

Abstract
During the perinatal period, the brain is highly vulnerable to hypoxia and inflammation, which often cause white matter injury and long-term neuronal dysfunction such as motor and cognitive deficits or epileptic seizures. We studied the effects of moderate hypoxia (HYPO), mild systemic inflammation (INFL), or the combination of both (HYPO+INFL) in mouse somatosensory cortex induced during the first postnatal week on network activity and compared it to activity in SHAM control animals. By performing in vitro electrophysiological recordings with multi-electrode arrays from slices prepared directly after injury (P8–10), one week after injury (P13–16), or in young adults (P28–30), we investigated how the neocortical network developed following these insults. No significant difference was observed between the four groups in an extracellular solution close to physiological conditions. In extracellular 8 mM potassium solution, slices from the HYPO, INFL, and HYPO+INFL group were more excitable than SHAM at P8–10 and P13–16. In these two age groups, the number and frequency of spontaneous epileptiform events were significantly increased compared to SHAM. The frequency of epileptiform events was significantly reduced by the NMDA antagonist D-APV in HYPO, INFL, and HYPO+INFL, but not in SHAM, indicating a contribution of NMDA receptors to this pathophysiological activity. In addition, the AMPA/kainate receptor antagonist CNQX suppressed the remaining epileptiform activity.
Electrical stimulation evoked prominent epileptiform activity in slices from HYPO, INFL and HYPO+INFL animals. Stimulation threshold to elicit epileptiform events was lower in these groups than in SHAM. Evoked events spread over larger areas and lasted longer in treated animals than in SHAM. In addition, the evoked epileptiform activity was reduced in the older (P28–30) group indicating that cortical dysfunction induced by hypoxia and inflammation was transient and compensated during early development.
Keywords: Hypoxia, systemic inflammation, Interleukin-1β, multi-electrode array, electrophysiology, epileptiform activity, barrel cortex, mouse, development, in vitro
Introduction
The perinatal period is a critical phase of development during which the brain is highly vulnerable to pathophysiological conditions, such as hypoxia or systemic inflammation, and can cause long-term neurological disabilities (Volpe, 2001; Back and Miller, 2014). Hypoxia during early development commonly leads to epileptic seizures (Garfinkle and Shevell, 2011) and these seizures may cause permanent cognitive and neurological deficits (Glass et al., 2009; Vesoulis et al., 2014). Besides hypoxia, systemic inflammation during early development also causes prominent structural changes such as white matter damage, and long-term cognitive and behavioral impairment (Dammann and Leviton, 2004; Glass et al., 2008). In addition, pro-inflammatory cytokines have been shown to disrupt not only myelination (Favrais et al., 2011), but also spontaneous neuronal activity and programmed cell death in the developing cortex (Nimmervoll et al., 2013).
In order to study the consequences of early developmental hypoxia and systemic inflammation, different animal models were investigated in the past that mimic these pathological events at diverse degrees of severity (Dingley et al., 2006; Salmaso et al., 2014). However, the effects of mild hypoxia were only rarely investigated (Zehendner et al., 2013a). We have previously shown that systemic inflammation induces changes in spontaneous network activity, which subsequently triggers apoptosis in the neonatal rat cerebral cortex in vitro and in vivo (Nimmervoll et al., 2013). Little is known on the effects of mild systemic inflammation in combination with moderate hypoxia, although this condition is clinically most relevant in preterm human neonates (Glass et al., 2008; Volpe, 2009). We addressed this question in the rodent somatosensory barrel cortex, which is organized into anatomically distinguishable functional layers and columns, thus making it a powerful model to investigate spatiotemporal alterations in neocortical activity following hypoxia and systemic inflammation (for review, see Feldmeyer et al., 2013).
To unravel the functional consequences of mild hypoxia and moderate systemic inflammation on neuronal excitability during early development, we examined the spatiotemporal properties of neuronal activity in somatosensory cortex slices using 60-channel 3D-microelectrode arrays (MEAs) (Sun and Luhmann, 2007). We used thalamocortical slices containing the primary somatosensory barrel cortex from mice of different ages treated with hypoxia and/or the pro-inflammatory cytokine Interleukin-1β (IL-1β) during the first postnatal week. Our results show that IL-1β treatment or moderate hypoxia transiently alters the neocortical network properties in young animals during the second postnatal week by increasing the firing activity and by inducing spontaneous epileptiform activity. These pathophysiological effects were only transient and could no longer be observed in one month old mice. Moreover, the combination of both treatments did not induce any further dysfunction, indicating that hypoxia and inflammation may act through the same pathways. Our data suggest that similar therapeutic approaches might be administered to counteract these injuries.
Materials and Methods
Animal treatments
All experiments were conducted in accordance with the national and European laws for the use of animals in research (86 / 609 / EEC) and were approved by the local ethical committee. Mice were kept under a 12:12 h light-dark cycle in a temperature-controlled room at 22 ± 2 °C. Standard food pellets and tap water were available ad libitum.
Hypoxia and systemic inflammation were performed on male and female C57BL/6 mice aged postnatal day (P) 1 to P7. For the induction of a systemic inflammation, animals were treated with 10 μl/g of phosphate-buffered saline (PBS) containing 10 μg/kg/injection of recombinant mouse Interleukin-1β (IL-1β, R&D Systems, Minneapolis, MN) injected intraperitoneally once per day from P1 to P6 as described elsewhere (Favrais et al., 2011). SHAM animals were treated in the same way except that they received injections of Ringer solution. For the induction of hypoxia, P7 mice were placed in an incubator (Labotect C16, Labotect, Göttingen, Germany) and exposed to either hypoxic (37°C, 8% O2, 0.04% CO2) or normoxic conditions (37°C, 21% O2, 0.04% CO2) for 50 minutes as previously described (Devoto et al., 2013; Dingley et al., 2006). Oxygen was replaced by nitrogen to achieve 8% O2, a level that was continuously controlled. Each animal received one out of four different treatment combinations: IL-1β and hypoxia (HYPO+INFL, N=21 mice), IL-1β and normoxia (INFL, N=41), Ringer-solution and hypoxia (HYPO, N=23), Ringer-solution and normoxia (SHAM animals, N=25). After treatments, animals were placed back to their mother. To track animal identity, animals of the same litter were marked differently every two days on one or several paws with ink.
In vitro experiments were performed on mice aged between P8 and P30. No significant difference was found in the body weight of animals between the four experimental conditions in the three age groups P8–10, P13–16, and P28–30 (Table 1). All experiments and data analyses were performed blinded. After all experiments were finished, the treatment of each animal was revealed and data obtained from animals with the same treatments were pooled together. The number of animals (N animals) and slices (N slices) studied at each age group and from each experimental condition are summarized in Table 1.
Table 1.
Number of animals (N) and slices (n) per age group and per experimental condition and average weight. Percentage of slices with spontaneous epileptiform activity is given for experiments performed in 3 mM and 8 mM extracellular K+.
| Age group | Experimental group | Weight (g) | N animals | n Slices | Slices with epileptiform events in 3 mM KCl ACSF | Slices with epileptiform events in 8 mM KCl ACSF | ||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| % | N | % | N | |||||
| P 8–10 | SHAM | 5.42 ± 0.42 | 6 | 7 | 29 | 2/7 | 43 | 3/7 |
| HYPO | 4.52 ± 0.20 | 14 | 18 | 44 | 8/18 | 83 * | 15/18 | |
| INFL | 4.33 ± 0.21 | 8 | 9 | 33 | 3/9 | 89 * | 8/9 | |
| HYPO+INFL | 4.64 ±0.2 | 8 | 12 | 50 | 6/12 | 83 * | 10/12 | |
|
| ||||||||
| P 13–16 | SHAM | 6.06 ± 0.46 | 5 | 7 | 29 | 2/7 | 57 | 4/7 |
| HYPO | 7.48 ± 0.3 | 14 | 21 | 52 | 11/21 | 62 | 13/21 | |
| INFL | 6.83 ± 0.44 | 6 | 8 | 88 | 7/8 | 100 * | 8/8 | |
| HYPO+INFL | 6.93 ± 0.41 | 10 | 17 | 65 | 11/17 | 94 * | 16/17 | |
|
| ||||||||
| P 28–30 | SHAM | 17.45 ± 0.47 | 10 | 10 | 10 | 1/10 | 10 | 1/10 |
| HYPO | 15.45 ± 0.78 | 13 | 17 | 12 | 2/17 | 29 | 5/17 | |
| INFL | 16.1 ± 0.81 | 9 | 11 | 18 | 2/11 | 18 | 2/11 | |
| HYPO+INFL | 14.41 ± 0.8 | 7 | 10 | 10 | 1/10 | 30 | 3/10 | |
Dunns test:
p<0.05
Slice preparation and in vitro electrophysiology
Thalamocortical slices including primary somatosensory cortex were prepared as described previously (Agmon and Connors, 1991). Animals were anaesthetized with isoflurane and decapitated. The brain was rapidly removed and transferred to oxygenated (95% O2 / 5% CO2), ice-cold artificial cerebrospinal fluid (ACSF) containing the following compounds (in mM): NaCl, 124; KCl, 3; CaCl2, 2; MgCl2, 2; NaHCO3, 26; NaH2PO4, 1.25; glucose, 20 (pH 7.4). Slices of 300–350 μm thickness were cut in oxygenated, ice-cold ACSF with a vibratome (Microm HM 650V, Thermo Scientific, United States) and transferred to an incubation chamber containing oxygenated ACSF at room temperature. Slices were briefly examined using a Zeiss Axioskop microscope (Carl Zeiss AG, Germany) and only slices with visible barrels in differential interference contrast microscopy were used for subsequent recordings. After an incubation period of at least one hour, slices were transferred to a 3D 60-channel multi-electrode array (MEA; inter-electrode spacing 200 μm, electrode impedance 450–650 kOhm, Ayanda Biosystems, Lausanne, Switzerland) on a MEA 1060-INV-BC interface (Multi Channel Systems [MCS], Reutlingen, Germany), which was mounted on an inverted microscope (Olympus, Tokyo, Japan) equipped with a digital camera. A picture of the slice on the MEA allowed subsequent identification of the different layers and their positions relative to the electrodes. Slices were perfused at a rate of 2–3 ml/min with the same ACSF solution used for slicing or with ACSF containing 8 mM KCl to increase excitability. Experiments were performed at 35°C held constant by a temperature control unit (TCO2 and TH01, MCS). Before starting recordings, slices recovered on the MEA for at least 15 min, which allowed the establishment of a stable contact between the tissue and the electrodes.
Spontaneous activity was recorded simultaneously with all 60 electrodes at a sampling rate of 25 kHz for a duration of 10 minutes. For recording of field potentials, a 10 Hz high-pass filter of the raw data was applied using the MC_Rack software (MCS). Spike detection was performed online by MC_Rack software using a band-pass filtering of the raw electrode signal (300–3000 Hz) and a threshold-based detector set to 7 standard deviations of the noise level. Monopolar electrical stimulations were performed in a barrel via a single microelectrode internal to the MEA. Biphasic voltage pulses of 2×100 μs duration were applied via a stimulator unit (STG 2004, MCS). By increasing the stimulation intensities from 250 mV to 4 V, we obtained input/output curves by measuring the relative amplitude of the evoked field potentials recorded in layer 2/3 of the stimulated cortical column. Synaptic fatigue was measured by applying a single train of 100 stimuli at 10 Hz.
For the detection of spontaneous epileptiform events (EEs), band-pass (300–3000 Hz) filtered data were analyzed. Cortical bursts were defined as a discharge consisting of at least four spikes with an interspike interval (ISI) shorter than 100 ms (Wagenaar et al., 2006). Spike datasets from all electrodes were imported into Matlab 7.7 (Mathworks, Natick, MA, USA) for analysis using a custom written routine. To be classified as an epileptiform event, a burst needed to last more than 200 ms and had to be detected on at least two neighboring cortical columns of at least 3 different electrodes monitoring activity in neocortical layers 2/3, 4, 5 and 6 (Chang et al., 2011).
Pharmacology and compounds
The different antagonists used in this study were bath applied in ACSF containing 8 mM KCl. Each compound was applied for at least 12 minutes before recording was started. 2-amino-5-phosphonopentanoic acid (D-APV, 50 mM, BIOTREND Chemikalien GmbH, Cologne, Germany), bicuculline bethiodide (Bic, 10 mM, BIOTREND Chemikalien GmbH, Cologne, Germany) stock solutions were prepared in double-distilled water; the stock solution of 7-nitro-2,3-dioxo-1,4-dihydroquinoxaline-6-carbonitrile (CNQX, 10 mM, BIOTREND Chemikalien GmbH, Cologne, Germany) was prepared in DMSO. Glucose, MgCl2.6H2O, KCl and Roti-Histofix, NaCl, NaHCO3 were purchased from Sigma (Sigma Aldrich Chemie GmbH, Steinheim, Germany). CaCl2.2H2O, NaH2PO2.H2O were purchased from Merck (Merck KGaA, Darmstadt, Germany).
Histology and immunochemistry
After the electrophysiological experiments, slices were carefully removed from the MEA and fixed for at least 24 h in 4% paraformaldehyde solution (Carl Roth GmbH + Co KG, Karlsruhe, Germany) at 4°C (pH 7.4). Digital photographs of the cortical slices on the MEA were used to align photographs of the Nissl-stained sections to the 60 recording sites. This procedure allowed layer-specific identification of each electrode. Slices were washed in 0.1 M phosphate-buffer (PB, pH 7.4) solution, incubated overnight in 0.1 M PB containing 30% sucrose and subsequently washed for 3×20 min in 0.1 M phosphate buffer saline (PBS). Slices were Nissl stained or stained against the vesicular glutamate transporter 2 (VGLUT2) and DAPI. For Nissl staining, free-floating sections were incubated in 0.5 ml staining solution before being mounted on microscope slides. Images were taken with a 20X objective at an Olympus IX81 epifluorescence microscope. For VGLUT2 staining, overnight staining was performed at 4°C with primary antibodies diluted in 2% bovine serum albumin with 0.05% azide and 0.1% triton. Then, slices were washed three times with PBS and incubated in DAPI and DyLight488 coupled secondary antibodies diluted in 2% bovine serum albumin with 0.05% azide in PBS at RT for 2 h. After washing in PBS, slices were mounted on microscope slides.
Statistical analysis
All data are presented as mean ± S.E.M. The number of animals (N) and the number of slices (n) in each age group and each experimental condition is given in Table 1. All statistical tests were performed using Prism 5 (GraphPad, La Jolla, CA, USA). The effects of the different treatments were analyzed by Kruskal-Wallis one-way ANOVA on ranks followed by Dunn’s method for posthoc comparisons. Significance was considered at p values of p<0.05 (*) and p<0.01 (**).
Results
The conditions of hypoxia and systemic inflammation used in the present work had no significant effect on the survival rate of animals (p=0.06666, Kruskal-Wallis ANOVA), since some animals from all four groups died during the period of treatments (P1–7). Five out of 63 HYPO animals (8%) and six out of 60 HYPO+INFL mice (10%) died during exposure to hypoxia. From 42 animals having received systemic inflammation (7%), three died during the period of daily administration of IL-1β and one out of 34 SHAM animals (3%) receiving saline solution died during the same period. This suggests that the mild treatments used in our experiments were not sufficient to induce lethal damage.
To test if hypoxia and/or inflammation altered the activity of neuronal populations we recorded neuronal activity in brain slices of primary somatosensory cortex using a 60 channel multielectrode array (MEA) (Figure 1A). We investigated whether spontaneous activity, but also evoked responses to local electrical stimulation in slices from HYPO, INFL, and HYPO+INFL animals differ from activity in SHAM animals.
Figure 1. Hypoxia and/or systemic inflammation increase the spontaneous firing rate.

A) Representative picture of a thalamocortical slice on a MEA prepared from a mouse of the P8–10 age group. B) Spike sorting of individual action potentials recorded over a 10 minute period from the slice in (A) on each of the 60 electrodes show low activity present in all cortical layers and in the hippocampus. Some electrodes could record the activity of more than one neuron. C), D), and E) Evolution of the MUA (in Hz) for neurons located in cortical layers 2 to 6 during 5 minutes recordings in ACSF with 8 mM KCl in representative slices from the P8–10 group (C), the P13–16 group (D), and the P28–30 group (E). For each condition, a raster plot shows the MUA on all MEA electrodes located in all layers of the Barrel cortex (bottom) with the total spike frequency of all these channels (bin: 1 sec, top). Note the peaks of activity corresponding to epileptiform events. Spikes occurring during these events were not counted in the spontaneous firing rate. F) Average firing rate per layers of the Barrel cortex recorded in thalamocortical slices from mice of P8–10 (left), 13–16 (middle) and P28–30 age groups (right) treated with all four conditions: SHAM (dark grey), HYPO (blue), INFL (red) and HYPO+INFL (green). G) The mean cortical firing rate in a ten minute recording period per slice and condition is plotted as box and whisker plots. For each age group, the mean firing rate is plotted for 3 mM (open boxes) and 8 mM (filled boxes) KCl ACSF. The numbers of slices recorded per condition are represented on the top of the graph.
Spontaneous activity
Hypoxia and inflammation transiently increase spontaneous firing rate
We first recorded spontaneous activity to explore whether differences in activity levels and patterns would emerge following treatments in absence of stimulation. The ongoing activity was composed of isolated action potentials recorded at a given electrode (Figure 1A and B) and epileptiform events (EEs), occurring simultaneously at many electrodes (Figure 1C, D and E). Spontaneous firing rate in layer (L) 2/3, 4, 5 and 6 of the barrel cortex was recorded for ten minutes in slices prepared from animals of the four experimental groups and in the three different age groups. In conditions mimicking a physiological extracellular solution (ACSF with 3 mM KCl), neurons in L2/3, L4, L5 and L6 displayed spike activity with a firing rate independent of the treatment conditions at P8–10 and P13–16 (Figure 1F). In P8–10 mice, the firing rate was similar between SHAM (0.16 ± 0.06 Hz, n=7 slices), HYPO (0.10 ± 0.03 Hz, n=18 slices), INFL (0.09 ± 0.02 Hz, n=8 slices) and HYPO+INFL (0.17 ± 0.04 Hz, n=11 slices, Figure 1G). Compared to P13–16 SHAM animals (0.22 ± 0.07 Hz, n=7 slices), the average firing rate in all cortical layers was lower in INFL animals (0.05 ± 0.01 Hz, n=7). In slices of the P28–30 group, the average neuronal activity was similar in all layers and only few neurons fired action potentials. These results suggest that inflammation alone depressed spontaneous firing rates in the P13–16 group.
Since steady state firing rates do not allow assessment of the dynamic abilities of the neuronal network, we tested the effects of increasing excitability on spontaneous activity. We increased excitability by using ACSF with 8 mM KCl. Despite these conditions, there was no change in the SHAM animals at P8–10. For the three other conditions, the firing rate increased in this high potassium solution in comparison to 3 mM KCl-ACSF. In SHAM animals, the firing rate was stable (0.28 ± 0.09 Hz, n=7), whereas it significantly increased by 208% (0.34 ± 0.08 Hz, n=18, p<0.01) in HYPO, 263% (0.48 ± 0.21 Hz, n=9, p<0.05) in INFL, and 175% (0.28 ± 0.05 Hz, n=12, p<0.05) in HYPO+INFL slices (Figure 1G). Similarly, this increase in firing rates was also present in P13–16 animals in HYPO (164%, 0.32 ± 0.08 Hz, n=20, p<0.05), INFL (260%, 0.41 ± 0.20 Hz, n=8, p<0.05) and HYPO+INFL (138%, 0.41 ± 0.09 Hz, n=16, p<0.01), but not in SHAM (38%, 0.25 ± 0.06 Hz, n=7). In P28–30 mice, the firing frequency remained stable in every condition. Overall, the firing rate increased more drastically in slices from HYPO, INFL and HYPO+INFL compared to SHAM after changing from 3 mM to 8 mM KCl-ACSF, suggesting that animals with injuries have a hyperexcitable neocortical network (Figure 1G). Interestingly, comparing the results of the INFL and HYPO condition with the HYPO+INFL condition shows that the changes are not additive, suggesting that similar pathways are activated by these two pathophysiological conditions.
Hypoxia and/or inflammation transiently increase spontaneous epileptiform activity
In addition to single action potentials, spontaneous EEs were present in slices from the 3 age groups of the 4 experimental groups (Table 1 and Figure 2). In 3 mM KCl-ACSF, synchronous epileptiform activity was observed in slices prepared from animals of all four conditions. No difference in the number of slices presenting spontaneous epileptiform activity could be observed between the 4 experimental conditions at any age. The presence of EEs in several SHAM slices with 3 mM KCl-ACSF might be explained by remaining spontaneous cortical oscillatory waves that normally occur during the first week of development (Garaschuk et al., 2000). This activity could still be present is some slices of all four treatment groups. The percentage of slices with EEs was ≤50% in all slices at P8–10 (SHAM: 2/7 slices, HYPO: 8/18 slices, INFL: 3/9 slices, HYPO + INFL: 6/12 slices, Table 1). The number of slices with EEs increased at P13–16. At this age, 29% of SHAM slices showed epileptiform activity (2/7 slices), which was less than in HYPO (11/21 slices, 52%), INFL (7/8 slices, 88%), and HYPO+INFL (11/17 slices, 65%; p=0.0246, Kruskal-Wallis ANOVA). The percentage of slices with EEs in all conditions in P28–30 slices did not exceed 18% (see Table 1).
Figure 2. Hypoxia and/or systemic inflammation significantly increase spontaneous epileptiform activity in slices from P8–10 and P13–16 animals.

A), B), and C) Representative MEA recordings of spontaneous epileptiform events occurring in the Barrel cortex of thalamocortical slices perfused with ACSF containing 8 mM KCl from a P8–10 mouse (A), P13–16 mouse (B), and P28–30 mouse (C) for all four treatment conditions. For each condition, a 30 s recording on all 60 electrodes is showed with epileptiform events occurring (top), the trace on one electrode marked by a dark gray square is magnified (middle), and one single epileptiform recorded from this electrode marked in light gray is further magnified (bottom). D) The mean frequencies of epileptiform events occurrence in a ten minute recording period per slice and condition is plotted as box and whisker plots. For each age group, the mean number of occurrence is plotted for 3 mM (open boxes) and 8 mM (filled boxes) KCl-ACSF for SHAM (dark grey), HYPO (blue), INFL (red) and HYPO+INFL (green). Dunns test: **, p<0.01; *, p<0.05.
In recordings performed with 8 mM KCl-ACSF, at P8–10, the number of slices with EEs was significantly higher in slices from HYPO (8/18 slices), INFL (3/9 slices) and HYPO+INFL (8/12 slices) animals compared to SHAM (2/7 slices) animals (p=0.0174, Kruskal-Wallis ANOVA, Table 1). Similarly, in P13–16 mice, the number of slices with EEs was higher for INFL (8/8 slices) and HYPO+INFL (16/17 slices) animals compared to SHAM (3/7 slices) animals (p=0.0246, Kruskal-Wallis ANOVA), but not for HYPO (13/21 slices). In P28–30 mice, only a limited number of slices (10–30 %) displayed epileptiform activity in every condition (see Table 1). These data indicate that animals that experienced mild hypoxia, systemic inflammation, or both treatments develop a transient neocortical dysfunction involving the same pathways.
Since all experimental groups showed epileptiform activity in our in vitro recording conditions, we next investigated whether the frequency of these spontaneous EEs would be higher in slices of animals that experienced systemic inflammation, hypoxia, or both treatments compared to slices from SHAM animals (Figure 2). In ACSF with 3 mM KCl, the number of EEs within a 10 minute recording period did not significantly differ between conditions at P8–10 or P13–16 (Figure 2D). At P28–30, the number of EEs was very low (<5 EEs per 10 minutes) independent of the condition (Figures 2C, D). The number of EEs over a 10 minute period increased in 8 mM KCl-ACSF for INFL, HYPO, and HYPO+INFL but not SHAM slices from P8–10 and P13–16 mice (Figure 2). Examples of single EEs for each treatment in these recording conditions are shown at ages P8–10, P13–16 and P28–30 in Figure 2A, 2B and 2C, respectively. In slices from P8–10 animals, spontaneous EEs spread over a large number of channels, mainly in L4 and L5 (Figure 2A). In slices from INFL, the number of EEs was significantly higher (19.8 ± 6.0 EE/10 min, n=9) than in SHAM animals (4.0 ± 2.6 EE/10 min, n=7; p=0.0128, Kruskal-Wallis ANOVA), but not in HYPO (10.3 ± 4.5 EE/10 min, n=18) or in HYPO+INFL groups (7.8 ± 2.7 EE/10 min, n=12). At P13–16, EEs spread over all cortical layers (Figure 2B). Notably, in animals of this age, slices from HYPO (8.3 ± 3.0 EE/10 min, n=21) and HYPO+INFL (8.7 ± 3.0 EE/10 min, n=17) had a significantly higher frequency of EEs than slices from SHAM (1.4 ± 1.9 EE/10 min, n=7; p=0.0337, Kruskal-Wallis ANOVA), but not from INFL (5.8 ± 7.3 EE/10 min, n=8, Figure 2D). In P28–30 mice, EEs were rare and spread only over few electrodes and no effect of treatments was found (Figure 2C and D). Statistical differences between each group at all ages are summarized in Table 2. Together these results suggest that after HYPO and/or INFL there is a transient period during which an increase in excitability causes a higher rate of epileptiform bursts.
Table 2. Statistical summary of the significant effects induced by the treatments on the different parameters investigated.
This table gives the type of changes (upward arrow, increase; downward arrow, decrease) and statistical significances obtained using Dunn’s method for posthoc comparison between groups of the effects induced by the different treatments in comparison to slices from SHAM animals in ACSF with 8 mM KCl for all age groups.
| SHAM vs | P 8–10 | P 13–16 | P 28–30 | |
|---|---|---|---|---|
| % of slices with EEs | HYPO |
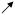 * *
|
ns | ns |
| INFL |
*
|
*
|
ns | |
| HYPO+ INFL |
*
|
*
|
ns | |
|
| ||||
| Number of EEs per 10 min | HYPO | ns |
*
|
ns |
| INFL |
*
|
ns | ns | |
| HYPO+ INFL | ns |
*
|
ns | |
|
| ||||
| Threshold | HYPO |
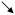 * *
|
*
|
ns |
| INFL | ns |
*
|
ns | |
| HYPO+ INFL |
*
|
*
|
ns | |
|
| ||||
| Duration EEs | HYPO |
*
|
ns | ns |
| INFL |
*
|
*
|
ns | |
| HYPO + INFL |
**
|
ns | ns | |
|
| ||||
| Number of Columns | HYPO | ns | ns | ns |
| INFL | ns |
*
|
ns | |
| HYPO + INFL |
*
|
**
|
ns | |
Dunns test:
p<0.05;
p<0.01; ns, not significant.
Increases in excitability could be due to increased synaptic excitation. We thus tested whether pharmacologically reducing excitatory neurotransmission would alter the rate of epileptiform bursts. We performed recordings of spontaneous activity in the presence of the NMDA receptor antagonist D-APV (50 μM). In SHAM animals of the P8–10 age group, D-APV reduced the frequency of EEs by 86% (0.6 ± 0.4 EE/10 min, n=7) in comparison to 8 mM KCl recording solution. Similar reductions in EE frequency were present in slices from HYPO (1.9 ± 1.4 EE/10 min, n=18, p<0.01; 81% reduction), INFL (5.22 ± 1.6 EE/10 min, n=7, p<0.05; 74% reduction) and HYPO+INFL condition (1.7 ± 1.1 EE/10 min, n=11, p<0.01; 79% reduction) (Figure 3B). No difference was observed between the different groups in the ability of D-APV to reduce the occurrence of EEs. The application of the AMPA/kainate antagonist CNQX (10 μM) together with D-APV blocked most epileptiform activity (Figure 3A). In P8–10 SHAM animals, epileptiform activity was maintained at a low level by addition of CNQX (0.7 ± 0.6 EE/10 min, n=6, p<0.05), whereas it was reduced by 96% (0.5 ± 0.3 EE/10 min, n=11, p<0.01) in HYPO, 98% (0.4 ± 0.3 EE/10 min, n=6, p<0.01) in INFL, and 96% (0.4 ± 0.4 EE/10 min, n=10, p<0.01) in HYPO+INFL animals of this age group (P8–10, Figure 3B). In animals of the P13–16 age group, EEs frequency remained unchanged in D-APV for SHAM (0.3 ± 0.3 EE/10 min, n=7, p>0.05) and INFL slices (2.6 ± 1.6 EE/10 min, n=8, p>0.05). However, D-APV significantly decreased EEs frequency in HYPO (88%, 1.0 ± 0.4 EE/10 min, n=18, p<0.01) and HYPO+INFL slices (84%, 1.4 ± 0.6 EE/10 min, n=15, p<0.01) of the P13–16 age group (Figure 3B). At this age group (P13–16), CNQX applied together with D-APV suppressed epileptiform activity in SHAM and reduced it further in HYPO to 30% (2.3 ± 1.7 EE/10 min, n=16, p<0.01), in INFL to 30% (1.9 ± 1.3 EE/10 min, n=7, p<0.05) and in HYPO+INFL to 38% (3.4 ± 2.2 EE/10 min, n=13, p<0.05). These results indicate that NMDA and AMPA/kainate glutamate receptors play a major role in epileptiform activity of P13–16 animals. In the P28–30 animals, APV applied alone or together with CNQX equally suppressed all EEs similarly in all conditions. In summary, these results indicate that NMDA receptors as well as AMPA/kainate receptors play a role (Figure 3) in the hyperexcitability of the neocortical network treated with hypoxia and/or inflammation.
Figure 3. Pharmacological properties of epileptiform events induced by hypoxia and inflammation.

A) Examples of EEs recorded from a slice prepared from a P13–16 HYPO animal with different recording conditions. For each of the recording solutions indicated, 500 ms recording traces show the activity recorded by each of the 60 electrodes of the MEA during a spontaneously occurring EE. The recording trace on the electrode marked by a grey square is magnified below. APV decreases the number of events compared to 8 mM KCl-ACSF alone while addition of CNQX suppresses it. B) The mean frequencies of EEs occurrence in a ten minute recording period per slice and per condition is plotted as box and whisker plots (SHAM, dark grey; HYPO, blue; INFL, red; HYPO+INFL, green). For each age group, the mean number of occurrence is plotted for 8 mM KCl ACSF (open boxes), APV alone (light filled boxes) or APV and CNQX together (dark filled boxes). Dunns test: **, p<0.01; *, p<0.05.
We next investigated if other mechanisms beyond glutamate contribute to the generation of EEs. Bicuculline (Bic) was applied in the bath solution to block GABAA inhibitory inputs and see whether inhibitory synapses were altered in treated groups, and whether there were differences between the treatments (Kilb et al., 2007). Figure 4A shows typical EEs in a slice from a HYPO mouse of the P8–10 age group in absence and presence of Bic. In P8–10 mice, in Bic the frequency of EEs occurrences was stable in slices of the SHAM group (4.8 ± 2.5 EE/10 min, n=4, p>0.05) as well as in other groups (HYPO: 3.8 ± 0.8 EE/10 min, n=4, p>0.05; INFL 5.0 ± 1.9 EE/10 min, n=4, p>0.05; HYPO+INFL: 3.8 ± 1.0 EE/10 min, n=5, p>0.05, Figure 4B). In P13–16 mice, Bic induced an increase in the frequency of EEs in slices of SHAM group (4.5 ± 1.7 EE/10 min, n=4, p<0.05), but not in the other groups (HYPO: 7.7 ± 3.0 EE/10 min, n=6, p>0.05; INFL 5.3 ± 3.0 EE/10 min, n=4, p>0.05; HYPO + INFL: 5.2 ± 2.5 EE/10 min, n=5, p>0.05, Figure 4B). In P28–30 mice, the frequency of EEs increased significantly in all conditions (SHAM: 5.0 ± 1.1 EE/10 min, n=4, p<0.01; HYPO: 3.8 ± 1.2 EE/10 min, n=6, p<0.01; INFL: 7.8 ± 4.2 EE/10 min, n=4, p<0.01; HYPO + INFL: 4.6 ± 2.3 EE/10 min, n=5, p<0.05; Figure 4B). These data demonstrate that in the P8–10 and P13–16 age group, pharmacological blockade of GABAA receptors does not induce a further increase in the expression of pathophysiological activity. Together these results show that while Bic affected EE frequency at older ages in all groups, mice from the P13–16 group showed a Bic induced increase in EEs frequency only for SHAM, but not HYPO, INFL, or HYPO+INFL.
Figure 4. Bicuculline increase EEs frequency and amplitude in all conditions.

Examples of EEs recorded from a slice prepared from a P8–10 HYPO animal in ACSF containing 8 mM KCl alone or together with bicuculline. For both recording solutions, 500 ms recording traces show the activity recorded by each of the 60 electrodes of the MEA during a spontaneous EE. The recording trace on the electrode marked by a grey square is magnified below. Note the larger spreading of the events with bicuculline, as well as the longer duration and higher amplitude. B) The mean frequencies of EEs occurrence in a ten minute recording period per slice and per condition is plotted as box and whisker plots (SHAM, dark grey; HYPO, blue; INFL, red; HYPO+INFL, green). For each age group, the mean number of occurrence is plotted for 8 mM KCl ACSF (open boxes), and 8 mM KCL-ACSF + Bic (filled boxes). C) Increase of the amplitude, D) duration, and E) number of electrodes with synchronous activity of epileptiform events in presence of Bic for each condition. Dunns test: **, p<0.01; *, p<0.05.
To further characterize the effects of Bic on EEs we compared the properties of EEs after Bic application in all conditions. In all age groups, the amplitude of EEs was significantly increased in the presence of Bic (Figure 4C). In P8–10 mice, the amplitude of EEs was increased threefold when changing from ACSF containing 8 mM KCl (SHAM: 31 ± 6 μV, n=7, HYPO: 32 ± 6 μV, n=18, INFL: 37 ± 3 μV, n=9, HYPO+INFL: 35 ± 6 μV, n=12) to 8 mM KCl ACSF + Bic (SHAM: 120 ± 15 μV, n=4, HYPO: 106 ± 12 μV, n=4, INFL: 108 ± 8 μV, n=4, HYPO+INFL: 114 ± 7 μV, n=5; p=0.0004, Kruskal-Wallis ANOVA). At P13–16, the amplitude of EEs in 8 mM KCl ACSF (SHAM: 28 ± 6 μV, n=7, HYPO: 37 ± 8 μV, n=20, INFL: 34 ± 5 μV, n=8, HYPO+INFL: 35 ± 2 μV, n=16) increased fourfold when Bic was applied to the solution (SHAM: 148 ± 11 μV, n=4, HYPO: 148 ± 26 μV, n=6, INFL: 145 ± 17 μV, n=4, HYPO+INFL: 145 ± 16 μV, n=5; p<0.0001, Kruskal-Wallis ANOVA). Similarly, at P28–30 the amplitude of EEs was increased fourfold by changing from 8 mM KCl ACSF (SHAM: 35 ± 3 μV, n=8, HYPO: 32 ± 3 μV, n=15, INFL: 34 ± 4 μV, n=11, HYPO+INFL: 37 ± 4 μV, n=10) to 8 mM KCl ACSF + Bic (SHAM: 157 ± 14 μV, n=4, HYPO: 130 ± 12 μV, n=6, INFL: 161 ± 14 μV, n=4, HYPO+INFL: 153 ± 9 μV, n=5; p=0.0004, Kruskal-Wallis ANOVA; Figure 4C).
At P8–10, the duration of EEs recorded in 8 mM KCl ACSF alone (SHAM: 148 ± 32 ms, n=7, HYPO: 111 ± 51 ms, n=18, INFL: 160 ± 30 ms, n=9, HYPO+INFL: 111 ± 28 ms, n=12) increased significantly when Bic was applied (SHAM: 372 ± 60 ms, n=4, HYPO: 352 ± 70 ms, n=4, INFL: 373 ± 62 ms, n=4, HYPO+INFL: 389 ± 99 ms, n=5; p=0.0021, Kruskal-Wallis ANOVA; Figure 4D). In P13–16 mice, the duration of EEs significantly increased by changing from 8 mm KCl ACSF (SHAM: 184 ± 17 ms, n=7, HYPO: 203 ± 18 ms, n=20, INFL: 185 ± 14 ms, n=8, HYPO+INFL: 178 ± 11 ms, n=16) to 8 mm KCl ACSF + Bic (SHAM: 332 ± 83 ms, n=4, HYPO: 283 ± 45 ms, n=6, INFL: 317 ± 84 ms, n=4, HYPO+INFL: 271 ± 26 ms, n=5; p=0.0175, Kruskal-Wallis ANOVA). At P28–30, the duration of EEs recorded in 8 mM KCl ACSF (SHAM: 169 ± 22 ms, n=8, HYPO: 154 ± 14 ms, n=15, INFL: 196 ± 13 ms, n=11, HYPO+INFL: 218 ± 34 ms, n=10) was also increased when Bic was applied (SHAM: 415 ± 102 ms, n=4, HYPO: 366 ± 86 ms, n=6, INFL: 349 ± 33 ms, n=4, HYPO+INFL: 368 ± 77 ms, n=5; p=0.0032, Kruskal-Wallis ANOVA; Figure 4D). At all ages, the increase of duration of EEs following application of Bic did not depend on the experimental treatment.
EEs induced by Bic spread over a large part of the cortical area. At P8–10, the number of electrodes recording synchronous activity in 8 mM KCl ACSF alone (SHAM: 12 ± 2 electrodes, n=7, HYPO: 18 ± 2 electrodes, n=18, INFL: 18 ± 2 electrodes, n=9, HYPO+INFL: 18 ± 3 electrodes, n=12) was increased after addition of Bic (SHAM: 27 ± 4 electrodes, n=4, HYPO: 24 ± 2 electrodes, n=4, INFL: 26 ± 3 electrodes, n=4, HYPO+INFL: 26 ± 3 electrodes, n=5; p=0.0044, Kruskal-Wallis ANOVA; Figure 4E). At P13–16, the number of electrodes recording simultaneously epileptiform events in 8 mM KCl ACSF (SHAM: 12 ± 2 electrodes, n=7, HYPO: 15 ± 2 electrodes, n=20, INFL: 16 ± 2 electrodes, n=8, HYPO+INFL: 15 ± 2 electrodes, n=16) was also increased by Bic (SHAM: 25 ± 3 electrodes, n=4, HYPO: 24 ± 3 electrodes, n=6, INFL: 24 ± 2 electrodes, n=4, HYPO+INFL: 25 ± 2 electrodes, n=5; p<0.0001, Kruskal-Wallis ANOVA). In P28–30 mice, EEs spread only to few electrodes in 8 mM KCl ACSF (SHAM: 3 ± 1 electrodes, n=8, HYPO: 4 ± 1 electrodes, n=15, INFL: 3 ± 1 electrodes, n=11, HYPO+INFL: 3 ± 1 electrodes, n=10) and propagated over a larger area when Bic was added (SHAM: 23 ± 2 electrodes, n=4, HYPO: 22 ± 3 electrodes, n=6, INFL: 23 ± 2 electrodes, n=4, HYPO+INFL: 20 ± 2 electrodes, n=5; p=0.0002, Kruskal-Wallis ANOVA; Figure 4E).
Altogether, blocking GABAA receptors affected the properties of EEs in SHAM, HYPO, INFL, and HYPO+INFL cortical slices similarly. The amplitude, duration and spreading of EEs were increased in all groups in a similar way. However, mice from the P13–16 group showed a Bic induced increase in EEs frequency only for SHAM, but not HYPO, INFL, or HYPO+INFL. Therefore, the inhibitory circuits involving GABAA receptors seem to be transiently impaired by the different treatments.
Evoked activity
Intact evoked synaptic transmission following hypoxia, inflammation or both
Our recordings of spontaneous activity indicate a higher level of excitability in animals treated with hypoxia and systemic inflammation in combination or separately. To investigate whether this increased excitability also manifested during evoked activity, we electrically stimulated L4 and recorded evoked responses in L2/3. Stimulation of a cortical column led to evoked field potentials in the principal and neighboring columns (Figure 5A). Although the input/output curves of all conditions showed slight variation with age, they had similar half-maximal stimulus amplitudes (Figure 5B). In ACSF with 3 mM KCl at P8–10, the half-maximum voltage did not vary between conditions (SHAM: 2.1 ± 0.1 V, n=6; HYPO: 2.1 ± 0.1 V, n=17; INFL: 1.8 ± 0.2 V, n=8; HYPO+INFL: 2.1 ± 0.2 V, n=10, p=0.3942, Kruskal-Wallis ANOVA). At P13–16, this voltage was lower but did not significantly change between groups (SHAM: 1.7 ± 0.1 V, n=7; HYPO: 1.8 ± 0.1 V, n=19; INFL: 1.8 ± 0.1 V, n=7; HYPO+INFL: 1.7 ± 0.1 V, n=16; p=0.1155, Kruskal-Wallis ANOVA). These values increased in the P28–30 age group and were similar in all experimental groups (SHAM: 2.0 ± 0.1 V, n=10; HYPO: 2.0 ± 0.1 V, n=17; INFL: 2.1 ± 0.2 V, n=11; HYPO+INFL: 2.0 ± 0.1 V, n=10; Figure 5B).
Figure 5. Evoked field response is not altered by hypoxia and/or systemic inflammation.

A) Example traces of the evoked field potentials recorded by 5 electrodes in the layers 2/3 and 4 surrounding the electrically stimulated electrode in layer 4 (2 V, channel without recording trace) for each age and treatment condition (SHAM, dark grey; HYPO, blue; INFL, red; HYPO+INFL, green) in ACSF with 3 mM KCl. The field potentials marked by a colored square, corresponding to the layer 2/3 of the stimulated column, were used to calculate the input-output curves. B) Input-output curves plotted for all four treatments and all three age groups. Slices were stimulated with a monopolar biphasic stimulus in layer 4 varying between 250 mV and 4 V. The half maximum voltage was similar for all condition in each age group. C) Amplitude of the evoked field potentials for each age group and each condition in 3 mM KCl-ACSF (open boxes), 8 mM KCl-ACSF (light filled boxes) and 8 mM KCl-ACSF + 50 μM APV (dark filled boxes). Dunns test: **, p<0.01; *, p<0.05.
We additionally measured the amplitude of the evoked field responses in L2/3 of the activated cortical column. The results showed high variations between the age groups in the trough-to-peak amplitude of the evoked response evoked at a 2 V stimulus intensity (Figure 5C). At P8–10, the response amplitudes were low, but similar for SHAM, HYPO, INFL, and HYPO+INFL animals (SHAM: 273 ± 88 μV, n=5, HYPO: 387 ± 70 μV, n=13; INFL: 267 ± 59 μV, n=8; HYPO+INFL: 233 ± 66 μV n=11; p=0.6644, Kruskal-Wallis ANOVA). At P13–16, the amplitudes were higher than P8–10, but similar between all four conditions (SHAM: 762 ± 209 μV, n=6; HYPO: 736 ± 123 μV, n=18; INFL: 872 ± 130 μV, n=5; HYPO+INFL: 846 ± 151 μV n=15; p=0.7456, Kruskal-Wallis ANOVA). At P28–30, response amplitudes were also similar in the four groups (SHAM: 490 ± 93 μV, n=10; HYPO: 616 ± 112 μV, n=17; INFL: 532 ± 93 μV, n=11; HYPO+INFL: 621 ± 132 μV n=10; p=0.9257, Kruskal-Wallis ANOVA). These data indicate that on a macroscopic level the synaptic connection between L4 and L2/3 at a given age is not impaired by the different experimental treatments.
Hypoxia and/or inflammation does not alter short-term synaptic plasticity
To investigate whether brain injury close to birth impairs short-term synaptic plasticity in our model, we investigated the paired-pulse ratio (PPR) while varying the time interval between both stimuli (10 to 200 ms). At P8–10, the amplitudes of the responses were similar for both stimuli for each of the four conditions and for each interval (Figure 6A). At P13–16, the PPRs were also similar between all groups, but the PPR for the 10 ms interval was lower than at P8–10 (SHAM: 0.66 ± 0.04, n=5; HYPO: 0.54 ± 0.05, n=13; INFL: 0.63 ± 0.04, n=8; HYPO+INFL: 0.49 ± 0.07 n=11, p=0.11, Kruskal-Wallis ANOVA; Figure 6B). At P28–30 again, no difference was observed between the PPRs for all treatment groups. Furthermore, synaptic fatigue investigated by repetitive stimulation was not changed by treatment with hypoxia, inflammation or both (Figure 6C). The average amplitude of the last 5 stimuli was similar in all groups (SHAM: 48 ± 12 μV; HYPO: 57 ± 18 μV; INFL: 53 ± 12 μV; HYPO+INFL: 52 ± 11 μV, Figure 6D) suggesting no differences in presynaptic release properties.
Figure 6. Synaptic activity between L4 and L2/3 is not altered by hypoxia and/or systemic inflammation.

A) Example traces of the evoked responses in L2/3 to paired-pulse stimulation in L4 (2 V) with 25 ms between both stimuli for each age and treatment condition (SHAM, dark grey; HYPO, blue; INFL, red; HYPO+INFL, green) recorded in ACSF with 3 mM KCl. B) Paired-pulse facilitation measured for each treatment condition in P13–16 mice in ACSF with 3 mM KCl. The graphs represent the average PPR obtained from all slices of the different ages and treatments at varying interstimulus intervals. C) Example traces of the first and last evoked responses in L2/3 to 100 pulses at 10 Hz in L4, recorded in ACSF with 8 mM KCl. D) Synaptic fatigue recorded for 100 stimuli at 10 Hz (2 V, 8 mM KCl-ACSF). The graphs represent the relative amplitude of the evoked responses in slices prepared from all treated animals relative to the response to the first stimulus.
Hypoxia and inflammation facilitate the propagation of evoked epileptiform activity
As our results showed that animals with mild systemic inflammation, hypoxia, or both display spontaneous epileptiform activity in vitro, we further investigated the functional properties of the cortical network in thalamocortical slices. The evoked EEs were mostly characterized by longer durations than spontaneous events and spread from the stimulation site over one or several columns, in some cases they propagated over the whole cortical area covered by the MEA.
If slices from HYPO, INFL and HYPO+INFL are more excitable, then epileptiform activity should be evoked by stimuli lower than from SHAM. We therefore investigated whether evoked EEs had different properties depending on the treatment administered to the mice. The maximum stimulation voltage used in this study (4 V) was sufficient to induce epileptiform events (EEs) in nearly all slices prepared from the treated animals at P8–10 (HYPO: 76% slices with evoked EEs; INFL: 87%; HYPO+INFL: 75%), except for four out of seven slices in the SHAM group (43% slices with EEs) where all stimuli applied failed to elicit EEs (Figure 7A). In P28–30 adult mice, only 12–18% of the slices showed evoked EEs. This suggests that we could reliably detect threshold values to evoke EEs. While low stimulation voltage did not induce epileptiform activity, the stimulus voltage needed to initiate an epileptiform response in physiological conditions (ACSF with 3 mM KCl) was similar in SHAM (2.8 ± 0.5 V, n=7), HYPO (2.1± 0.3 V, n=17), INFL (2.2 ± 0.4 V, n=9) and HYPO+INFL animals (2.7 ± 0.3 V, n=12) of the P8–10 age group (Figure 7B). P13–16 slices and P28–30 slices also did not show significant differences in the threshold voltages eliciting EEs between conditions.
Figure 7. Increase neuronal excitability in hypoxia and/or IL-1β treated animals.

A) Representative 500 ms recordings of the evoked responses to monopolar stimulations in the layer 4 at 2 V for each treatment in slices prepared from P8–10 mice (top), P13–16 (middle), and P28–30 mice (top). The position of the cortical layers is indicated. The electrodes used for stimulation and calculation of evoked EEs durations are located above the stimulation electrode, marked by the absence of recording, as it was switched off during voltage application. B) Average minimum stimulation voltage needed to initiate an epileptiform response in each treatment (SHAM, dark grey; HYPO, blue; INFL, red; HYPO+INFL, green) in 3 mM KCl- (open bars) or 8 mM KCl-ACSF (filled bars). C) Duration of the evoked responses recorded in layer 2/3 following a 2.5 V stimulation in layer 4 presented as box-and-whisker plots representing the median as a solid line with a box to indicate the interquartile range; the whiskers represent the 10th and 90th percentiles. D) Similar box and whiskers plots representing the median number of barrel columns over which the evoked response was propagating following stimulation in layer 4. Dunns test: **, p<0.01; *, p<0.05.
When the recording solution was changed to 8 mM KCl-ACSF, the voltage threshold that elicits EEs at P8–10 was significantly lower in HYPO (1.4 ± 0.2 V, n=18) and HYPO+INFL (1.8 ± 0.3 V, n=12) than in SHAM (2.5 ± 0.6 V, n=7; p=0.0194, Kruskal-Wallis ANOVA). Similar results were obtained in P13–16 mice, where the voltage threshold necessary to elicit EEs was also lower in HYPO (2.0 ± 0.3 V, n=19), INFL (1.7 ± 0.5 V, n=8) and HYPO+INFL animals (1.4 ± 0.2 V, n=16; p=0.0149; Kruskal-Wallis ANOVA) as compared to SHAM (2.8 ± 0.6 V, n=7) (Figure 7B). The threshold voltage necessary to evoke epileptiform activity did not significantly change between conditions in P28–30 mice (Figure 7B). These results suggest that there is a transient period of hyperexcitability during which EEs can be evoked by low stimulation after hypoxia, inflammation, or both.
Besides stimulation thresholds we also measured duration of EEs. The duration of evoked EEs in ACSF with 3 mM ACSF did not vary between HYPO (234 ± 33 ms), INFL (122.9 ± 33 ms) and HYPO+INFL (176 ± 37 ms) compared to SHAM (116 ± 52 ms) animals at P8–10 (Figure 7C). Similarly, the duration of evoked EEs did not differ between groups in P13–16 or P28–30 animals. In ACSF with 8 mM KCl at P8–10, the duration of evoked EEs was significantly longer in slices from INFL (305 ± 67 ms), HYPO (288 ± 80 ms) and HYPO+INFL (313 ± 63 ms) compared to SHAM (178 ± 80 ms; p=0.0049; Kruskal-Wallis ANOVA, Figure 7C). Similarly at P13–16, the duration of evoked EEs in SHAM (123 ± 83 ms) differed significantly from INFL (232 ± 91 ms; p=0.0279, Kruskal-Wallis ANOVA) but not from HYPO (217 ± 101 ms) or HYPO+INFL (272 ± 95 ms; Figure 7). Additionally, evoked EEs had similar duration in HYPO, INFL, and HYPO+INFL. No difference in the durations of evoked EE was observed between treatments in P28–30 mice. These data further indicate that hypoxia and/or inflammation causes a transient hyperexcitability in the two younger age groups.
Furthermore we measured the spatial extent of propagation of evoked EEs in HYPO, INFL, and HYPO+INFL slices and compared it with the extent of evoked responses in SHAM slices. In 3 mM KCl-ACSF, no significant difference was observed between the groups, for each age investigated. In 8 mM KCl-ACSF, at P8–10, evoked EEs spread over a significantly larger area in HYPO+INFL slices (5.7 ± 0.4 columns, n=12) than in SHAM slices (4.0 ± 0.7 columns, n=6; p=0.0482; Kruskal-Wallis ANOVA), but not in HYPO (4.9 ± 0.4 columns, n=18) or INFL (4.9 ± 0.7 columns, n=9, Figure 7D and Table 2). Similarly in P13–16 animals, EEs spread over a smaller area in slices from SHAM group (3.0 ± 0.4 columns, n=7) than in slices from INFL (4.9 ± 0.5 columns, n=8) and HYPO+INFL (5.1 ± 0.3 columns, n=16; p=0.0091, Kruskal-Wallis ANOVA) but not than in HYPO (4.2 ± 0.4 columns, n=21; Figure 7D and Table 2). At P28–30, no effects of the treatments were observed on the spreading area of EEs. Indeed, within the very low number of slices with evoked epileptiform activity, EEs occurred only locally and did not spread over more than two columns (see Figure 2C).
Discussion
In the present work, we used in vitro MEA recording techniques to address the question how early moderate hypoxia and systemic inflammation influence neuronal network activity in the developing cerebral cortex and how these brain injuries affect the further development of the cortical network. The main results can be summarized as follows: (1) Animals with hypoxia, systemic inflammation, or both display hyperexcitability with high frequency epileptiform activity, (2) evoked EEs in these three groups occur with lower stimulation intensity, last longer, and spread across a larger area than in SHAM animals, (3) the combination of both treatments failed to induce a stronger hyperexcitability than hypoxia or inflammation alone, (4) NMDA and AMPA/kainate receptor antagonists were both effective in reducing epileptiform activity in treated animals, and (5) these pathophysiological changes are most prominent at P8–10 and P13–16 and disappear in adult mice. These results indicate that early hypoxia and/or inflammation causes a transient period of NMDA- and AMPA/kainate dependent hyperexcitability.
Mild treatments allow the investigation of moderate injury evolution
As opposed to more severe treatments used in previous studies (Eklind et al., 2001; Murray et al., 2015), the degree of severity of hypoxia and systemic inflammation used in the present work was moderate. Mild treatments applied elsewhere still led to brain inflammation associated with necrosis in young animals (Brochu et al., 2011), a loss of pericytes within the developing cortex in mice (Zehendner et al., 2013b), or to a blood-brain barrier breakdown (Zehendner et al., 2013a), but with less severe consequences. Our results similarly show that even at a moderate level, hypoxia and inflammation can alter the cortical neuronal network even if the consequences of stronger treatments are more striking (Salmaso et al., 2014). The mild treatments used here were associated with high survival rates. Dohlen et al. (2005) reported a mortality rate of 27% following their protocol for hypoxia (4% O2) but 0% when they decreased the hypoxic conditions, which was however severe enough to induce brain injury. Similarly, our results show that although treatments did not significantly affect the survival rate of animals, they were strong enough to induce dysfunction in the cortical network.
Hypoxia and inflammation induce dysfunction of excitatory and inhibitory neurotransmission
Hypoxia and inflammation, alone or in combination, induce hyperexcitability of the cortical network characterized by a high frequency of spontaneous EEs. The amplitudes of the EEs match values from studies in which similar methods were used to discriminate EEs (Chang et al., 2011), confirming the validity of our method. The lower stimulation thresholds needed to evoke epileptiform activity in all treated animals suggest a more sensitive and thus more excitable neuronal network. Our results suggest that the increased excitability after both types of pathophysiological events was caused by an increase in NMDA and AMPA/kainate receptor dependent glutamatergic neurotransmission. Enhancement of NMDA (Battaglia et al., 2013, Telfeian and Connors, 1999) and AMPA (Alefeld et al., 1998) receptors neurotransmission are well described causes of epileptogenicity in young animals. The AMPA antagonist CNQX applied with D-APV allowed the suppression of virtually all events and this confirms that both NMDA and AMPA/kainate receptors are largely responsible for the incidence of epileptiform activity as already described independently of the treatment (Sun et al., 2004). Interestingly, this enhanced excitatory activity involving those types of receptors is not present in adult animals.
We also observed that in all treated groups evoked EEs spread across the slices. Importantly at P8–10 and P13–16, the number of cortical columns over which evoked EEs spread is greater in HYPO+INFL animals than in SHAM and individual treatments (see Figure 7 and Table 2). The increased spread of electrically evoked EEs likely involves GABAA receptor mediated mechanisms, consistent with prior studies (Jimbo and Robinson, 2000). By blocking GABAA receptors the frequency of EEs increased in P13–16 SHAM mice, but not in injured animals. This suggests that a component of the inhibitory circuitry involving GABAA receptors controlling the timing of EEs is already impaired by hypoxia and inflammation. However, blocking GABAA receptors similarly increased the duration, amplitude, and spreading area of spontaneous EEs. This supports the idea that hypoxia and systemic inflammation induces a weakening of another component of inhibitory activity in the cortical network controlling the properties of EEs by decreasing the strength of GABAA neurotransmission. Thus, hypoxia and systemic inflammation might induce a complex rewiring of the network that affects differently diverse component of GABAergic signaling (Cossart et al., 2005).
Improvement during neurodevelopment: a neuroprotection mechanism?
We find a low amount of EEs in slices from adult mice (P28–30) indicating that as the brain matures, there seems to be a stabilization of excitability to reduce EE occurrence. Multiple processes could contribute to such stabilization, e.g. mild hypoxia might prevent the degradation of tissue by inducing the transient expression of proteins that protect nerve terminals (Manzur et al., 2001) or hypoxia might trigger neuroprotective mechanisms that avoid further insults within the brain (Zhang et al., 2006; Baranova et al., 2007). Systemic inflammation may also have a similar ability in that it protects against subsequent insults caused by hypoxia, probably by enhancing oligodendrocyte progenitors survival (Suryana and Jones, 2014).
The disappearance of EEs in adult mice might be the consequence of neuroprotective mechanisms that permit a recovery of the brain to a more physiological state during development. Interestingly, the weakly impaired inhibitory neurotransmission fully recovers in adult mice (see Figure 4). Moreover glutamatergic neurotransmission, which is impaired in P8–10 and P13–16 treated animals, also recovers in the adult as no difference is found between all groups. Our results are consistent with the existence of a homeostatic regulation of excitatory and inhibitory synapses to bring neurons to levels of activity closer to physiological values (Galarreta and Hestrin, 1998).
Neuroprotection prevents severe damage from combined treatments
Our mild hypoxia or systemic inflammation induce brain injuries that result in epilepsy similar to those seen in preterm newborns that often undergo complications (Glass et al., 2009). It is a critical point of our results that the combination of both injuries, which causes the highest mortality, causes impairments that are not stronger than the individual insults. One explanation for the lack of an additive effect is that the cortical outcomes we measure are independent of the damages that affect survival. An alternative explanation is that hypoxia and inflammation affect similar cellular mechanisms. Hypoxia results in the damage of neurons expressing hypoxia inducible 1α (HIF-1α) transcription factor responsible for an increase in brain injury (Baranova et al., 2007). Interestingly, the release of IL-1β induced by HIF-1α further activates HIF-1α, causing a positive feedback loop, that increases the effect of hypoxia (Kaluz and Van Meir, 2011). Since systemic inflammation involves similar cascades of events to mild hypoxia (Zhang et al., 2006), and the latter is known to be protective against more severe hypoxia, it is possible that a neuroprotective mechanism allowed a partial recovery of the cortical network. In a similar way, mild hypoxia following a severe hypoxic event protects against further damage (Rybnikova et al., 2012). Therefore mechanisms of neuroprotection could be triggered in animals that suffer successively inflammation and hypoxia, which are not triggered in animals suffering from hypoxia or inflammation only.
Contradictory to our results, Yang et al. (2004) showed that the combination of both inflammation and hypoxia increases the neuronal damage caused by single treatments separately, but the treatment used in their study was more severe. While we observe transient effects in our recordings, preterm newborns that underwent such traumatic events have motor and cognitive deficits as they mature to the adult stage (Volpe, 2009). A reason for these discrepancies might be that the moderate treatments that we used did not induce injuries reaching a critical threshold to become permanent or that our electrophysiological MEA recordings are not sensitive to detect subtle network changes that can cause perceptual or cognitive dysfunction.
Conclusion
In order to better understand how to prevent the frequent brain injuries caused by hypoxia and neuronal inflammation especially for preterm birth, the present work investigates conditions of hypoxia and systemic inflammation that mimic mild injuries existing in human neonates. Our results show that both mild hypoxia and systemic inflammation, either separated or in combination, induce similar hyperexcitability leading to frequent EEs in the somatosensory cortex. Our results suggest that these two types of injuries involve similar cascades of events. Therefore, we speculate that one therapy could be used to counteract the consequences of these injuries.
Acknowledgments
We thank Beate Krumm and Sigrid Stroh-Kaffei for their excellent technical assistance. A.S. was supported by a UMD International Graduate Fellowship, POK is supported by NIH RO1DC009607, HJL by DFG and the German Federal Ministry of Education and Research (KMU-innovativ: Medizintechnik, project TENECOR). CMZ was supported by a start-up grant by the University Medical Center of Mainz.
Footnotes
Conflict of interest: The authors declare no competing financial interests.
Reference list
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (Barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Alefeld M, Sutor B, Luhmann HJ. Pattern and pharmacology of propagating epileptiform activity in mouse cerebral cortex. Experimental Neurology. 1998;153:113–122. doi: 10.1006/exnr.1998.6837. [DOI] [PubMed] [Google Scholar]
- Back SA, Miller SP. Brain injury in premature neonates: a primary cerebral dysmaturation disorder? Annals of Neurology. 2014;75:469–486. doi: 10.1002/ana.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. Journal of Neuroscience. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Colciaghi F, Finardi A, Nobili P. Intrinsic epileptogenicity of dysplastic cortex: Converging data from experimental models and human patients. Epilepsia. 2013;54:33–36. doi: 10.1111/epi.12272. [DOI] [PubMed] [Google Scholar]
- Brochu ME, Girard S, Lavoie K, Sebire G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: An experimental study. Journal of Neuroinflammation. 2011:8. doi: 10.1186/1742-2094-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Wu JS, Lee CM, Vogt BA, Shyu BC. Spatiotemporal organization and thalamic modulation of seizures in the mouse medial thalamic-anterior cingulate slice. Epilepsia. 2011;52:2344–2355. doi: 10.1111/j.1528-1167.2011.03312.x. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Inflammatory brain damage in preterm newborns - dry numbers, wet lab, and causal inferences. Early Human Development. 2004;79:1–15. doi: 10.1016/j.earlhumdev.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Devoto VMP, Bogetti ME, de Plazas SF. Developmental and hypoxia-induced cell death share common ultrastructural and biochemical apoptotic features in the central nervous system. Neuroscience. 2013;252:190–200. doi: 10.1016/j.neuroscience.2013.07.065. [DOI] [PubMed] [Google Scholar]
- Dingley J, Tooley J, Porter H, Thoresen M. Xenon provides short-term neuroprotection in neonatal rats when administered after hypoxia-ischemia. Stroke. 2006;37:501–506. doi: 10.1161/01.STR.0000198867.31134.ac. [DOI] [PubMed] [Google Scholar]
- Dohlen G, Carlsen H, Blomhoff R, Thaulow E, Saugstad OD. Reoxygenation of hypoxic mice with 100% oxygen induces brain nuclear factor-kappa B. Pediatric Research. 2005;58:941–945. doi: 10.1203/01.PDR.0000182595.62545.EE. [DOI] [PubMed] [Google Scholar]
- Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, Hagberg H. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. European Journal of Neuroscience. 2001;13:1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelievre V, Gressens P. Systemic inflammation disrupts the developmental program of white matter. Annals of Neurology. 2011;70:550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Brecht M, Helmchen F, Petersen CCH, Poulet JFA, Staiger JF, Luhmann HJ, Schwarz C. Barrel cortex function. Progress in Neurobiology. 2013;103:3–27. doi: 10.1016/j.pneurobio.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nature Neuroscience. 1998;1:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Linn J, Eilers J, Konnerth A. Large-scale oscillatory calcium waves in the immature cortex. Nat Neurosci. 2000;3:452–459. doi: 10.1038/74823. [DOI] [PubMed] [Google Scholar]
- Garfinkle J, Shevell MI. Prognostic factors and development of a scoring system for outcome of neonatal seizures in term infants. European Journal of Paediatric Neurology. 2011;15:222–229. doi: 10.1016/j.ejpn.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Glass HC, Bonifacio SL, Chau V, Glidden D, Poskitt K, Barkovich AJ, Ferriero DM, Miller SP. Recurrent postnatal infections are associated with progressive white matter injury in premature infants. Pediatrics. 2008;122:299–305. doi: 10.1542/peds.2007-2184. [DOI] [PubMed] [Google Scholar]
- Glass HC, Glidden D, Jeremy RJ, Barkovich AJ, Ferriero DM, Miller SP. Clinical neonatal seizures are independently associated with outcome in infants at risk for hypoxic-ischemic brain injury. Journal of Pediatrics. 2009;155:318–323. doi: 10.1016/j.jpeds.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbo Y, Robinson HPC. Propagation of spontaneous synchronized activity in cortical slice cultures recorded by planar electrode arrays. Bioelectrochemistry. 2000;51:107–115. doi: 10.1016/s0302-4598(99)00083-5. [DOI] [PubMed] [Google Scholar]
- Kaluz S, Van Meir EG. At the crossroads of cancer and inflammation: Ras rewires an HIF-driven IL-1 autocrine loop. Journal of Molecular Medicine-Jmm. 2011;89:91–94. doi: 10.1007/s00109-010-0706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in-vitro intact hippocampus of the newborn mouse. Neuropharmacology. 2007;53:524–533. doi: 10.1016/j.neuropharm.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Manzur A, Sosa M, Seltzer AM. Transient increase in rab 3A and synaptobrevin immunoreactivity after mild hypoxia in neonatal rats. Cellular and Molecular Neurobiology. 2001;21:39–52. doi: 10.1023/A:1007169228329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KN, Parry-Jones AR, Allan SM. Interleukin-1 and acute brain injury. Frontiers in Cellular Neuroscience. 2015:9. doi: 10.3389/fncel.2015.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmervoll B, White R, Yang JW, An SM, Henn C, Sun JJ, Luhmann HJ. LPS-induced microglial secretion of TNF alpha increases activity-dependent neuronal apoptosis in the neonatal cerebral cortex. Cerebral Cortex. 2013;23:1742–1755. doi: 10.1093/cercor/bhs156. [DOI] [PubMed] [Google Scholar]
- Rybnikova E, Vorobyev M, Pivina S, Samoilov M. Postconditioning by mild hypoxic exposures reduces rat brain injury caused by severe hypoxia. Neuroscience Letters. 2012;513:100–105. doi: 10.1016/j.neulet.2012.02.019. [DOI] [PubMed] [Google Scholar]
- Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, Gallo V. Neurobiology of premature brain injury. Nature Neuroscience. 2014;17:341–346. doi: 10.1038/nn.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JJ, Luhmann HJ. Spatio-temporal dynamics of oscillatory network activity in the neonatal mouse cerebral cortex. European Journal of Neuroscience. 2007;26:1995–2004. doi: 10.1111/j.1460-9568.2007.05819.x. [DOI] [PubMed] [Google Scholar]
- Sun QQ, Huguenard SR, Prince DA. Reorganization of barrel circuits leads to thalamically-evoked cortical epileptiform activity. Epilepsia. 2004;45:359–360. doi: 10.1017/S1472928807000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryana E, Jones NM. The effects of hypoxic preconditioning on white matter damage following hypoxic-ischaemic injury in the neonatal rat brain. International Journal of Developmental Neuroscience. 2014;37:69–75. doi: 10.1016/j.ijdevneu.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Telfeian AE, Connors BW. Epileptiform propagation patterns mediated by NMDA and non-NMDA receptors in rat neocortex. Epilepsia. 1999;40:1499–1506. doi: 10.1111/j.1528-1157.1999.tb02032.x. [DOI] [PubMed] [Google Scholar]
- Vesoulis ZA, Inder TE, Woodward LJ, Buse B, Vavasseur C, Mathur AM. Early electrographic seizures, brain injury, and neurodevelopmental risk in the very preterm infant. Pediatric Research. 2014;75:564–569. doi: 10.1038/pr.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev. 2001;7:56–64. doi: 10.1002/1098-2779(200102)7:1<56::AID-MRDD1008>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurology. 2009;8:110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. An extremely rich repertoire of bursting patterns during the development of cortical cultures. Bmc Neuroscience. 2006:7. doi: 10.1186/1471-2202-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Sameshima H, Ikeda T, Ikenoue T. Lipopolysaccharide administration enhances hypoxic-ischemic brain damage in newborn rats. Journal of Obstetrics and Gynaecology Research. 2004;30:142–147. doi: 10.1111/j.1447-0756.2003.00174.x. [DOI] [PubMed] [Google Scholar]
- Zehendner CM, Librizzi L, Hedrich J, Bauer NM, Angamo EA, de Curtis M, Luhmann HJ. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. Plos One. 2013a:8. doi: 10.1371/journal.pone.0082823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehendner CM, Wedler HE, Luhmann HJ. A novel in vitro model to study pericytes in the neurovascular unit of the developing cortex. PLoS One. 2013b;8:e81637. doi: 10.1371/journal.pone.0081637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WD, Petrovie JM, Callaghan D, Jones A, Cui H, Howlett C, Stanimirovic D. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1 beta (IL-1 beta) in astrocyte cultures. Journal of Neuroimmunology. 2006;174:63–73. doi: 10.1016/j.jneuroim.2006.01.014. [DOI] [PubMed] [Google Scholar]