

SUMMARY
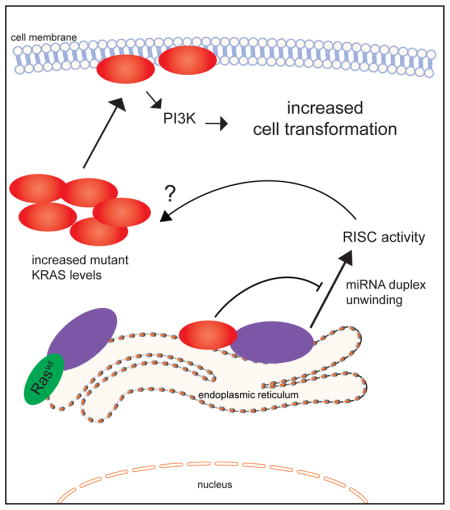
Oncogenic mutations in RAS provide a compelling yet intractable therapeutic target. Using co-immunoprecipitation mass spectrometry, we uncovered an interaction between RAS and Argonaute 2 (AGO2). Endogenously, RAS and AGO2 co-sediment and co-localize in the endoplasmic reticulum. The AGO2 N-terminal domain directly binds the Switch II region of KRAS, agnostic of nucleotide (GDP/GTP) binding. Functionally, AGO2 knockdown attenuates cell proliferation in mutant KRAS-dependent cells, and AGO2 overexpression enhances KRASG12V-mediated transformation. Using AGO2−/− cells, we demonstrate that the RAS-AGO2 interaction is required for maximal mutant KRAS expression and cellular transformation. Mechanistically, oncogenic KRAS attenuates AGO2 mediated gene silencing. Overall, the functional interaction with AGO2 extends KRAS function beyond its canonical role in signaling.
Keywords: cancer, KRAS, EIF2C2, Argonaute 2, RNA silencing
Graphical Abstract
INTRODUCTION
Approximately one third of human cancers harbor an oncogenic mutation in HRAS, KRAS, or NRAS (Balmain and Pragnell, 1983; Karnoub and Weinberg, 2008; Pylayeva-Gupta et al., 2011). The tumor types most frequently harboring RAS mutations, predominantly in KRAS, include pancreatic, lung, and colon carcinoma, among others (COSMIC, 2013; Hand et al., 1984; Karachaliou et al., 2013; Lauchle et al., 2006; Lohr et al., 2005). RAS genes encode a family of small GTPases (Sweet et al., 1984) that transduce extracellular growth signals by cycling between an active GTP-bound state and an inactive GDP-bound state (Karnoub and Weinberg, 2008; Schubbert et al., 2007). Oncogenic Ras proteins exhibit reduced intrinsic GTPase activity and are resistant to negative regulation by GTPase activating proteins (GAPs) such as p120GAP and neurofibromin (Cichowski and Jacks, 2001). Constitutively elevated levels of Ras-GTP aberrantly activate downstream effector pathways that promote neoplastic transformation (Karnoub and Weinberg, 2008; Shaw and Cantley, 2006; Trahey and McCormick, 1987). Despite extensive characterization of the Ras/GAP molecular switch(es) and downstream signaling axes, therapeutic targeting of RAS driven cancers remains elusive (Baines et al., 2011; Downward, 2003; Stephen et al., 2014).
The oncogenic activity of RAS-GTP is mediated through canonical effectors including RAF, PI3 kinase (PI3K) and Ral-GDS (Cox and Der, 2010; Karnoub and Weinberg, 2008); and other effectors have been described in various contexts (Gysin et al., 2011). RAS effectors bind through the conserved Switch I and Switch II domains, and drive cellular transformation by activating downstream kinases and GTPase signaling modules, the best known of which are the RAF/MEK/ERK (Mitogen Activation Protein (MAP) kinase) and the PI3K/Akt signaling cascades. RAS interactors have been identified using conventional approaches of ectopically expressed epitope-tagged RAS constructs (Goldfinger et al., 2007; Vasilescu et al., 2004). Here, we employed co-immunoprecipitation followed by mass spectrometry (co-IP MS) to analyze the endogenous interactome of RAS in a panel of lung and pancreatic cancer cell lines representing the spectrum of both KRAS mutation and dependency status. Surprisingly, the most prominent interacting protein, across all cell lines analyzed, was EIF2C2, commonly known as Argonaute 2 (AGO2), a key effector of the RNA silencing pathway. Interestingly, a role for AGO2 in RAS induced senescence has been described recently (Benhamed et al., 2012; Yang et al., 2014). Also, phosphorylation of AGO2 by MAPK/PI3K pathway activators has been shown to alter its microRNA related function through different mechanisms (Horman et al., 2013; Rudel et al., 2011; Shen et al., 2013; Zeng et al., 2008), portending a broader, direct interface between intracellular signaling and RNA silencing mechanisms (Paroo et al., 2009). Considering the potential functional implications of RAS-AGO2 interaction, here we corroborated and characterized this interaction in detail.
RESULTS
Endogenous RAS and AGO2 Interaction
To analyze RAS-interacting proteins in an endogenous setting, we first used the pan-RAS antibody RAS10 (Cheng et al., 2011), which efficiently immunoprecipitates RAS proteins by binding to the Switch I domain (amino acids, aa, 32–40) (Figure S1A–C). Co-immunoprecipitation of RAS followed by tandem mass spectrometry (RAS co-IP MS) was performed as outlined in Figure S1D, using a panel of ten lung and pancreatic cancer cell lines of known KRAS mutation status (Table S1), as well as NIH3T3 cells ectopically overexpressing human KRAS wild-type (KRASWT) or mutant (KRASG12V) proteins. Peptide fragments deduced from MS analyses spectral counts revealed robust detection of the bait protein (RAS) in all the 12 cell lines, as expected (Table S2). To minimize individual cell specific observations in the endogenous system employed, we focused on observations common across different cell lines. Intriguingly, peptides spanning EIF2C2 protein, commonly known as Argonaute 2 (AGO2), the catalytic component of the RNA-induced silencing complex (RISC), were observed in the RAS co-IP MS of all of cancer cell lines (n=10) tested, as well as in NIH3T3 cells expressing KRASWT or KRASG12V (Figure 1A). Remarkably, only the RAS and AGO2 peptides were detected in every cell line tested, with cumulative spectral counts of 576 and 229 respectively. Other interactors detected in 5 or more of the 12 cell lines are tabulated in Table S2. The notable absence of known RAS effectors like RAF/PI3K in the mass spectrometric analysis is due to the RAS10 antibody binding the Switch I domain preventing effector binding (Figure S1C). The lack of other RAS regulators like SOS1 and NF1 that associate with RAS through the Switch II domain may be due to their transient association and plasma membrane localized/cell specific expression. Interestingly, we did not detect peptides spanning AGO2 in our earlier mass spectrometric based studies involving ERG, PRC complex protein EED (Brenner et al., 2011; Cao et al., 2014), and at least 4 other protein pull down datasets (data not shown), indicating the specificity of AGO2 co-IP with RAS. Analyzing the RAS co-IP MS data further, we noted peptides mapping uniquely to all three RAS family members namely KRAS, NRAS and HRAS were readily detected across the cell line panel (Figure 1B). In contrast, almost all uniquely mapping peptides to AGO family proteins were specific to AGO2 in all of the 12 cell lines (except for a single unique peptide that mapped to AGO1 in one sample) (Figure 1B).
Figure 1. Identification of the RAS-AGO2 interaction.

Spectral counts of RAS and AGO2 peptides detected in RAS co-immunoprecipitation mass spectrometric (co-IP MS) analysis of NIH3T3 cells expressing KRASWT and KRASG12V (A) and indicated cancer cell lines (B). (C) Distribution of peptides mapping to RAS and AGO gene families from RAS co-IP MS based on ClustalW alignments. Representative experiment from H358 cells is shown. Blue boxes indicate peptides mapping to multiple gene family members, and red boxes indicate peptides mapping uniquely to a protein. (D) Immunoprecipitation (IP) of RAS or AGO2 in H358 (left) and H460 (right) lung cancer cells followed by immunoblot analysis using multiple distinct antibodies, as indicated. (E) IP of RAS from a panel of benign and cancer cells with differing mutational status of KRAS (as indicated) followed by immunoblot analysis of AGO2 or RAS. RAS10 mAb was used for both IP and IB. See also Figure S1.
The putative endogenous interaction between RAS and AGO2 was corroborated by reciprocal IPs using two different antibodies for each, in two different lung cancer cell lines, H358 and H460, harboring distinct KRAS mutations (Figure 1C). Further, consistent with the co-IP MS analyses (Figure 1A), the RAS-AGO2 interaction was readily detected by co-IP followed by immunoblot analysis in two cell lines with wild-type KRAS and representative lung and pancreatic cancer cells harboring various activating mutations of KRAS (Figure 1D). The observed RAS-AGO2 interaction was maintained even under highly stringent conditions of 1 M NaCl (Figure S1E). The RAS-AGO2 co-IP was maintained in the presence of RNase, suggesting that the interaction is independent of AGO2 interaction with RNA (Figure S1F–G). To demonstrate further specificity of this interaction, we overexpressed FLAG-tagged AGO2 construct in HEK293 cells and detected RAS in FLAG immunoprecipitates (Figure S1H). We also performed the co-IP analysis in genetically engineered “RASless” mouse embryonic fibroblast cells (Drosten et al., 2010) and failed to detect this interaction upon ablation of KRAS expression (Figure S1I), further establishing the specificity of the RAS-AGO2 interaction.
Co-localization of RAS and AGO2 in the membrane component of endoplasmic reticulum
RAS proteins are known to localize to the plasma membrane and membranes of various intracellular organelles like the endoplasmic reticulum (ER), Golgi, and mitochondria with distinct signaling outputs (Bivona et al., 2006; Prior and Hancock, 2012). AGO2 is known to assemble in the endoplasmic reticulum (Kim et al., 2014; Stalder et al., 2013), cytoplasm (Hock et al., 2007), and nucleus (Dudley and Goldstein, 2003; Gagnon et al., 2014). Consistent with this observation, cell fractionation of H358 cells revealed that RAS was restricted mainly to the membrane (plasma membrane and endomembrane) fraction along with AGO2, which was also detected in the cytoplasmic and nuclear extracts (Figure 2A). Sedimentation analyses using sucrose density gradient showed that total RAS and mutant KRAS predominantly co-sedimented with AGO2 in smaller molecular weight fractions (Complex I; Figure 2B) as defined by a previous study (Hock et al., 2007).
Figure 2. Co-sedimentation and co-localization of RAS and AGO2 in the endoplasmic reticulum.

(A) Cell fractionation analysis of H358 cells to show enrichment of distinct proteins in the cytosolic/membrane or organelle/nuclear fractions. GAPDH was used as a cytosolic marker while SAM68 and H3 histone were used as nuclear markers. (B) Sucrose density gradient fractionation of cell lysates from H358 cells followed by immunoblot detection of total RAS, KRAS, AGO1 and AGO2 proteins. (C) Representative images of immunofluorescence analysis of RAS (red) and AGO2 (green) in H358, MIA PaCa-2 and DLD-1 cells. Yellow spots in merged images indicate perinuclear co-localization of RAS and AGO2. The nucleus was visualized by DAPI staining (blue). Dotted boxes highlight plasma membrane regions predominantly localized by RAS. Manders overlap coefficient, in the intracellular regions of the cells are indicated on the right. An overlap coefficient of 0 suggests no co-localization, whereas a value of 1 indicates complete co-localization. The inset shows a magnified 5.3 μm x 5.3 μm view of the areas marked. Images are pseudocolored maximum intensity projections (across 2.5 μm), obtained from 3D imaging. Scale bar, 5 μm. (D) Representative images of immunofluorescence analysis of AGO2 (green), RAS (red) and ER marker, PDI, (blue) in MiaPaCa-2 cells. White spots indicate co-localization signals for RAS/AGO2/PDI in each panel. Pairwise Manders overlap coefficients are shown on the right. The inset shows a magnified 6.7 μm x 6.7 μm view of the areas marked. Scale bar, 5 μm. See also Figure S2.
Next, to assess co-localization of endogenous RAS and AGO2 we performed indirect immunofluorescence using RAS10 and AGO211A9 (Rudel et al., 2008) antibodies in different cells. To ascertain the specificity of RAS10 Ab, antigenic peptides were used for competition prior to immunofluorescence analysis (Figure S2A); in addition, AGO211A9 has been demonstrated to be a highly specific, validated monoclonal antibody for immunofluorescence detection of AGO2 (Rudel et al., 2008). In H358, MIA PaCa-2 and DLD-1 cells (Figure 2C), RAS staining was visible both at the plasma membrane and intracellular regions, while only cytoplasmic staining was detected for AGO2. Manders coefficient analysis indicative of signal overlap between the two proteins was determined to be 0.53, 0.65 and 0.60 in H358, MIA PaCa-2 and DLD-1 cells respectively (where 1 is considered complete overlap while 0 is considered no overlap). These findings suggest significant co-localization of RAS and AGO2 predominantly in the intracellular perinuclear regions of cells (Figure 2C).
Given that cytoplasmic RAS is restricted to the endomembrane bound organelles, we performed a three-color immunofluorescence staining for RAS and AGO2 along with specific protein markers of different organelles in MIA PaCa-2 cells. While we observed a significant signal overlap between RAS, AGO2 and ER-marker (PDI) (Figures 2D and S2B) Manders coefficient values were minimal for Golgi (RCAS1), endosomal (Rab5/7/11) or mitochondrial (COX4) markers (Figure S2B–C). This suggests that endogenous RAS and AGO2 are predominantly found in the endoplasmic reticulum (Manders coefficient for both RAS in ER and AGO2 in ER was 0.62 each) where they co-localize. Hence along with the cell fractionation analyses, this immunofluorescence data suggests that a subset of RAS proteins co-localizes with AGO2 in the endomembranous components of the ER.
AGO2 binds RAS through its N-terminal wedge domain
To identify specific region(s) in AGO2 involved in the interaction with RAS, we employed a panel of FLAG-epitope tagged AGO2 expression constructs (summarized in the schematic in Figure 3A). RAS co-IP analysis of the FLAG tagged AGO2 deletion constructs showed that the N-terminal domain of AGO2 was necessary (Figure 3B) and sufficient (Figure S3A) for RAS binding. Further analysis of a panel of deletion constructs spanning the N-terminal domain suggested that the region spanning 50–139 amino acids (aa) was critical for RAS binding (Figure S3B). Interestingly, this amino acid stretch was recently shown to be part of the “wedging” domain, important for microRNA duplex unwinding prior to RISC assembly (Kwak and Tomari, 2012). To further define AGO2 residues critical for interaction with RAS, we focused on the 50–139 aa stretch that are uniquely present in AGO2 (and not in AGO1, 3 or 4) based on the fact that, amongst the Argonaute family proteins, AGO2 was almost singularly represented in the RAS co-IP MS data. ClustalW alignment of all human Argonaute proteins (AGO1–4) identified 10 residues unique to AGO2 in this region (Figure S3C). Alanine substitution of each of the 10 residues was followed by RAS co-IP analysis, and amino acids K112 and E114 of AGO2 were found to be critical for a direct association with RAS (Figure 3C).
Figure 3. The N-terminal domain of AGO2 interacts with RAS.

(A) Schematic summary of FLAG tagged AGO2 deletion and mutant constructs used for RAS co-IP analyses (B) Expression of FLAG tagged N-terminal, PAZ, or PIWI domains of AGO2 in HEK293 cells (left panel), followed by RAS IP (right panel). Immunoblot analysis shows that deletion of (1-226aa) N terminal domain in AGO2 abrogates RAS interaction. (C) Expression of indicated AGO2 N terminal point mutant constructs within the wedge domain (50-139aa) in HEK293 cells, followed by RAS co-IP analysis. See also Figure S3.
Y64 residue within the Switch II domain of KRAS is critical for direct AGO2 binding
In a parallel analyses aiming to define the residues in RAS critical for AGO2 association, we first employed two RAS antibodies that bind exclusively either to the Switch I (RAS10 mAb) or the Switch II (Y13–259) domains (summarized in Figure 4A). While both antibodies efficiently immunoprecipitated RAS in H358 cell lysates, AGO2 was present only in IPs with Switch I specific RAS10 Ab, and not in Switch II specific Y13–259 Ab (Figure 4B), suggesting that the Switch II domain in RAS is critical for AGO2 interaction. Next, we hypothesized that if the RAS-AGO2 interaction is restricted through contacts with the Switch II domain we may be able to detect AGO2 in RAS-GTP complexed with RAF, on RAS binding domain (RBD) agarose beads. As predicted, we were able to detect AGO2 on RAS-GTP bound to RBD-agarose in H358 (KRASG12C) cells (Figure S4A), further supporting that AGO2 binds to the Switch II domain of GTP bound KRAS.
Figure 4. The Switch II domain of RAS interacts with AGO2.

(A) Schematic summary of the antibodies and recombinant proteins used for RAS-AGO2 co-IP analysis to identify residues in RAS, critical for AGO2 interaction. (B) RAS co-IP using antibodies that bind switch I domain (RAS10 Ab) or switch II domain (Y13-259 Ab), followed by immunoblot analysis for RAS and AGO2. (C–E) Characterization of direct RAS-AGO2 interaction, in vitro. (C) Immunoblot analysis following in vitro co-IP of recombinant KRASG12V (top panel) and KRASWT (bottom panel) in the presence of varying concentrations of recombinant AGO2. (D) In vitro co-IP analysis of KRAS-AGO2 interaction using a panel of KRAS mutant proteins spanning amino acid residues 62–65 in the switch II domain. (E) Immunoblot analysis following His-AGO2 pull down assay using Ni-NTA beads upon incubation with different KRAS mutant proteins. See also Figure S4.
Next, we sought to determine the specific residues in the Switch II region of KRAS involved in its interaction with AGO2, using in vitro co-IP assays. Purified recombinant KRASG12V or KRASWT proteins were incubated with varying concentrations of AGO2 protein followed by RAS immunoprecipitation. We observed a concentration dependent, direct interaction between recombinant AGO2 and both the wild type and mutant KRAS proteins (Figure 4C). Further, in vitro co-IP of recombinant AGO2 protein with the panel of Switch II mutant KRAS proteins showed that altering the Y64 residue (but not the neighboring amino acids) significantly reduced KRAS binding to AGO2 (Figure 4D). To further substantiate this observation, and to obviate potential technical concerns inherent in antibody based co-IP, we carried out an antibody-independent pull down assay using recombinant His-tagged AGO2 protein bound to Ni-NTA beads. Consistent with the in vitro co-IP analyses, the His-tagged AGO2 pull down assay also showed specific dependency of AGO2-RAS binding on the Y64 residue (Figure 4E).
To assess if GDP/GTP loading of KRAS may influence the AGO2 interaction in vitro, we carried out in vitro co-IP analyses using KRASWT and KRASG12V proteins loaded with GDP/GTPγS, and as seen in Figure S4B. Our results showed that AGO2 binding was agnostic to nucleotide loading status of KRAS. Similarly, both the KRASWT and KRASG12V proteins were observed to bind to His-tagged AGO2, independent of the nucleotide loading on KRAS (Figure S4C). To validate the efficiency and specificity of nucleotide loading onto KRAS proteins, we performed RAF-RBD pull down assays and observed the expected differential between GDP and GTP bound KRAS with respect to RAF-RBD binding (Figure S4D). Thus, these data define the amino acids in RAS (Y64) and AGO2 (K112/E114) as critical for the RAS-AGO2 interaction.
Reduced RISC activity elevates oncogenic KRAS levels, making AGO2 essential for mutant KRAS dependent cell proliferation
Next, we set out to analyze functional implications of the RAS-AGO2 interaction, particularly in the context of KRAS driven transformation. To this end, we first carried out knockdown of AGO2 in H358 lung cancer cells that harbor a homozygous KRAS mutation and are known to be KRAS-dependent (Symonds et al., 2011). Whereas the microRNA let-7/AGO2 axis is reported to negatively regulate wild-type RAS levels (Diederichs and Haber, 2007; Johnson et al., 2005), we observed a remarkable reduction in mutant KRAS protein levels in H358 cells with AGO2 knockdown(Figure 5A, left panel). Conversely, overexpression of AGO2 in the same cells led to elevated levels of KRAS, implying a positive regulation of mutant KRAS levels by AGO2 (Figure 5A, right panel). Consistent with these observations, knockdowns of AGO2 and/or KRAS in H358 cells (using two independent shRNAs, Figure S5A–B) showed reduced rates of cell proliferation while AGO2 overexpression resulted in increased cell proliferation (Figure 5B). Furthermore, AGO2 knockdown reduced the ability of H358 cells to form colonies in colony formation assays (Figure 5C) and resulted in a marked reduction in levels of known mediators of KRAS signaling, including p-Akt, p-mTOR and p-RPS6 based on our analysis with Pathscan intracellular signaling array (Cell Signaling) (Figure 5D and Figure S5C–D). Interestingly, similar AGO2 depletion experiments (using the same shRNAs described above) in KRAS independent H460 lung cancer cells, which also harbors a mutant KRAS, did not affect cell proliferation, colony formation (Figure 5E) or intracellular signaling (Figure 5F and Figure S5E). Phenotypic effects upon AGO2 knockdown in the context of KRAS dependency were also observed in pancreatic cancer cell lines where knockdown of either KRAS or AGO2 dramatically reduced cell proliferation in mutant KRAS-dependent MIA PaCa-2 cells, but not in mutant KRAS-independent PANC-1 cells (Figure 5G and Figure S5A–B). Further, AGO2 depleted MIA PaCa-2 cells failed to establish xenografts in SCID mice (Figure 5H) with a concomitant reduction in KRAS protein levels (Figure 5H, inset). These data suggest that KRAS dependent cancer cells manifest a coincident dependence on AGO2 to maintain oncogenic KRAS protein levels and support a functional role for AGO2 in potentiating the oncogenic activities of mutant KRAS.
Figure 5. AGO2 is essential for mutant KRAS dependent cell proliferation.

(A) Immunoblot analysis of AGO2 and KRAS after knockdown or overexpression of AGO2. (B) Growth curves and (C) colony formation assays of mutant KRAS dependent H358 lung cancer cells, following either knockdown of KRAS/AGO2 using shRNA or AGO2 overexpression. Error bars are based on standard error of the mean. *(P<0.05) and **(P<0.005) denote significant differences in growth at the indicated times compared to either scrambled or vector control. Data were obtained from three independent experiments. (D) Pathscan intracellular signaling arrays probed with lysates from H358 cells following AGO2 knockdown. (E) Growth curves (left) and colony formation assays (right) of mutant KRAS independent H460 lung cancer cells, following knockdown of KRAS/AGO2. Data obtained from three independent experiments are shown. Inset, immunoblot analysis of AGO2 and KRAS upon AGO2 knockdown. (F) Intracellular signaling array probed with lysates from H460 following AGO2 knockdown. (G) Growth curves of pancreatic cancer cells, MIA PaCa-2 (mutant KRAS dependent) (left) and PANC-1 (mutant KRAS independent) (right) following knockdown of KRAS or AGO2, as indicated. *(P<0.05) and **(P<0.005) denote significant differences in growth at the indicated times compared to scrambled control. Data were obtained from three independent experiments. (H) In vivo growth of Mia PaCa-2 cells transiently treated with either scrambled shRNA or shRNA targeting AGO2 prior to injecting in nude mice. For each group (n=8), one million cells were injected and average tumor volume (in mm3) was plotted on y-axis and days after injection on the x-axis. Right, immunoblot analysis of AGO2 and RAS following AGO2 knockdown in Mia PaCa-2 cells. Indicated P-value was calculated using two sided student t-test for the two groups. (I) Top, Schematic of the labeled let-7 microRNA used in the intracellular strand unwinding assays. Straight lines and double dots represent, Watson-Crick and Wobble pairs respectively. The thermodynamically unstable end (highlighted in yellow), promotes asymmetric loading of the guide strand. Bottom left, representative images of the guide strand (green) and passenger strand (red) of let-7 microRNA, 30 minutes post intracellular injections in DLD-1 isogenic lines, expressing wild type KRAS (− / WT) or KRASG12C (MUT / WT). Numbers represent the Guide: Passenger strand ratio. Ratio of 1:1 indicates attenuation of let-7 dsRNA unwinding while a higher guide:passenger strand ratio indicates efficient unwinding and functional RISC. Scale bar, 10 μm. Bottom right, native acrylamide gel electrophoresis of let-7 unwinding assay and immunoblot analysis of DLD-1 isogenic cell line extracts. M1 and M2 represent double (ds) and single stranded (ss) markers respectively. (J) Box plot representing the Guide: Passenger strand ratio in the indicated cell lines with varying KRAS mutation status. Whiskers represent minimum and maximum values and line represents median of the data set (n ≥ 2, # cells ≥ 13, ***p < 0.0005). Red asterisk indicates cells expressing mutant KRAS. See also Figure S5.
To directly address the consequence of mutant KRAS binding at the N-terminal of AGO2, critical for microRNA duplex unwinding (Kwak and Tomari, 2012; Wang et al., 2009), we performed let-7 unwinding assays in isogenic colorectal cancer cells, DLD-1, harboring heterozygous KRASG13D (MUT/WT) alleles or wildtype KRAS (−/WT). Dually labeled double stranded let-7a (Figure 5I schematic) was injected and assessed for the extent of single strand formation. Quantitation of the guide-to-passenger strand ratio was estimated 30 min after injection, where a 1:1 ratio was considered as no unwinding while higher ratios indicate active unwinding. As seen Figure 5I (left), the formation of single-stranded (ss) RNA molecules from double-stranded (ds) let-7 substrates, a key step in the formation of active RISC, was attenuated in DLD-1 MUT/WT cells (ratio=1.3). Duplex unwinding was restored in isogenic cells lacking mutant KRAS (−/WT) (ratio=5.7). Biochemical assays using cellular lysates followed by gel electrophoresis also showed reduced let-7 unwinding in DLD-1 MUT/WT cells (Figure 5I, right), even though the RAS-AGO2 interaction was detected in both DLD-1 isogenic cells (Figure S5F). Additionally, the let-7 unwinding assay was performed in mouse embryonic fibroblasts lacking AGO2 (MEFAGO2−/−) and MEFAGO2−/− reconstituted with AGO2 (MEFAGO2−/− + AGO2) (Broderick et al., 2011) to demonstrate that the microRNA unwinding assay is AGO2 dependent (Figure S5G–H). Further, to circumvent any artificial effects of gene knockout models, we subjected multiple cancer cells, naturally harboring different KRAS alleles, to the same let-7 unwinding assay. As seen in Figures 5J and S5I, only oncogenic KRAS expressing cells showed reduced let-7 unwinding, indicative of diminished AGO2 function in cells harboring mutations in KRAS.
Mutant KRAS-AGO2 interaction promotes cellular transformation
To address the mechanistic underpinnings of the phenotypic effects associated with the mutant KRAS-AGO2 interaction, we employed the classic NIH3T3 experimental model system to ectopically express human KRASWT or KRASG12V (Qiu et al., 1995; Shih et al., 1981), with or without AGO2, and carried out transient foci formation assays. As expected, no foci were observed in cells transfected with KRASWT, as well as in cells with KRASWT±AGO2. However, NIH3T3 cells transfected with KRASG12V generated characteristic foci of transformed cells. Remarkably, co-transfection of KRASG12V with AGO2 enhanced the number of foci by approximately five-fold, compared to the vector control (Figure 6A). In contrast, AGO2 overexpression did not enhance BRAFV600E driven focus formation (Figure S6A), suggesting that AGO2 specifically potentiates RAS-mediated oncogenesis, most likely as a result of its direct interaction with RAS. In vivo experiments using a mouse xenograft model also showed a significant increase in tumor growth with cells expressing KRASG12V+AGO2 compared to KRASG12V+vector control (Figure S6B). As expected, in these experiments cells expressing either KRASWT or AGO2 alone did not develop tumors. Consistent with AGO2 overexpression in H358 cells (Figure 5A), immunoblot analysis of NIH3T3 cells overexpressing AGO2 showed an increase in KRAS protein levels (Figure 6B).
Figure 6. Mutant KRAS-AGO2 interaction promotes transformation.

(A) Representative images of foci formation assays using NIH3T3 cells co-transfected with KRASWT or KRASG12V and AGO2 (left panel). Quantitation of foci from two technical replicate experiments (right panel). Foci assays were performed at least three times with similar results. P-value, calculated using two-sided student t-test between the two groups. (B) Immunoblot analysis shows increased levels of oncogenic KRAS levels in the presence of AGO2. (C) Intracellular signaling arrays probed with lysates from NIH3T3 cells stably expressing vector, AGO2, or KRASG12V±AGO2. The colored circles mark duplicate spots corresponding to p-AKT (S473), p-RPS6 (S235/236) and p-mTOR (S2448). (D) Representative images of foci formation assays using NIH3T3 cells co-transfected with KRASG12V or KRASG12VY64G. Quantitation of foci from two independent experiments (right). Indicated P-value was calculated using two-sided student t-test. (E) KRAS immunoprecipitation (using sc-521 pAb) followed by immunoblot analysis (RAS10 Ab) showing low levels of oncogenic KRAS protein expression in NIH3T3 cells stably expressing KRASG12VY64G. RAS-GTP levels were assessed using RBD agarose beads. Signaling through phospho-Akt and phospho-ERK activation was performed after serum starvation by immunoblot analysis. Lower panel shows morphology of indicated stable lines grown in 10% serum upon crystal violet staining. (F) In vivo growth of NIH3T3 cells stably overexpressing KRASG12V and KRASG12VY64G in nude mice. For each group (n=8), 500,000 cells were injected and average tumor volume (in mm3) was plotted on y-axis and days after injection on the x-axis. (G) Left, Representative 3.14 x 3.14 μm2 regions from NIH3T3 (top, left), NIH3T3-KRASWT (top, right) NIH3T3-KRASG12V (bottom, left) and NIH3T3-KRASG12V,Y64G cells (bottom, right) that were imaged 4 hours (h) after microinjection of let-7-a1-Cy5. Individual particle tracks (colored) and their net displacements (white arrow) over a 5s period (time, color bar) are shown. Shorter displacement vectors indicate let-7 assembly in larger mRNP complexes with less mobility, whereas longer white arrows indicate let-7 assembly in smaller mRNP complexes with high mobility. Right, Graphical representation of ratio of “Slow” moving complexes (particles with diffusion coefficients < 0.06 μm2/s) and “Fast” moving complexes (particles with diffusion coefficients > 0.06 μm2/s), normalized to the first hour time point, are plotted as a function of time. See also Figure S6.
To understand the effects of AGO2 on the RAS signaling pathways, we analyzed protein lysates from NIH3T3 cells stably expressing KRASG12V+vector or KRASG12V+AGO2, using the Pathscan intracellular signaling arrays. Cells expressing KRASG12V+AGO2 showed a marked increase in the levels of p-Akt, p-mTOR, p-RPS6 and p-BAD, but not phospho-ERK (Figure 6C, right panel and Figure S5C–D), suggesting that the increased levels of oncogenic KRASG12V protein signals largely through PI3K activation.
Exploiting the NIH3T3 overexpression model to probe the reciprocal effects of mutant KRAS on AGO2 function, we profiled microRNAs from foci obtained from KRASG12V+vector and KRASG12V+AGO2 using high-throughput sequencing. While AGO2 overexpression is known to elevate levels of mature microRNAs (Diederichs and Haber, 2007), we observed a marked reduction in microRNA levels (214/781) in KRASG12V+AGO2 expressing foci, including most of the let-7 family members (Figure S6E). Interestingly, a small proportion (27/781) of microRNAs were elevated and included known ‘oncomiRs’ miR-221 and miR-222. microRNA qPCR analysis of NIH3T3 cells expressing AGO2 alone, or KRASWT/KRASG12V ± AGO2 also showed reduced let-7 levels only in the KRASG12V +AGO2 expressing cells (Figure S6F), suggesting an inhibition of AGO2 function in oncogenic KRAS expressing cells.
To further investigate a requirement for an AGO2 interaction in KRASG12V driven transformation, we first performed in vitro RAS co-IP assays using mutant KRASG12D and the double mutant KRASG12DY64G, which has previously been shown to have limited oncogenic potential (Shieh et al., 2013). While KRASG12D binds AGO2, KRASG12DY64G failed to bind AGO2 (Figure S6G). Transfecting a retroviral vector encoding KRASG12VY64G double mutant into NIH3T3 cells failed to generate foci (Figure 6D). As an important corollary to our hypothesis that mutant KRAS-AGO2 interaction leads to elevated mutant KRAS protein levels, the KRASG12VY64G stably expressing cells also showed much lower levels of KRAS protein than KRASG12V expressing cells (Figure 6E, top panel). An independent construct encoding KRASG12VY64G showed similar results despite high levels of KRAS transcript expression (Figure S6H–J). Curiously, RBD assays suggest that expressed KRASG12VY64G was GTP loaded and activated phospho-Akt and phospho-ERK similar to KRASG12V, suggesting that although KRASG12VY64G levels are low, it is GTP loaded and likely signaling at the membrane. Yet, despite expressing activated RAS, NIH3T3 stable cells expressing KRASG12VY64G failed to show the characteristic morphology of KRASG12V cells (Figure 6E, bottom panel). In vivo, these cells also failed to establish tumors in the xenograft mouse model (Figure 6F), supporting a critical role for Switch II region (Y64) in KRAS driven transformation, including its association with AGO2. While NIH3T3 cells stably expressing KRASG12V showed reduced let-7 levels, KRASG12VY64G expressing cells, which do not allow for the mutant KRAS-AGO2 interaction, showed no change in let-7 expression, providing evidence for a direct role of mutant KRAS in the modulation of microRNA levels in this model (Figure S6K). Cognate analysis of the levels of let-7 target transcripts (Lee and Dutta, 2007) showed an almost log-fold change in the mRNA levels of HMGA1 and HMGA2 only in KRASG12V expressing cells (Figure S6L). Together, our data using the KRASG12VY64G mutant and let-7 levels as readout of AGO2 function broadly support the conclusion that mutant KRAS, through its direct association, inhibits AGO2 activity.
To more directly explore the potential effect of KRASG12V on functional messenger ribonucleoprotein particles (mRNPs), we exploited a recently described method for intracellular single-molecule, high-resolution localization and counting (iSHiRLoC) of microRNAs (Pitchiaya et al., 2012; Pitchiaya et al., 2013). Diffusion coefficients of microinjected fluorophore labeled let-7a molecules suggest that, in NIH3T3 cells expressing KRASWT, let-7a assembles into both ‘fast’ (low molecular weight) and ‘slow’ (high molecular weight) mRNA-protein complexes (mRNPs; Figure 6G and S6M). By contrast, in cells expressing KRASG12V, let-7a manifested predominantly in fast moving complexes, suggesting that let-7a is unable to accumulate in larger mRNPs (known to be functional RISC; (Pitchiaya et al., 2012; Pitchiaya et al., 2013)) in an oncogenic KRAS setting. Importantly, in cells expressing KRASG12VY64G, let-7a accumulates in both fast and slow mRNPs, further implicating that a direct interaction between mutant KRAS and AGO2 is essential to prevent functional RISC assembly. Thus, the NIH3T3 overexpression model suggests that through its interaction with AGO2, mutant KRAS modulates levels of mature microRNAs likely due to its ability to inhibit an early step of RISC assembly.
AGO2 interaction is required to maximize oncogenic potential of mutant KRAS
To further underscore the role of AGO2 in KRASG12V driven oncogenesis, we generated NIH3T3 cells with AGO2 knockout (NIH3T3 AGO2−/−) using the CRISPR/Cas9 methodology (Ran et al., 2013) (Figure S7A). Validation of AGO2 knockout in NIH3T3 AGO2−/− cells was performed at the DNA, RNA and protein levels (Figure S7B–D). Sucrose density sedimentation analysis of NIH3T3 AGO2−/− showed that, in contrast to NIH3T3 parental cells, RAS is restricted largely to the first four fractions of the gradient with minimal overlap with AGO1 complexes, indicating that RAS associates with higher molecular weight fractions through its interaction with AGO2 (Figure 7A). NIH3T3 AGO2−/− cells had lower levels of let-7 family microRNAs (Figure S7E), consistent with previous studies demonstrating that a loss of AGO2 results in reduction of absolute levels of all microRNAs (Diederichs and Haber, 2007). In NIH3T3 AGO2−/− cells, the reduction of let-7 family microRNA levels also resulted in a concomitant increase in let-7 target (HMGA1/HMGA2) transcript levels (Figure S7F).
Figure 7. AGO2 interaction is required for maximal oncogenic potential of mutant KRAS.

(A) Sucrose density gradient fractionation of parental NIH3T3, NIH3T3 KRASG12V and NIH3T3 AGO2−/− cell lysates followed by immunoblot detection of RAS, AGO1 and AGO2 proteins. (B) Left, representative images of KRASG12V driven foci in NIH3T3 and NIH3T3 AGO2−/− cells upon co-transfection with various AGO2 constructs. Right, quantitation of foci from two replicate experiments. Error bars show standard error of mean and asterisks indicate P values less than 0.005 for the indicated conditions compared to vector control. (C) Upper panel, shows crystal violet staining of indicated stable lines grown in 10% serum. Lower panel, in vivo growth of NIH3T3 or NIH3T3 AGO2−/− cells stably expressing KRASG12V in nude mice. For each group (n=8), 500,000 cells were injected and average tumor volume (in mm3) was plotted on y-axis and days after injection on the x-axis. Error bars are standard error of mean * P<0.05 and ** P<0.005 at the indicated times. (D) Immunoblot analysis showing reduced expression of oncogenic KRAS in KRAS AGO2−/− stably expressing KRASG12V and the extent of phospho-ERK and phospho-AKT activation in these cells. (E) Schematic representation of the N-terminal domain of AGO2 interacting with the switch II domain in RAS. See also Figure S7.
Despite reduced levels of microRNAs, KRASG12V expression in the NIH3T3 AGO2−/− cells showed a markedly reduced ability to generate foci compared to parental NIH3T3 (Figure 7B and Figure S7G). Partial rescue of the ability to establish foci in these cells was achieved by overexpression of AGO2 or AGO2K98A (which permits RAS interaction) but not the AGO2K112A mutant (which does not bind RAS) (Figure 3C). These observations also support the notion that a direct association of oncogenic KRAS and AGO2 is required for mutant KRAS driven transformation. In addition, NIH3T3 AGO2−/− cells stably expressing KRASG12V did not display the characteristic morphology of NIH3T3 KRASG12V cells (Figure 7C, top panel). In vivo experiments in a mouse xenograft model also showed significantly decreased tumor growth with NIH3T3 AGO2−/− cells expressing KRASG12V compared to parental NIH3T3 cells expressing KRASG12V, further demonstrating a requirement for AGO2 in KRAS driven transformation (Figure 7C, lower panel). At the protein level, NIH3T3 AGO2−/− cells stably expressing KRASG12V showed reduced expression of mutant KRAS compared to that of NIH3T3 cells stably expressing KRASG12V (Figure 7D). Reduced activation of phospho-Akt signaling by mutant KRAS and a slight increase in phospho-ERK signaling in NIH3T3 AGO2−/− cells suggests that AGO2 plays an essential role in modulating the signaling output of mutant KRAS.
Taken together, we have established AGO2 as a critical regulator of RAS-GTP in cells and our study posits an essential role for the KRAS-AGO2 interaction in oncogenic KRAS driven cellular transformation.
DISCUSSION
RAS, one of the first proto-oncogenes identified (DeFeo et al., 1981), has emerged as one of the genes with most frequent recurrent mutations in a broad spectrum of human cancers. In recent years, there is a renewed interest in targeting RAS to alter its status from an undruggable to druggable candidate (Burns et al., 2014; Ostrem et al., 2013; Spiegel et al., 2014; Stephen et al., 2014; Sun et al., 2012). In this context, discovery of novel endogenous interactors of RAS could potentially advance our understanding of RAS biology and provide additional therapeutic avenues.
Here, we identify the interaction of RAS with AGO2, a key mediator of RNA-based gene silencing (Czech and Hannon, 2011; Peters and Meister, 2007; Wilson and Doudna, 2013). Like guanine nucleotide exchange factors (GEFs), AGO2 binds RAS-GTP and RAS-GDP, and likely interacts functionally with both wild-type and oncogenic RAS proteins (Jeng et al., 2012; Margarit et al., 2003). Furthermore, AGO2 and RAS co-localize in the endoplasmic reticulum, known sites for both RAS trafficking and AGO2 RISC activity. The KRAS-AGO2 interaction involves Y64 in the Switch II domain of KRAS, and K112–E114 residues in the N-terminal Wedge domain of AGO2 (Figure 7E). Functionally, the mutant KRAS-AGO2 interaction is critical for KRAS mediated oncogenesis. Mechanistically, mutant KRAS binding attenuates AGO2 N-terminal dependent microRNA duplex unwinding, critical for functional RISC assembly. Reciprocally, AGO2 modulates mutant KRAS mediated signaling output, particularly the AKT-mTOR pathway.
Our study focused on analyzing endogenous interactors of RAS, common across a panel of cancer cells spanning the spectrum of KRAS aberrations. To the best of our knowledge, this is the first study using endogenous RAS as bait for mass spectrometric analyses as all previous co-IP MS analyses used N-terminal epitope-tagged-HRAS, -MRAS, or -RRAS ectopically expressed in NIH3T3 cells (Goldfinger et al., 2007; Vasilescu et al., 2004). Studies using tagged AGO2 as bait for mass spectrometry have also been reported (MacRae et al., 2008; Meister et al., 2005), and as a 25-kDa cutoff was employed for analyses, may have missed the detection of the 21 kDa RAS protein. In this study, the pull-down of AGO2 using multiple independent antibodies consistently co-precipitated RAS (Figure 1C), and we found that this interaction is direct, as assessed using purified components (Figure 4). Endogenously, the RAS-AGO2 interaction is readily detected in both cancer and benign cells, independent of KRAS mutation status (Figure 1D), portending a fundamental role for this interaction in the cell.
KRAS interacts with AGO2 through the Switch II domain, the Y64 residue being critical for its AGO2 association. The Switch II domain and particularly Y64 was recently demonstrated to be critical in hematopoietic malignancies, where KRASG12DY64G mutant expressed at lower levels compared to KRASG12D (Shieh et al., 2013), much like we observed in our NIH3T3 model, extending a role for the KRAS-AGO2 interaction in models other than lung and pancreas. It should be noted that the Switch II domain in RAS is the site for allosteric regulation through its binding to various regulators and may contribute to the biological effects observed in these studies. Yet, this study provides a first instance where the mutant KRAS Switch II domain (and Y64) has a direct bearing on RISC assembly through its association with AGO2.
The AGO2 N-terminal domain represents the most distinct region in the highly conserved AGO protein family. A recent report (Kwak and Tomari, 2012) suggests that the region we identified in AGO2 as critical for RAS binding (i.e., the ‘wedge domain’), is important for small RNA duplex unwinding, a prerequisite for RISC assembly. Using isogenic lines we demonstrate that mutant KRAS, but not wild type KRAS, interaction with AGO2 attenuates microRNA duplex unwinding function with a direct bearing on AGO2-RISC assembly. Inhibition of RISC assembly by mutant KRAS may be the critical step that likely contributes to global loss of microRNA levels and downstream effects on increased protein translation of target mRNAs, features of human tumors (Lu et al., 2005). Since we have used mutant KRAS constructs that do not have 3′UTR regions that can bind microRNAs, it remains unclear how AGO2 elevates mutant KRAS levels (Figure 5A, 6B) to increase its transformation potential.
Recent studies have shown that KRAS, but not HRAS translation, is tightly regulated by rare synonymous codons of the KRAS transcript (Lampson et al., 2013; Pershing et al., 2015), suggesting a significant role for KRAS regulation at a level prior to its better characterized post-translational modifications. An association of mutant KRAS with the RNA machinery through binding to HNRNPA2B1 was also reported (Barcelo et al., 2014), supporting a likely interface of RAS with the RNA processing machinery, including the hub protein AGO2 as observed in our study. The EGFR kinase was also recently shown to phosphorylate AGO2 in response to hypoxia, leading to inhibition of AGO2-mediated microRNA processing (McCarthy, 2013; Shen et al., 2013). Similarly, Akt was shown to phosphorylate AGO2 to inhibit AGO2-mediated mRNA endonucleolytic activity (Horman et al., 2013). Interestingly, AGO2 phosphorylation also leads to inhibition of microRNA loading into RISC complexes in the presence of mutant HRASG12V (Yang et al., 2014). The identification of AGO2 as a critical partner of RAS further provides a direct mechanistic link between RAS oncogenic signaling and RNA silencing. Further illumination of such integral effector mechanisms of RAS may inform novel approaches to therapeutically target this frequently mutated cancer pathway.
Experimental Procedures
Coimmunoprecipitation and Tandem Mass Spectrometric analysis
Methods used for immunoprecipitation with RAS/control IgG followed by Tandem Mass Spectrometric analysis and database searching are schematically outlined in Supplemental Figure S1D. Complete data of the peptides represented in the RAS co-IP mass spectrometric analysis from the different cell lines are provided in Supplemental Table S5.
Immunoprecipitation (IP) and Western blot Analysis
Routine methods to immunoprecipitate proteins were employed and detailed in Supplemental Experimental Procedures. Antibodies used in the study are detailed in Table S3.
RAS-GTP pull down assay
The RAS-RAF interaction was studied using the RBD agarose beads as per manufacturer’s instructions (Millipore) and detailed in Supplemental Experimental Procedures.
Focus formation assay
Foci formation assays were performed by transfecting/co-transfecting (the indicated constructs) 150,000 early passage NIH3T3 cells in 6 well dishes using Fugene HD (Promega). After two days, cells were trypsinized and plated onto 150 mm dishes containing 4–5% calf serum. The cells were maintained under low serum conditions and medium was refreshed every two days. After 21 days in culture the plates were stained for foci using crystal violet.
Generation of NIH3T3 AGO2−/− line
AGO2-knockout NIH3T3 cells were generated by CRISPR-Cas9-mediated genome engineering (Ran et al., 2013). Genomic regions in murine AGO2 between exons 8 and 9, and between exons 11and 12 were targeted for deletion using primers TCCTTGGTTACCCGATCCTGG and AGAGACTATCTGCAACTATGG, respectively (PAM motif underlined). Selection of clones is detailed in Supplemental Experimental Procedures.
iSHiRLoC analyses
Oligos (let-7-a1 guide: P-UGA GGU AGU AGG UUG UAU AGU U-Cy5; let-7-a1-passenger: P-CUA UAC AAU CUA CUG UCU UUC C) were microinjected in cells were incubated in phenol red-free DMEM containing 2% (v/v) CS in the presence of a 5% CO2 atmosphere at 37 °C for the indicated amounts of time prior to imaging. Details of microinjection and imaging are provided in Supplemental Experimental Procedures.
Extract preparation and in vitro miRNA unwinding assay
Cell extracts were prepared as described (Kwak et al, Nat. Struct. Mol. Boil. 2012 and Rakotondrafara et al, Nature protocols, 2011), with minor modifications detailed in Supplemental Experimental Procedures.
Further details on other methods are provided in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
RAS interacts with AGO2 in the membrane component of the endoplasmic reticulum
The N-terminus of AGO2 directly binds the Switch II domain of RAS
Oncogenic KRAS association inhibits AGO2 mediated microRNA duplex unwinding
AGO2 interaction elevates oncogenic KRAS levels to enhance cellular transformation
Acknowledgments
We acknowledge the work of Shanker Kalyan-Sundaram, Krishnapriya Chinnaswamy, Vijaya L. Dommeti, Matthew Shuler, Anton Poliakov, Xiaoju Wang and Vishalakshi Krishnan who helped with analysis and experimentation. We thank Mariano Barbacid for KRAS-only expressing mouse embryonic fibroblast cells. We thank Eric Fearon for helpful discussions, Joseph Mierzwa, Kevin Eid and Jincheng Pan for technical assistance, Bushra Ateeq and Rachell Stender for help with the xenograft studies, William Brown for His-AGO2 protein preparation and Robin Kunkel for assistance with schematic representations. We also thank Ingrid Apel, Xiaojun Jing, and David O. Apiyo (Pall Life Sciences) for carrying out additional experiments that were not used for the final manuscript. We also benefited from discussions with Denzil Bernard (structure-function) and John O’Bryan (University of Illinois; nucleotide loading). We thank Ester Fernandez-Salas for her inputs on the manuscript. We thank the University of Michigan Xenograft Core and Dr. Diane Simeone for providing PDX1319 cell line. S.P. was supported by IFOM Fondazione Istituto FIRC di Oncologia Molecolare, Milan, Italy (Sponsor: Fabrizio D’Adda di Fagagna). R.M. is supported by Prostate Cancer Foundation Young Investigator Award. Y.W. was supported by a Howard Hughes Medical Institute (HHMI) Medical Student Research Fellowship., and A.F. by a Damon Runyon Foundation Fellowship. A.M.C. is supported by the Alfred A. Taubman Institute and the HHMI. A.M.C. and K.S. are American Cancer Society Research Professors. This project is supported in part by NIH grants NIH 1R21 AI109791 (PI: N.G.W), RO1 CA154365 and R37 CA40046, and the Prostate Cancer Foundation (PI: A.M.C).
Footnotes
AUTHOR CONTRIBUTIONS
S.S. performed RAS/AGO2 immunoprecipitation, in vitro binding, cellular fractionation, immunoblot analyses, RAS/AGO2 knockdown and overexpression cell biology based experiments. S.S. jointly conceived the study with C.K.S. and A.M.C. let-7 based assays were carried out by S.P.; immunofluorescence analyses, S.P. and R.M.; sucrose density fractionation, R.M.; cloning and construct generation, V.K.; NIH3T3 AGO2−/− cells using CRISPR/Cas9, Y.H.; IP-mass spectrometric analysis, A.K.Y.; experimental assistance and replication of studies, H.G.; microRNA sequencing and analysis, X.C. and S.M.D.; Recombinant protein preparation and purification, J.A.S.; KRAS reagents and advice on KRAS biology Y.W., A.F., K.S., and G.B.; Single molecule analysis S.P. and N.G.W.; Manuscript preparation, S.S., C.K-S. and A.M.C.; Funding and overall supervision of the study, A.M.C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983;303:72–74. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- Barcelo C, Etchin J, Mansour MR, Sanda T, Ginesta MM, Sanchez-Arevalo Lobo VJ, Real FX, Capella G, Estanyol JM, Jaumot M, et al. Ribonucleoprotein HNRNPA2B1 Interacts With and Regulates Oncogenic KRAS in Pancreatic Ductal Adenocarcinoma Cells. Gastroenterology. 2014 doi: 10.1053/j.gastro.2014.06.041. [DOI] [PubMed] [Google Scholar]
- Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol. 2012;14:266–275. doi: 10.1038/ncb2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick JA, Salomon WE, Ryder SP, Aronin N, Zamore PD. Argonaute protein identity and pairing geometry determine cooperativity in mammalian RNA silencing. RNA. 2011;17:1858–1869. doi: 10.1261/rna.2778911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MC, Sun Q, Daniels RN, Camper D, Kennedy JP, Phan J, Olejniczak ET, Lee T, Waterson AG, Rossanese OW, Fesik SW. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc Natl Acad Sci U S A. 2014;111:3401–3406. doi: 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, Poliakov A, Yocum AK, Li Y, Chen W, et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 2014;5:3127. doi: 10.1038/ncomms4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CM, Li H, Gasman S, Huang J, Schiff R, Chang EC. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol. 2011;31:983–997. doi: 10.1128/MCB.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- COSMIC. Catalog of Somatic Mutations in Cancer, COSMIC Release v66. Wellcome Trust Sanger Institute; 2013. [Google Scholar]
- Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFeo D, Gonda MA, Young HA, Chang EH, Lowy DR, Scolnick EM, Ellis RW. Analysis of two divergent rat genomic clones homologous to the transforming gene of Harvey murine sarcoma virus. Proc Natl Acad Sci U S A. 1981;78:3328–3332. doi: 10.1073/pnas.78.6.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091–1104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley NR, Goldstein B. RNA interference: silencing in the cytoplasm and nucleus. Curr Opin Mol Ther. 2003;5:113–117. [PubMed] [Google Scholar]
- Gagnon KT, Li L, Chu Y, Janowski BA, Corey DR. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014;6:211–221. doi: 10.1016/j.celrep.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfinger LE, Ptak C, Jeffery ED, Shabanowitz J, Han J, Haling JR, Sherman NE, Fox JW, Hunt DF, Ginsberg MH. An experimentally derived database of candidate Ras-interacting proteins. J Proteome Res. 2007;6:1806–1811. doi: 10.1021/pr060630l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand PH, Thor A, Wunderlich D, Muraro R, Caruso A, Schlom J. Monoclonal antibodies of predefined specificity detect activated ras gene expression in human mammary and colon carcinomas. Proc Natl Acad Sci U S A. 1984;81:5227–5231. doi: 10.1073/pnas.81.16.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock J, Weinmann L, Ender C, Rudel S, Kremmer E, Raabe M, Urlaub H, Meister G. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007;8:1052–1060. doi: 10.1038/sj.embor.7401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horman SR, Janas MM, Litterst C, Wang B, MacRae IJ, Sever MJ, Morrissey DV, Graves P, Luo B, Umesalma S, et al. Akt-mediated phosphorylation of argonaute 2 downregulates cleavage and upregulates translational repression of MicroRNA targets. Mol Cell. 2013;50:356–367. doi: 10.1016/j.molcel.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng HH, Taylor LJ, Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun. 2012;3:1168. doi: 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Karachaliou N, Mayo C, Costa C, Magri I, Gimenez-Capitan A, Molina-Vila MA, Rosell R. KRAS mutations in lung cancer. Clin Lung Cancer. 2013;14:205–214. doi: 10.1016/j.cllc.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Maizel A, Chen X. Traffic into silence: endomembranes and post-transcriptional RNA silencing. EMBO J. 2014;33:968–980. doi: 10.1002/embj.201387262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak PB, Tomari Y. The N domain of Argonaute drives duplex unwinding during RISC assembly. Nat Struct Mol Biol. 2012;19:145–151. doi: 10.1038/nsmb.2232. [DOI] [PubMed] [Google Scholar]
- Lampson BL, Pershing NL, Prinz JA, Lacsina JR, Marzluff WF, Nicchitta CV, MacAlpine DM, Counter CM. Rare codons regulate KRas oncogenesis. Curr Biol. 2013;23:70–75. doi: 10.1016/j.cub.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauchle JO, Braun BS, Loh ML, Shannon K. Inherited predispositions and hyperactive Ras in myeloid leukemogenesis. Pediatr Blood Cancer. 2006;46:579–585. doi: 10.1002/pbc.20644. [DOI] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Ma E, Zhou M, Robinson CV, Doudna JA. In vitro reconstitution of the human RISC-loading complex. Proc Natl Acad Sci U S A. 2008;105:512–517. doi: 10.1073/pnas.0710869105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras. GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- McCarthy N. MicroRNA: lacking in maturity. Nat Rev Cancer. 2013;13:377. doi: 10.1038/nrc3534. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Luhrmann R, Tuschl T. Identification of novel argonaute-associated proteins. Curr Biol. 2005;15:2149–2155. doi: 10.1016/j.cub.2005.10.048. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139:112–122. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershing NL, Lampson BL, Belsky JA, Kaltenbrun E, MacAlpine DM, Counter CM. Rare codons capacitate Kras-driven de novo tumorigenesis. J Clin Invest. 2015;125:222–233. doi: 10.1172/JCI77627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L, Meister G. Argonaute proteins: mediators of RNA silencing. Mol Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Pitchiaya S, Androsavich JR, Walter NG. Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO Rep. 2012;13:709–715. doi: 10.1038/embor.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Krishnan V, Custer TC, Walter NG. Dissecting non-coding RNA mechanisms in cellulo by Single-molecule High-Resolution Localization and Counting. Methods. 2013;63:188–199. doi: 10.1016/j.ymeth.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Hancock JF. Ras trafficking, localization and compartmentalized signalling. Semin Cell Dev Biol. 2012;23:145–153. doi: 10.1016/j.semcdb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel S, Flatley A, Weinmann L, Kremmer E, Meister G. A multifunctional human Argonaute2-specific monoclonal antibody. RNA. 2008;14:1244–1253. doi: 10.1261/rna.973808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel S, Wang Y, Lenobel R, Korner R, Hsiao HH, Urlaub H, Patel D, Meister G. Phosphorylation of human Argonaute proteins affects small RNA binding. Nucleic Acids Res. 2011;39:2330–2343. doi: 10.1093/nar/gkq1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497:383–387. doi: 10.1038/nature12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh A, Ward AF, Donlan KL, Harding-Theobald ER, Xu J, Mullighan CG, Zhang C, Chen SC, Su X, Downing JR, et al. Defective K-Ras oncoproteins overcome impaired effector activation to initiate leukemia in vivo. Blood. 2013;121:4884–4893. doi: 10.1182/blood-2012-05-432252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature. 1981;290:261–264. doi: 10.1038/290261a0. [DOI] [PubMed] [Google Scholar]
- Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H. Small-molecule modulation of Ras signaling. Nat Chem Biol. 2014;10:613–622. doi: 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet P, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RW, Yokoyama S, Kamata T, Feramisco JR, Rosenberg M, Gross M. The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature. 1984;311:273–275. doi: 10.1038/311273a0. [DOI] [PubMed] [Google Scholar]
- Symonds JM, Ohm AM, Carter CJ, Heasley LE, Boyle TA, Franklin WA, Reyland ME. Protein kinase C delta is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res. 2011;71:2087–2097. doi: 10.1158/0008-5472.CAN-10-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987;238:542–545. doi: 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- Vasilescu J, Guo X, Kast J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics. 2004;4:3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- Wang B, Li S, Qi HH, Chowdhury D, Shi Y, Novina CD. Distinct passenger strand and mRNA cleavage activities of human Argonaute proteins. Nat Struct Mol Biol. 2009;16:1259–1266. doi: 10.1038/nsmb.1712. [DOI] [PubMed] [Google Scholar]
- Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu Rev Biophys. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Haase AD, Huang FK, Coulis G, Rivera KD, Dickinson BC, Chang CJ, Pappin DJ, Neubert TA, Hannon GJ, et al. Dephosphorylation of tyrosine 393 in argonaute 2 by protein tyrosine phosphatase 1B regulates gene silencing in oncogenic RAS-induced senescence. Mol Cell. 2014;55:782–790. doi: 10.1016/j.molcel.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Sankala H, Zhang X, Graves PR. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem J. 2008;413:429–436. doi: 10.1042/BJ20080599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.