

SUMMARY
The ataxia telangiectasia-mutated and Rad3-related (ATR) kinase checkpoint pathway maintains genome integrity; however, the role of the sirtuin 2 (SIRT2) acetylome in regulating this pathway is not clear. We found that deacetylation of ATR-interacting protein (ATRIP), a regulatory partner of ATR, by SIRT2 potentiates the ATR checkpoint. SIRT2 interacts with and deacetylates ATRIP at lysine 32 (K32) in response to replication stress. SIRT2 deacetylation of ATRIP at K32 drives ATR autophosphorylation and signaling and facilitates DNA replication fork progression and recovery of stalled replication forks. K32 deacetylation by SIRT2 further promotes ATRIP accumulation to DNA damage sites and binding to replication protein A-coated single-stranded DNA (RPA-ssDNA). Collectively, these results support a model in which ATRIP deacetylation by SIRT2 promotes ATR-ATRIP binding to RPA-ssDNA to drive ATR activation and thus facilitate recovery from replication stress, outlining a mechanism by which the ATR checkpoint is regulated by SIRT2 through deacetylation.
Keywords: sirtuin, metabolism, SIRT2, ATR, ATRIP, replication stress, DNA replication, DNA damage response, DNA repair, acetylome, cell cycle, checkpoint
ETOC BLURB
Zhang et al. demonstrate that ATRIP deacetylation at conserved lysine 32 by SIRT2 promotes ATR-ATRIP binding to RPA-ssDNA to drive ATR activation and thus facilitate recovery from replication stress.
INTRODUCTION
The precise replication of the genome and the continuous surveillance of its integrity are essential for cell survival and the avoidance of various diseases, including cancer and premature aging. The genome is constantly exposed to environmental and endogenous genotoxic insults that challenge DNA replication. To cope with this challenge, the replication stress response (RSR), a subset of the DNA damage response (DDR), coordinates diverse DNA repair and cell cycle checkpoint signaling pathways necessary to maintain genome integrity. The ataxia telangiectasia mutated and Rad3 related (ATR) checkpoint kinase and its regulatory partner, ATR interacting protein (ATRIP), function at the apex of the RSR (Cimprich and Cortez, 2008; Marechal and Zou; Zeman and Cimprich). ATR deficiency causes embryonic lethality (Brown and Baltimore, 2000) and premature aging in adult mice (Ruzankina et al., 2007), and hypomorphic mutations in ATR and ATRIP are associated with Seckel syndrome (O’Driscoll et al., 2003; Ogi et al., 2012). Furthermore, cells lacking ATR have defects of DNA replication (Brown and Baltimore, 2000; Cortez et al., 2001), chromosomal instability (Casper et al., 2002), and expression of fragile sites (Casper et al., 2002). The ATR checkpoint pathway promotes genome integrity following replication stress through a kinase signaling cascade that mobilizes DNA repair, causes cell cycle arrest, or induces apoptosis or senescence; however, the precise mechanisms by which the pathway is regulated, including through acetylation, are not well understood.
ATR is activated by single-stranded DNA (ssDNA), resulting from stalled replication forks (Byun et al., 2005; Sogo et al., 2002) or processing of DNA double-strand breaks (DSB) (Garcia-Muse and Boulton, 2005; Jazayeri et al., 2006), which is then bound by the single-stranded binding protein replication protein A (RPA) (Costanzo et al., 2003). RPA-ssDNA recruits ATR-ATRIP through direct and indirect interactions with ATRIP mediated by RPA-ssDNA ubiquitylation (Marechal et al., 2014; Zou and Elledge, 2003), leading to the autophosphorylation of ATR at Thr-1989 (Liu et al., 2011; Nam et al., 2011). RPA-ssDNA also recruits the RAD17 clamp loader (Zou et al., 2003), which loads the RAD9-HUS1-RAD1 (9-1-1) clamp complex onto DNA (Bermudez et al., 2003) and recruits the MRE11-RAD50-NBS1 (MRN) complex (Oakley et al., 2009; Olson et al., 2007). Both the 9-1-1 complex and MRN interact with topoisomerase II beta binding protein 1 (TopBP1), which enables it to activate ATR and stimulate checkpoint signaling (Delacroix et al., 2007; Duursma et al.; Kumagai et al., 2006; Lee et al., 2007). The activation of ATR is facilitated by the interaction of TopBP1 with ATRIP (Kumagai et al., 2006; Mordes et al., 2008). ATR activation is also potentiated by the sumoylation of ATRIP, which promotes multiple interactions in the ATR pathway (Wu et al., 2014). Once activated, ATR phosphorylates numerous downstream substrates including the CHK1 kinase, which helps to disperse the signal.
Sirtuin 2 (SIRT2) is a member of the sirtuin family of NAD+ dependent deacetylases, which regulate multiple biological processes, including genome maintenance, aging, tumorigenesis and metabolism (Choi and Mostoslavsky, 2014; Finkel et al., 2009; Guarente; Saunders and Verdin, 2007). There are seven mammalian homologues of S. cerevisiae silent information regulator 2 (Sir2). Significantly, Sirt2 overexpression prolongs longevity in M. muscularis hypomorphic for BubR1 (North et al., 2014), and mice deficient in Sirt2 develop breast, liver, and other cancers (Kim et al., 2011; Serrano et al., 2013), suggesting that SIRT2 functions in both aging and tumor suppression. We and others have shown that SIRT2 promotes genome integrity (Kim et al., 2011; Serrano et al., 2013; Zhang et al., 2013). In particular, we found that SIRT2 directs the RSR at least in part through deacetylation of cyclin-dependent kinase 9 (CDK9) (Zhang et al., 2013), a protein required for recovery from replication stress (Yu and Cortez, 2011; Yu et al., 2010; Zhang et al., 2013). As sirtuins regulate networks of proteins, it may be possible that SIRT2 directs the functions of other RSR proteins. Indeed, a number of DDR and DNA repair proteins have been shown to be regulated by SIRT1 and SIRT6 (Choi and Mostoslavsky, 2014). Both ATM and DNA dependent protein kinase, catalytic subunit (DNA-PKcs), members of the phosphatidylinositol 3-kinase-related kinase (PIKK) family, which includes ATR, are regulated by acetylation (McCord et al., 2009; Sun et al., 2005). TopBP1 was also recently identified as a SIRT1 substrate (Liu et al., 2014; Wang et al., 2014). however, whether the SIRT2 acetylome is important for the ATR checkpoint pathway is unclear.
In this study, we show that deacetylation of ATRIP by SIRT2 drives ATR checkpoint activation by promoting binding to RPA-ssDNA, revealing a mechanism by which the ATR checkpoint pathway is regulated by SIRT2 deacetylation, and more generally the acetylome.
RESULTS
SIRT2 Interacts in a Complex with and Deacetylates ATRIP in vitro and in Cells
ATRIP was identified in a proteomic analysis for proteins that interact with SIRT2 (data not shown). To validate the interaction of SIRT2 and ATRIP, we performed co-immunoprecipitation (co-IP) of HA-ATRIP and SIRT2-FLAG expressed in 293T cells. IP of HA-ATRIP pulled down SIRT2-FLAG and endogenous ATR (Figure 1A), and reverse IP using an anti-FLAG antibody pulled down SIRT2-FLAG, HA-ATRIP, and endogenous ATR (Figure 1B), suggesting that the proteins interact in a complex. IP of FLAG-ATR also pulled down SIRT2-GFP expressed in 293T cells (Supplemental Figure S1A), confirming their interaction. In contrast, we failed to detect an interaction of FLAG-SIRT1 with ATRIP or ATR by co-IP (Supplemental Figure S1B,C), providing evidence that the interaction between ATR-ATRIP and SIRT2 is specific. The endogenous interaction of SIRT2 and ATRIP was validated by a reciprocal co-IP in HeLa cells (Figure 1C,D). Finally, this interaction showed no significant change in response to hydroxyurea (HU) treatment (Figure 1C,D), suggesting that it is not regulated by replication stress.
Figure 1. SIRT2 Interacts in a Complex with and Deacetylates ATRIP in vitro and in Cells.
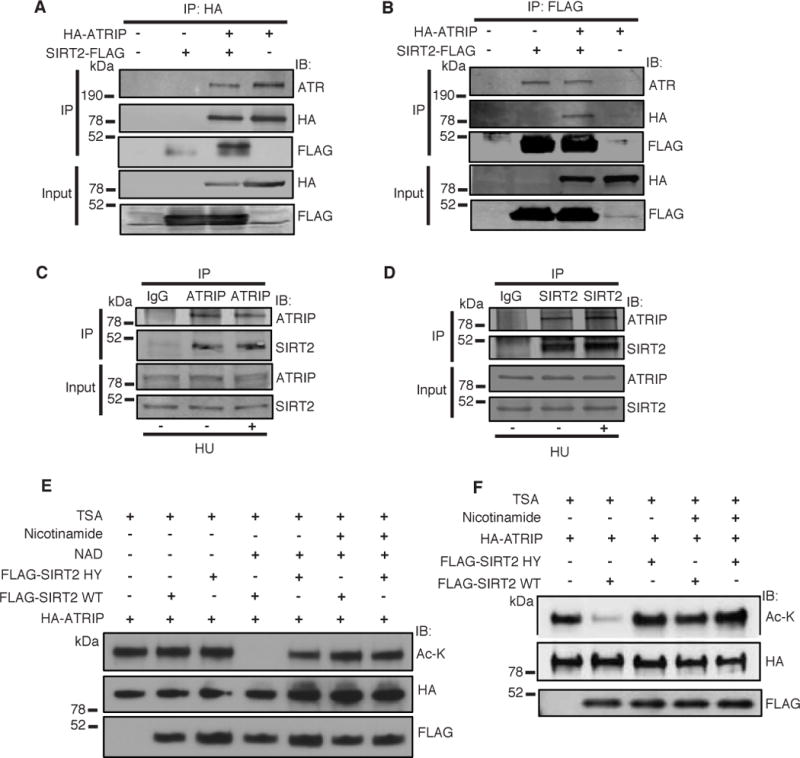
(A, B) 293T cells were transfected with HA-ATRIP and/or SIRT2-FLAG, harvested and immunoprecipitated with either anti-HA agarose beads (A) or anti-Flag M2 beads (B). Bound proteins were washed, separated by SDS–PAGE and immunoblotted with antibodies against HA, Flag and ATR. (C, D) Endogenous ATRIP or SIRT2 was immunoprecipitated from HeLa cell lysates treated with or without HU. Immunocomplexes were washed, separated by SDS-PAGE, and immunoblotted with antibodies against ATRIP and SIRT2. (E) Acetylated ATRIP was isolated from 293T cells transfected with HA-ATRIP and histone acetyltransferases, and incubated in an in vitro deacetylation assay with FLAG-SIRT2 WT or H187Y isolated from 293T cells, and in the presence of TSA with or without NAD and nicotinamide. The reaction mixtures were separated by SDS-PAGE and immunoblotted with pan-acetyl lysine, HA and FLAG antibodies. (F) HeLa cells were transfected with HA-ATRIP and wild-type FLAG-SIRT2 or deacetylase-inactive FLAG-SIRT2 HY in the presence of TSA with or without nicotinamide, immunoprecipitated with an anti-HA antibody, separated by SDS-PAGE, and immunoblotted with antibodies against HA, acetyl-lysine, and FLAG. See also Figure S1.
To determine if SIRT2 deacetylates ATRIP, we immunopurified acetylated HA-ATRIP from 293T cells and performed an in vitro deacetylation assay with purified wild-type FLAG-SIRT2 or FLAG-SIRT2 H187Y, a deacetylase inactive mutant (North et al., 2003), in the presence and absence of NAD+, nicotinamide, an inhibitor of sirtuin activity, and TSA, a non-sirtuin HDAC inhibitor. Wild-type FLAG-SIRT2 but not FLAG-SIRT2 H187Y deacetylated HA-ATRIP in a NAD+ dependent manner (Figure 1E, lanes 2 and 5 versus 4), and deacetylation was inhibited by nicotinamide (lane 6). To determine if SIRT2 deacetylates ATRIP in cells, we transfected HeLa cells with HA-ATRIP and wild-type FLAG-SIRT2 or FLAG-SIRT2 H187Y. Transfection with wild-type FLAG-SIRT2 but not FLAG-SIRT2 H187Y deacetylated HA-ATRIP in a similar manner as our cell-free biochemical system (Figure 1F, lanes 1 and 3 versus 2), confirming ATRIP as a legitimate SIRT2 downstream deacetylation target.
SIRT2 Deacetylates ATRIP at Lysine 32 in Response to Replication Stress
To determine the specific SIRT2 downstream acetyl-lysine target, mass spectrometry analysis was performed from immunoprecipitated purified HA-ATRIP expressed in 293T cells. Lysines 32, 96, and 101, located around the amino-terminal RPA-ssDNA binding domain (Ball et al., 2007; Ball et al., 2005; Namiki and Zou, 2006), were identified as potential reversible acetyl-lysines (Figure 2A and Supplemental Figure S2A–C). To determine whether ATRIP is acetylated at these sites, we generated ATRIP mutants where the lysines (K) are replaced by arginines (R), which prevent acetylation. The ATRIP K32R and K101R mutants and to a lesser extent the K96R mutant were acetylated at lower levels than wild-type ATRIP as determined by western blot analysis with an anti-acetyl-lysine antibody (Figure 2B), suggesting that lysines K32, K96, and K101 all contribute to acetylation of ATRIP. Moreover, overexpresssion of FLAG-SIRT2 in cells resulted in decreased acetylation more noticeably in the K96R and K101R mutants compared with the K32R mutant (Figure 2B), implying that K32 may be a significant SIRT2 downstream deacetylation target.
Figure 2. SIRT2 Deacetylates ATRIP at K32 in Response to Replication Stress.
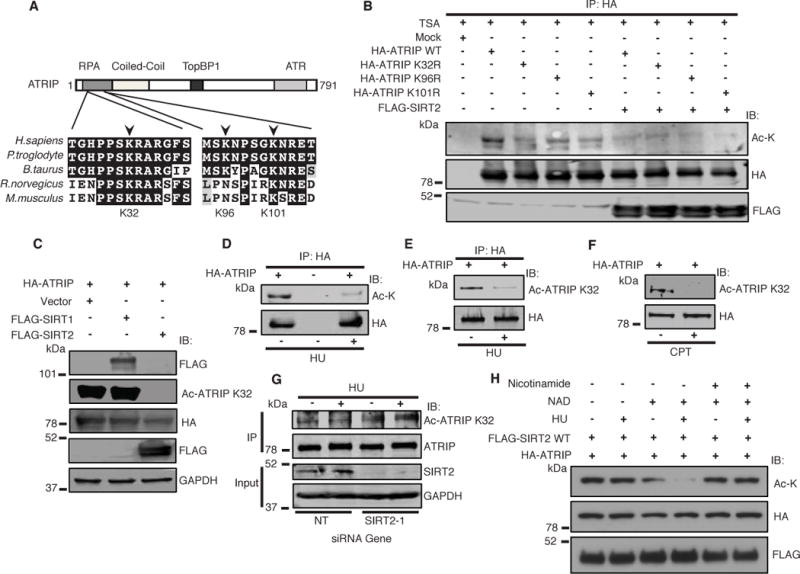
(A) Clustal Omega and BoxShade were used to align ATRIP protein sequences from the indicated organisms. The positions of the acetylated residues K32, K96 and K101 are depicted relative to the domains of ATRIP. (B) 293T cells were transfected with wild-type ATRIP or deacetylated mutants ATRIP K32R, ATRIP K96R, or ATRIP K101R and histone acetyltransferases with or without FLAG-SIRT2 in the presence of TSA, immunoprecipitated with anti-HA beads, separated by SDS-PAGE, and immunoblotted with antibodies against HA, acetyl-lysine. and FLAG. (C) 293T cells were transfected with HA-ATRIP and histone acetyltransferases, together with vector or FLAG-SIRT1 or FLAG-SIRT2 in the presence of TSA, harvested, separated by SDS-PAGE, and immunoblotted with antibodies against Ac-ATRIP K32, HA, Flag and GAPDH. (D, E, F) 293T cells were transfected with HA-ATRIP and histone acetyltransferases, treated with or without HU or CPT in the presence of TSA, harvested for whole cell lysates (F), or immunoprecipitated with anti-HA beads (D, E), separated by SDS-PAGE, and immunoblotted with antibodies against acetyl-lysine (D) or anti-Ac-ATRIP K32 (E, F) and HA. (G) HCT116 transfected with SIRT2 OR NT siRNA were treated with or without HU in the presence of TSA, harvested, immunoprecipitated with an anti-ATRIP antibody, separated by SDS-PAGE, and immunoblotted with antibodies against Ac-ATRIP K32, ATRIP, SIRT2 and GAPDH. (H) Acetylated ATRIP was isolated from 293T cells transfected with HA-ATRIP and histone acetyltransferases (p300/CBP and pCAF), and incubated in an in vitro deacetylation assay with FLAG-SIRT2 WT isolated from 293T cells treated with or without HU, in the presence of TSA with or without NAD and nicotinamide. The reaction mixtures were separated by SDS-PAGE and immunoblotted with pan-acetyl lysine, HA and FLAG antibodies. See also Figure S2.
Since K32 and its surrounding region is the most evolutionarily conserved amongst the acetyl-lysine sites we identified (Figure 2A) as well as independently identified as an acetyl-lysine site by proteomic analysis (Hornbeck et al., 2012), we generated and validated a rabbit monoclonal anti-acetyl ATRIP K32 antibody. This antibody recognizes acetylated endogenous ATRIP and exogenous tagged wild-type HA-ATRIP, but not HA-ATRIP K32R expressed in HCT-116 cells where these bands are also recognized by an anti-ATRIP antibody, as determined by western blot analysis (Supplemental Figure S2D). The specificity of the anti-acetyl ATRIP K32 antibody for endogenous ATRIP in HCT-116 cells was also validated by siRNA knockdown (Supplemental Figure S2E). To determine if ATRIP is acetylated at K32 in cells and further regulated by sirtuin or HDAC deacetylation, HCT-116 cells stably transfected with HA-ATRIP were treated with nicotinamide or TSA. Lysine 32 was acetylated in the absence of treatment, and the level of acetylation was increased to a similar extent with nicotinamide or TSA treatment (Supplemental Fig S2F), suggesting that K32 is acetylated in untreated cells and can be regulated by both sirtuin and HDAC deacetylation. Consistent with the results of our interaction studies (Supplemental Figure S1B,C), FLAG-SIRT2, but not FLAG-SIRT1, deacetylated HA-ATRIP expressed in 293T cells at K32 (Figure 2C), implying that SIRT2 deacetylates ATRIP at K32.
To determine if ATRIP deacetylation is regulated by replication stress, we analyzed the acetylation of HA-ATRIP expressed in 293T cells treated with or without HU. Western blot analysis with an anti-acetyl-lysine antibody of HA-ATRIP immunoprecipitated from 293T cells (Figure 2D) and co-immunoprecipitated with SIRT2-FLAG from cells (Supplemental Figure S2G) showed a significant decrease in acetylation of ATRIP from HU-treated cells compared to non-treated cells. HA-ATRIP from 293T cells were also deacetylated specifically at K32 in response to HU and camptothecin (CPT) treatment (Figure 2E and F). Moreover, a mild decrease in K32 acetylation was observed following ionizing radiation (IR) treatment of 293T cells synchronized predominantly in S-phase but not G1 phase (Supplemental Figure S2H) suggesting that ATRIP is deacetylated at K32 after IR in cells that support DNA end resection, RPA binding at ATR activation.
To determine if endogenous ATRIP K32 is deacetylated by SIRT2 in response to replication stress, we analyzed the acetylation of ATRIP in HCT-116 cells stably transfected with SIRT2 or non-targeting (NT) shRNA controls and treated with or without HU. Endogenous ATRIP was acetylated at K32 in the absence of treatment and deacetylated at K32 after HU treatment, and importantly, the HU regulated deacetylation of ATRIP at K32 was alleviated by SIRT2 depletion (Figure 2G), suggesting that ATRIP is present in the acetylated form at K32 in cells, and is subsequently deacetylated by SIRT2 in response to replication stress.
To further investigate what triggers SIRT2-dependent deacetylation of ATRIP K32, we performed an in vitro deacetylation assay using acetylated-ATRIP as a substrate and SIRT2 purified from 293T cells treated with or without HU. Increased deacetylation of the substrate using SIRT2 purified from cells treated with HU compared with no treatment was observed (Figure 2H), suggesting that SIRT2 deacetylase activity increases in response to replication stress.
SIRT2 Promotes ATR Activation
To determine if SIRT2 directs ATR activity, we examined HCT-116 cells transfected with SIRT2 or a non-targeting (NT) siRNA and treated with or without HU for ATR autophosphorylation at Thr1989, a marker for ATR activation (Liu et al., 2011; Nam et al., 2011). SIRT2 depletion significantly reduced ATR autophosphorylation, but not total ATR levels, in response to HU treatment (Figure 3A), suggesting that SIRT2 promotes ATR activation in response to replication stress. To determine if SIRT2 functions in ATR-dependent signaling, we examined HCT-116 cells for phosphorylation of a panel of ATR substrates in response to HU treatment. SIRT2 depletion reduced the HU-induced phosphorylation of RAD17 Ser645, CHK1 Ser317, and p53 Ser15 (Figure 3B). No significant difference in percentage of cells in S-phase was observed following SIRT2 depletion (Supplemental Figure S3A), suggesting that the effects of SIRT2 depletion on ATR activation are likely not due to changes in cell cycle. The effects of SIRT2 depletion on ATR Thr1989 and CHK1 Ser317 phosphorylation following HU treatment were also observed with an independent siRNA targeting the 3′ untranslated region (UTR) of SIRT2 (Supplemental Figure S3B), and significantly, expression of FLAG-SIRT2 rescued the impairment in CHK1 Ser317 phosphorylation following depletion of endogenous SIRT2 (Figure 3C).
Figure 3. SIRT2 Deacetylation of ATRIP at K32 Promotes ATR Activation.
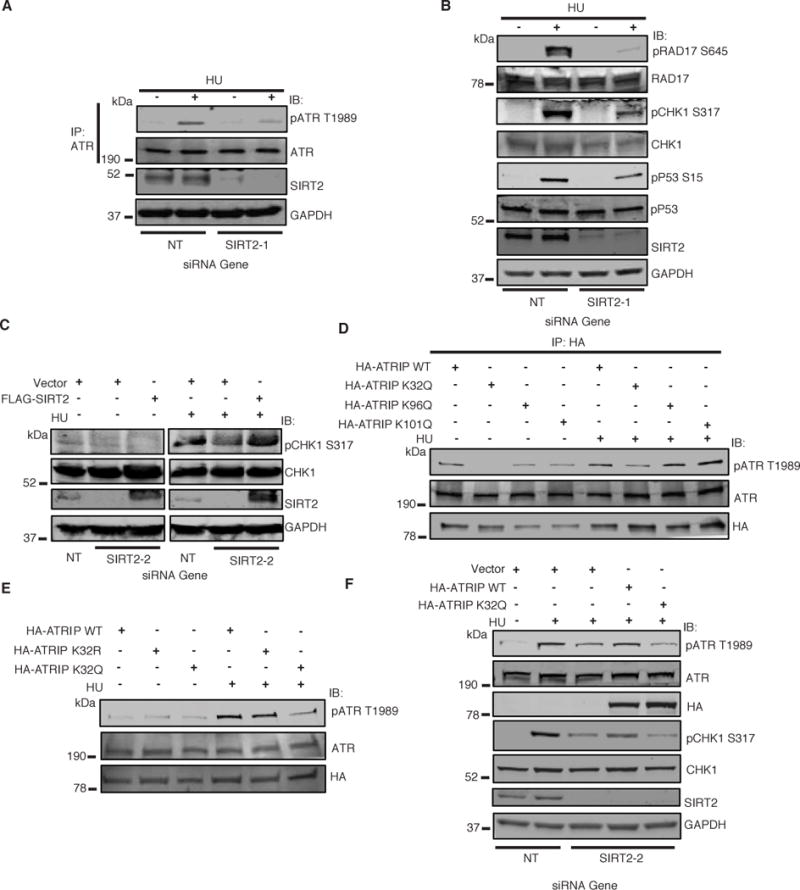
(A) HCT116 cells were transfected with NT or SIRT2 siRNA, treated with or without HU, harvested, immunoprecipitated with an anti-ATR antibody, separated by SDS-PAGE, and immunoblotted with antibodies against P-ATR T1989, ATR, SIRT2 and GAPDH. (B) HCT116 cells were transfected with NT or SIRT2 siRNA, treated with or without HU, harvested, separated by SDS-PAGE, and immunoblotted with indicated antibodies. (C) Cells stably expressing an empty vector or FLAG-SIRT2 and transfected with NT or SIRT2 siRNA targeting the 3′UTR, were treated with or without HU, harvested, separated by SDS-PAGE, and immunoblotted with indicated antibodies. (D) 293T cells were transfected with ATRIP WT or K32Q, K96Q, or K101Q, treated with or without HU, immunoprecipitated with anti-HA beads, separated by SDS-PAGE, and immunoblotted with antibodies against HA, P-ATR T1989 and ATR. (E) 293T cells were transfected with ATRIP WT or K32R, or K32Q, treated with or without HU, and extracted for nuclear extraction. The nuclear extraction was separated by SDS-PAGE, and immunoblotted with antibodies against HA, P-ATR T1989 and ATR. (F) HCT-116 cells stably expressing an empty vector or HA-ATRIP WT or K32Q and transfected with NT or SIRT2 siRNA, were treated with or without HU, harvested, separated by SDS-PAGE, and immunoblotted with indicated antibodies. See also Figure S3.
ATRIP Acetylation at Lysine 32 Impairs ATR Autophosphorylation and Signaling
To assess the functional significance of ATRIP acetylation, we generated ATRIP mutants in which K32, K96, or K101 was replaced by glutamine to mimic an acetylated lysine state. 293T cells expressing HA-ATRIP K32Q, at least to a greater degree than K96Q or K101Q, showed an impairment in ATR autophosphorylation, as compared to wild-type HA-ATRIP following HU treatment (Figure 3D), suggesting that the acetylation status of ATRIP at K32, but not lysine 96 or 1ysine 101, is most critical for ATR activation. Indeed, significant impairment in ATR autophosphorylation was observed only in cells expressing HA-ATRIP K32Q, but not HA-ATRIP K32R or wild-type HA-ATRIP (Figure 3E). Moreover, cells expressing HA-ATRIP K32Q but not wild-type HA-ATRIP showed an impairment in ATR-dependent phosphorylation of RAD17 at Ser645 in response to HU treatment (Supplemental Figure S3C). No significant difference in percentage of S-phase cells was observed in these cells (Supplemental Figure S3D), suggesting that the effects of K32 acetylation on ATR activation are not likely cell cycle related. Of further note, HA-ATRIP K32Q co-immunoprecipitated a similar amount of ATR as wild-type HA-ATRIP (Figure 3D), suggesting that ATRIP acetylation at K32 does not impair its interaction with ATR. Finally, expression of HA-ATRIP WT but not K32Q alleviated the impairment in HU-induced ATR autophosphorylation and CHK1 Ser317 phosphorylation following SIRT2 depletion (Figure 3F), strongly suggesting that the effects of SIRT2 depletion on ATR activation are mediated through K32 deacetylation. Collectively, these findings suggest that acetylation of ATRIP at K32 impairs ATR activation and deacetylation of ATRIP at K32 by SIRT2 promotes ATR activation in response to replication stress.
ATRIP Acetylation at Lysine 32 Impairs DNA Replication Fork Progression and Recovery of Stalled Replication Forks
To determine if ATRIP acetylation at K32 is critical for recovery from replication stress, we examined U2OS cells stably expressing vector or siRNA resistant HA-ATRIP wild-type, K32Q, K96Q, or K101Q and transfected with NT or ATRIP siRNA (Supplemental Figure S4A,B) for cell cycle recovery following HU treatment. Cells expressing HA-ATRIP K32Q showed the greatest impairment in HU recovery compared with cells expressing wild-type HA-ATRIP WT or HA-ATRIP K96Q and K101Q and silenced for endogenous ATRIP (Supplemental Figure S4C,D), suggesting that the acetylation status of K32 is the most critical for recovery from replication stress. The impairment in HU recovery of cells expressing HA-ATRIP K32Q was present even in the absence of endogenous ATRIP depletion, suggesting that the K32Q mutant can act in a dominant negative manner.
To further determine if K32 acetylation is important for replication fork progression, we used DNA fiber labeling to directly measure DNA replication fork dynamics. Newly synthesized DNA was labeled with 5′iododeoxyuridine (IdU) for 20 minutes followed by 5′-chlorodeoxyuridine (CldU) for 20 minutes in cells before DNA fiber spreads were processed by indirect immunofluorescence. Consistent with findings using an ATR inhibitor (Couch et al., 2013), cells expressing HA-ATRIP K32Q demonstrated decreased CldU fiber length, as compared with cells expressing wild-type HA-ATRIP or HA-ATRIP-K32R (Figure 4A,B), implying that K32 acetylation decreases replication fork elongation. To determine if K32 acetylation is involved in recovery of DNA synthesis after replication fork arrest, we labeled cells with IdU, blocked DNA replication with HU, washed out the drug, and labeled the cells with CldU. Cells expressing HA-ATRIP-K32Q demonstrated a significant decrease in CldU fiber length (Figure 4C,D) as well as a significant increase in replication tracks with no CldU uptake after HU release (Figure 4E), indicative of collapsed replication forks that cannot restart, compared with cells expressing wild-type HA-ATRIP or HA-ATRIP-K32R, suggesting that K32 acetylation impairs DNA replication fork restart following replication arrest. Similar to findings with an ATR inhibitor (Couch et al., 2013), cells expressing HA-ATRIP-K32Q but not wild-type or K32R also showed an increase in replication origin initiation (Figure 4F). Collectively, these findings demonstrate that ATRIP K32 acetylation impairs DNA replication fork progression and recovery of stalled replication forks.
Figure 4. ATRIP Acetylation at K32 Impairs DNA Replication Fork Progression and Recovery from Replication Arrest.
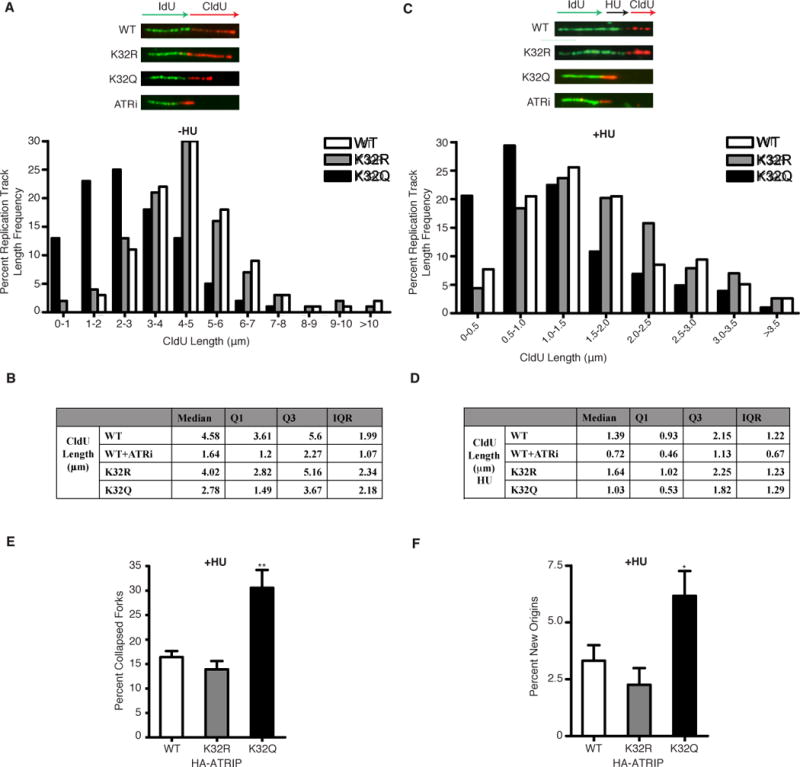
(A, B) 293T cells transfected with HA-ATRIP WT, K32R or K32Q were labeled with IdU for 20 min and then with CldU for 20 min in the presence of DMSO or 5 uM ATRi during both labeling periods before harvesting for DNA fiber labeling. (A) Representative replication tracks and distribution of the lengths of CldU (red) tracks in dual-labeled tracks are shown. p < 0.01 WT compared to K32Q by Mann-Whitney U test. (B) Median, 1st quartile, 3rd quartile, and interquartile range of the lengths of CldU tracks in dual-labeled tracks is shown. (C–F) 293T cells transfected with WT, K32R or K32Q ATRIP were labeled with IdU for 20 min treated with 2 mM HU for 1 h in the presence of DMSO or 5 uM ATRi and then labeled with CldU for 20 min before harvesting for fiber staining. (C) Representative replication tracks and distribution of the lengths of CldU tracks in dual-labeled tracks are shown. p < 0.01 WT compared to K32Q by Mann-Whitney U test. (D) Median, 1st quartile, 3rd quartile, and interquartile range of the lengths of CldU tracks in dual-labeled tracks is shown. (E) Percentage of collapsed forks (green-only tracks) following HU treatment is compared. (F) Percentage of newly initiated origins (red-only tracks) following HU treatment is compared. In all experiments, data was collected from several replicates with high-quality DNA fibers with at least 100 fibers counted for lengths and 300 fibers counted for collapsed fork and origins. *p < 0.05, **p < 0.01. See also Figure S4.
To determine whether K32 acetylation is important for mediating sensitivity following replication stress, we examined for cell viability following a course of CPT treatment. Expression of wild-type HA-ATRIP and HA-ATRIP K32R but not K32Q significantly increased the percent viability of U2OS cells silenced for endogenous ATRIP by siRNA following CPT treatment (Supplemental Figure S4E), suggesting that ATRIP K32 deacetylation promotes response to replication stress.
Lysine 32 Acetylation Impairs ATRIP Accumulation to Sites of DNA Damage
To determine how ATRIP acetylation affects ATR activation, we investigated if K32 acetylation is critical for localization of ATRIP to sites of DNA damage. Both wild-type GFP-ATRIP and GFP-ATRIP-K32R expressed in HeLa cells localized to DNA damage foci in response to HU and ionizing radiation (IR) treatment (Figure 5A–C). In contrast, GFP-ATRIP-K32Q showed a significant decrease in DNA damage foci formation (Figure 5A–C), suggesting that K32 acetylation impairs ATRIP localization to sites of DNA damage. In addition, cells expressing GFP-ATRIP-K32Q showed a decrease in formation of DNA damage foci that co-localize with RPA70, as compared with cells expressing wild-type GFP-ATRIP or GFP-ATRIP-K32R (Figure 5D–F), despite similar formation of RPA70 foci following DNA damage in GFP-ATRIP expressing cells (Supplemental Figure S5A,B) and similar cell cycle profiles (Supplemental Figure S5C), suggesting that K32 acetylation impairs the recruitment of ATRIP to DNA damage sites marked by RPA70. Western blot analysis confirmed expression of wild-type GFP-ATRIP and the GFP-ATRIP-K32R and -K32Q mutants (Supplemental Figure S5D).
Figure 5. K32 Acetylation Impairs ATRIP Accumulation to Sites of DNA Damage.
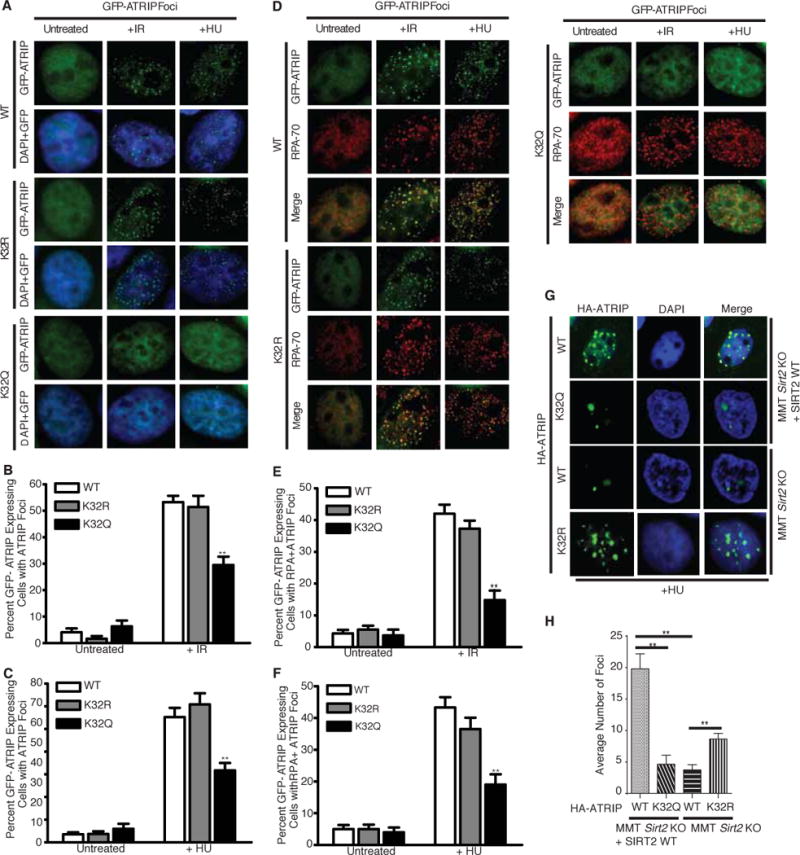
HeLa cells were transfected with WT, K32R or K32Q GFP-ATRIP respectively. Cells were either treated with 10 Gy IR and allowed 4 h to recover or cultured with 3mM HU for 24 h before RPA70 staining for immunofluorescence analysis. (A) Representative cell nuclei with GFP-ATRIP foci formation in response to IR and HU treatment are shown. The percentage of GFP-ATRIP foci positive cells from three replicate experiments are separately shown for IR treatment (B) and HU treatment (C). (D) Representative cell nuclei with both RPA and GFP-ATRIP foci formation in response to IR and HU treatment are shown. The percentage of RPA+GFP-ATRIP foci double positive cells from three replicate experiments are separately shown for IR treatment (E) and HU treatment (F). (G) Representative cell nuclei with HA-ATRIP foci formation in response to HU treatment in MMT Sirt2 KO cells complemented with or without SIRT2 WT. (H) The average number of HA-ATRIP foci following HU treatment from three replicate experiments are shown. **p < 0.01. See also Figure S5.
To determine whether SIRT2 is required for the localization of ATRIP to sites of DNA damage, we examined for the localization of HA-ATRIP expressed in mouse mammary tumor Sirt2 knockout (MMT S2KO) cells (Kim et al., 2011) complemented with or without wild-type SIRT2 and treated with or without HU. MMT S2KO cells expressing wild-type HA-ATRIP showed a significant impairment in foci formation in response to HU treatment compared with MMT S2KO cells expressing HA-ATRIP and complemented with wild-type SIRT2 (Figure 5G, compare rows 1 and 3, and 5H and Supplemental Figure 5E). The impairment in HA-ATRIP foci formation in MMT S2KO cells was comparable to that of HA-ATRIP K32Q expressed in MMT S2KO cells complemented with wild-type SIRT2 (Figure 5G, compare rows 3 and 2), and moreover was partially rescued by the expression of HA-ATRIP K32R in MM2 S2KO cells (Figure 5G, row 4 and 5H), suggesting that K32 deacetylation by SIRT2 in response to replication stress promotes ATRIP accumulation to sites of DNA damage.
SIRT2 Deacetylation of ATRIP at Lysine 32 Promotes Binding to RPA-ssDNA
The efficient activation of ATR requires its association with RPA-ssDNA at sites of DNA damage through ATRIP. Our observation that K32 acetylation impairs the recruitment of ATRIP to sites of DNA damage that co-localize with RPA70, as well as the location of K32 near the amino-terminal RPA-ssDNA binding domain of ATRIP, suggest that K32 acetylation directs its interaction with RPA-ssDNA. As such, co-IP of HA-ATRIP with endogenous RPA70 and RPA32 in HeLa cells was significantly decreased following SIRT2 depletion (Figure 6A), suggesting that SIRT2 enhances the interaction of ATRIP with RPA in cells. In contrast, we observed no significant change in the amount of HA-ATRIP co-immunoprecipitated with ATR, TopBP1, or NBS1 following SIRT2 depletion (Figure 6A), consistent with our finding that acetylation of ATRIP does not alter its interaction with ATR (Figure 3D). In a reciprocal manner, the co-IP of HA-ATRIP with RPA70 and RPA32, but not ATR, in 293T cells was significantly increased following SIRT2 overexpression (Supplemental Figure S6A). To determine if acetylation at K32 is critical for the interaction of ATRIP with RPA, we performed co-IP of HA-ATRIP K32R and K32Q in HeLa cells. A significant decrease in interaction of RPA70 but not ATR with HA-ATRIP K32Q compared with K32R was observed (Supplemental Figure S6B), suggesting that acetylation at K32 impairs the interaction of ATRIP with RPA.
Figure 6. SIRT2 Deacetylation of ATRIP at K32 Promotes Binding to RPA-ssDNA.
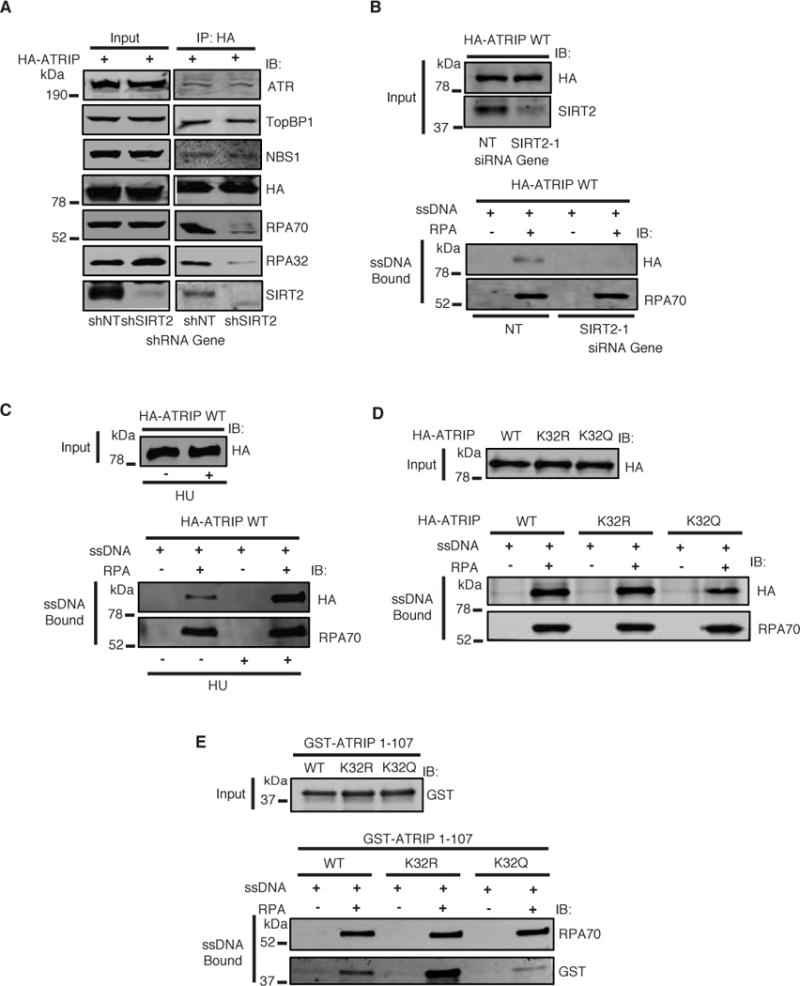
(A) HeLa cells stably expressing SIRT2 or control shRNA were transfected with HA-ATRIP and treated with HU in the presence of TSA, harvested, immunoprecipitated with anti-HA beads, separated by SDS-PAGE, and immunoblotted with antibodies against ATR, TopBP1, NBS1, HA, RPA70, RPA32 and SIRT2. (B–D) HA-ATRIP expressed in 293T cells was incubated with biotinylated ssDNA bound to streptavidin beads with or without recombinant RPA for 2 hours. Bound proteins were washed, separated by SDS-PAGE, and immunoblotted with the indicated antibodies. (B) RPA-ssDNA binding assay for HA-ATRIP WT expressed in 293T cells and transfected with SIRT2 or control siRNA. (C) RPA-ssDNA binding assay for HA-ATRIP WT purified from 293T cells treated with or without HU. (D) RPA-ssDNA binding assay for HA-ATRIP WT, K32R, or K32Q purified from 293T cells. (E) Recombinant GST-ATRIP 1–107 WT, K32R, or K32Q was incubated with biotinylated ssDNA bound to streptavidin beads with or without recombinant RPA for 2 hours. Bound proteins were washed, separated by SDS-PAGE, and immunoblotted with the indicated antibodies. See also Figure S6.
To determine if SIRT2 specifically affects the binding of ATRIP to RPA-ssDNA, we performed an in vitro RPA-ssDNA binding assay with biotinylated ssDNA coated with or without bacterial purified recombinant trimeric RPA (Supplemental Figure S6C) and wild-type HA-ATRIP expressed in 293T cells transfected with SIRT2 or control siRNA. The binding of wild-type HA-ATRIP to RPA-ssDNA was significantly decreased following SIRT2 depletion, (Figure 6B), suggesting that SIRT2 promotes the binding of ATRIP to RPA-ssDNA. The binding of wild-type HA-ATRIP purified from 293T cells to RPA-ssDNA was also significantly enhanced following treatment of the cells with HU (Figure 6C), which correlated with a decrease in acetylation of K32 (Figure 2E). To determine if K32 acetylation affects the binding of ATRIP to RPA-ssDNA, we examined wild-type HA-ATRIP, HA-ATRIP-K32R, or HA-ATRIP-K32Q expressed in cells treated with or without HU with RPA-ssDNA. The binding of HA-ATRIP-K32Q to RPA-ssDNA was significantly impaired compared with wild-type HA-ATRIP and HA-ATRIP-K32R both in the absence and presence of HU treatment (Figure 6D and Supplemental Figure S6D), suggesting that K32 acetylation impairs the binding of ATRIP with RPA-ssDNA. A conserved acidic checkpoint recruitment domain of ATRIP mapped to amino acids 54–70 has previously been shown to interact with a basic cleft in RPA (Ball et al., 2007; Feldkamp et al.; Souza-Fagundes et al.). As K32 lies just outside this domain, whether the acetylation of ATRIP at K32 is critical for mediating a direct or indirect interaction, i.e. through another protein, with RPA-ssDNA is not clear. Thus, we examined the binding of recombinant wild-type and mutant ATRIP aa 1–107 purified from bacteria (Supplemental Figure S6E) with recombinant RPA-ssDNA. ATRIP 1–107 has been shown to be sufficient for binding to RPA-ssDNA (Ball et al., 2007). Similar to full-length ATRIP K32Q, recombinant ATRIP 1–107 K32Q purified from bacteria showed a significant impairment in binding with RPA-ssDNA compared with ATRIP 1–107 wild-type and K32R (Figure 6E), suggesting that K32 deacetylation likely promotes a direct interaction with RPA-ssDNA. No significant difference in interaction of recombinant ATRIP 1–107 K32Q with ssDNA alone compared ATRIP 1–107 wild-type or K32R was observed (Figure 6E). Collectively, these findings imply that deacetylation of ATRIP at K32 by SIRT2 in response to replication stress promotes its direct binding to RPA-ssDNA.
DISCUSSION
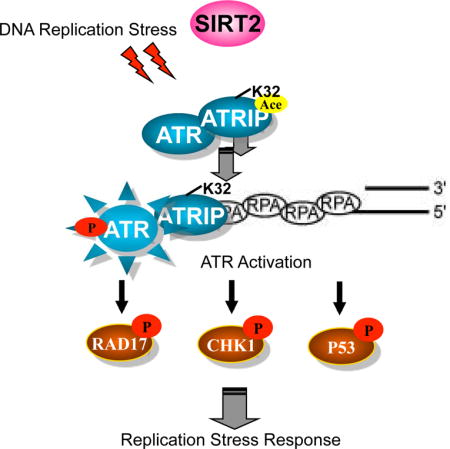
Our results describe a mechanism for regulation of the ATR checkpoint pathway whereby ATRIP deacetylation by SIRT2 drives ATR checkpoint activation by promoting binding to RPA-ssDNA. In this regard, we found that SIRT2 interacts with and deacetylates ATRIP at K32 in response to replication stress, providing evidence that ATR-ATRIP is regulated by SIRT2 deacetylation and indicating that ATRIP is an interacting partner and substrate for SIRT2. We further found that SIRT2 deacetylation of ATRIP at K32 drives ATR autophosphorylation and signaling and facilitates DNA replication fork progression and recovery of stalled replication forks following replication stress, demonstrating that SIRT2 functions as an important regulator of ATR activity and helping to explain why Sirt2 deficiency results in genomic instability and cancer development. Finally, we found that K32 deacetylation by SIRT2 promotes ATRIP accumulation to sites of DNA damage and binding to RPA-ssDNA, providing a mechanistic model for how ATRIP deacetylation by SIRT2 drives ATR activation. In this model, replication stress activates SIRT2 and triggers SIRT2 deacetylation of ATRIP at K32, which promotes its recruitment and binding to RPA-ssDNA thus driving ATR activation and facilitating DNA replication fork progression and recovery from replication stress (Figure 7).
Figure 7. Model for ATR Checkpoint Activation Following SIRT2 Deacetylation.
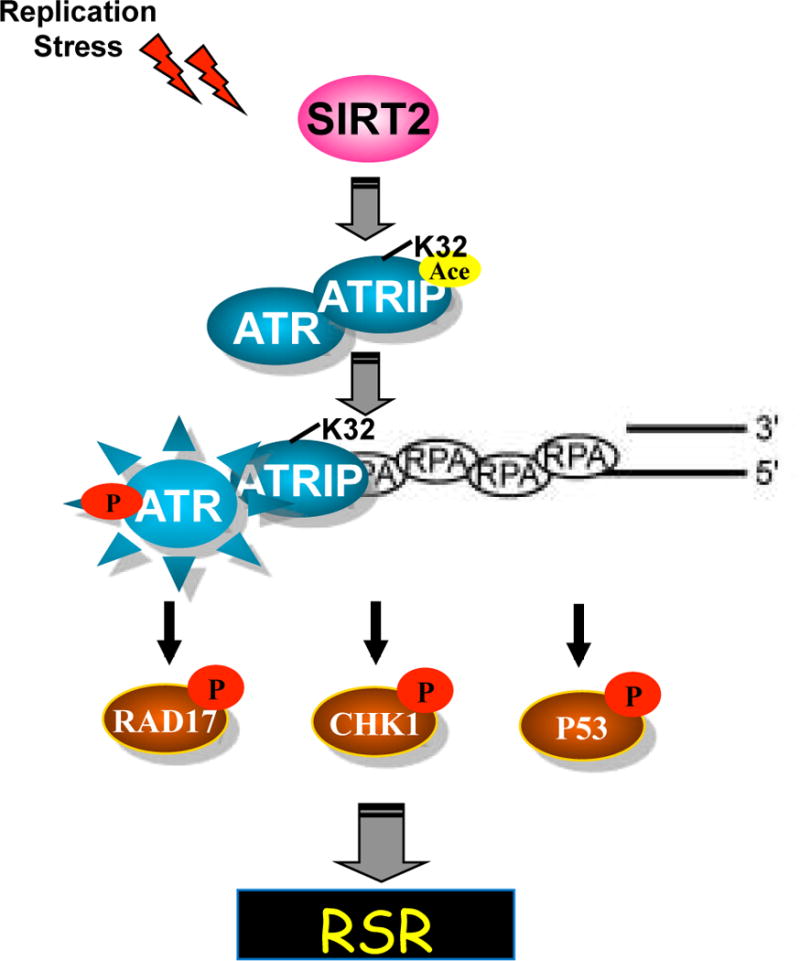
ATRIP deacetylation at K32 by SIRT2 promotes its binding to RPA-ssDNA, driving ATR activation, thus facilitating recovery from replication stress.
Our finding that SIRT2 promotes the interaction of ATRIP with RPA but not with ATR, TopBP1, or NBS1 suggests that ATRIP deacetylation by SIRT2 is a distinct mechanism for mediating the interaction of ATRIP specifically with RPA in contrast to the sumoylation of ATRIP at K234 and K289, which promotes its interaction with multiple partners in the ATR signaling pathway (Wu et al., 2014). We also found that ATRIP is deacetylated in response to HU, CPT, and IR treatment whereas ATRIP sumoylation does not increase in response to HU treatment, suggesting that ATRIP acetylation and sumoylation may be complementary mechanisms for regulation of ATR activation. In this regard, it is important to note that ATRIP acetylation is also critical for mediating its binding specifically to RPA-ssDNA, which is induced by DNA damage and replication stress and may have distinct binding properties from free RPA.
A conserved acidic checkpoint recruitment domain for ATRIP, mapped to amino acids 54–70, has previously been shown to interact with a basic cleft in RPA(Ball et al., 2007; Feldkamp et al.; Souza-Fagundes et al.). Since K32 lies just outside this domain, our observation that acetylation of K32 in a recombinant fragment of ATRIP 1–107 impairs binding to RPA-ssDNA suggest that K32 deacetylation may promote a direct interaction with RPA-ssDNA through a different interface or possibly by inducing a conformational change. Indeed, the interaction of RPA with associated proteins often occurs through at least two interfaces and can be influenced by conformational changes induced by ssDNA binding (Bochkareva et al., 2001). Importantly, K32 deacetylation as a mechanism for promoting the interaction of ATRIP with RPA-ssDNA is regulated by replication stress, consistent with a previous observation of an increase in the binding of ATRIP with RPA-ssDNA following UV treatment (Namiki and Zou, 2006).
Our observation that K32 acetylation only partially impairs ATRIP localization to RPA foci and binding with RPA-ssDNA, suggests that there may be additional mechanisms for regulation of ATRIP localization and binding with RPA. While the amino-terminal domain of ATRIP is required for its localization to DNA damage foci (Ball et al., 2005; Itakura et al., 2004), ATRIP also makes additional contacts with RPA-ssDNA outside its amino-terminal domain (Namiki and Zou, 2006). Thus, it is possible that these sites may contribute to the stabilization of the interaction of ATRIP with RPA-ssDNA, perhaps facilitated by ATRIP sumoylation. In addition, the relatively modest defect in ATRIP localization compared with ATR activation with K32 acetylation suggests that the defect in ATR activation with ATRIP acetylation cannot be fully accounted for by a defect in ATRIP localization and may be more closely linked with the defect in binding of ATRIP with RPA-ssDNA. A similar discrepancy between ATRIP localization with ATR activation has also previously been reported(Ball et al., 2005; Wu et al.). Thus, these data provide further support for the notion that ATR activity is more closely linked with RPA-ssDNA binding than with ATR-ATRIP localization.
Although we found that ATRIP is also acetylated at K96 and K101, we did not observe a significant impairment in ATR autophosphorylation or CHK1 phosphorylation in response to replication stress with K96 or K101 acetylation compared with K32 acetylation, suggesting that K32, but not K96 or K101, directs ATR activation. However, K96 and K101 acetylation causes a partial impairment in cell cycle recovery following replication stress, suggesting that they may also regulate important biological functions in the RSR in parallel or downstream of ATR activation. Interestingly, both sites are adjacent to the coiled coil domain of ATRIP as well as a region of ATRIP that is capable of binding ssDNA by itself in the absence of RPA. While our data identify SIRT2 is a major driver of K32 deacetylation in response to replication stress, an increase in acetylation of K32 was also observed following TSA treatment, suggesting that K32 may also be regulated by HDACs. The near complete deacetylation of HA-ATRIP K32 by SIRT2 overexpression in cells even in the presence of HDAC inhibition with TSA suggests that SIRT2 is sufficient for K32 deacetylation, and the alleviation of the HU-regulated deacetylation of endogenous ATRIP K32 by SIRT2 depletion even in the presence of HDAC inhibition with TSA, suggests that the HU-regulated deacetylation of ATRIP at K32 occurs in an HDAC independent manner, and that SIRT2 plays a major role in deacetylating K32 in response to replication stress, although we cannot exclude a role for HDACs in also regulating ATRIP activity in this context. The interplay of multiple HDACs and sirtuins or even of different sirtuins in regulating a single substrate is poorly understood but may provide an additional layer of control of ATR-ATRIP function through regulation of acetylation dynamics.
We previously found that SIRT2 directs the RSR at least in part through deacetylation of CDK9 (Zhang et al., 2013). Our identification of ATRIP as a binding partner and substrate of SIRT2 provides further validation for SIRT2 as an important regulator of the RSR and ATRIP as an additional downstream SIRT2 substrate in promoting the RSR. Although CDK9 overexpression can fully complement the HU hypersensitivity of SIRT2 depletion in cells, the HU-regulated deacetylation of CDK9 that requires SIRT2 is alleviated by ATR depletion (Zhang et al., 2013), suggesting that the effects of SIRT2 on CDK9 activity may be both direct through deacetylating CDK9 and indirect through deacetylating ATRIP. We speculate that SIRT2 likely regulates acetylome networks involved in maintaining genome integrity in response to replication stress. What might trigger SIRT2-dependent deacetylation of its downstream substrates in response to replication stress? One mechanism may be through regulating its deacetylase activity, which we found increases in response to replication stress. Sirt2 deficiency in mice leads to breast, liver, and other cancers (Kim et al., 2011; Serrano et al., 2013). Our identification of the ATR checkpoint signaling pathway as a SIRT2 target provides one possible explanation for how SIRT2 dysregulation leads to genomic instability and ultimately cancer development. In summary, our results support a model in which ATRIP deacetylation by SIRT2 promotes ATR-ATRIP recruitment and binding to RPA-ssDNA to drive ATR activation and thus facilitate recovery from replication stress, revealing a unique mechanism by which the ATR checkpoint is regulated through deacetylation by SIRT2.
EXPERIMENTAL PROCEDURES
Cell Culture
Human HEK 293T, HeLa, HCT116, and U2OS, and MM2 S2KO cells were grown in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 7.5% fetal bovine serum. Stably transfected HCT116, HeLa, and U2OS cells were maintained in standard medium containing 1ug/ml puromycin (Fisher).
Transfections, In vitro Deacetylation Assay, Cellular Deacetylation Assay, Multi-Sequence Alignment, Immunofluorescence Analysis, DNA Fiber Labeling Assay, Immunoblot, Immunoprecipitation, Cell Cycle Recovery, Nuclear Extraction, RPA-ssDNA Pull Down, LC-MS/MS, Brdu Labeling
Details of these methodologies can be found in Supplemental Experimental Procedures.
Supplementary Material
HIGHLIGHTS.
SIRT2 interacts with and deacetylates ATRIP at K32 in vitro and in cells
ATRIP deacetylation by SIRT2 promotes ATR activation and replication stress recovery
ATRIP deacetylation by SIRT2 promotes accumulation to sites of DNA damage
SIRT2 deacetylation of ATRIP at K32 promotes its direct binding to RPA-ssDNA
Acknowledgments
We thank members of the Yu lab for helpful discussion. We thank Dr. Akira Matsuura for ATRIP-GFP and David Cortez for HA-ATRIP, His-RPA, and GST-ATRIP 1–107 plasmids. This work was supported by National Institutes of Health (NIH)/National Cancer Institute (NCI) K08CA143902, R01CA178999, and R01CA178999S1 to D.S.Y; Emory University Research Committee (URC) Pilot Award to D.S.Y.; and Georgia Research Alliance (GRA) Cancer Scientist 11072 to D.S.Y.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
P.E.H, W.D., X.L., and S.P. contributed substantially and equally to this manuscript. Conceptualization, H.Z., M.A.T., Y.W., W.S.D., P.W.D., Z.D., N.T.S., D.G., D.S.Y; Investigation, H.Z., P.E.H., W.D., S.P., X.L., Y.P., M.Z.M., D.M.D., M.X., B.Y., M.D.W., E.A.L., V.R.D., C.L., I.P., M.A.T.; Writing – Original Draft, H.Z., D.S.Y., Writing – Review and Editing, H.Z., P.E.H., W.D., S.P., X.L., Y.P., M.Z.M., D.M.D., M.X., B.Y., M.D.W., E.A.L., V.R.D., C.L., I.P., M.A.T., Y.W., W.S.D., P.W.D., X.D., N.T.S., D.G., D.S.Y.; Supervision, M.A.T., Y.W., W.S.D., P.W., X.D., N.T.S., D.G., D.S.Y.; Funding Acquisition, D.S.Y.
References
- Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol. 2007;27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Molecular biology of the cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J, Sancar A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc Natl Acad Sci U S A. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva E, Belegu V, Korolev S, Bochkarev A. Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. EMBO J. 2001;20:612–618. doi: 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- Choi JE, Mostoslavsky R. Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev. 2014;26C:24–32. doi: 10.1016/j.gde.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nature reviews Molecular cell biology. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Molecular cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes & development. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Molecular cell. 50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkamp MD, Frank AO, Kennedy JP, Patrone JD, Vangamudi B, Waterson AG, Fesik SW, Chazin WJ. Surface reengineering of RPA70N enables cocrystallization with an inhibitor of the replication protein A interaction motif of ATR interacting protein. Biochemistry. 52:6515–6524. doi: 10.1021/bi400542z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Muse T, Boulton SJ. Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J. 2005;24:4345–4355. doi: 10.1038/sj.emboj.7600896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L, Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 364:2235–2244. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- Herskowitz JH, Seyfried NT, Duong DM, Xia Q, Rees HD, Gearing M, Peng J, Lah JJ, Levey AI. Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in frontotemporal lobar degeneration. J Proteome Res. 2010;9:6368–6379. doi: 10.1021/pr100666c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Takai KK, Umeda K, Kimura M, Ohsumi M, Tamai K, Matsuura A. Amino-terminal domain of ATRIP contributes to intranuclear relocation of the ATR-ATRIP complex following DNA damage. FEBS Lett. 2004;577:289–293. doi: 10.1016/j.febslet.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer cell. 2011;20:487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. The Journal of biological chemistry. 2007;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell. 2011;43:192–202. doi: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Lin YH, Leng W, Jung SY, Zhang H, Deng M, Evans D, Li Y, Luo K, Qin B, et al. A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Molecular cell. 56:681–695. doi: 10.1016/j.molcel.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, Zou L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol Cell. 2014;53:235–246. doi: 10.1016/j.molcel.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 5 doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 2009;1:109–121. doi: 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes & development. 2008;22:1478–1489. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB, Cortez D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J Biol Chem. 2011;286:28707–28714. doi: 10.1074/jbc.M111.248914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiki Y, Zou L. ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc Natl Acad Sci U S A. 2006;103:580–585. doi: 10.1073/pnas.0510223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- North BJ, Rosenberg MA, Jeganathan KB, Hafner AV, Michan S, Dai J, Baker DJ, Cen Y, Wu LE, Sauve AA, et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. Embo J. 2014;33:1438–1453. doi: 10.15252/embj.201386907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- Oakley GG, Tillison K, Opiyo SA, Glanzer JG, Horn JM, Patrick SM. Physical interaction between replication protein A (RPA) and MRN: involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry. 2009;48:7473–7481. doi: 10.1021/bi900694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogi T, Walker S, Stiff T, Hobson E, Limsirichaikul S, Carpenter G, Prescott K, Suri M, Byrd PJ, Matsuse M, et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet. 2012;8:e1002945. doi: 10.1371/journal.pgen.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Lee AY, Chen L, Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. The Journal of biological chemistry. 2007;282:22939–22952. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- Serrano L, Martinez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB, Kane-Goldsmith N, Tong Q, Rabanal RM, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013;27:639–653. doi: 10.1101/gad.211342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Souza-Fagundes EM, Frank AO, Feldkamp MD, Dorset DC, Chazin WJ, Rossanese OW, Olejniczak ET, Fesik SW. A high-throughput fluorescence polarization anisotropy assay for the 70N domain of replication protein A. Anal Biochem. 421:742–749. doi: 10.1016/j.ab.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RH, Lahusen TJ, Chen Q, Xu X, Jenkins LM, Leo E, Fu H, Aladjem M, Pommier Y, Appella E, et al. SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. Int J Biol Sci. 10:1193–1202. doi: 10.7150/ijbs.11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Ouyang J, Mori E, Nguyen HD, Marechal A, Hallet A, Chen DJ, Zou L. SUMOylation of ATRIP potentiates DNA damage signaling by boosting multiple protein interactions in the ATR pathway. Genes & development. 28:1472–1484. doi: 10.1101/gad.238535.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Ouyang J, Mori E, Nguyen HD, Marechal A, Hallet A, Chen DJ, Zou L. SUMOylation of ATRIP potentiates DNA damage signaling by boosting multiple protein interactions in the ATR pathway. Genes Dev. 2014;28:1472–1484. doi: 10.1101/gad.238535.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DS, Cortez D. A role for CDK9-cyclin K in maintaining genome integrity. Cell Cycle. 2011;10:28–32. doi: 10.4161/cc.10.1.14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, Shyr Y, Cortez D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010;11:876–882. doi: 10.1038/embor.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Park SH, Pantazides BG, Karpiuk O, Warren MD, Hardy CW, Duong DM, Park SJ, Kim HS, Vassilopoulos A, et al. SIRT2 directs the replication stress response through CDK9 deacetylation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13546–13551. doi: 10.1073/pnas.1301463110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.