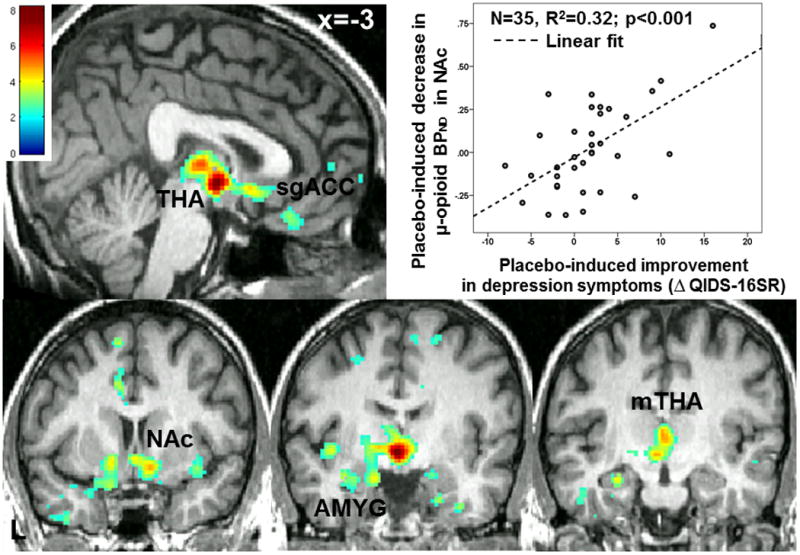
(displayed at p<0.01). mTHA: medial Thalamus; sgACC: subgenual Anterior Cingulate Cortex; NAc: Nucleus Accumbens; AMY: Amygdala.
Last, we examined the relationship between i.v. placebo activation of endogenous opioid neurotransmission and depression improvement after 10 weeks of antidepressant. Reductions in QIDS-16SR scores (n=25) after open-label trial were significantly associated with placebo-induced MOR system activation in the same network of regions associated with placebo antidepressant effects: sgACC, NAc, AMY and mid THA (Fig.4, Table 1, E).
Figure 4. Voxel-by-voxel placebo-induced activation of MOR mediated neurotransmission is associated with open-label antidepressant treatment response.
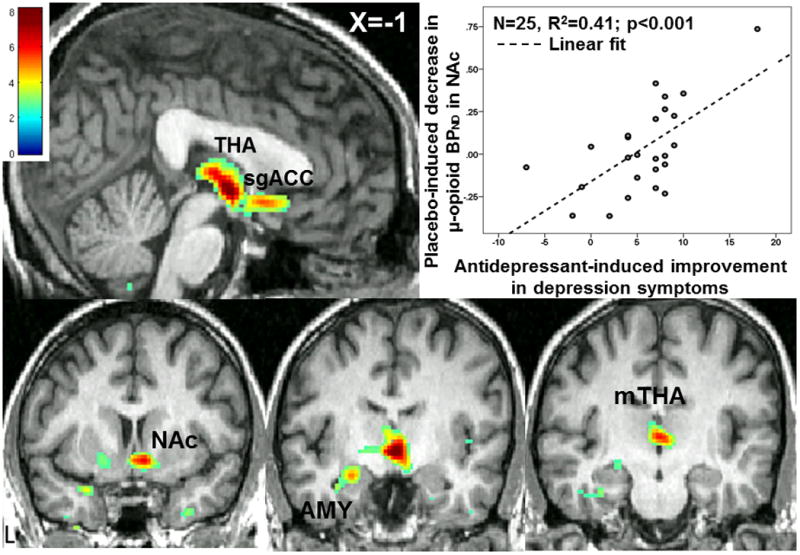
(Displayed at p<0.01, abbreviated as in Fig.3).
Placebo-induced Δ in QIDS-16SR, Δ PIDS, and Δ in μ-opioid BPND as Predictors of Antidepressant Treatment Response
The mixed model analyses revealed that sustained oral placebo responses were associated with significant reductions in QIDS-16SR scores during antidepressant treatment over time, but not acute i.v. placebo responses (eTable 1).
The categorical analysis showed that the sustained oral placebo responder group showed larger reductions in QIDS-16SR scores during antidepressant treatment compared to non-responders, but this effect was only present after 4 weeks of its administration (eFig. 2). By weeks 8 and 10, the mean QIDS-16SR score was roughly twice as high among placebo non-responders compared to placebo responders. Achievement of remission (QIDS-16SR ≤ 5) was also significantly higher in placebo responders, with 60% of those categorized as placebo responders and only 20% of non-responders being considered in remission (χ2=3.9; p = 0.048).
Furthermore, the capacity to activate the MOR system during placebo administration was associated with greater reductions in QIDS-16SR over the 10-week trial (Table 1, Fig. 4). A simple regression model that included objectively measured placebo-induced opioid release in the sgACC, NAc, THA and AMY as regressors, accounted for 43% of the variance in the response to open-label antidepressant (adjusted r2= 0.43). Similarly, subjective clinical placebo responsiveness itself predicted 46% of the variance in the response to 10-weeks of antidepressant treatment (adjusted r2= 0.46), while the combination of both the clinical and the opioid release measures predicted 57% of the variance in the response to 10-weeks of antidepressant treatment (adjusted r2= 0.57).
Discussion
The present study is the first direct demonstration of the role of a specific neurotransmitter system, namely MOR-mediated neurotransmission, in the formation of placebo effects in MDD, and explaining variability in antidepressant treatment responses.
Substantial evidence supports the possible implication of the endogenous opioid system in the modulation and regulation of emotional states as well as in the pathophysiology of various psychiatric illnesses, including MDD 36,37. Here we described that in patients with MDD, higher baseline MOR BPND in the NAc is associated with both, higher depression symptomatology and antidepressant, but not placebo, responsiveness. Alterations in MOR BPND and function have been previously described in MDD, and linked to both dysfunctions in the neuroendocrine hypothalamic-pituitary adrenal axis and treatment non-responsiveness 38.
The activation of the MOR system has also been implicated in the formation of placebo effects in pain 9,12,17,18,39,40, suggesting that similar neurobiological mechanisms can contribute to the formation of clinical placebo effects across pathologies. By comparison, one single previous neuroimaging study aimed to define the neuroanatomy of placebo responses in MDD 22 using metabolic PET imaging in a group of depressed men during a RCT with an SSRI. This study showed overlapping metabolic changes with both SSRI and placebo at 6 weeks and early (1 week) increases in activity in the NAc and orbitofrontal cortex regardless of treatment. Here we observed a similar pattern of activation in the NAc, but also the sgACC, midline THA and AMY, in response to 1-week of placebo, but within a specific neurochemical system, the endogenous opioid and MORs. While not a priory hypothesized, the midline THA has strong and specific connections with the AMY, NAc and sgACC as shown in rodent and nonhuman primate studies 41. These connections represent pathways by which the midline THA, known to be strongly activated by a wide variety of stressors, may influence structures that regulate motivation and mood 41.
Importantly, placebo-induced MOR system activation in stress and emotion regulatory regions (sgACC, THA, NAc, AMY) 42-46 predicted 43% of the variance in the response to antidepressant treatment after 10 weeks. Similarly, subjective clinical placebo responsiveness itself predicted 46% of the response to antidepressant treatment, while the combination of both predicted 57% of the total antidepressant response. Still, by weeks 8 and 10, depression severity scores were roughly twice as high among placebo non-responders compared to placebo responders. This observation may indicate that the endogenous opioid system, through MORs, reinforces treatment responses over time, a form of positive reward learning, as has been suggested by data acquired in the field of pain and Parkinson Disease 15,47,48 and in animal models of reward learning 49. Alternatively, it could be possible that this effect is explained by the patient expectations of delayed response to common antidepressant treatments, disclosed prior to the initiation of the active treatment. Furthermore, achievement of remission was also significantly higher among placebo responders compared to non-responders, an observation that potentially challenges a common tenant that eliminating placebo responders in clinical trials with placebo-lead-in phases or novel sequential parallel comparison designs 50 would help to more clearly interpret RCT results.
Several hypotheses could explain these findings. First, it is possible that mechanisms involved in placebo responding are also engaged during antidepressant treatment. In this regard, a number of studies have shown that the analgesic effects of tricyclic antidepressants (TCAs) are reversed by opioid receptor antagonists 51-55 and that TCAs potentiate morphine-induced analgesia both in animals 56 and in humans 57. From another perspective, compounds such as buprenorphine, a partial μ-opioid agonist, exhibit antidepressant properties in treatment refractory depressed patients 58, leading to recent studies examining the modulation of opioid mechanisms in MDD 59.If aminergic and opioid interactions synergistically improve depressive symptomatology, greater placebo-like (e.g., opioid-mediated) responses would be expected within active treatment arms, compared to the placebo arm, compromising the interpretation of RCTs. A different possibility would be that the overall reduction of depressive symptoms in an open label treatment could be explained by a combination of specific and non-specific effects, which, in addition to placebo neurobiological effects, may include variations in the natural history of illness, regression to the mean, reporting biases, or lack of adherence to the treatment (which was not assessed in this study beyond patient's report). However, these non-specific effects are not likely to be linked to placebo-activated neurotransmission, or be represented differentially in placebo responders or non-responders.
Our results show that placebo administration impacts homeostatic, resiliency mechanisms that can facilitate recovery from illness, and could be seen as a probe for the development of new therapeutic targets that regulate those biological processes. In clinical trials, this evidence could help inform decisions regarding patient stratification and drug-“specific” or “non-specific” effects. In clinical practice, placebo-responsiveness could potentially indicate the likelihood of responsiveness to enhanced patient-clinician interactions or psychosocial or cognitive approaches.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by the NIH R01 MH086858, (JKZ), the Phil F. Jenkins Foundation, the Michigan Institute for Clinical & Health Research grant support (CTSA: UL1RR024986).
We would also like to acknowledge the contribution of the technologists of the PET Center and the Department of Radiology at the University of Michigan.
Footnotes
Role of the Funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflict of Interest Disclosures: The authors have no interests to disclose that are or might be perceived to be in conflict with the work reported in this study.
Access to Data and Data Analysis: Drs. Peciña and Zubieta had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Agid O, Siu CO, Potkin SG, et al. Meta-regression analysis of placebo response in antipsychotic trials, 1970-2010. The American journal of psychiatry. 2013 Nov;170(11):1335–1344. doi: 10.1176/appi.ajp.2013.12030315. [DOI] [PubMed] [Google Scholar]
- 2.Meissner K, Fassler M, Rucker G, et al. Differential effectiveness of placebo treatments: a systematic review of migraine prophylaxis. JAMA internal medicine. 2013 Nov 25;173(21):1941–1951. doi: 10.1001/jamainternmed.2013.10391. [DOI] [PubMed] [Google Scholar]
- 3.Mestre TA, Shah P, Marras C, Tomlinson G, Lang AE. Another face of placebo: the lessebo effect in Parkinson disease: meta-analyses. Neurology. 2014 Apr 22;82(16):1402–1409. doi: 10.1212/WNL.0000000000000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stolk P, Ten Berg MJ, Hemels ME, Einarson TR. Meta-analysis of placebo rates in major depressive disorder trials. The Annals of pharmacotherapy. 2003 Dec;37(12):1891–1899. doi: 10.1345/aph.1D172. [DOI] [PubMed] [Google Scholar]
- 5.Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression: variable, substantial, and growing. JAMA : the journal of the American Medical Association. 2002 Apr 10;287(14):1840–1847. doi: 10.1001/jama.287.14.1840. [DOI] [PubMed] [Google Scholar]
- 6.Cressey D. Psychopharmacology in crisis. Nature. 2011 http://www.nature.com/news/2011/110614/full/news2011.367.html. (June 14)
- 7.Nutt D, Goodwin G. ECNP Summit on the future of CNS drug research in Europe 2011: report prepared for ECNP by David Nutt and Guy Goodwin. European Neuropsychopharmacology. 2011;21:495–499. doi: 10.1016/j.euroneuro.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Levine JD, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet. 1978 Sep 23;2(8091):654–657. doi: 10.1016/s0140-6736(78)92762-9. [DOI] [PubMed] [Google Scholar]
- 9.Petrovic P, Kalso E, Petersson KM, Ingvar M. Placebo and opioid analgesia-- imaging a shared neuronal network. Science. 2002;295(5560):1737–1740. doi: 10.1126/science.1067176. [DOI] [PubMed] [Google Scholar]
- 10.Scott DJ, Stohler CS, Egnatuk CM, Wang H, Koeppe RA, Zubieta JK. Individual differences in reward responding explain placebo-induced expectations and effects. Neuron. 2007 Jul 19;55(2):325–336. doi: 10.1016/j.neuron.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Wager TD, Rilling JK, Smith EE, et al. Placebo-induced changes in FMRI in the anticipation and experience of pain. Science. 2004 Feb 20;303(5661):1162–1167. doi: 10.1126/science.1093065. [DOI] [PubMed] [Google Scholar]
- 12.Zubieta JK, Bueller JA, Jackson LR, et al. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J Neurosci. 2005 Aug 24;25(34):7754–7762. doi: 10.1523/JNEUROSCI.0439-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellingsen DM, Wessberg J, Eikemo M, et al. Placebo improves pleasure and pain through opposite modulation of sensory processing. Proc Natl Acad Sci U S A. 2013 Oct 29;110(44):17993–17998. doi: 10.1073/pnas.1305050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wanigasekera V, Lee MC, Rogers R, et al. Baseline reward circuitry activity and trait reward responsiveness predict expression of opioid analgesia in healthy subjects. Proc Natl Acad Sci U S A. 2012 Oct 23;109(43):17705–17710. doi: 10.1073/pnas.1120201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pecina M, Stohler CS, Zubieta JK. Neurobiology of placebo effects: expectations or learning? Social cognitive and affective neuroscience. 2013 Jul 25; doi: 10.1093/scan/nst079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pecina M, Stohler CS, Zubieta JK. Role of mu-opioid system in the formation of memory of placebo responses. Molecular psychiatry. 2013 Feb;18(2):135–137. doi: 10.1038/mp.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott DJ, Stohler CS, Egnatuk CM, Wang H, Koeppe RA, Zubieta JK. Placebo and nocebo effects are defined by opposite opioid and dopaminergic responses. Archives of general psychiatry. 2008 Feb;65(2):220–231. doi: 10.1001/archgenpsychiatry.2007.34. [DOI] [PubMed] [Google Scholar]
- 18.Wager TD, Scott DJ, Zubieta JK. Placebo effects on human mu-opioid activity during pain. Proc Natl Acad Sci U S A. 2007 Jun 26;104(26):11056–11061. doi: 10.1073/pnas.0702413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall KT, Lembo AJ, Kirsch I, et al. Catechol-O-methyltransferase val158met polymorphism predicts placebo effect in irritable bowel syndrome. PloS one. 2012;7(10):e48135. doi: 10.1371/journal.pone.0048135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pecina M, Martinez-Jauand M, Hodgkinson C, Stohler CS, Goldman D, Zubieta JK. FAAH selectively influences placebo effects. Molecular psychiatry. 2014 Mar;19(3):385–391. doi: 10.1038/mp.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pecina M, Martinez-Jauand M, Love T, et al. Valence-specific effects of BDNF Val66Met polymorphism on dopaminergic stress and reward processing in humans. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014 Apr 23;34(17):5874–5881. doi: 10.1523/JNEUROSCI.2152-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayberg HS, Silva JA, Brannan SK, et al. The functional neuroanatomy of the placebo effect. The American journal of psychiatry. 2002 May;159(5):728–737. doi: 10.1176/appi.ajp.159.5.728. [DOI] [PubMed] [Google Scholar]
- 23.Benedetti F, Mayberg HS, Wager TD, Stohler CS, Zubieta JK. Neurobiological mechanisms of the placebo effect. J Neurosci. 2005 Nov 9;25(45):10390–10402. doi: 10.1523/JNEUROSCI.3458-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kreek MJ, Koob GF. Drug dependence: stress and dysregulation of brain reward pathways. Drug and alcohol dependence. 1998 Jun-Jul;51(1-2):23–47. doi: 10.1016/s0376-8716(98)00064-7. [DOI] [PubMed] [Google Scholar]
- 25.Vaccarino AL, Kastin AJ. Endogenous opiates: 1999. Peptides. 2000 Dec;21(12):1975–2034. doi: 10.1016/s0196-9781(00)00345-4. [DOI] [PubMed] [Google Scholar]
- 26.Benedetti F. The opposite effects of the opiate antagonist naloxone and the cholecystokinin antagonist proglumide on placebo analgesia. Pain. 1996 Mar;64(3):535–543. doi: 10.1016/0304-3959(95)00179-4. [DOI] [PubMed] [Google Scholar]
- 27.Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. The Journal of neuropsychiatry and clinical neurosciences. 1997 Summer;9(3):471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- 28.Phan KL, Wager T, Taylor SF, Liberzon I. Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. NeuroImage. 2002 Jun;16(2):331–348. doi: 10.1006/nimg.2002.1087. [DOI] [PubMed] [Google Scholar]
- 29.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biological psychiatry. 2003 Sep 1;54(5):515–528. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 30.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: The neural basis of normal emotion perception. Biological psychiatry. 2003 Sep 1;54(5):504–514. doi: 10.1016/s0006-3223(03)00168-9. [DOI] [PubMed] [Google Scholar]
- 31.Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2010 Jan;35(1):192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price JL, Drevets WC. Neural circuits underlying the pathophysiology of mood disorders. Trends in cognitive sciences. 2012 Jan;16(1):61–71. doi: 10.1016/j.tics.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biological psychiatry. 2003 Sep 1;54(5):573–583. doi: 10.1016/s0006-3223(02)01866-8. [DOI] [PubMed] [Google Scholar]
- 34.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982 Dec;38(4):963–974. [PubMed] [Google Scholar]
- 35.Peduzzi P, Henderson W, Hartigan P, Lavori P. Analysis of randomized controlled trials. Epidemiologic reviews. 2002;24(1):26–38. doi: 10.1093/epirev/24.1.26. [DOI] [PubMed] [Google Scholar]
- 36.Agren H, Terenius L, Wahlstrom A. Depressive phenomenology and levels of cerebrospinal fluid endorphins. Annals of the New York Academy of Sciences. 1982;398:388–398. doi: 10.1111/j.1749-6632.1982.tb39510.x. [DOI] [PubMed] [Google Scholar]
- 37.Tejedor-Real P, Mico JA, Maldonado R, Roques BP, Gibert-Rahola J. Implication of endogenous opioid system in the learned helplessness model of depression. Pharmacology, biochemistry, and behavior. 1995 Sep;52(1):145–152. doi: 10.1016/0091-3057(95)00067-7. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy SE, Koeppe RA, Young EA, Zubieta JK. Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Archives of general psychiatry. 2006 Nov;63(11):1199–1208. doi: 10.1001/archpsyc.63.11.1199. [DOI] [PubMed] [Google Scholar]
- 39.Amanzio M, Benedetti F. Neuropharmacological dissection of placebo analgesia: expectation-activated opioid systems versus conditioning-activated specific subsystems. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999 Jan 1;19(1):484–494. doi: 10.1523/JNEUROSCI.19-01-00484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zubieta JK, Yau WY, Scott DJ, Stohler CS. Belief or Need? Accounting for individual variations in the neurochemistry of the placebo effect. Brain Behav Immun. 2006 Jan;20(1):15–26. doi: 10.1016/j.bbi.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 41.Hsu DT, Kirouac GJ, Zubieta JK, Bhatnagar S. Contributions of the paraventricular thalamic nucleus in the regulation of stress, motivation, and mood. Frontiers in behavioral neuroscience. 2014;8:73. doi: 10.3389/fnbeh.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drevets WC, Ongur D, Price JL. Reduced glucose metabolism in the subgenual prefrontal cortex in unipolar depression. Molecular psychiatry. 1998 May;3(3):190–191. doi: 10.1038/sj.mp.4000380. [DOI] [PubMed] [Google Scholar]
- 43.Gotlib IH, Sivers H, Gabrieli JD, et al. Subgenual anterior cingulate activation to valenced emotional stimuli in major depression. Neuroreport. 2005 Nov 7;16(16):1731–1734. doi: 10.1097/01.wnr.0000183901.70030.82. [DOI] [PubMed] [Google Scholar]
- 44.Hamani C, Mayberg H, Snyder B, Giacobbe P, Kennedy S, Lozano AM. Deep brain stimulation of the subcallosal cingulate gyrus for depression: anatomical location of active contacts in clinical responders and a suggested guideline for targeting. Journal of neurosurgery. 2009 Dec;111(6):1209–1215. doi: 10.3171/2008.10.JNS08763. [DOI] [PubMed] [Google Scholar]
- 45.Hamani C, Mayberg H, Stone S, Laxton A, Haber S, Lozano AM. The subcallosal cingulate gyrus in the context of major depression. Biological psychiatry. 2011 Feb 15;69(4):301–308. doi: 10.1016/j.biopsych.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 46.Mayberg HS. Positron emission tomography imaging in depression: a neural systems perspective. Neuroimaging clinics of North America. 2003 Nov;13(4):805–815. doi: 10.1016/s1052-5149(03)00104-7. [DOI] [PubMed] [Google Scholar]
- 47.Buchel C, Geuter S, Sprenger C, Eippert F. Placebo analgesia: a predictive coding perspective. Neuron. 2014 Mar 19;81(6):1223–1239. doi: 10.1016/j.neuron.2014.02.042. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt L, Braun EK, Wager TD, Shohamy D. Mind matters: placebo enhances reward learning in Parkinson's disease. Nature neuroscience. 2014 Dec;17(12):1793–1797. doi: 10.1038/nn.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Ree JM, Gerrits MA, Vanderschuren LJ. Opioids, reward and addiction: An encounter of biology, psychology, and medicine. Pharmacological reviews. 1999 Jun;51(2):341–396. [PubMed] [Google Scholar]
- 50.Fava M, Evins AE, Dorer DJ, Schoenfeld DA. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychotherapy and psychosomatics. 2003 May-Jun;72(3):115–127. doi: 10.1159/000069738. [DOI] [PubMed] [Google Scholar]
- 51.Benbouzid M, Choucair-Jaafar N, Yalcin I, et al. Chronic, but not acute, tricyclic antidepressant treatment alleviates neuropathic allodynia after sciatic nerve cuffing in mice. European journal of pain. 2008 Nov;12(8):1008–1017. doi: 10.1016/j.ejpain.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 52.Benbouzid M, Gaveriaux-Ruff C, Yalcin I, et al. Delta-opioid receptors are critical for tricyclic antidepressant treatment of neuropathic allodynia. Biological psychiatry. 2008 Mar 15;63(6):633–636. doi: 10.1016/j.biopsych.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 53.Biegon A, Samuel D. Interaction of tricyclic antidepressants with opiate receptors. Biochemical pharmacology. 1980 Feb;29(3):460–462. doi: 10.1016/0006-2952(80)90531-6. [DOI] [PubMed] [Google Scholar]
- 54.Gray AM, Spencer PS, Sewell RD. The involvement of the opioidergic system in the antinociceptive mechanism of action of antidepressant compounds. British journal of pharmacology. 1998 Jun;124(4):669–674. doi: 10.1038/sj.bjp.0701882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marchand F, Alloui A, Chapuy E, et al. Evidence for a monoamine mediated, opioid-independent, antihyperalgesic effect of venlafaxine, a non-tricyclic antidepressant, in a neurogenic pain model in rats. Pain. 2003 Jun;103(3):229–235. doi: 10.1016/S0304-3959(03)00168-4. [DOI] [PubMed] [Google Scholar]
- 56.Hamon M, Gozlan H, Bourgoin S, et al. Opioid receptors and neuropeptides in the CNS in rats treated chronically with amoxapine or amitriptyline. Neuropharmacology. 1987 Jun;26(6):531–539. doi: 10.1016/0028-3908(87)90144-4. [DOI] [PubMed] [Google Scholar]
- 57.Mico JA, Ardid D, Berrocoso E, Eschalier A. Antidepressants and pain. Trends in pharmacological sciences. 2006 Jul;27(7):348–354. doi: 10.1016/j.tips.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Emrich HM, Vogt P, Herz A. Possible antidepressive effects of opioids: action of buprenorphine. Annals of the New York Academy of Sciences. 1982;398:108–112. doi: 10.1111/j.1749-6632.1982.tb39483.x. [DOI] [PubMed] [Google Scholar]
- 59.Ehrich E, Turncliff R, Du Y, et al. Evaluation of Opioid Modulation in Major Depressive Disorder. Neuropsychopharmacology. 2014 Dec 18; doi: 10.1038/npp.2014.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.