

Abstract
Among various gene therapy methods for cancer, suicide gene therapy attracts a special attention because it allows selective conversion of non-toxic compounds into cytotoxic drugs inside cancer cells. As a result, therapeutic index can be increased significantly by introducing high concentrations of cytotoxic molecules to the tumor environment while minimizing impact on normal tissues. Despite significant success at the preclinical level, no cancer suicide gene therapy protocol has delivered the desirable clinical significance yet. This review gives a critical look at the six main enzyme/prodrug systems that are used in suicide gene therapy of cancer and familiarizes readers with the state-of-the-art research and practices in this field. For each enzyme/prodrug system, the mechanisms of action, protein engineering strategies to enhance enzyme stability/affinity and chemical modification techniques to increase prodrug kinetics and potency are discussed. In each category, major clinical trials that have been performed in the past decade with each enzyme/prodrug system are discussed to highlight the progress to date. Finally, shortcomings are underlined and areas that need improvement in order to produce clinical significance are delineated.
Keywords: enzyme prodrug, bystander effect, thymidine kinase, gene therapy, nitroreductase, cytosine deaminase, GDEPT, ganciclovir, CB1954, 5-FC
Graphical abstract
1. Introduction
Dysregulation of proliferation in cells can lead to overgrowth and production of tumor masses with aberrant leaky blood vessels, hypoxic environment and elevated interstitial fluid pressure due to poor lymphatic drainage[1, 2]. This complex tumor pathophysiology therefore demands sophisticated therapeutic modalities for effective treatment. Early attempts to cure cancer were based on using agents that can inhibit cell growth. Unfortunately, severe side effects on normal high proliferating cells such as those in hematopoietic and immune system significantly limited the use of anti-metabolite agents. Despite these challenging adverse effects, the proliferative features of cancer remained an attractive focus for rational design of “targeted therapeutics” in second half of 20th century. Concurrently, advances in cancer biology and genetics opened new horizons and rapidly translated into targeted drug design in which the molecular aspects of cancer became as important as its proliferative features. Approved in 1996, Imatinib mesylate (Gleevec®) is the first targeted chemotherapy agent designed based on advances in genetics [3]. One year later, FDA approved bevacizumab (Avastin®) which is the first monoclonal antibody against vascular endothelial growth factor receptor. These two drugs among others are the milestones of molecular targeted cancer therapy where the basic biological differences between cancer and normal cells are exploited to effectively target cancer. Unfortunately, these distinctive features are not always available to be exploited. The rationally designed drug may enter other organs which share the same biological features with the tumors [4]. For instance, Imatinib’s off-target effects such as hypophosphatemia and hypocalcemia are caused by its inhibitory effect on c-fms tyrosine kinase in osteoclasts and osteoblasts. In general, mere use of active targeting strategy has not been sufficient to effectively eradicate cancer.
In parallel to actively targeted antibody-based therapeutics, numerous nanomedicines have been developed in an attempt to not only enhance drug localization at the tumor site and increase drug efficacy, but also decrease chances of multidrug resistance and toxicity [5]. Nanomedicines are designed to take advantage of tumor leaky vessels in order to passively target and accumulate in tumor tissues. Doxil® is among the first FDA approved nanomedicines that is mainly used for the treatment of Kaposi sarcoma where tumor vessels are very leaky. This PEGylated liposomal formulation of doxurubicine passively targets and accumulates in the tumors through enhanced permeability and retention effect (EPR) and then releases the drug [6]. Due to its small size, the released doxorubicin can then diffuse throughout the tumor tissue via concentration gradient and significantly impact tumor growth. Although such passively targeted formulations enhance the concentration of drug in tumor interstitial fluid but still a significant number of the liposomal particles are picked up by the reticuloendothelial system. Furthermore, the efficacy of such nanomedicines that rely solely on passive targeting is also limited by the degree of leakiness of tumor blood vessels which varies by cancer type and tumor size. As a result, there is a significant probability to observe toxicity in non-target tissues before the drug concentration in tumors reach the therapeutic level. Hence, passive targeting by itself may not be sufficient to render an effective and safe therapeutic outcome. Published data in the past decades suggest that more refined approaches may be necessary in order to overcome the obstacles mentioned above.
2. Cancer Gene Therapy
In recent years, more sophisticated approaches have emerged that combine passive and active targeting strategies in order to maximize efficacy at the target tumor site while minimizing the potential for off target toxicity. Targeted-shielded nanomedicines (viral and non-viral) carrying gene therapy agents (RNAi or DNA) are newer generation of targeted therapeutics that first accumulate in tumors passively via EPR effect and then due to the presence of ligands can bind to specific antigens on the surface of cancer cells and internalize [7–9]. This approach is especially useful for several gene therapy-based nanomedicines where the target site is inside the cancer cells. Cancer gene therapy is the treatment that is based on the transfer of therapeutic genes into cancer cells in order to slow down or cease the progress of malignancy. Cancer gene therapy can be classified into three categories: corrective gene therapy, toxin/apoptosis-inducing gene therapy and suicide gene therapy. Cancer corrective gene therapy is the approach that applies therapeutic genes into cancer cells to adjust the deranged gene profile and consequently moderate or stop cell proliferation. Tumor suppressor genes such as p53 or genetic interference agents that interfere with cancer cell proliferation (eg., siRNA or miRNA) are two prominent examples of this approach [10–13]. Toxin/apoptosis-inducing cancer gene therapy is a more straightforward method where the delivered transgene results in production of a toxic protein (e.g., diphtheria toxin or TNF-α) that in turn induces cell death. The main weakness of corrective gene therapy and toxin/apoptosis-inducing gene therapy is that only the cancer cells that have received the therapeutic gene get affected and those that have not received the therapeutic gene continue to proliferate. This becomes especially problematic for nanomedicines that rely solely on these two gene therapy strategies because they cannot penetrate deep into the tumor tissues due to tumor’s dense physiological environment and elevated interstitial fluid pressure [14]. As a result, not all cancer cells in tumors can be eliminated and this significantly increases the probability of cancer recurrence. Overall, it appears that off-target toxicity and lack of access to all cancer cells in the tumor environment are among the major obstacles to successful treatment of cancer. Because of these reasons, no passively and actively targeted nucleic acid-based nanomedicine has reached the clinic yet.
2.1. Suicide Gene Therapy
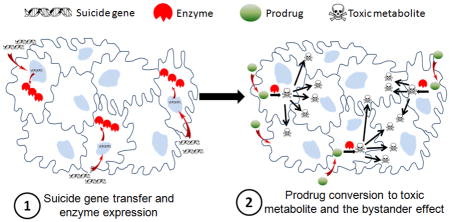
One idea that has gained significant attraction for cancer therapy with potential to overcome the discussed obstacles is targeted suicide gene therapy. In literature, this approach is also known as gene directed enzyme prodrug therapy (GDEPT). It allows us to combine passive, active, and transcriptional targeting strategies to maximize anticancer activity at the tumor site while minimizing impact on normal tissues. By definition, GDEPT is a two step process where the cancer cells are first transduced by a gene coding for a non-toxic enzyme (suicide gene) followed by administration of a non toxic prodrug [15]. Cell death occurs as a result of prodrug conversion to its toxic metabolite by transduced cells which actively express the suicide gene. In the context of GDEPT, therapeutic index increases by reducing side effects and restricting the toxicity of a chemotherapy agent only to target cancer cells. This approach provides two distinct advantages over the conventional cancer therapeutic strategies such as chemotherapy and radiation therapy.
The first advantage is the ability for transcriptional targeting where the suicide gene is put under the control of a tumor-specific promoter so that the gene expression occurs only in tumor cells but not in normal cells [16]. Consequently, the prodrug gets activated only in tumor environment reducing the probability of observing off-target toxicity. The suicide genes can then be loaded onto targeted vectors (viral or non-viral) and delivered to the tumor cells first passively and then actively [17]. Until early 2000s, variety of cancer/tissue specific promoters such as human telomerase reverse transcriptase (hTERT) promoter, carcinoembryonic antigen (CEA) promoter, osteocalcin promoter (OC), and hypoxia and radiation responsive elements had been developed [18–21]. The application, advantages and disadvantages of using these promoters are eloquently reviewed by several groups [16, 22, 23], and summarized in a book chapter by our group [24]. Over the past decade, several new promising promoters have been developed which attracted significant attention (Table 1). Although all these promoters have shown promise, hTERT is the only one that has successfully entered clinical trials [25]. The major obstacle preventing transcriptional targeting strategy moving into the clinic is that the cancer-specific promoters most often do not have enough transcriptional power. Accordingly, low level expression of the suicide gene may not be sufficient to activate significant number of prodrug molecules into the toxic forms. To overcome these challenges, construction of chimeric promoters and artificial promoters are currently being investigated which are discussed elsewhere [26, 27] [28, 29].
Table 1.
This table highlights the list of recently discovered cancer-specific promoters.
| Name | In Vitro Targets | In Vivo Model | Ref. |
|---|---|---|---|
| ARF | U-2 OS, Saos-2 (osteosarcoma), C-33 A, HeLa (cervix) H1299, A549 (lung) |
NA | [163] |
| CgA | SH-SY-5Y, SK-N-DZ(neuroblastoma), BON (pancreas), T47D(breast), T24(bladder) | BON (pancreas) Xenografts | [164] |
| GRP78 | NA | U87 (glioma) Xenografts | [165] |
| CXCR4 | MDA-MB-361(breast), OVCAR-3, SKOV3(ovary) Melanoma cells from patients A498, SN12C, SN12C-MMP, 786-0, and CAKI-1 (renal cell carcinoma) |
Colorectal Tumors | [166–168] |
| HMGB2 | U87, U251 (glioma), HeLa(cervix) H1299(lung) | U87 (glioma) Xenografts | [169] |
| INSM1 | NCI-H69, NCI-H1155, NCI-H727, DMS53, U87MG (lung), IMR-32, SK-N-SH, SK-N-BE, D283 Med (brain), Y79, WERI-Rb1(eye), HeLa(cervix), PANC1, RIN(pancreas), HepG2(liver) | D283Med (brain) Xenografts | [170] |
| Mesothelin | SK-OV-3, OV-3, OV-4, Hey (ovary) HeLa (cervix) |
SK-OV-3 (ovary) Xenografts | [171] |
| OPN | HTB-1, HTB-5, MB49 (bladder) PC-3 (prostate) |
NA | [28] |
| RAD51 | HCC1954, T47-D MCF-7 (Breast) Hela (cervix) |
HeLa (cervix) Xenografts | [172, 173] |
| TETP | M000301, M21-L4, MeWo, SK-Mel23 (melanoma), A549 (lung), HeLa,(cervix) SW480, DLD-1 (colon) |
NA | [174] |
The second advantage and critical element that governs the success of cancer suicide gene therapy over corrective gene therapy and toxin/apoptosis inducing gene therapy is the bystander effect. The success of GDEPT is heavily dependent on the bystander effect which is defined as the extension of drug cytotoxic effect from transduced cells to surrounding non-transduced cells, making a killing zone around the transduced ones (Figure 1). Unlike other gene therapy approaches where only the cells which receive the therapeutic genes undergo apoptosis or necrosis, in GDEPT the therapeutic effect and cytotoxicity can spread from affected cells to neighboring cells. Because the activated forms of prodrugs are small molecules, they can simply diffuse toward tumor core due to concentration gradient. This phenomenon can explain the observations that transduction of even less than 10% of cells could be sufficient to eradicate the whole cancer cell population [30, 31]. The main suggested mechanism for bystander effect is through diffusion of toxic metabolites from transduced cells to non-transduced ones, either passive or active. For example, ganciclovir triphosphate (GCV-TP), the final toxic metabolite of herpes simplex virus thymidine kinase/ganciclovir (HSVTK/GCV) system is a charged molecule. The bystander effect induced by this metabolite is entirely dependent on active transport via gap junctional intercellular communication (GJIC) [32]. This can be a drawback for such systems because compared to normal tissue, tumor tissues usually lack highly ordered cell junctions.
Figure 1.

Schematic representation of the two step process in suicide gene therapy. In step 1, with the help of a vector the cancer cells are transduced by suicide genes resulting in expression of an enzyme. In step 2, prodrug is administered which can be converted into its cytotoxic form by the enzyme and kill not only the transduced cells but also the neighboring ones.
Beside diffusion of toxic metabolites through GJIC, another interesting mechanism is endocytosis of apoptotic bodies released from dying cells by neighboring untransfected ones. It has been shown that by preventing the release of such apoptotic vesicles bystander effect could effectively be shut down [33]. The bystander effect either through GJIC or apoptotic bodies is effective only in vicinity of transduced cells where cells are connected through cell to cell communication or located in a short distance to receive the apoptotic bodies or toxic metabolite of prodrug. Another type of bystander effect, namely distant bystander effect, involves the activation of immune system [34]. This phenomenon has been observed when CD4+ and CD8+ T cells and NK cells are attracted into the primary tumor site followed by systemic immune response to the metastatic sites which are anatomically far from the primary suicide gene/prodrug treated tumors [35]. Distant bystander effect can be quite effective in preventing formation of secondary tumors [36]. It is believed that death of tumor cells causes the release of tumor-associated antigens which are stimulators for an anti-tumor immune response [37]. Both necrosis and apoptosis can trigger immune response; however, the immune reaction after necrosis has shown to be stronger [38]. In general, GDEPT provides this opportunity to significantly limit off-target toxicity, effectively reach and kill more cancer cells in the tumor tissue and remain relatively unaffected by tumor heterogeneity.
In the following section, we will discuss the important factors that play roles in the success of suicide gene therapy. The purpose of this review is to give a critical look at the six main enzyme/prodrug systems that are used in suicide gene therapy of cancer and discuss the forte and weaknesses associated with the efficacy and safety of each system at both preclinical and clinical levels. By examining the protein engineering strategies that are utilized to enhance enzyme stability/affinity and the chemical modification techniques to increase prodrug kinetics and potency, we intend to underline the deficiencies and make suggestions in the areas that need improvements. Therefore, the coverage of the literature in this critical review is not encyclopedic; rather, select examples have been chosen to highlight certain important points.
3. Pillars of Suicide Gene Therapy
As of January 2015, 157 out of 2076 clinical trials (7.7%) conducted worldwide focused on suicide gene therapy, indicating that it is considered one of the major approaches for cancer gene therapy. The success of suicide gene therapy of cancer is highly dependent on each of its three components, i.e. enzyme, prodrug and the delivery system (vector). The choice of each component becomes especially critical at the clinical level because only a few numbers of vectors are deemed safe and efficient to elicit a significant therapeutic response.
Enzymes that are used in GDEPT can be categorized into two groups. The first group includes enzymes that can be found in normal human cells such as cytochrome P450. This group of enzymes is less probable to induce any immune response; however, the presence of enzymes in normal cells could result in off-target toxicity. The second group of enzymes usually originate from viruses or bacteria [39]. Examples of this group include thymidine kinase (TK, Viral), cytosine deaminase (CD, bacterial and yeast), carboxypeptidase G2 (CPG2, bacterial) and nitroreductase (NTR, bacterial) [40]. In contrast to the first group, enzymes in this group are more likely to be immunogenic but there is less probability for observing toxicity in non-target tissues [24]. In general, the expression of enzyme should not be a metabolic burden for the cell housekeeping activities and should show a higher affinity to prodrug than enzyme’s natural substrate. Otherwise, high concentration of prodrug will be needed to reach desirable therapeutic effects which then bring about the probability of prodrug related side effects. In the past decades, many attempts such as site directed mutagenesis and protein recombination techniques have been made to introduce new versions of enzymes with not only higher affinity to prodrug, but also more stability and less immunogenicity. Besides the enzymes, the prodrugs should also possess several important characteristics in order to help maximize therapeutic response. The prodrug should be stable under physiological conditions, show low toxicity profile before activation and high toxicity to cancer cells after activation. In addition, the activated prodrug should possess a high bystander effect in order to overcome the deficiencies related to vectors’ low transduction rates. Based on the activation mechanism, the prodrugs are categorized into two groups; direct-linked and self-immolative [39, 41]. Direct-linked prodrugs become activated in one straightforward reaction to produce the active component. GCV and CB1954 are examples of prodrugs in this group which are targets for TK and NTR enzymes, respectively. In Self-immolative mechanism, the prodrug is converted to an intermediate form which spontaneously changes to active form through fragmentation process. The most well-known example of this category is doxorubicin prodrug which can break into doxorubicin by CPG2 enzyme [42]. Self-immolative mechanism makes it possible to use many different well-known anticancer drugs as prodrugs for GDEPT [43, 44].
The third important component of suicide cancer gene therapy is the vector. The desirable features of a suitable vector for clinical applications include low cytotoxicity/immunogenicity, high transfection efficiency, tissue specificity and cost-effectiveness [45]. In general, gene delivery systems can be divided into three major groups including synthetic (e.g., polymeric and lipid based) [46, 47], microorganism–based (viral, bacterial, yeast) and cell-based (e.g., stem cells or dendritic cells) [48]. To date, most vectors used for suicide gene therapy have been viral and amongst them adenovirus occupies the highest share for gene delivery in clinical trials [49]. In spite of several advantages such as ability to infect both dividing and non-dividing cells and inducing gene expression without integration into the host genome, the clearance of adenoviral particles by reticuloendothelial system (e.g., liver), potential for significant immune response to the viral capsid proteins, non-specific binding to all cells that overexpress coxsackie virus and adenovirus receptor (CAR), high costs of production and the fact that the majority of cancer cells do not express CAR have limited success of this vector in cancer gene therapy trials [50, 51]. Integrating vectors such as retrovirus and lentivirus are the second preferred vectors for suicide gene therapy. Unlike adenoviral vectors, these viruses trigger less abrupt immune response and are able to integrate their genome into host cell’s genome. As useful as this feature is for a long term expression of transgenes, the risk of insertional mutagenesis remains to be addressed [52]. Because the focus of this article is not on vectors but on enzyme/prodrug systems, we will not discuss them further here. However, for more information on vector development for cancer gene therapy, we would like to invite the readers refer to a recent review by Mohit et al. (2013) [48].
4. Enzyme/Prodrug Systems: From Bench to Bed
In the following sections, we take a close look at the most commonly used enzyme/prodrug systems and highlight their most significant accomplishments as well as shortcomings at both preclinical and clinical levels. To remain focused, we have refrained from elaboration on the history, use of less common enzyme/prodrug systems and also application of enzyme/prodrug systems in cancer imaging which can be found elsewhere [24, 53]. An overview of the most widely used enzyme/prodrug systems is presented in Table 2.
Table 2.
This table summarizes the most important features of six main enzyme/prodrug systems that are used in GDEPT.
| Enzyme | Prodrug | Toxic Metabolite | Mechanism of Action | Bystander Effect | Distant Bystander Effect |
|---|---|---|---|---|---|
| Herpes Simplex Virus Thymidine Kinase | Ganciclovir (GCV) | Ganciclovir Triphosphate (GCV-TP) | Blocks DNA synthesis. S and G2 phase arrest. Mitochondrial damage. Active in dividing cells. |
High, when GJIC exists Low, when GJIC doesn’t exist |
Yes |
| Cytosine Deaminase | 5-Fluorocytosine (5-FC) | 5-Fluorouracil (5-FU) | Blocks DNA and RNA synthesis. Active mostly in dividing cells, but at high concentrations can inhibit growth of both dividing and non-dividing cells. |
High, independent of GJIC | Yes |
| Nitroreductase | CB1954 and analogues | 2-hydroxylamine and 4-hydroxylamine derivatives | DNA interstrand cross linker. Active in both dividing and non-dividing cells |
Very High, independent of GJIC | Yes |
| Carboxypeptidase G2 | CMDA; ZD-2767P | N-[4-[(2-Chloroethyl)(2- (mesyloxyethyl)amino)benzoic acid (CMBA); Bis-iodophenol mustard | DNA interstrand cross linker. Active in both dividing and non-dividing cells. |
High, independent of GJIC | Yes |
| Purine Nucleoside Phosphorylase | 6-methylpurine deoxyriboside | 6-methylpurine | Inhibits DNA, RNA and protein synthesis. Active in both dividing and non-dividing cells. |
High, independent of GJIC | Yes |
| Cytochrome P450 | Cyclophosphamide; Ifosfamide | phosphoramide mustard; acrolein | DNA interstrand crosslinking agent. Active mostly in dividing cells. |
Medium, independent of GJIC | Unknown |
4.1. Thymidine Kinase/Ganciclovir System
Thymidine kinase plays an important role in reactivation of herpes simplex virus-1 (HSV-1) from latent phase [54]. TK initiates the phosphorylation of deoxythymidine to deoxythymidine monophosphate which is then turned into deoxythymidine triphosphate by cell’s endogenous kinases [55]. This mechanism, as necessary as it is for viral DNA replication in infected cells, has been used for activation of guanosine analogue, GCV, to its toxic metabolite GCV-TP by endogenous kinases [56]. The incorporation of GCV-TP in DNA structure leads to single-strand breaks and eventually inhibition of cellular DNA polymerase (Figure 2) [56]. In comparison to cytosolic TK, the affinity of HSVTK is 1000-fold higher to GCV. Therefore, the first step of GCV phosphorylation occurs predominantly by viral TK [57].
Figure 2.

Mechanism of action for ganciclovir (GCV). HSVTK phosphorylates thymidine to thymidine monophosphate (thymidine-MP) which undergoes more phosphopylation steps by cell endogenous kinases. The final product, thymidine triphosphate (thymidine-TP) is one of the building blocks of DNA structure. GCV competes with thymidine and gets phosphorylated to ganciclovir-MP first and then ganciclovir-TP. Ganciclovir-TP blocks DNA elongation by inhibiting DNA polymerase.
HSVTK/GCV system is the most abundantly used enzyme/prodrug in preclinical suicide gene therapy studies. The mechanism of GCV mediated cell death is involved with necrosis and apoptosis rather than direct chemical toxicity [58]. In spite of great literature on applications of HSVTK/GCV in cancer therapy, there are a number of shortcomings which are highlighted by several groups [59, 60]. Firstly, as mentioned earlier, the cytotoxic bystander effect of GCV is heavily dependent on the GJIC; thereby, its anticancer activity is significantly limited as GJIC is considerably compromised in many tumor tissues [59, 61]. The second concern is the relatively high affinity (indicated by Km) of HSVTK for its natural substrate thymidine. Although Km of HSVTK for GCV is 100 folds higher than it is for thymidine, but the dose of GCV required to win the competition for active site is still too high. This subsequently generates nonspecific toxicity such as severe bone marrow and immune system suppression, slow cancer cell killing kinetics and to some extent incomplete killing [31, 62]. In order to alleviate this problem, Black et al. (2001) modified HSVTK’s active site at five different residues generating a mutant, namely SR39. This mutant showed 294-fold decrease in the dose of prodrug that was needed to render the same effect as wild type HSVTK (wt-HSVTK) (Table 3) [63]. Ardiani et al. (2010), later developed a construct made of HSVTK mutants fused to the second enzyme in GCV phosphorylation pathway, guanylate kinase. In the presence of GCV (as low as 0.1 mg/Kg), the combination of SR39 with guanylate kinase showed improved tumor growth inhibition in comparison to SR39 alone [64].
Table 3.
Sequence differences among wild type HSVTK, SR39 and TK007 mutants. In comparison to wild-type, SR39 has five amino acid mutations at positions 159, 160, 161, 168 and 169, whereas TK007 bears only one mutation at position 168.
| Amino Acid Position | 159 | 160 | 161 | 168 | 169 |
|---|---|---|---|---|---|
| wt-HSVTK | L | I | F | A | L |
| SR39 | I | F | L | F | M |
| TK007 | L | I | F | H | L |
Other groups have also made attempts to enhance the affinity of HSVTK for GCV. Balzarini et al. (2006), reported a mutant TK (A168H mutation) which showed significantly lower kinase activity towards enzyme’s natural substrate thymidine, but with 4-fold increase in activity towards GCV [65]. Preuss et al. (2011) splice corrected the A168H mutant to make another mutant, namely TK007 (Table 3). The in vitro results showed an improved cancer cell killing efficiency and significantly higher bystander effect over the wt-HSVTK. The in vivo studies also showed complete remission of glioblastoma xenograft tumors in the presence of GCV with doses as low as 10 mg/kg [60].
Considering the outstanding cancer cell killing efficiency of HSVTK/GCV system at the in vitro level, it has been studied for its anticancer efficacy against leukemia[66], glioma[67], bladder cancer[68], ovarian cancer [69], oral cancer, as well as others in various animal models. The promising preclinical results encouraged a number of clinical trials in the past decade, in which HSVTK/GCV system been used as an adjunct therapy in patients with glioblastoma, prostate, hepatocarcinoma, head and neck carcinoma and others (Table 4) [70–72]. Most of the clinical trials so far have been in phase I/II with the focus on the safety of suicide gene therapy treatment. This is mostly due to the use of adenoviral vectors in these clinical trials. Review of the data show a number of such studies that have addressed the safety of adenoviral vectors after local administration [71]. For example, Nasu et al. (2007) injected escalating doses from 109 to 1010 of viral particles directly to prostate of patients with prostate cancer. Their study showed neither viral DNA in systemic circulation nor any change in phenotype distribution in peripheral blood samples [71]. Later, Xu et al. (2009), tried intratumoral injection with dose escalation from 2.5 × 1011 to 1 × 1012 virus particles in patients with head and neck carcinoma. In spite of minor side effects, the authors demonstrated the safety of the study and also reported a partial response in the loci of injection [73]. In a more recent study by Sangro et al. (2010), patients with hepatocellular carcinoma received different intratumoral doses of first generation adenoviral vector bearing HSVTK followed by systemic administration of GCV. Even at the high dose of 2 × 1012 viral particles per patient no serious side effects was observed. In patients who received high doses, partial tumor stabilization and intratumoral necrosis was also reported. The authors confirmed the safety and feasibility of such local therapy in patients with hepatocellular carcinoma [74]. As the first in this category, Cerepro® (Sitimagene ceradenovec) is an adenoviral vector-based HSVTK/GCV system with the potential for the treatment of high grade glioma. Cerepro® (Ark Therapeutics; UK and Finland) has passed preclinical and phase I/II clinical trials (2010) in patients with operable high-grade glioma [75, 76]. Although significant increase in survival rate has been observed in patients who received intra-cavity injection of Cerepro® after tumor resection, but the efficacy results so far have not been satisfactory to receive approval by the European Committee for Medicinal Products for Human Use [76]. So far, in none of the clinical trials based on localized HSVTK suicide gene therapy any serious vector related toxicity has been reported.
Table 4.
The list of clinical trials performed with the major enzyme/prodrug systems in the past decade. Clinical trials before year 2003 are not shown.
| Enzyme/Prodrug System | Clinical Stage | Tumor Type | Vector | Reference |
|---|---|---|---|---|
| HSVTK/GCV | I/II | Glioblastoma multiform | Liposomal | [70] |
| HSVTK/GCV | I | Prostate cancer | Adenoviral | [71] |
| HSVTK/GCV | I | Head and neck cancer and other malignant tumors | Adenoviral | [73] |
| HSVTK/GCV | I | Prostate cancer | Adenoviral | [82] |
| HSVTK/GCV | I | Hepatocellular carcinoma | Adenoviral | [74] |
| HSVTK/GCV | I | Recurrent Gynecologic Cancer | Adenoviral | [81] |
| CD/5-FC and HSVTK/GCV | I | Prostate cancer | Adenoviral | [98] |
| CD/5-FC and HSVTK/GCV | I | Prostate cancer | Adenoviral | [101] |
| CD/5-FC and HSVTK/GCV | I | Prostate cancer | Adenoviral | [100] |
| NTR/CB1954 | I | Gastrointestinal | Adenoviral | [107] |
| NTR/CB1954 | I | Liver cancer | Adenoviral | [118] |
| NTR/CB1954 | I/II | Prostate cancer | Adenoviral | [113] |
| CPG2/Nitrogen Mustard | I | Colorectal carcinoma | ADEPT | [120] |
| P450/Oxazaphosphorine | I/II | Pancreatic carcinoma | Encapsulated Allogeneic Cells | [128] |
| P450/Oxazaphosphorine | I | Breast cancer or Melanoma | MetXia-P450 (Retroviral) | [129] |
Overall, it seems that the efficiency of therapy and not the safety is the main problem with this enzyme/prodrug system that should be addressed. The bottleneck of the most studies is the low percentage of transduced tumor cells by adenoviral vectors. To address this problem, redirecting the tropism of adenoviral vectors has been suggested to be a potential solution for higher rate of transduction, especially in cancer tissues which barely express CAR [77]. Redirecting the tropism of adenoviral vector through changing the surface ligands has been reported in various studies [52, 78, 79]. Different strategies have been suggested such as the incorporation of new targeting moiety in the body of viral surface protein or using a bispecific adaptor which can facilitate the internalization of virus by attaching to both viral and cell surface proteins. Molecules such as single chain variable fragment (ScFv), Fab fragment or IgG binding domain have been used for viral tropism modifications [80]. The pros and cons of these molecules is beyond the scope of this review and the reader is invited to refer to the most updated review paper by Verheije and Rottier [80]. In a recent phase I clinical study by Kim et al. (2012), the application of this technique has been demonstrated by using a tropism modified adenovirus carrying RGD sequence in the fiber knob[81]. As a result, the transfection of adenovirus depended on integrin receptors rather than CAR. Although the safety profile of this adenoviral vector was tested before the clinical trial and deemed satisfactory [77], but the efficacy results have not been posted and still in progress.
In parallel to the strategies mentioned above for the enhancement of vectors’ transduction efficiency and therapeutic efficacy, tissue- or cell-specific promoters with elevated activity in tumor tissue have also been utilized to increase vectors’ safety as they tend to accumulate in non-target tissues such as liver and lungs. The most common promoters which have been used to enhance the expression of suicide genes at the target tumor site are human telomerase reverse transcriptase (hTERT) promoter, carcinoembryonic antigen (CEA) promoter, osteocalcin promoter (OC), and hypoxia and radiation responsive elements [18–21, 24]. The last reported clinical trial using this approach was conducted by Kubo et al. which goes back to year 2003. A DNA construct with HSVTK under the control of osteocalcin promoter was designed in an adenoviral vector backbone for local suicide gene therapy of prostate cancer [82]. Although the treatment was well-tolerated but due to the low expression of CAR in prostate tumors of the patients, this study did not produce significant results in terms of efficacy and did not move forward.
In summary, it is apparent that the therapeutic efficacy of HSVTK/GCV system in the clinic has stagnated mostly due to the low efficiency of vectors and dependency of GCV-TP on GJIC. Therefore, to overcome these obstacles there is a need for the development of a potent GCV derivative that works independent of GJIC and/or new targeted vectors with extremely high rate of transduction efficiency.
4.2. Cytosine Deaminase/5-Fluorocytosine System
The anticancer drug, 5-flourouracil (5-FU) has been used over four decades for the treatment of colorectal, breast and head and neck solid tumors. Its side effects include myelosuppression, mucositis, dermatitis, diarrhea and cardiac toxicity among others [83]. A precursor of 5-FU, 5-FC is shown to be converted to 5-FU by bacterial or fungal CD which does not exist in mammalian cells [84]. Once converted to 5-FU, it can easily diffuse to neighboring cells thanks to its small size and neutral charge [85]. 5-FU is converted by intracellular enzymes to several metabolites which lead to formation of fraudulent 5FU-RNA and 5FU-DNA, inhibition of thymidylate synthase and eventually apoptosis (Figure 3). At high 5-FU concentrations, the level of mRNA inhibition is reported to be so high that can even lead to protein starvation in non-dividing cancer cells [86]. CD/5-FC system has a number of advantages over HSVTK/GCV including gap junction independent bystander effect and induction of significant distant bystander effect [87]. 5-FC, but not 5-FU, is able to diffuse through blood brain barrier; hence, many studies have focused on using this property for treating hard-to-reach tumors such as glioblastoma [87]. The other advantage of CD/5-FC system is the radiosensitizing ability of 5-FU which can enhance its tumor killing efficiency in combination with radiotherapy [88]. It is feasible to detect the conversion of 5-FC to 5-FU in patients using fluorine magnetic resonance spectroscopy (19F MRS) and fluorine magnetic resonance spectroscopic imaging (19F MRSI) which makes the system suitable for clinical translation [89]. It has also been reported that the immune reaction is directed toward the cells expressing CD and is independent of 5-FC treatment [90]. This feature might be beneficial in terms of vaccination but in the context of suicide gene therapy it might lead to the early removal of CD expressing cells.
Figure 3.

Deamination of 5-FC by cytosine deaminase (CD). The product, 5-FU is converted to 5-FUMP and eventually 5-FUTP which blocks RNA synthesis. Other byproducts such as 5-FdUMP and 5-FdUTP block DNA synthesis after incorporation into DNA structure.
In spite of the above mentioned advantages, there are few drawbacks which overshadow the clinical usefulness of CD/5-FC system. One is that 5-FC might be converted to 5-FU by normal flora in the gut resulting in side effects. The other is the high affinity of bacterial cytosine deaminase (bCD) for its natural substrate, cytosine, which necessitates the higher dose of 5-FC. Several attempts have been made to improve bCD kinetics characteristics such as higher affinity and lower IC50 for 5-FC. Kaliberova et al. (2008), reported the sequence of a mutant, bCD-D314A, which showed a significant specificity towards 5-FC with a lower IC50 in comparison to wt-bCD. This mutant has been successfully used in combination with low dose radiation to reduce tumor sizes in various cancer models [91, 92]. Another mutant, reported by Fuchita et al. (2009), has showed the most desirable features for suicide gene therapy in terms of significant shift to 5-FC, decrease in IC50 and an outstanding bystander effect compared to wt-CD[93].
While many studies have been focused on improving bCD toxicity profile, cytosine deaminase from the yeast saccharomyces cerevisiae (yCD), possesses the natural characteristics which overtakes bCD in enzyme kinetics. It comparison to bCD, yCD has significantly higher affinity for 5-FC. However, the thermolability of yCD and its short half life in vivo has limited its use at the clinical level [31]. New mutants of yCD, namely yCD double and yCD triple seem to overcome these problems, imparting more thermostability and in case of yCD triple, even greatest enhancement in sensitivity [94, 95]. In another approach, the combination of yCD with uracil phosphoribosyltransferase (UPRT) gene which converts 5-FU to 5-fluorouridine 5′-monophosphate produces a higher sensitivity to 5-FC in comparison to yCD alone [96, 97]. While CD/5-FC alone has been tested successfully on a variety of tumors in preclinical settings, there are studies that show the significant advantages of using of CD along with HSVTK/GCV, or as a radiation sensitizer in combination with radiotherapy [98, 99].
Despite the vast use of CD/5-FC system at the preclinical level, examples of clinical studies are more limited than HSVTK/GCV system (Table 4). Freytag et al. (2003 and 2007), in two separate studies examined the combination of CD/HSVTK in phase I clinical trials against prostate cancer using first and second generation adenoviral vectors [100, 101]. The expression of transgene was detectable up to three weeks after injection and no serious side effects related to adenoviral vector or prodrug were reported. It was observed that PSA doubling time increased from 17 to 31 months in these patients, delaying androgen salvage therapy for 2 years [101, 102]. These findings established adenoviral mediated double suicide gene therapy as a potentially safe and effective treatment for prostate cancer. These studies also raised the possibility that it may be used to improve the outcome of conformal radiotherapy. Recently (2012), a commercialized viral vector (Toca 511) for heat stabilized yCD suicide gene therapy was introduced by Tocagen Inc. Toca 511 is a non-lytic retroviral vector armed with yCD which has been deemed suitable for clinical trials. Tocagen Inc. is currently conducting a phase I/II clinical trial in the United States on patients with recurrent or progressive Grade III or Grade IV gliomas after tumor resection (clinicaltrials.gov; see NCT01156584 and NCT01470794). Although no results has been posted yet but their protocol was based on in vivo data demonstrating the safety and effectiveness of Toca 511 therapy followed by oral dose of extended-release 5-FC in animals [103, 104].
To date, significant progress has been made in stabilizing and optimizing the cytosine deaminase enzyme. Given that the 5-FU has significant bystander effect, is able to effectively kill dividing cells and at high concentrations even the non-dividing cancer cells, this enzyme/prodrug system emerges with great potential to succeed in the clinic. The major limitation at this point comes from the low transduction efficiency of the targeted vectors.
4.3. Nitroreductase/CB1954 System
CB1954 is a DNA alkylating agent which becomes activated after conversion to its toxic metabolite by E. coli NfsB nitroreductase (NTR)[105]. After reduction by cellular thioesterase, CB1954 becomes a potent DNA chelating agent which can freely diffuse to surrounding cells and trigger extensive DNA damage and a P53 and cell cycle independent apoptosis in both replicating and non-replicating cells (Figure 4) [106]. Because NTR enzyme needs NADPH or NADH as an electron donor, the activation of prodrug happens only intracellularly; therefore, side effects are limited to modified cells and the cells in vicinity. The major drawback of NTR/CB1954 system is the low activation rate of prodrug as CB1954 is not the natural substrate of NTR. Chung-Faye et al. (2001), reported that CB1954 shows a dose-dependent hepatotoxicity [107], which provoked others to focus on the improvement of enzyme kinetics [108]. In a study by Grohmann et al. (2009), the bacterial NTR was codon optimized to enhance and stabilize its expression in mammalian cells [109]. Other groups have also worked to assess other prokaryotic enzymes which are able to activate CB1954 or design nitroaromatic derivatives which are potentially better prodrugs for nitroreductase [106, 108, 110]. For example, Prosser et al. (2010), evaluated all E. coli nitroreductases with the ability to activate CB1954 and reported few new NTRs such as AzoR, NFsA and NemA. In comparison to NfsB NTR, NfsA in the form of purified protein was 10 fold more efficient in CB1954 activation [108]. In addition, they observed wild type and mutated version of other nitroreductase enzymes such as flavin reductase FRase I from Vibrio Fischeri are able to activate CB1954 and its analogues [106, 111]. In parallel, others have developed new generation of nitroaromatic prodrugs such as nitro-CBI-DEI and PR-104A which can be activated both by hypoxia and bacterial nitroreductases [112]. Under hypoxic conditions (e.g., tumor environment), these two compounds undergo a one-electron transfer reduction reaction by human endogenous oxidoreductases but in normal cells they rapidly get reoxidazed [110]. Bacterial nitroreductases catalyze the same reaction through a two step electron transfer in an oxygen-independent fashion which results in toxic metabolites [110]. Therefore, these two mechanisms induce synergistic cytotoxic effects.
Figure 4.

Activation of CB1954 by bacterial nitroreductase. After activation, both 4-hydroxylamine and 2-hydroxylamine metabolites cause DNA damage.
So far, a limited number of clinical trials have been conducted with NTR/CB1954 system (Table 4). In a phase I/II clinical trial conducted in 2009, replication-defective adenoviruses encoding nitroreductase and systemic administration of prodrug CB1954 were used for the treatment of patients with localized prostate cancer [113]. The trial assessed the effect of dose, route of administration and volume of injection on biodistribution of viral vectors. Although immune response against adenoviral vectors was detected, but signs of decrease in tumor sizes were also observed in some patients (decrease in PSA >10% and in two patients >50%) which lasted for six months. Another piece of evidence which supported the effectiveness of therapy was a delayed PSA progression. In a 2012 phase I clinical study which has conditionally been approved in UK, a tumor specific promoter (human telomerase) has been used in an adenoviral vector along with NTR/CB1954 system. The system was injected to the patients with advanced intra-abdominal cancer followed by the injection of prodrug CB1954 [114]. This clinical trial (UK-0125) is still in progress and no data has been released so far.
This enzyme/prodrug system overall has the advantage of being able to kill both dividing and non-dividing cancer cells with significant distant bystander effect; however, concerns related to CB1954 conversion rate and dose-dependent hepatotoxicity has slowed down its progress. In a study by our group using genetically-modified theranostic mesenchymal stem cells as gene delivery vehicles, we also observed significant limitations with the use of NTR/CB1954 system [115]. Our comparative studies using quantitative imaging technique clearly showed CB1954 prodrug’s slow conversion rate in vivo resulting in superior anticancer activity with yCD:UPRT/5-FC system over NTR/CB1954. Although, new generation of prodrugs such as PR-104A (Proacta Inc.) and nitro-CBI-DEI in recent years have revived hope and several new studies are underway to evaluate their therapeutic potential in clinical trials [116], but the limited availability of these prodrugs through commercially available sources has restricted the number of studies that are necessary for comprehensive preclinical evaluation.
4.4. Carboxypeptidase G2/Nitrogen Mustard System
In all other enzyme/prodrug systems discussed so far, the prodrug undergoes more than one step to get activated. If one of these steps is dependent on intracellular enzymes, then the activity of the enzyme imposes rate limit which could decelerate the prodrug activation reaction. Bacterial enzyme Carboxypeptidase G2 (CPG2) from Pseudomonas RS-16 with no mammalian equivalent cleaves glutamic acid from nitrogen mustard-based drugs to release the cognate drug which unlike the product of other systems such as HSVTK/GCV or CD/5-FC is active by itself and does not require further modification (Figure 5). The final alkylating metabolite is lipophilic which freely diffuses from cell to cell, independent from gap junctions and eventually makes inter- and intra-strand DNA linkage. This enzyme/prodrug system affects both dividing and quiescent cells and potent bystander effect has been observed both in vitro and in vivo [117, 118]. CMDA (4-[(2-chloroethyl)(2-mesyloxyethyl)amino]benzoyl-L-glutamicacid) is among the first developed nitrogen mustard-based prodrugs which can be hydrolyzed to glutamic acid and DNA alkylating agent 4-[(2-chloroethyl)(2-mesyloxyethyl)amino]benzoic[105]. Sribbling et al. (2000), reported that after treating xenograft breast tumors that expressed CPG2 with prodrug CMDA, most of the tumor cells were in apoptotic phase, even though the percentage of transduced cells was low [119]. Other nitrogen mustard (NM) derivates such as ZD2767P with at least 300 times more potency than CMDA have also been introduced (Figure 5) [120]. Friedlos et al. (2002), reported the design of three new prodrugs for CPG2 which showed much greater (19 to 27 folds) cytotoxicity to cancer cells as compared to CMDA. Surprisingly, these prodrugs were more effective when the lower percentage of cells (10% to 50%) expressed CPG2 enzyme [121]. In another attempt to develop a new prodrug for CPG2, Mancini et. al (2009) designed a new non-toxic derivative suitable for evaluating gene delivery and expression of enzyme. The conversion of compounds to their cleaved byproducts was detectable by 19F magnetic resonance spectroscopy, thanks to the fluorine groups [122]. One of the drawbacks of CPG2/NM system is the toxicity caused by secretion of the enzyme from transduced cells into blood circulation. In addition, due to the bacterial origin of the enzyme, a potent immune reaction might be triggered. In order to prevent these problems, the secretory tag has been cut off from the enzyme to restrict its localization to cytosol while maintaining its activity [119].
Figure 5.

Activation of CMDA and ZD2767P to their toxic metabolites by bacterial carboxypeptidase G2 (CPG2).
The main application of CPG2/NM system in clinical studies has been in combination with antibodies in Antibody-Directed Enzyme Prodrug Therapy (Table 4). In this approach, the enzyme is attached to a tumor-specific antibody via chemical reaction or recombinant protein fusion technology [123]. Early clinical trials with this kind showed the feasibility of technique, with the evidence of localized CPG2 activation only in tumor tissues [124]. The main reported toxicity was myelosuppression due to long half life of the drug and its leakage to the blood. Later, in another dose-escalating phase I clinical trial, Francis et al. (2002) used A5CP, a conjugation of bacterial carboxypeptidase to a Fab2 fragment of a mouse monoclonal antibody for carcinoembryonic antigen (CEA) in 27 patients with advanced colorectal carcinoma. The prodrug ZD2767P was used because it had higher potency than CMDA and shorter half life after conversion to its toxic metabolite [120]. The maximum tolerated dose of ZD2767P was reached at 15.5 mg/m−2 × three administrations with a serum CPG2 level of 0.05 U ml−1. Overall, no significant therapeutic effect was observed because of an inadequate localization of enzyme/antibody conjugate and a humoral immune reaction against CPG2 and mouse antibody. However, the prodrug had a fast clearance and no active form of drug was detectable in blood which resulted in less Myelosuppression [120].
From the review of literature, it appears that success in the clinic may be achieved when a NM-based prodrug with suitable half-life (e.g., ZD2767P) in combination with non-secretory CPG2 and an efficient targeted vector are used. Unfortunately, there have been no clinical trials using this system in the past decade to discuss at this point.
4.5. Cytochrome P450/Oxazaphosphorine System
P450 enzymes are the most important super family in metabolism of xenobiotics. They are located mostly in liver; however, their activity in other organs and to some extent in tumors has been detected. Although the role of P450 enzymes in metabolism and inactivation of chemotherapy agents is well-understood [125], but the effect of P450-mediated metabolism on oxazaphosphorine drugs such as cyclophosphamide (CPA) and ifosfomide (IFO) is quite different. While most of the chemotherapeutics are deactivated by P450 enzymes, these enzymes convert CPA and IFO to their unstable but active metabolites (Figure 6). The metabolites also show a strong bystander effect quite independent of gap junctions [125]. With a high level of P450 activity in liver, the goal of GDEPT has been set on inhibition of hepatic P450 while localizing the activity of recombinant P450 enzymes in tumors so that the systemic side effects of oxazaphosphorine drugs such as neurotoxicity, nephrotoxicity, and urotoxicity could be avoided. Huang et al. (2001) used CYP2C inhibitors to inhibit the conversion of CPA in liver while gliosarcoma xenografts were able to express CYP2B1. Unfortunately, the results were not significant indicating that the inhibitors were not specific for hepatic P450 [126]. Another approach to shut down hepatic P450 is co-administration of cytochrome P450 reductase (CPR) inhibitors and anti-thyroid drugs such as Methimazole since thyroid hormone controls the expression of CPR [127]. Utilizing this strategy, a number of clinical trials have been carried out worldwide for P450/oxazaphosphorine system (Table 4). In one of the most successful phase I/II trials conducted in 2003 with this system, fourteen patients with pancreatic tumor were treated with genetically modified allogenic cells to express CYP2B1. The cells were delivered to the tumors via tumor vasculature followed by administration of low dose IFO two days after cell injection. In four patients the tumor regression was complete whereas other 10 patients had a stabilized tumor size. The median survival time was doubled from 22 weeks to 44 weeks [128]. In another phase I/II clinical trial (2005), human CYP2B6 a commercial retroviral vector, MetXia®, was used to deliver CYP2B6 (human cytochrome P450 type 2B6 gene) [129]. A total of nine patients with breast cancer and three with melanoma received MetXia® followed by oral administration of cyclophosphamide. The results demonstrated safety and moderate efficiency of MetXia supporting the idea of using P450 isoforms/CPA in future clinical trials. However, no other clinical trial with this enzyme/prodrug system has been reported in recent years. At present, several groups are working to engineer new mutants of CYP2B6 in order to improve the affinity towards CPA. For example, Touati et al. (2014), reported the structure of a triple mutant CYP2B6 in fusion with NADPH cytochrome P450 reductase that showed superior ability in converting CPA into its cytotoxic form [130]. While this system has shown great promise at the preclinical level, its safety and efficacy in clinical trials remains to be determined.
Figure 6.

The two-step process which produces the toxic metabolites, phosphoramide mustard and acrolein from cyclophosphamide. CYP enzyme super family catalyzes the first step whereas the second step is a self-immolative reaction.
4.6. Purine Nucleoside Phosphorylase/6-Methylpurine Deoxyriboside (PNP/MEP) System
The E. coli purine nucleoside phosphorylase (PNP) is a hexameric enzyme, catalyzing the glycosidic cleavage of purine ribonucleoside prodrugs such as 6-methylpurine 2-deoxyriboside and fludarabine to 2-deoxyribose-1-phosphate (or arabinose-1-phosphate) and free base compounds such as 6-methylpurine and 2-fluoroadenine, respectively (Figure 7). Both compounds are freely diffusible across cell membranes, allowing their spread from PNP transduced to untransduced cells and are toxic to both proliferating and non-proliferating cells; thereby, achieving a potent bystander effect [131, 132]. The ability of the activated prodrug in killing both proliferating and non-proliferating cancer cells is attributed to its mechanism of action which is independent of DNA synthesis [132]. The bystander activity is facilitated by the nucleotide and nucleobase transporters across membranes in both directions not requiring cell-cell contact or gap junctions. Initial in vivo studies on prostate tumors suggested that PNP/MEP system is superior to HSVTK/GCV system [133, 134]. Metabolites from PNP are incorporated during RNA synthesis and hence eventually block protein synthesis. Limitations related to immunogenicity from the bacterial PNP led investigators to develop human PNP (hPNP) mutants that can cleave adenosine based prodrugs which are not recognized by wild-type hPNP [135]. These mutants are an attractive target because the endogenous wild-type hPNP cannot use (deoxy) adenosine-based prodrugs as substrate, whereas the mutants are highly effective in cleaving (deoxy) adenosine-based prodrugs and generating high levels of cytotoxic drugs in preclinical models [136]. Preclinical studies with PNP have shown that the enzyme activity from PNP transfected tumor cells are more than that of endogenous APRT possibly causing MEP to diffuse away from the tumor. However, unlike CD/5-FC system where UPRT plays a role in increasing sensitivity of the prodrugs, APRT overexpression did not improve the efficiency of the PNP system [137]. In an effort to improve anti-tumor activity, designer nucleosides have been reported in combination with a structurally modified PNP enzyme [138, 139]. Unfortunately, this approach has not yet been successful in identifying prodrug/enzyme combinations that demonstrate better in vivo anti-tumor activity. In addition to MEP, the PNP gene is able to metabolize a series of other prodrug substrates such as fludarabine phosphate (F-araAMP) and 2-fluoro-2-deoxyadenosine (F-dAdo) [140–142]. To date, there has been only one phase I clinical trial with this system which started in 2011 by PNP Therapeutics®. This trial investigates the safety of E. coli PNP/fludarabine phosphate in patients with head and neck cancers or other solid tumors (clinicaltrials.gov; NCT01310179). This is an ongoing clinical trial and no data has been released yet.
Figure 7.

The conversion of 6-methylpurine deoxyriboside to 6 -methylpurine by bacterial purine nucleoside phosphorylase.
4.7. Other Enzyme/Prodrug Systems
In addition to the six main enzyme/prodrug systems that were discussed so far, a few other potentially effective systems have also been investigated. One enzyme/prodrug system that has been studied is the horseradish peroxidase/indole-3-acetic acid (HRP/IAA) system, which is especially active under hypoxic conditions [143]. In combination with radiation therapy, this system has shown significant effectiveness against solid tumors [144]. Unfortunately, this enzyme/prodrug combination has not reached clinical trials yet. Another enzyme/prodrug system worth mentioning is the carboxylesterase/irinotecan (CE/Irinotecan) system. The irinotecan (CPT-11) prodrug can be converted into cytotoxic SN38, which is able to inhibit DNA topoisomerase I, an essential component for DNA relaxation machinery [145]. In an interesting approach, Metz et al. (2013), engineered secretary form of CE which could be transported outside of the cell; thereby, expanding the prodrug activation radius [146]. One safety concern with this system is that after systemic administration of irinotecan, it could get activated by CE that exist in normal tissues (e.g., liver) resulting in off-target toxicity [147, 148]. Currently there is one ongoing study in Phase I clinical trials with CE/Irinotecan using neural stem cells as delivery vehicle for the treatment of glioma (clinicaltrials.gov; NCT02192359).
4.8. Combination Therapy
To maximize the therapeutic efficacy and safety, several studies have examined combination of the suicide gene therapy with other therapeutic modalities. For example, suicide gene therapy has been used in combination with conventional cancer therapy approaches such as surgery, radiation therapy, cell therapy, immunotherapy and chemotherapy. To minimize the possibility of cancer recurrence, suicide gene therapy could also be utilized as the follow-up strategy after surgery (tumor excision) in order to eradicate the leftover cancer cells [149, 150]. In an attempt to achieve more precise targeting, radiation responsive promoters are also used to switch on or enhance the expression of suicide gene thorough a remote and non-invasive radiation control [151, 152]. As a result, less dose of radiation is required and fewer side effects are produced. Suicide gene therapy can also be combined with immunotherapy to enhance the safety of treatment protocol. Adoptive T cells, especially chimeric antigen receptors (CARs) T cells have shown great potential in treating a broad range of malignancies [153–155]. A recent report has shown that complete remission can be achieved by applying this strategy to treat acute lymphoid leukemia [156]. Since unexpected T cell proliferation could induce several severe adverse effects [157], T cells are engineered to express a suicide gene that could serve as an artificial kill switch [158]. Combining suicide gene therapy with chemotherapy could also provide superior efficacy over each therapeutic modality individually [104, 159, 160]. For this purpose, over the past decades various delivery systems such as micelles, liposomes and inorganic particles have been developed and used for multimodal deliveries [161]. Duarte et al. (2012), have recently discussed the use of such biomaterials for combination therapy of cancer [162]. Given the broad therapeutic potential of suicide genes and our current understanding of the mechanisms of cancer resistance, we expect to observe an exponential growth in the use of biomaterials for multimodal drug/gene delivery.
5. Conclusions and Future Direction
Significant progress has been made in the field of suicide gene therapy of cancer since its introduction in 1984. While promising, the overall progress appears to be somewhat slow as only 45 clinical trials have reached phase III, one in phase IV and none approved for clinical use. One of the major contributing factors to this slow progress has been the low transduction efficiency of the vectors, especially in solid and dense tumors where the cells at the inner layers of tumor are not easily accessible. In order to transition ideas effectively and efficiently from bench to bedside, issues such as lack of an appropriate delivery method, short-term and low expression of transgenes, and prodrug’s conversion rate and bystander effect need to be addressed. Therefore, more assets need to be diverted towards enzyme engineering and prodrug development to accelerate the development of stable/high affinity enzymes in combination with safe prodrugs with potent bystander effect. Given that clinical trials are extremely costly, development of a reliable method for screening a large number of enzyme mutants or prodrugs with different levels of bystander effects at the preclinical level has also become a necessity. The success and failure stories in the past decades clearly show that cancer is a complex and evolving disease; therefore, a combination of conventional and newly emerging strategies are required to effectively deal with this health issue. These altogether signify that to win the war against cancer, a coherent approach backed by appropriate investment in this field are needed.
Acknowledgments
This work was supported in part by grants from the Department of Defense Prostate Cancer Program (PC081055) and National Institutes of Health to A. Hatefi.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baban DF, Seymour LW. Control of tumour vascular permeability. Adv Drug Deliv Rev. 1998;34:109–119. doi: 10.1016/s0169-409x(98)00003-9. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81–89. doi: 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeVita VT, Chu E. A History of Cancer Chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 4.Vandyke K, Fitter S, Dewar AL, Hughes TP, Zannettino AC. Dysregulation of bone remodeling by imatinib mesylate. Blood. 2010;115:766–774. doi: 10.1182/blood-2009-08-237404. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Y, Kopecek J. Biological rationale for the design of polymeric anti-cancer nanomedicines. J Drug Target. 2013;21:1–26. doi: 10.3109/1061186X.2012.723213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barenholz Y. Doxil(R)--the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Yao XL, Yoshioka Y, Ruan GX, Chen YZ, Mizuguchi H, Mukai Y, Okada N, Gao JQ, Nakagawa S. Optimization and internalization mechanisms of PEGylated adenovirus vector with targeting peptide for cancer gene therapy. Biomacromolecules. 2012;13:2402–2409. doi: 10.1021/bm300665u. [DOI] [PubMed] [Google Scholar]
- 8.Yao X, Yoshioka Y, Morishige T, Eto Y, Watanabe H, Okada Y, Mizuguchi H, Mukai Y, Okada N, Nakagawa S. Systemic administration of a PEGylated adenovirus vector with a cancer-specific promoter is effective in a mouse model of metastasis. Gene Ther. 2009;16:1395–1404. doi: 10.1038/gt.2009.95. [DOI] [PubMed] [Google Scholar]
- 9.Shah V, Taratula O, Garbuzenko OB, Taratula OR, Rodriguez-Rodriguez L, Minko T. Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: an optimal delivery of siRNA and anticancer drug. Clin Cancer Res. 2013;19:6193–6204. doi: 10.1158/1078-0432.CCR-13-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356:197–203. doi: 10.1016/j.canlet.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, Nunan R, Pirollo KF, Rait A, Chang EH. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013;21:1096–1103. doi: 10.1038/mt.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, Paz-Ares L, Cho DC, Infante JR, Alsina M, Gounder MM, Falzone R, Harrop J, White AC, Toudjarska I, Bumcrot D, Meyers RE, Hinkle G, Svrzikapa N, Hutabarat RM, Clausen VA, Cehelsky J, Nochur SV, Gamba-Vitalo C, Vaishnaw AK, Sah DW, Gollob JA, Burris HA., 3rd First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406–417. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Gao DY, Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv Drug Deliv Rev. 2015;81:128–141. doi: 10.1016/j.addr.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65:1284–1298. doi: 10.1016/j.addr.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Portsmouth D, Hlavaty J, Renner M. Suicide genes for cancer therapy. Mol Aspects Med. 2007;28:4–41. doi: 10.1016/j.mam.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Dorer DE, Nettelbeck DM. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv Drug Deliv Rev. 2009;61:554–571. doi: 10.1016/j.addr.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Yao XL, Nakagawa S, Gao JQ. Current targeting strategies for adenovirus vectors in cancer gene therapy. Curr Cancer Drug Targets. 2011;11:810–825. doi: 10.2174/156800911796798896. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Hou J, Liu Z, Yu H, Sun W, Xiong J, Liao Z, Zhou F, Xie C, Zhou Y. Gene therapy with tumor-specific promoter mediated suicide gene plus IL-12 gene enhanced tumor inhibition and prolonged host survival in a murine model of Lewis lung carcinoma. J Transl Med. 2011;9:39. doi: 10.1186/1479-5876-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dabrowska A, Szary J, Kowalczuk M, Szala S, Ugorski M. CEA-negative glioblastoma and melanoma cells are sensitive to cytosine deaminase/5-fluorocytosine therapy directed by the carcinoembryonic antigen promoter. Acta Biochim Pol. 2004;51:723–732. [PubMed] [Google Scholar]
- 20.Greco O, Marples B, Joiner MC, Scott SD. How to overcome (and exploit) tumor hypoxia for targeted gene therapy. J Cell Physiol. 2003;197:312–325. doi: 10.1002/jcp.10374. [DOI] [PubMed] [Google Scholar]
- 21.Scott SD, Greco O. Radiation and hypoxia inducible gene therapy systems. Cancer Metastasis Rev. 2004;23:269–276. doi: 10.1023/B:CANC.0000031766.58614.f1. [DOI] [PubMed] [Google Scholar]
- 22.Robson T, Hirst DG. Transcriptional Targeting in Cancer Gene Therapy. J Biomed Biotechnol. 2003;2003:110–137. doi: 10.1155/S1110724303209074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papadakis ED, Nicklin SA, Baker AH, White SJ. Promoters and control elements: designing expression cassettes for gene therapy. Curr Gene Ther. 2004;4:89–113. doi: 10.2174/1566523044578077. [DOI] [PubMed] [Google Scholar]
- 24.Karjoo Z, Ganapathy V, Hatefi A. Gene Directed Enzyme Prodrug Cancer Therapy. In: Lattime ES, Gerson S, editors. Gene Therapy of Cancer, Translational Approaches from preclinical Studies to Clinical Implemantation. Elsevier; 2013. pp. 77–91. [Google Scholar]
- 25.Nemunaitis J, Tong AW, Nemunaitis M, Senzer N, Phadke AP, Bedell C, Adams N, Zhang YA, Maples PB, Chen S, Pappen B, Burke J, Ichimaru D, Urata Y, Fujiwara T. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther. 2010;18:429–434. doi: 10.1038/mt.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L, Matherly J, Smallwood A, Adams JY, Billick E, Belldegrun A, Carey M. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Ther. 2001;8:1416–1426. doi: 10.1038/sj.gt.3301549. [DOI] [PubMed] [Google Scholar]
- 27.Kanegae Y, Terashima M, Kondo S, Fukuda H, Maekawa A, Pei Z, Saito I. High-level expression by tissue/cancer-specific promoter with strict specificity using a single-adenoviral vector. Nucleic Acids Res. 2011;39:e7. doi: 10.1093/nar/gkq966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen XG, Godbey WT. The potential of the human osteopontin promoter and single-nucleotide polymorphisms for targeted cancer gene therapy. Curr Gene Ther. 2015;15:82–92. doi: 10.2174/1566523214666141127094033. [DOI] [PubMed] [Google Scholar]
- 29.Chuah MK, Petrus I, De Bleser P, Le Guiner C, Gernoux G, Adjali O, Nair N, Willems J, Evens H, Rincon MY, Matrai J, Di Matteo M, Samara-Kuko E, Yan B, Acosta-Sanchez A, Meliani A, Cherel G, Blouin V, Christophe O, Moullier P, Mingozzi F, VandenDriessche T. Liver-specific transcriptional modules identified by genome-wide in silico analysis enable efficient gene therapy in mice and non-human primates. Mol Ther. 2014;22:1605–1613. doi: 10.1038/mt.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber BE, Austin EA, Richards CA, Davis ST, Good SS. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc Natl Acad Sci U S A. 1994;91:8302–8306. doi: 10.1073/pnas.91.17.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ardiani A, Johnson AJ, Ruan H, Sanchez-Bonilla M, Serve K, Black ME. Enzymes to die for: exploiting nucleotide metabolizing enzymes for cancer gene therapy. Curr Gene Ther. 2012;12:77–91. doi: 10.2174/156652312800099571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao J, Zhang G, Qiu P, Liu X, Wu Y, Du B, Li J, Zhou J, Tan Y. Tanshinone IIA Increases the Bystander Effect of Herpes Simplex Virus Thymidine Kinase/Ganciclovir Gene Therapy via Enhanced Gap Junctional Intercellular Communication. PLoS One. 2013;8:e67662. doi: 10.1371/journal.pone.0067662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freeman SM, Abboud CN, Whartenby KA, Packman CH, Koeplin DS, Moolten FL, Abraham GN. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274–5283. [PubMed] [Google Scholar]
- 34.Agard C, Ligeza C, Dupas B, Izembart A, El KC, Moullier P, Ferry N. Immune-dependent distant bystander effect after adenovirus-mediated suicide gene transfer in a rat model of liver colorectal metastasis. Cancer Gene Ther. 2001;8:128–136. doi: 10.1038/sj.cgt.7700281. [DOI] [PubMed] [Google Scholar]
- 35.Kuriyama S, Tsujinoue H, Yoshiji H. Immune response to suicide gene therapy. Methods Mol Med. 2004;90:353–369. doi: 10.1385/1-59259-429-8:353. [DOI] [PubMed] [Google Scholar]
- 36.Pierrefite-Carle V, Baque P, Gavelli A, Brossette N, Benchimol D, Bourgeon A, Saint Paul MC, Staccini P, Rossi B. Subcutaneous or intrahepatic injection of suicide gene modified tumour cells induces a systemic antitumour response in a metastatic model of colon carcinoma in rats. Gut. 2002;50:387–391. doi: 10.1136/gut.50.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aguilar LK, Guzik BW, Aguilar-Cordova E. Cytotoxic immunotherapy strategies for cancer: mechanisms and clinical development. J Cell Biochem. 2011;112:1969–1977. doi: 10.1002/jcb.23126. [DOI] [PubMed] [Google Scholar]
- 38.Agard C, Ligeza C, Dupas B, Izembart A, El Kouri C, Moullier P, Ferry N. Immune-dependent distant bystander effect after adenovirus-mediated suicide gene transfer in a rat model of liver colorectal metastasis. Cancer Gene Ther. 2001;8:128–136. doi: 10.1038/sj.cgt.7700281. [DOI] [PubMed] [Google Scholar]
- 39.Niculescu-Duvaz I, Springer CJ. Introduction to the background, principles, and state of the art in suicide gene therapy. Mol Biotechnol. 2005;30:71–88. doi: 10.1385/MB:30:1:071. [DOI] [PubMed] [Google Scholar]
- 40.Yoon KJ, Potter PM, Danks MK. Development of prodrugs for enzyme-mediated, tumor-selective therapy. Curr Med Chem Anticancer Agents. 2005;5:107–113. doi: 10.2174/1568011053174837. [DOI] [PubMed] [Google Scholar]
- 41.Niculescu-Duvaz D, Niculescu-Duvaz I, Springer CJ. Design of prodrugs for suicide gene therapy. Methods Mol Med. 2004;90:161–202. doi: 10.1385/1-59259-429-8:161. [DOI] [PubMed] [Google Scholar]
- 42.Niculescu-Duvaz I, Niculescu-Duvaz D, Friedlos F, Spooner R, Martin J, Marais R, Springer CJ. Self-immolative anthracycline prodrugs for suicide gene therapy. J Med Chem. 1999;42:2485–2489. doi: 10.1021/jm980696v. [DOI] [PubMed] [Google Scholar]
- 43.Tranoy-Opalinski I, Legigan T, Barat R, Clarhaut J, Thomas M, Renoux B, Papot S. beta-Glucuronidase-responsive prodrugs for selective cancer chemotherapy: An update. Eur J Med Chem. 2014;74:302–313. doi: 10.1016/j.ejmech.2013.12.045. [DOI] [PubMed] [Google Scholar]
- 44.Tranoy-Opalinski I, Fernandes A, Thomas M, Gesson JP, Papot S. Design of self-immolative linkers for tumour-activated prodrug therapy. Anticancer Agents Med Chem. 2008;8:618–637. [PubMed] [Google Scholar]
- 45.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 46.Halama A, Kulinski M, Librowski T, Lochynski S. Polymer-based non-viral gene delivery as a concept for the treatment of cancer. Pharmacol Rep. 2009;61:993–999. doi: 10.1016/s1734-1140(09)70160-4. [DOI] [PubMed] [Google Scholar]
- 47.McCarthy HO, Wang Y, Mangipudi SS, Hatefi A. Advances with the use of bio-inspired vectors towards creation of artificial viruses. Expert Opin Drug Deliv. 2010;7:497–512. doi: 10.1517/17425240903579989. [DOI] [PubMed] [Google Scholar]
- 48.Mohit E, Rafati S. Biological delivery approaches for gene therapy: strategies to potentiate efficacy and enhance specificity. Mol Immunol. 2013;56:599–611. doi: 10.1016/j.molimm.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 - an update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 50.Beatty MS, Curiel DT. Chapter two--Adenovirus strategies for tissue-specific targeting. Adv Cancer Res. 2012;115:39–67. doi: 10.1016/B978-0-12-398342-8.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen Y, Nemunaitis J. Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther. 2006;13:975–992. doi: 10.1038/sj.cgt.7700946. [DOI] [PubMed] [Google Scholar]
- 52.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 53.Penet MF, Chen Z, Li C, Winnard PT, Jr, Bhujwalla ZM. Prodrug enzymes and their applications in image-guided therapy of cancer: tracking prodrug enzymes to minimize collateral damage. Drug Deliv Transl Res. 2012;2:22–30. doi: 10.1007/s13346-011-0052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilcox CL, Crnic LS, Pizer LI. Replication, latent infection, and reactivation in neuronal culture with a herpes simplex virus thymidine kinase-negative mutant. Virology. 1992;187:348–352. doi: 10.1016/0042-6822(92)90326-k. [DOI] [PubMed] [Google Scholar]
- 55.Chen SH, Pearson A, Coen DM. Failure of thymidine kinase-negative herpes simplex virus to reactivate from latency following efficient establishment. J Virol. 2004;78:520–523. doi: 10.1128/JVI.78.1.520-523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55:459–472. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gallois-Montbrun S, Veron M, Deville-Bonne D. Antiviral nucleoside analogs phosphorylation by nucleoside diphosphate kinase. Mini Rev Med Chem. 2004;4:361–369. doi: 10.2174/1389557043403990. [DOI] [PubMed] [Google Scholar]
- 58.Beltinger C, Fulda S, Kammertoens T, Meyer E, Uckert W, Debatin KM. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci U S A. 1999;96:8699–8704. doi: 10.1073/pnas.96.15.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nicholas TW, Read SB, Burrows FJ, Kruse CA. Suicide gene therapy with Herpes simplex virus thymidine kinase and ganciclovir is enhanced with connexins to improve gap junctions and bystander effects. Histol Histopathol. 2003;18:495–507. doi: 10.14670/HH-18.495. [DOI] [PubMed] [Google Scholar]
- 60.Preuss E, Muik A, Weber K, Otte J, von Laer D, Fehse B. Cancer suicide gene therapy with TK.007: superior killing efficiency and bystander effect. J Mol Med (Berl) 2011;89:1113–1124. doi: 10.1007/s00109-011-0777-8. [DOI] [PubMed] [Google Scholar]
- 61.Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: role of gap-junctional intercellular communication. Cancer Res. 2000;60:3989–3999. [PubMed] [Google Scholar]
- 62.Dachs G, Hunt M, Syddall S, Singleton D, Patterson A. Bystander or No Bystander for Gene Directed Enzyme Prodrug Therapy. Molecules. 2009;14:4517–4545. doi: 10.3390/molecules14114517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001;61:3022–3026. [PubMed] [Google Scholar]
- 64.Ardiani A, Sanchez-Bonilla M, Black ME. Fusion enzymes containing HSV-1 thymidine kinase mutants and guanylate kinase enhance prodrug sensitivity in vitro and in vivo. Cancer Gene Ther. 2010;17:86–96. doi: 10.1038/cgt.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balzarini J, Liekens S, Solaroli N, El Omari K, Stammers DK, Karlsson A. Engineering of a single conserved amino acid residue of herpes simplex virus type 1 thymidine kinase allows a predominant shift from pyrimidine to purine nucleoside phosphorylation. J Biol Chem. 2006;281:19273–19279. doi: 10.1074/jbc.M600414200. [DOI] [PubMed] [Google Scholar]
- 66.Blumenthal M, Skelton D, Pepper KA, Jahn T, Methangkool E, Kohn DB. Effective suicide gene therapy for leukemia in a model of insertional oncogenesis in mice. Mol Ther. 2007;15:183–192. doi: 10.1038/sj.mt.6300015. [DOI] [PubMed] [Google Scholar]
- 67.Barzon L, Pacenti M, Franchin E, Colombo F, Palu G. HSV-TK/IL-2 gene therapy for glioblastoma multiforme. Methods Mol Biol. 2009;542:529–549. doi: 10.1007/978-1-59745-561-9_28. [DOI] [PubMed] [Google Scholar]
- 68.Pan JG, Zhou X, Luo R, Han RF. The adeno-associated virus-mediated HSV-TK/GCV suicide system: a potential strategy for the treatment of bladder carcinoma. Med Oncol. 2012;29:1938–1947. doi: 10.1007/s12032-011-0091-x. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y, Canine BF, Hatefi A. HSV-TK/GCV cancer suicide gene therapy by a designed recombinant multifunctional vector. Nanomedicine. 2011;7:193–200. doi: 10.1016/j.nano.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voges J, Reszka R, Gossmann A, Dittmar C, Richter R, Garlip G, Kracht L, Coenen HH, Sturm V, Wienhard K, Heiss WD, Jacobs AH. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54:479–487. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 71.Nasu Y, Saika T, Ebara S, Kusaka N, Kaku H, Abarzua F, Manabe D, Thompson TC, Kumon H. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol Ther. 2007;15:834–840. doi: 10.1038/sj.mt.6300096. [DOI] [PubMed] [Google Scholar]
- 72.Li N, Zhou J, Weng D, Zhang C, Li L, Wang B, Song Y, He Q, Lin D, Chen D, Chen G, Gao Q, Wang S, Xu G, Meng L, Lu Y, Ma D. Adjuvant adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of liver transplantation in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2007;13:5847–5854. doi: 10.1158/1078-0432.CCR-07-0499. [DOI] [PubMed] [Google Scholar]
- 73.Xu F, Li S, Li XL, Guo Y, Zou BY, Xu R, Liao H, Zhao HY, Zhang Y, Guan ZZ, Zhang L. Phase I and biodistribution study of recombinant adenovirus vector-mediated herpes simplex virus thymidine kinase gene and ganciclovir administration in patients with head and neck cancer and other malignant tumors. Cancer Gene Ther. 2009;16:723–730. doi: 10.1038/cgt.2009.19. [DOI] [PubMed] [Google Scholar]
- 74.Sangro B, Mazzolini G, Ruiz M, Ruiz J, Quiroga J, Herrero I, Qian C, Benito A, Larrache J, Olague C, Boan J, Penuelas I, Sadaba B, Prieto J. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010;17:837–843. doi: 10.1038/cgt.2010.40. [DOI] [PubMed] [Google Scholar]
- 75.Langford G, Dayan A, Yla-Herttuala S, Eckland D. A preclinical assessment of the safety and biodistribution of an adenoviral vector containing the herpes simplex virus thymidine kinase gene (Cerepro) after intracerebral administration. J Gene Med. 2009;11:468–476. doi: 10.1002/jgm.1328. [DOI] [PubMed] [Google Scholar]
- 76.van Putten EH, Dirven CM, van den Bent MJ, Lamfers ML. Sitimagene ceradenovec: a gene-based drug for the treatment of operable high-grade glioma. Future Oncol. 2010;6:1691–1710. doi: 10.2217/fon.10.134. [DOI] [PubMed] [Google Scholar]
- 77.Matthews K, Noker PE, Tian B, Grimes SD, Fulton R, Schweikart K, Harris R, Aurigemma R, Wang M, Barnes MN, Siegal GP, Hemminki A, Zinn K, Curiel DT, Alvarez RD. Identifying the safety profile of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in anticipation of a phase I clinical trial in patients with recurrent ovarian cancer. Clin Cancer Res. 2009;15:4131–4137. doi: 10.1158/1078-0432.CCR-08-3354. [DOI] [PubMed] [Google Scholar]
- 78.Witlox MA, Van Beusechem VW, Grill J, Haisma HJ, Schaap G, Bras J, Van Diest P, De Gast A, Curiel DT, Pinedo HM, Gerritsen WR, Wuisman PI. Epidermal growth factor receptor targeting enhances adenoviral vector based suicide gene therapy of osteosarcoma. J Gene Med. 2002;4:510–516. doi: 10.1002/jgm.308. [DOI] [PubMed] [Google Scholar]
- 79.Nicklin SA, Baker AH. Tropism-modified adenoviral and adeno-associated viral vectors for gene therapy. Curr Gene Ther. 2002;2:273–293. doi: 10.2174/1566523023347797. [DOI] [PubMed] [Google Scholar]
- 80.Verheije MH, Rottier PJ. Retargeting of viruses to generate oncolytic agents. Adv Virol. 2012;2012:798526. doi: 10.1155/2012/798526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim KH, Dmitriev I, O’Malley JP, Wang M, Saddekni S, You Z, Preuss MA, Harris RD, Aurigemma R, Siegal GP, Zinn KR, Curiel DT, Alvarez RD. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin Cancer Res. 2012;18:3440–3451. doi: 10.1158/1078-0432.CCR-11-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kubo H, Gardner TA, Wada Y, Koeneman KS, Gotoh A, Yang L, Kao C, Lim SD, Amin MB, Yang H, Black ME, Matsubara S, Nakagawa M, Gillenwater JY, Zhau HE, Chung LW. Phase I dose escalation clinical trial of adenovirus vector carrying osteocalcin promoter-driven herpes simplex virus thymidine kinase in localized and metastatic hormone-refractory prostate cancer. Hum Gene Ther. 2003;14:227–241. doi: 10.1089/10430340360535788. [DOI] [PubMed] [Google Scholar]
- 83.Papanastasopoulos P, Stebbing J. Molecular Basis of 5-Fluorouracil-related Toxicity: Lessons from Clinical Practice. Anticancer Res. 2014;34:1531–1535. [PubMed] [Google Scholar]
- 84.Duarte S, Carle G, Faneca H, Lima MCPd, Pierrefite-Carle V. Suicide gene therapy in cancer: Where do we stand now? Cancer Lett. 2012;324:160–170. doi: 10.1016/j.canlet.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 85.Kuriyama S, Masui K, Sakamoto T, Nakatani T, Kikukawa M, Tsujinoue H, Mitoro A, Yamazaki M, Yoshiji H, Fukui H, Ikenaka K, Mullen CA, Tsujii T. Bystander effect caused by cytosine deaminase gene and 5-fluorocytosine in vitro is substantially mediated by generated 5-fluorouracil. Anticancer Res. 1998;18:3399–3406. [PubMed] [Google Scholar]
- 86.Garcia-Sanchez F, Pizzorno G, Fu SQ, Nanakorn T, Krause DS, Liang J, Adams E, Leffert JJ, Yin LH, Cooperberg MR, Hanania E, Wang WL, Won JH, Peng XY, Cote R, Brown R, Burtness B, Giles R, Crystal R, Deisseroth AB. Cytosine deaminase adenoviral vector and 5-fluorocytosine selectively reduce breast cancer cells 1 million-fold when they contaminate hematopoietic cells: a potential purging method for autologous transplantation. Blood. 1998;92:672–682. [PubMed] [Google Scholar]
- 87.Ostertag D, Amundson KK, Lopez Espinoza F, Martin B, Buckley T, Galvao da Silva AP, Lin AH, Valenta DT, Perez OD, Ibanez CE, Chen CI, Pettersson PL, Burnett R, Daublebsky V, Hlavaty J, Gunzburg W, Kasahara N, Gruber HE, Jolly DJ, Robbins JM. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012;14:145–159. doi: 10.1093/neuonc/nor199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaliberov SA, Buchsbaum DJ. Chapter seven--Cancer treatment with gene therapy and radiation therapy. Adv Cancer Res. 2012;115:221–263. doi: 10.1016/B978-0-12-398342-8.00007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tong X, Chen X, Li C. Imaging beyond the diagnosis: image-guided enzyme/prodrug cancer therapy. Acta Biochim Biophys Sin (Shanghai) 2011;43:4–12. doi: 10.1093/abbs/gmq113. [DOI] [PubMed] [Google Scholar]
- 90.Haack K, Linnebacher M, Eisold S, Zoller M, von Knebel Doeberitz M, Gebert J. Induction of protective immunity against syngeneic rat cancer cells by expression of the cytosine deaminase suicide gene. Cancer Gene Ther. 2000;7:1357–1364. doi: 10.1038/sj.cgt.7700245. [DOI] [PubMed] [Google Scholar]
- 91.Kaliberova LN, Della Manna DL, Krendelchtchikova V, Black ME, Buchsbaum DJ, Kaliberov SA. Molecular chemotherapy of pancreatic cancer using novel mutant bacterial cytosine deaminase gene. Mol Cancer Ther. 2008;7:2845–2854. doi: 10.1158/1535-7163.MCT-08-0347. [DOI] [PubMed] [Google Scholar]
- 92.Deng LY, Wang JP, Gui ZF, Shen LZ. Antitumor activity of mutant bacterial cytosine deaminase gene for colon cancer. World J Gastroenterol. 2011;17:2958–2964. doi: 10.3748/wjg.v17.i24.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fuchita M, Ardiani A, Zhao L, Serve K, Stoddard BL, Black ME. Bacterial cytosine deaminase mutants created by molecular engineering show improved 5-fluorocytosine-mediated cell killing in vitro and in vivo. Cancer Res. 2009;69:4791–4799. doi: 10.1158/0008-5472.CAN-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stolworthy TS, Korkegian AM, Willmon CL, Ardiani A, Cundiff J, Stoddard BL, Black ME. Yeast cytosine deaminase mutants with increased thermostability impart sensitivity to 5-fluorocytosine. J Mol Biol. 2008;377:854–869. doi: 10.1016/j.jmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi DZ, Hu WX, Li LX, Chen G, Wei D, Gu PY. Pharmacokinetics and the bystander effect in CD::UPRT/5-FC bi-gene therapy of glioma. Chin Med J (Engl) 2009;122:1267–1272. [PubMed] [Google Scholar]
- 97.Richard C, Duivenvoorden W, Bourbeau D, Massie B, Roa W, Yau J, Th’ng J. Sensitivity of 5-fluorouracil-resistant cancer cells to adenovirus suicide gene therapy. Cancer Gene Ther. 2007;14:57–65. doi: 10.1038/sj.cgt.7700980. [DOI] [PubMed] [Google Scholar]
- 98.Freytag SO, Khil M, Stricker H, Peabody J, Menon M, DePeralta-Venturina M, Nafziger D, Pegg J, Paielli D, Brown S, Barton K, Lu M, Aguilar-Cordova E, Kim JH. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 2002;62:4968–4976. [PubMed] [Google Scholar]
- 99.Freytag SO, Barton KN, Brown SL, Narra V, Zhang Y, Tyson D, Nall C, Lu M, Ajlouni M, Movsas B, Kim JH. Replication-competent adenovirus-mediated suicide gene therapy with radiation in a preclinical model of pancreatic cancer. Mol Ther. 2007;15:1600–1606. doi: 10.1038/sj.mt.6300212. [DOI] [PubMed] [Google Scholar]
- 100.Freytag SO, Stricker H, Pegg J, Paielli D, Pradhan DG, Peabody J, DePeralta-Venturina M, Xia X, Brown S, Lu M, Kim JH. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63:7497–7506. [PubMed] [Google Scholar]
- 101.Freytag SO, Movsas B, Aref I, Stricker H, Peabody J, Pegg J, Zhang Y, Barton KN, Brown SL, Lu M, Savera A, Kim JH. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther. 2007;15:1016–1023. doi: 10.1038/mt.sj.6300120. [DOI] [PubMed] [Google Scholar]
- 102.Freytag SO, Stricker H, Peabody J, Pegg J, Paielli D, Movsas B, Barton KN, Brown SL, Lu M, Kim JH. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol Ther. 2007;15:636–642. doi: 10.1038/sj.mt.6300068. [DOI] [PubMed] [Google Scholar]
- 103.Perez OD, Logg CR, Hiraoka K, Diago O, Burnett R, Inagaki A, Jolson D, Amundson K, Buckley T, Lohse D, Lin A, Burrascano C, Ibanez C, Kasahara N, Gruber HE, Jolly DJ. Design and selection of Toca 511 for clinical use: modified retroviral replicating vector with improved stability and gene expression. Mol Ther. 2012;20:1689–1698. doi: 10.1038/mt.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stedt H, Samaranayake H, Pikkarainen J, Maatta AM, Alasaarela L, Airenne K, Yla-Herttuala S. Improved therapeutic effect on malignant glioma with adenoviral suicide gene therapy combined with temozolomide. Gene Ther. 2013;20:1165–1171. doi: 10.1038/gt.2013.46. [DOI] [PubMed] [Google Scholar]
- 105.Greco O, Dachs GU. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol. 2001;187:22–36. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1060>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 106.Green LK, Storey MA, Williams EM, Patterson AV, Smaill JB, Copp JN, Ackerley DF. The Flavin Reductase MsuE Is a Novel Nitroreductase that Can Efficiently Activate Two Promising Next-Generation Prodrugs for Gene-Directed Enzyme Prodrug Therapy. Cancers (Basel) 2013;5:985–997. doi: 10.3390/cancers5030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chung-Faye G, Palmer D, Anderson D, Clark J, Downes M, Baddeley J, Hussain S, Murray PI, Searle P, Seymour L, Harris PA, Ferry D, Kerr DJ. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: a phase I and pharmacokinetic study of its prodrug, CB1954. Clin Cancer Res. 2001;7:2662–2668. [PubMed] [Google Scholar]
- 108.Prosser GA, Copp JN, Syddall SP, Williams EM, Smaill JB, Wilson WR, Patterson AV, Ackerley DF. Discovery and evaluation of Escherichia coli nitroreductases that activate the anti-cancer prodrug CB1954. Biochem Pharmacol. 2010;79:678–687. doi: 10.1016/j.bcp.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 109.Grohmann M, Paulmann N, Fleischhauer S, Vowinckel J, Priller J, Walther DJ. A mammalianized synthetic nitroreductase gene for high-level expression. BMC Cancer. 2009;9:301. doi: 10.1186/1471-2407-9-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Prosser GA, Copp JN, Mowday AM, Guise CP, Syddall SP, Williams EM, Horvat CN, Swe PM, Ashoorzadeh A, Denny WA, Smaill JB, Patterson AV, Ackerley DF. Creation and screening of a multi-family bacterial oxidoreductase library to discover novel nitroreductases that efficiently activate the bioreductive prodrugs CB1954 and PR-104A. Biochem Pharmacol. 2013;85:1091–1103. doi: 10.1016/j.bcp.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 111.Swe PM, Copp JN, Green LK, Guise CP, Mowday AM, Smaill JB, Patterson AV, Ackerley DF. Targeted mutagenesis of the Vibrio fischeri flavin reductase FRase I to improve activation of the anticancer prodrug CB1954. Biochem Pharmacol. 2012;84:775–783. doi: 10.1016/j.bcp.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 112.Green LK, Syddall SP, Carlin KM, Bell GD, Guise CP, Mowday AM, Hay MP, Smaill JB, Patterson AV, Ackerley DF. Pseudomonas aeruginosa NfsB and nitro-CBI-DEI--a promising enzyme/prodrug combination for gene directed enzyme prodrug therapy. Mol Cancer. 2013;12:58. doi: 10.1186/1476-4598-12-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Patel P, Young JG, Mautner V, Ashdown D, Bonney S, Pineda RG, Collins SI, Searle PF, Hull D, Peers E, Chester J, Wallace DM, Doherty A, Leung H, Young LS, James ND. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954 [correction of CB1984] Mol Ther. 2009;17:1292–1299. doi: 10.1038/mt.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barar J, Omidi Y. Translational Approaches towards Cancer Gene Therapy: Hurdles and Hopes. Bioimpacts. 2012;2:127–143. doi: 10.5681/bi.2012.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nouri FS, Wang X, Hatefi A. Genetically engineered theranostic mesenchymal stem cells for the evaluation of the anticancer efficacy of enzyme/prodrug systems. J Control Release. 2015;200:179–187. doi: 10.1016/j.jconrel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Donate F, Wilson WR, Patterson AV. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70:1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- 117.Hedley D, Ogilvie L, Springer C. Carboxypeptidase-G2-based gene-directed enzyme-prodrug therapy: a new weapon in the GDEPT armoury. Nat Rev Cancer. 2007;7:870–879. doi: 10.1038/nrc2247. [DOI] [PubMed] [Google Scholar]
- 118.Palmer DH, Mautner V, Mirza D, Oliff S, Gerritsen W, van der Sijp JR, Hubscher S, Reynolds G, Bonney S, Rajaratnam R, Hull D, Horne M, Ellis J, Mountain A, Hill S, Harris PA, Searle PF, Young LS, James ND, Kerr DJ. Virus-directed enzyme prodrug therapy: intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J Clin Oncol. 2004;22:1546–1552. doi: 10.1200/JCO.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 119.Stribbling SM, Friedlos F, Martin J, Davies L, Spooner RA, Marais R, Springer CJ. Regressions of established breast carcinoma xenografts by carboxypeptidase G2 suicide gene therapy and the prodrug CMDA are due to a bystander effect. Hum Gene Ther. 2000;11:285–292. doi: 10.1089/10430340050016021. [DOI] [PubMed] [Google Scholar]
- 120.Francis RJ, Sharma SK, Springer C, Green AJ, Hope-Stone LD, Sena L, Martin J, Adamson KL, Robbins A, Gumbrell L, O’Malley D, Tsiompanou E, Shahbakhti H, Webley S, Hochhauser D, Hilson AJ, Blakey D, Begent RH. A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours. Br J Cancer. 2002;87:600–607. doi: 10.1038/sj.bjc.6600517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Friedlos F, Davies L, Scanlon I, Ogilvie LM, Martin J, Stribbling SM, Spooner RA, Niculescu-Duvaz I, Marais R, Springer CJ. Three new prodrugs for suicide gene therapy using carboxypeptidase G2 elicit bystander efficacy in two xenograft models. Cancer Res. 2002;62:1724–1729. [PubMed] [Google Scholar]
- 122.Mancini L, Davies L, Friedlos F, Falck-Miniotis M, Dzik-Jurasz AS, Springer CJ, Leach MO, Payne GS. A novel technique to monitor carboxypeptidase G2 expression in suicide gene therapy using 19F magnetic resonance spectroscopy. NMR Biomed. 2009;22:561–566. doi: 10.1002/nbm.1375. [DOI] [PubMed] [Google Scholar]
- 123.Mahato R, Tai W, Cheng K. Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev. 2011;63:659–670. doi: 10.1016/j.addr.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Martin J, Stribbling SM, Poon GK, Begent RH, Napier M, Sharma SK, Springer CJ. Antibody-directed enzyme prodrug therapy: pharmacokinetics and plasma levels of prodrug and drug in a phase I clinical trial. Cancer Chemother Pharmacol. 1997;40:189–201. doi: 10.1007/s002800050646. [DOI] [PubMed] [Google Scholar]
- 125.Chiocca EA, Waxman DJ. Cytochrome p450-based gene therapies for cancer. Methods Mol Med. 2004;90:203–222. doi: 10.1385/1-59259-429-8:203. [DOI] [PubMed] [Google Scholar]
- 126.Huang Z, Waxman DJ. Modulation of cyclophosphamide-based cytochrome P450 gene therapy using liver P450 inhibitors. Cancer Gene Ther. 2001;8:450–458. doi: 10.1038/sj.cgt.7700325. [DOI] [PubMed] [Google Scholar]
- 127.Chen L, Waxman DJ. Cytochrome P450 gene-directed enzyme prodrug therapy (GDEPT) for cancer. Curr Pharm Des. 2002;8:1405–1416. doi: 10.2174/1381612023394566. [DOI] [PubMed] [Google Scholar]
- 128.Salmons B, Lohr M, Gunzburg WH. Treatment of inoperable pancreatic carcinoma using a cell-based local chemotherapy: results of a phase I/II clinical trial. J Gastroenterol. 2003;38(Suppl 15):78–84. [PubMed] [Google Scholar]
- 129.Braybrooke JP, Slade A, Deplanque G, Harrop R, Madhusudan S, Forster MD, Gibson R, Makris A, Talbot DC, Steiner J, White L, Kan O, Naylor S, Carroll MW, Kingsman SM, Harris AL. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin Cancer Res. 2005;11:1512–1520. doi: 10.1158/1078-0432.CCR-04-0155. [DOI] [PubMed] [Google Scholar]
- 130.Touati W, Tran T, Seguin J, Diry M, Flinois JP, Baillou C, Lescaille G, Andre F, Tartour E, Lemoine FM, Beaune P, de Waziers I. A suicide gene therapy combining the improvement of cyclophosphamide tumor cytotoxicity and the development of an anti-tumor immune response. Curr Gene Ther. 2014;14:236–246. doi: 10.2174/1566523214666140424152734. [DOI] [PubMed] [Google Scholar]
- 131.Gadi VK, Alexander SD, Waud WR, Allan PW, Parker WB, Sorscher EJ. A long-acting suicide gene toxin, 6-methylpurine, inhibits slow growing tumors after a single administration. J Pharmacol Exp Ther. 2003;304:1280–1284. doi: 10.1124/jpet.102.044743. [DOI] [PubMed] [Google Scholar]
- 132.Hong JS, Waud WR, Levasseur DN, Townes TM, Wen H, McPherson SA, Moore BA, Bebok Z, Allan PW, Secrist JA, 3rd, Parker WB, Sorscher EJ. Excellent in vivo bystander activity of fludarabine phosphate against human glioma xenografts that express the escherichia coli purine nucleoside phosphorylase gene. Cancer Res. 2004;64:6610–6615. doi: 10.1158/0008-5472.CAN-04-0012. [DOI] [PubMed] [Google Scholar]
- 133.Martiniello-Wilks R, Garcia-Aragon J, Daja MM, Russell P, Both GW, Molloy PL, Lockett LJ, Russell PJ. In vivo gene therapy for prostate cancer: preclinical evaluation of two different enzyme-directed prodrug therapy systems delivered by identical adenovirus vectors. Hum Gene Ther. 1998;9:1617–1626. doi: 10.1089/hum.1998.9.11-1617. [DOI] [PubMed] [Google Scholar]
- 134.Lockett LJ, Molloy PL, Russell PJ, Both GW. Relative efficiency of tumor cell killing in vitro by two enzyme-prodrug systems delivered by identical adenovirus vectors. Clin Cancer Res. 1997;3:2075–2080. [PubMed] [Google Scholar]
- 135.Afshar S, Asai T, Morrison SL. Humanized ADEPT comprised of an engineered human purine nucleoside phosphorylase and a tumor targeting peptide for treatment of cancer. Mol Cancer Ther. 2009;8:185–193. doi: 10.1158/1535-7163.MCT-08-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Voeks D, Martiniello-Wilks R, Madden V, Smith K, Bennetts E, Both GW, Russell PJ. Gene therapy for prostate cancer delivered by ovine adenovirus and mediated by purine nucleoside phosphorylase and fludarabine in mouse models. Gene Ther. 2002;9:759–768. doi: 10.1038/sj.gt.3301698. [DOI] [PubMed] [Google Scholar]
- 137.Parker WB, Allan PW, Waud WR, Hong JS, Sorscher EJ. Effect of expression of adenine phosphoribosyltransferase on the in vivo anti-tumor activity of prodrugs activated by E. coli purine nucleoside phosphorylase. Cancer Gene Ther. 2011;18:390–398. doi: 10.1038/cgt.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bennett EM, Anand R, Allan PW, Hassan AE, Hong JS, Levasseur DN, McPherson DT, Parker WB, Secrist JA, 3rd, Sorscher EJ, Townes TM, Waud WR, Ealick SE. Designer gene therapy using an Escherichia coli purine nucleoside phosphorylase/prodrug system. Chem Biol. 2003;10:1173–1181. doi: 10.1016/j.chembiol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 139.Parker WB, Allan PW, Ealick SE, Sorscher EJ, Hassan AE, Silamkoti AV, Fowler AT, Waud WR, Secrist JA., 3rd Design and evaluation of 5′-modified nucleoside analogs as prodrugs for an E. coli purine nucleoside phosphorylase mutant. Nucleosides Nucleotides Nucleic Acids. 2005;24:387–392. doi: 10.1081/ncn-200059807. [DOI] [PubMed] [Google Scholar]
- 140.Xie X, Guo J, Kong Y, Xie GX, Li L, Lv N, Xiao X, Tang J, Wang X, Liu P, Yang M, Xie Z, Wei W, Spencer DM, Xie X. Targeted expression of Escherichia coli purine nucleoside phosphorylase and Fludara(R) for prostate cancer therapy. J Gene Med. 2011;13:680–691. doi: 10.1002/jgm.1620. [DOI] [PubMed] [Google Scholar]
- 141.Chen ZH, Huang GL, Tu YQ, Jiang Y, Dai WX. Dual specific antitumor effects of Semliki Forest virus-based DNA vector carrying suicide Escherichia coli purine nucleoside phosphorylase gene via Salmonella. Int J Oncol. 2013;42:2009–2018. doi: 10.3892/ijo.2013.1900. [DOI] [PubMed] [Google Scholar]
- 142.Sorscher EJ, Hong JS, Allan PW, Waud WR, Parker WB. In vivo antitumor activity of intratumoral fludarabine phosphate in refractory tumors expressing E. coli purine nucleoside phosphorylase. Cancer Chemother Pharmacol. 2012;70:321–329. doi: 10.1007/s00280-012-1908-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Greco O, Rossiter S, Kanthou C, Folkes LK, Wardman P, Tozer GM, Dachs GU. Horseradish peroxidase-mediated gene therapy: choice of prodrugs in oxic and anoxic tumor conditions. Mol Cancer Ther. 2001;1:151–160. [PubMed] [Google Scholar]
- 144.Greco O, Tozer GM, Dachs GU. Oxic and anoxic enhancement of radiation-mediated toxicity by horseradish peroxidase/indole-3-acetic acid gene therapy. Int J Radiat Biol. 2002;78:173–181. doi: 10.1080/09553000110097983. [DOI] [PubMed] [Google Scholar]
- 145.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 146.Metz MZ, Gutova M, Lacey SF, Abramyants Y, Vo T, Gilchrist M, Tirughana R, Ghoda LY, Barish ME, Brown CE, Najbauer J, Potter PM, Portnow J, Synold TW, Aboody KS. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem cells Transl Med. 2013;2:983–992. doi: 10.5966/sctm.2012-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) Clin Cancer Res. 2001;7:2182–2194. [PubMed] [Google Scholar]
- 148.Swami U, Goel S, Mani S. Therapeutic targeting of CPT-11 induced diarrhea: a case for prophylaxis. Curr Drug Targets. 2013;14:777–797. doi: 10.2174/1389450111314070007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chiocca EA, Aguilar LK, Bell SD, Kaur B, Hardcastle J, Cavaliere R, McGregor J, Lo S, Ray-Chaudhuri A, Chakravarti A, Grecula J, Newton H, Harris KS, Grossman RG, Trask TW, Baskin DS, Monterroso C, Manzanera AG, Aguilar-Cordova E, New PZ. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29:3611–3619. doi: 10.1200/JCO.2011.35.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Finocchiaro LM, Glikin GC. Cytokine-enhanced vaccine and suicide gene therapy as surgery adjuvant treatments for spontaneous canine melanoma: 9 years of follow-up. Cancer Gene Ther. 2012;19:852–861. doi: 10.1038/cgt.2012.72. [DOI] [PubMed] [Google Scholar]
- 151.Xiong J, Sun WJ, Wang WF, Liao ZK, Zhou FX, Kong HY, Xu Y, Xie CH, Zhou YF. Novel, chimeric, cancer-specific, and radiation-inducible gene promoters for suicide gene therapy of cancer. Cancer. 2012;118:536–548. doi: 10.1002/cncr.26289. [DOI] [PubMed] [Google Scholar]
- 152.Liao Z, Huang C, Zhou F, Xiong J, Bao J, Zhang H, Sun W, Xie C, Zhou Y. Radiation enhances suicide gene therapy in radioresistant laryngeal squamous cell carcinoma via activation of a tumor-specific promoter. Cancer Lett. 2009;283:20–28. doi: 10.1016/j.canlet.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 153.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tey SK. Adoptive T-cell therapy: adverse events and safety switches. Clin Transl Immunology. 2014;3:e17. doi: 10.1038/cti.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mailly L, Leboeuf C, Tiberghien P, Baumert T, Robinet E. Genetically engineered T-cells expressing a ganciclovir-sensitive HSV-tk suicide gene for the prevention of GvHD. Curr Opin Investig Drugs. 2010;11:559–570. [PubMed] [Google Scholar]
- 159.Faneca H, Faustino A, Pedroso de Lima MC. Synergistic antitumoral effect of vinblastine and HSV-Tk/GCV gene therapy mediated by albumin-associated cationic liposomes. J Control Release. 2008;126:175–184. doi: 10.1016/j.jconrel.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 160.Boucher PD, Shewach DS. In vitro and in vivo enhancement of ganciclovir-mediated bystander cytotoxicity with gemcitabine. Mol Ther. 2005;12:1064–1071. doi: 10.1016/j.ymthe.2005.07.643. [DOI] [PubMed] [Google Scholar]
- 161.Zhang H. Multifunctional nanomedicine platforms for cancer therapy. J Nanosci Nanotechnol. 2012;12:4012–4018. doi: 10.1166/jnn.2012.6160. [DOI] [PubMed] [Google Scholar]
- 162.Duarte S, Carle G, Faneca H, de Lima MC, Pierrefite-Carle V. Suicide gene therapy in cancer: where do we stand now? Cancer Lett. 2012;324:160–170. doi: 10.1016/j.canlet.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 163.Kurayoshi K, Ozono E, Iwanaga R, Bradford AP, Komori H, Ohtani K. Cancer cell specific cytotoxic gene expression mediated by ARF tumor suppressor promoter constructs. Biochem Biophys Res Commun. 2014;450:240–246. doi: 10.1016/j.bbrc.2014.05.102. [DOI] [PubMed] [Google Scholar]
- 164.Leja J, Dzojic H, Gustafson E, Oberg K, Giandomenico V, Essand M. A novel chromogranin-A promoter-driven oncolytic adenovirus for midgut carcinoid therapy. Clin Cancer Res. 2007;13:2455–2462. doi: 10.1158/1078-0432.CCR-06-2532. [DOI] [PubMed] [Google Scholar]
- 165.Kia A, Przystal JM, Nianiaris N, Mazarakis ND, Mintz PJ, Hajitou A. Dual systemic tumor targeting with ligand-directed phage and Grp78 promoter induces tumor regression. Mol Cancer Ther. 2012;11:2566–2577. doi: 10.1158/1535-7163.MCT-12-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Haviv YS, van Houdt WJ, Lu B, Curiel DT, Zhu ZB. Transcriptional targeting in renal cancer cell lines via the human CXCR4 promoter. Mol Cancer Ther. 2004;3:687–691. [PubMed] [Google Scholar]
- 167.Zhu ZB, Makhija SK, Lu B, Wang M, Kaliberova L, Liu B, Rivera AA, Nettelbeck DM, Mahasreshti PJ, Leath CA, 3rd, Yamamoto M, Alvarez RD, Curiel DT. Transcriptional targeting of adenoviral vector through the CXCR4 tumor-specific promoter. Gene Ther. 2004;11:645–648. doi: 10.1038/sj.gt.3302089. [DOI] [PubMed] [Google Scholar]
- 168.Rajendran S, O’Sullivan GC, O’Hanlon D, Tangney M. Adenovirus-mediated transcriptional targeting of colorectal cancer and effects on treatment-resistant hypoxic cells. Clin Colorectal Cancer. 2013;12:152–162 e151. doi: 10.1016/j.clcc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 169.Balani P, Boulaire J, Zhao Y, Zeng J, Lin J, Wang S. High mobility group box2 promoter-controlled suicide gene expression enables targeted glioblastoma treatment. Mol Ther. 2009;17:1003–1011. doi: 10.1038/mt.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Akerstrom V, Chen C, Lan MS, Breslin MB. Adenoviral insulinoma-associated protein 1 promoter-driven suicide gene therapy with enhanced selectivity for treatment of neuroendocrine cancers. Ochsner J. 2013;13:91–99. [PMC free article] [PubMed] [Google Scholar]
- 171.Tsuruta Y, Pereboeva L, Breidenbach M, Rein DT, Wang M, Alvarez RD, Siegal GP, Dent P, Fisher PB, Curiel DT. A fiber-modified mesothelin promoter-based conditionally replicating adenovirus for treatment of ovarian cancer. Clin Cancer Res. 2008;14:3582–3588. doi: 10.1158/1078-0432.CCR-07-5053. [DOI] [PubMed] [Google Scholar]
- 172.Hine CM, Seluanov A, Gorbunova V. Rad51 promoter-targeted gene therapy is effective for in vivo visualization and treatment of cancer. Mol Ther. 2012;20:347–355. doi: 10.1038/mt.2011.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hine CM, Seluanov A, Gorbunova V. Use of the Rad51 promoter for targeted anti-cancer therapy. Proc Natl Acad Sci USA. 2008;105:20810–20815. doi: 10.1073/pnas.0807990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fontecedro AC, Lutschg V, Eichhoff O, Dummer R, Greber UF, Hemmi S. Analysis of adenovirus trans-complementation-mediated gene expression controlled by melanoma-specific TETP promoter in vitro. Virol J. 2010;7:175. doi: 10.1186/1743-422X-7-175. [DOI] [PMC free article] [PubMed] [Google Scholar]