

Summary
Pathogens that evade adaptive immunity typically exhibit antigenic variation. By contrast, it appears that although the chronic human tuberculosis (TB)-causing pathogen Mycobacterium tuberculosis needs to counter host T cell responses, its T cell epitopes are hyperconserved. Here we present an extensive analysis of the T cell epitopes of M. tuberculosis. We combined population genomics with experimental immunology to determine the number and identity of T cell epitope sequence variants in 216 phylogenetically diverse strains of M. tuberculosis. Antigen conservation is indeed a hallmark M. tuberculosis. However, our analysis revealed a set of 7 variable antigens that were immunogenic in subjects with active TB. These findings suggest that M. tuberculosis uses mechanisms other than antigenic variation to evade T cells. T cell epitopes that exhibit sequence variation may not be subject to the same evasion mechanisms and hence vaccines that include such variable epitopes may be more efficacious.
Introduction
Microbial pathogens must adapt to host environments to establish replicative niches and counter innate and adaptive immune responses. The need to adapt is especially marked for obligate pathogens that must infect, replicate, and be transmitted to new hosts to survive and propagate. In the case of pathogens that encounter adaptive immune responses, a common mechanism of adaptation is antigenic variation, in which pathogen targets that are recognized by antibodies and T lymphocytes develop escape mutations that allow them to evade recognition and elimination by the host immune system (Deitsch et al., 2009; Goulder et al., 2001; Heaton et al., 2013; Palmer et al., 2009). As pathogens evolve to evade host immunity by antigenic variation, hosts respond by developing antibodies and T cells with specificity to the newly-selected antigens. The cycle of antigenic variation and host response can be continuous (Dawkins and Krebs, 1979; Woolhouse et al., 2002). The consequences of antigenic variation can include persistent viral (Dustin and Rice, 2007), parasitic, or bacterial infection (Deitsch et al., 2009; Palmer et al., 2009), pandemics of diseases such as influenza (Itoh et al., 2009), and reinfection after recovery (Henderson et al., 1979). Antigenic variation also impacts vaccine development, by compelling the use of multivalent vaccines (containing multiple antigenic variants) such as for poliovirus and Streptococcus pneumoniae, or by requiring new vaccines at regular intervals, such as for influenza. Extreme antigenic variation confounds development of HIV vaccines that can protect against diverse viral strains (Walker and Korber, 2001).
Mycobacterium tuberculosis and other members of the Mycobacterium tuberculosis complex (MTBC) cause tuberculosis (TB) (Coscolla and Gagneux, 2014), a chronic infection transmitted by the aerosol route that remains a major global health problem despite the availability of drug treatment (WHO, 2014). M. tuberculosis is an obligate human pathogen, as it has no ecological niche other than its human hosts, with which it has coevolved for thousands of years (Bos et al., 2014; Comas et al., 2013). The success of M. tuberculosis as a pathogen is due to its efficiency of transmission and its ability to evade elimination by adaptive immune responses (Ernst, 2012). Immunity to M. tuberculosis depends on T lymphocytes, as T cell-deficient humans, nonhuman primates, and mice are susceptible to rapidly-progressive disease (Kwan and Ernst, 2011; Lin et al., 2009; North and Jung, 2004). Among T lymphocytes, CD4 T cells are essential for protective immunity to TB: in HIV-infected humans, loss of CD4 T cells greatly increases susceptibility to TB, and reconstitution of CD4 T cells by antiretroviral therapy reduces susceptibility to TB (Kwan and Ernst, 2011). Humans also generate CD8 T cell responses to M. tuberculosis antigens during infection, but the contribution of CD8 T cells to protection is less clear (Lancioni et al., 2012). Despite development of CD4 and CD8 T cell responses directed against M. tuberculosis antigens, the bacteria can persist for the life of an individual, allowing for reactivation, progression, and transmission of TB. The mechanisms that allow persistence and progression of M. tuberculosis infection are incompletely understood, but they include modulation of bacterial antigen expression, inefficient antigen presentation, and bacterial interference with T cell effector mechanisms (Ernst, 2012). The contribution of antigenic variation to immune evasion in TB is also incompletely understood.
For T cells to contribute to immunity, they must recognize specific peptides (termed epitope peptides), generated by proteolysis of pathogen proteins, bound to Major Histocompatibility Complex (MHC) molecules on the surface of antigen presenting cells. CD4 T cells recognize epitope peptides bound to MHC (Human Leukocyte Antigen (HLA) in humans) class II molecules, while CD8 T cells recognize peptides bound to MHC/HLA class Ia molecules. For a given epitope to be immunogenic, it must bind with sufficient affinity to one or more MHC proteins, and the peptide-MHC complex must be recognized by a clonotypic T cell antigen receptor. Variation in the sequence of an epitope can result in loss of MHC binding and recognition by T cells (Corradin and Chiller, 1979), indicating that sequence variation of pathogen molecules can result in escape from control of infection by T cells (Farci et al., 2000; Kawashima et al., 2009).
To determine whether antigenic (epitope peptide) sequence variation contributes to immune evasion in human TB, we previously characterized the sequences of 491 human T cell epitopes in the genomes of 21 phylogenetically-diverse strains of the MTBC and unexpectedly found that the T cell epitopes of the MTBC are hyperconserved (Comas et al., 2010). In that study, 95% of the T cell epitopes studied contained no amino acid substitutions, even in bacterial strains that diverged from a common ancestor thousands of years ago. This observation implied that human T cell recognition of the known human T cell epitopes of M. tuberculosis is not sufficiently detrimental to the pathogen to select for escape variants. If the persistence and evolutionary success of M. tuberculosis do not involve antigenic variation, it will be necessary to formulate new models for understanding the biology of TB and certain other infectious diseases, especially those that have been refractory to development of efficacious vaccines.
Although the results of our previous study established that T cell epitopes are hyperconserved, M. tuberculosis clearly adapts to antimicrobial drug pressure by development of resistance through chromosomal mutations, and not through horizontal gene exchange (Borrell and Gagneux, 2011; Goldberg et al., 2012; Muller et al., 2013). This indicates that M. tuberculosis can respond to selection pressure with adaptive mutations, and the finding of a very low frequency of epitope variation in our previous study may have been influenced by the approach commonly employed to discover T cell epitopes. Indeed, most studies to date have used derivatives (native or recombinant antigens, or synthetic peptides) based on the M. tuberculosis reference strain H37Rv, a member of the MTBC Lineage 4 (Gagneux and Small, 2007). Use of those derivatives to identify epitopes recognized by T cells of individuals who were infected by strains from MTBC lineages other than Lineage 4 may bias the results toward discovery of conserved epitopes. In addition, since the total number of antigens and T cell epitopes encoded by the MTBC genome is unknown, it is possible that are epitopes that have not been identified with usual approaches and that are under diversifying selection from T cell recognition.
To better understand the evolutionary impact of human T cell recognition, and to determine the extent of M. tuberculosis antigenic variation, we first extended our analysis to an expanded set of experimentally-validated epitopes and a larger number of bacterial strains. We then analyzed the genomes of 216 strains representative of the seven main human-adapted phylogenetic lineages of the MTBC (Coscolla and Gagneux, 2014), and used an innovative strategy based on a combination of population genomics and computational and experimental immunology to determine the number and the identity of human T cell epitopes with naturally-occurring sequence variants. Our findings reinforce the finding of epitope conservation, reveal a small number of epitopes with sequence variants, and indicate that antigenic variation is not a major mechanism of immune evasion in M. tuberculosis.
Results
Expanded analysis of epitopes and bacterial strains confirms conservation of human T cell epitopes in M. tuberculosis
Our initial finding of hyperconservation of the epitopes of M. tuberculosis recognized by human T cells was based on the analysis of 21 genomes and 491 T cell epitopes; this may have limited the likelihood of finding epitopes with sequence variants. To better define the frequency of conserved and of variable epitopes, we analyzed the genomes of 216 bacterial strains (Comas et al., 2013) representative of the seven known human-adapted phylogenetic lineages of the MTBC (Coscolla and Gagneux, 2014). Recent epitope discovery efforts also facilitated our analysis of conservation and diversity of a larger number of human T cell epitopes.
We first identified 1,730 epitopes catalogued in the Immune Epitope Database (www.iedb.org). After excluding epitopes in repetitive regions (see supplemental methods), we used 1,226 of these for analysis of their sequence diversity in the 216 phylogenetically diverse bacterial strains (Comas et al., 2013). This revealed that 953 (78%) of the 1,226 epitopes contained no amino acid variants in the 216 strains, while we found 1 amino acid substitution in 232 (19%) of the epitopes (Figure 1A). Notably, of the epitopes in which we found 1 amino acid substitution, 57% were found in a single isolate. No epitope exhibited more than three distinct amino acid variants. To further explore the degree of T cell epitope conservation, we compared the ratios of non-synonymous to synonymous substitution rates (dN/dS) in epitopes and other elements of the MTBC genome (Figure 1B). Consistent with our previous observation (Comas et al, 2010), we found that essential genes showed a significantly lower dN/dS than nonessential genes (Wilcoxon Signed-Rank Test p = 5.6−15). This was expected, as essential genes are known to be under stronger purifying selection than nonessential genes. Again consistent with our previous findings (Comas et al, 2010), we found that all epitope regions combined showed a significantly lower dN/dS compared to the corresponding nonepitope regions of the same proteins, or to essential genes (Wilcoxon Signed-Rank Test p = p < 2.2−16 and p = 4.4−14, respectively) (Figure 1B). Since inclusion of epitopes in the IEDB does not require that peptides meet preset criteria (Ernst et al., 2008), we separately analyzed a subset of 163 peptides (included in the initial set of 1,226) identified in a recent T cell epitope screen as the most immunodominant among 20,610 peptides examined (Arlehamn et al., 2013). After filtering out epitopes found in repetitive loci (58/163), we found that 9 of the 105 immunodominant epitopes (8.6%) contained sequence variants in more than one strain, while 11 additional epitopes contain variants in a single strain (singletons) (Table S1). Since singletons may be transient and destined to be removed by purifying selection, their biological significance is uncertain. Thus, expanded analyses of a larger number of epitopes in a larger number of bacterial strains confirmed that sequence conservation dominates in the known T cell epitopes of M. tuberculosis.
Figure 1.

Genetic diversity of experimentally-verified human T cell epitopes of M. tuberculosis. (A) Frequency distribution of the number of epitopes (total n = 1,226) with the stated number of sequence variants. (B) Comparison of dN/dS of epitopes considered in panel A; nonepitope domains of the epitope-containing proteins; essential proteins, and nonessential proteins of M. tuberculosis.
Since the present experimental approaches to M. tuberculosis epitope discovery may be biased toward discovery of conserved epitopes (by studying responses to antigens or epitope peptides derived from bacteria from a single phylogenetic lineage), we used an alternative approach to determine the existence and frequency of human T cell epitopes with sequence variants in the MTBC. We reasoned that effective immune recognition would drive a higher level of sequence diversity than the genome average in genes that encode antigens containing variable epitopes, and that variable epitopes could be discovered by use of comparative genomics.
Identification of variable regions in the M. tuberculosis genome
To identify and quantitate variable candidate antigens in the MTBC genome, we analyzed 3,774 coding regions in whole genome sequences of 216 human-adapted MTBC strains representative of the seven main lineages (Comas et al., 2013) (Figure 2). First, we calculated the nucleotide diversity (π, defined as the average number of nucleotide differences per site between any two DNA sequences chosen randomly). This revealed a median π=0.0002 for all coding regions, ranging from 0 to 0.0046, compared to π=0.0003 for the whole genome (Figure 2, Table S2). Because only nSNPs lead to amino acid changes and potential antigenic variation, we further focused our analyses on coding regions exhibiting high rates of nSNPs. To this end, we selected the most variable 5% of the genes (N=189; Table S2) and determined that the dN/dS in these genes ranged from 0 to 4.21, and 47% (88/189) of the genes showed a dN/dS>1 (Table S2). Because 19 genes showed only nSNPs, a dN/dS value could not be calculated for these genes and was indicated as ‘infinite’ (Table S2).
Figure 2.

Visualization of M. tuberculosis genetic diversity on a circular map of the chromosome of the H37Rv reference strain. The first ring from the outside shows the scale of the chromosome in nucleotides. The spikes in the second ring (red) illustrate the 7 highly variable genes identified in the present study. The third ring (blue) denotes genes encoding previously reported T cell epitopes (Vita et al., 2010). The spikes radiating from the innermost circle represent a histogram of the mean pairwise nucleotide diversity (π) per gene shown in Table S2, ranging from 0 to 0.002. Average π for the whole genome is indicated by the green circle. Surrounding the circular map, insets show the genomic regions containing the selected genes in this work highlighted in yellow, and the π values of these genes are shown in the accompanying bar graphs. See also Table S2.
Next, we selected genes for further characterization based on the three following criteria: i) ranking in the 5% of genes with the highest π (Table S2), ii) a dN/dS >1, and iii) harboring at least one nSNP present in an entire lineage (Table S2). To assess the possibility that the impact of a given nSNP on immune response depends on its distribution in the MTBC phylogeny, we also selected candidates with variants in distinct lineages. A total of 7 genes were chosen for further analyses. These 7 genes were associated with several functional categories (Lew et al., 2011), including cell wall and cell processes (3 genes), intermediary metabolism (2 genes), lipid metabolism (1 gene) and information pathways (1 gene) (Table S3). The protein products encoded by 5 of the 7 genes have been detected in proteomics studies which have revealed the presence of 3 of them in the bacterial membrane fraction (de Souza et al., 2011; Mawuenyega et al., 2005; Rosenkrands et al., 2000; Schubert et al., 2013). None of the proteins were predicted or determined to be secreted.
Epitope prediction
To explore whether sequence diversity in the 7 genes of interest could be related to human T cell recognition, we first computationally predicted human CD4 and CD8 T cell epitopes in the protein products of these genes using HLA class I and HLA class II alleles that are prevalent in diverse human populations (Hoof et al., 2009; Nielsen et al., 2006) (Table S4). This identified a mean of 207 potential high-affinity epitopes per protein for HLA class I and 150 epitopes per protein for HLA class II.
We then examined the relationship between the predicted human T cell epitopes and DNA sequence variation in the 7 genes of interest. Comparative analysis of the 216 whole genomes revealed 56 nSNPs in these 7 genes (Table S5). Moreover, we found that 91% (51/56) of these nSNPs coincided with predicted CD4 and/or CD8 T cell epitopes (Figure 3). We then evaluated whether the corresponding amino acid changes altered the results of the epitope predictions. We found that on average, a given amino acid substitution led to 5% fewer predicted CD4 T cell epitopes (range, 0-8%) and 18% fewer predicted CD8 T cell epitopes, ranging from 11% (10 of 88 for RimJ) to 53% (9 of 17 for TB7.3) (Table S6A). To test whether this reduction in the number of predicted CD4 and CD8 epitopes encoded by these genes was statistically significant, we selected a control set of 100 predicted T cell epitopes from random regions of the MTBC genome that contained nSNPs (Table S6B) and evaluated the consequences of these nSNPs on the predicted binding affinity of this other set of epitopes. We found that the impact of nSNPs on predicted CD4 T cell epitopes was similar regardless of whether the epitopes were derived from the 7 genes of interest or elsewhere in the genome. In contrast, for the predicted CD8 T cell epitopes, nSNPs in the 7 genes of interest were ~5 times more likely to affect the predicted epitope binding than nSNPs in predicted epitopes encoded in the random proteins (16% versus 3.3%; χ = 8.233, P < 0.005). These results suggest that the 7 genes of interest identified here are under distinct selection pressures compared with random genes in the MTBC genome, and that this difference could be due to CD8 T cell recognition.
Figure 3.

Location of 82 naturally occuring amino acid substitutions and predicted CD4 and CD8 T cell epitopes in 7 candidate M. tuberculosis antigens. For each protein, a length scale in amino acids is shown. The red rectangles highlight regions containing predicted CD4 and/or CD8 T cell epitopes for which predicted HLA binding affinity was affected by amino acid substitutions. The number of affected predicted T cell epitopes per protein is indicated in the white boxes. T cell epitope predictions were done using HLA molecules representing major global human populations (Table S4 and accompanying map).
Next, we examined the impact of naturally-occurring sequence variation on the predicted capacity of putative epitope peptides to bind to selected HLA alleles with high affinity. We found that on average, a nSNP decreased the binding affinity of 25% (7 of 33) and 18% (9 of 51) of the predicted peptide:HLA class I and peptide:HLA class II interactions, respectively (Table S6A). We also found that the naturally-occurring sequence variation could lead to an increase in the number of HLA alleles capable of high-affinity binding of the corresponding epitope peptide. Overall, epitope variants were predicted to form between 4 to 13 new HLA class I and 2 to 17 new HLA class II high affinity binding interactions (Table S6A). In summary, our results indicate that the naturally occurring sequence variation in epitopes predicted from our 7 genes of interest can result in either loss or gain of recognition by human T cells.
The different phylogenetic lineages of the MTBC have been associated with different human populations (Brites and Gagneux, 2015), and some MTBC lineages are more globally widespread than others (Coscolla and Gagneux, 2014). Because 53 of the 56 (95%) nSNPs found in the 7 genes of interest were specific to individual MTBC lineages, we assessed the possibility that the impact of a given nSNP on T cell epitope recognition depends on its distribution in the MTBC phylogeny and the corresponding human population. We initially concentrated on nSNPs specific to Lineage 4 (also known as the Euro-American lineage), because it is the most successful MTBC lineage worldwide and also well represented in our strain collection. Moreover, HLA allele frequencies are best characterized in the Caucasian populations where Lineage 4 strains are prevalent. This analysis revealed that the 14 nSNPs specific to Lineage 4 strains led to a net decrease of 17% of the predicted T cell epitopes (Table S6C). By contrast, we found that nSNPs in Lineages 1, 3 or 6 increase the number of predicted epitopes by an average of 37% (Table S6C). The latter finding may be related to the relative lack of success of Lineage 1, 3 and 6 compared to Lineage 4 in spreading globally (Hershberg et al., 2008).
Immunogenicity of peptides representing candidate variable CD4 and CD8 T cell epitopes
To experimentally validate our epitope predictions, we assessed the immunogenicity of the predicted T cell epitopes and their respective naturally-occurring sequence variants by assaying immune responses from 88 sputum smear-positive, HIV-seronegative adults with newly diagnosed TB in The Gambia. For this analysis, we focused on variable T cell epitopes with high affinities for the HLA alleles that are most prevalent in The Gambia (Table S4). A total of 9 CD4 and 5 CD8 candidate T cell epitopes were selected, containing 16 amino acid changes predicted to alter the interactions with some or all selected HLA alleles (Figure 4). In total, 30 peptides corresponding to 14 ancestral (present in the MTBC common ancestor) and 16 variant (any departure from the ancestral) sequences were synthesized from the 7 genes of interest (Figure 4). Each peptide was used to stimulate fresh diluted whole blood samples followed by ELISA quantitation of interferon gamma (IFN-γ) in supernatants. Cells of six of the subjects did not respond to any of three positive controls, therefore the IFN-γ responses of 82 subjects were used for further analysis (Table S7).
Figure 4.

Localization of the peptides representing candidate variable CD4 and CD8 T cell epitopes predicted using HLA molecules prevalent in the Gambian population. For each protein, a length scale in amino acids is shown. The rectangles depict the positions of the candidate CD4 (yellow) and CD8 (blue) T cell epitopes within the protein sequences. Each epitope is also represented by an arrow and associated with a candidate epitope number (from CE1 to CE14). The amino acid sequences in black are the sequences of the candidate epitopes in the inferred most recent common ancestor of the MTBC. The sequences in white font represent the naturally-occurring variants identified in this work; the amino acid changes compared with the ancestral sequence are in bold. Each variant sequence is color-coded indicated according to its distribution in the MTBC phylogeny. The phylogenetic lineages and the number of strains containing the variant version of each epitope sequence are also indicated. See also Table S2, S3 and S5.
Each of the candidate epitope peptides was immunogenic in subjects with active TB, as defined by the ability to stimulate IFN-γ release to a level at least 2-fold higher than the individual subject's unstimulated control sample. When considered together, the ancestral and variant sequences of each peptide induced responses from the cells of an average of 12 subjects (Figure 5A, Table S7). By summing the responses to all peptides from one protein, we found that epitopes in RimJ and Rv0010c induced responses in ≥ 30% of the subjects (Figure 5B). Considered separately, the HLA class I and class II candidate epitopes stimulated the cells of an average of 12% and 16% of subjects (10/82 and 13/82), respectively. No statistically significant difference was found between these percentages, indicating that the accuracy of predicting immunogenic HLA class I and class II epitopes did not differ.
Figure 5.

Frequency of immune responses to the candidate T cell epitopes selected in this work. The results were derived from the diluted whole blood assay using cells from 82 human subjects with active TB, stimulated with synthetic epitope peptides, followed by quantitation of secreted IFN-γ. Number of subjects (of 82 total) responding to (A) the ancestral or variant sequences of each candidate epitope or (B) to all peptides derived from each antigenic protein. Responses were defined as a stimulation index >2, as defined in Methods. Each bar represents the cumulative responses to the ancestral and variant sequences of each candidate epitope. See also Table S7.
Fifty-two of the 82 (63%) subjects with TB responded to at least one candidate epitope peptide. Cells from individual subjects responded to an average of 3 of the 30 (10%) candidate epitope peptides (ancestral and/or variant form), although cells of some subjects responded to as many as 22 (73.3%). The amount of IFN-γ secreted in response to peptide stimulation of cells from certain subjects was in a range similar to that observed when a pool of overlapping peptides from ESAT-6 and CFP10 was used, indicating that the candidate epitope peptides were immunogenic in this population (Figure 6A). Of note, epitope peptides from Rv0010c stimulated responses from the highest fraction of the subjects, and also induced responses with the highest magnitude (Figure 6A). Evidence that responses to the peptides were attributable to infection with M. tuberculosis was obtained by analysis of responses obtained after 2 months and 6 months of drug treatment for TB. With the exception of Candidate Epitope 3 (CE3; from Rv2719c), the magnitude of the responses decreased significantly with successful treatment and resolution of TB (Figure 6B).
Figure 6.
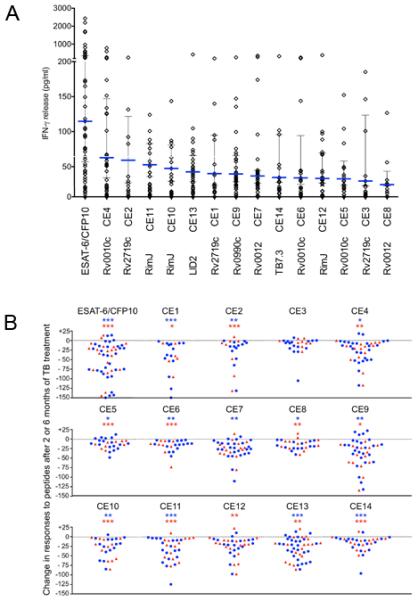
Quantitation of IFN-γ from diluted whole blood samples stimulated with synthetic peptides representing ancestral sequences of the candidate epitopes. (A) Each diamond represents the response of a single subject; the horizontal blue line represents the median response calculated using data from responders only. The vertical line represents the interquartile range. Each value is the net concentration after subtraction of background determined with an unstimulated sample from each subject assayed simultaneously (see also Table S7) (B) Comparison of responses (as IFN-γ release in pg/ml) after 2 and 6 months of TB treatment, compared with responses before treatment. Results were obtained by subtracting response values measured after 2 months (blue circles) or 6 months (red triangle) of treatment from those before treatment in the same subject. Statistical analysis was done by Wilcoxon signed-rank test; * p<0.05, ** p<0.01 or *** p<0.001; blue asterisks represent analysis of differences after 2 months, and red asterisks represent analysis of differences after 6 months of treatment.
To assess the impact of the amino acid substitutions on the responses to the candidate epitope peptides, we compared the responses (assayed as IFN-γ secretion) induced by the ancestral or the variant sequences of each of the 14 candidate epitopes. We found that an average of 72% of the responding subjects for a given candidate epitope exhibited differential responses to the ancestral compared with the variant sequences of each of the 14 candidate epitopes (designated CE1-CE14) (Table S7 and S8). Amino acid substitutions in 10 of the 14 candidate epitopes altered T cell responses in the majority of the responding subjects (ranging from 63% of the responding subjects for CE14 to 87% for CE3, Bayesian p<0.05) (Table S7 and S8, Figure 7A and 7B). Amino acid substitutions in the 4 other candidate epitopes influenced T cell responses in a smaller fraction of the subjects, although all of them still altered the responses in >40% of the subjects (Table S7). Notably, amino acid changes in CE9 completely abrogated T cell responses in 8 individuals (Figure 7A). CE12, contained in RimJ, was distinct from the other candidate epitopes, as more of the subjects responded to the variant peptides than to the ancestral peptide (Figure 7B). Whether this reflects a difference in the sequence of this epitope in the bacterial strains infecting these subjects, or a difference in binding of the variant peptides to the subjects' HLA alleles, will require further investigation. Together, our results demonstrate that naturally-occurring sequence variation in these candidate T cell epitopes affects host recognition, suggesting that T cell recognition is a factor driving variation in the 7 genes identified in this study.
Figure 7.

Naturally-occurring amino acid variants alter immune recognition of candidate epitopes. Comparison of the magnitude of IFN-γ secretion from cells of individual subjects induced by the ancestral and the variant sequences of individual candidate epitopes. (A) Responses of individuals whose cells responded to the ancestral but not the variant sequence peptide. (B) Responses of individuals whose cells responded to the variant but not the ancestral sequence of each candidate. Each connecting line represents responses of a single subject. Responses were defined as a stimulation index >2, as defined in Methods. See also Table S8.
Discussion
The most significant findings of the present study are the strong evidence that antigen and epitope conservation dominate in M. tuberculosis, and the discovery of human T cell epitopes that exhibit sequence variation and evidence of diversifying selective pressure. The combination of two distinct approaches to determining the frequency of conserved and variable T cell epitopes in the genome of M. tuberculosis yielded incontrovertible evidence that epitope sequence conservation is the rule, and not the exception, in this highly successful human pathogen. First, we examined 1,226 experimentally-verified peptide epitopes for sequence variants in 216 phylogenetically diverse strains of the MTBC and confirmed that the vast majority (78%) showed no amino acid variation. Moreover, we confirmed that T cell epitopes in the MTBC are significantly more evolutionarily conserved than non-epitope regions in the same antigens. Second, we used a comparative genomics strategy which avoids the potential discovery bias of the former approach to determine the frequency and identity of human T cell epitopes with sequence variants and confirmed their recognition by cells of humans with pulmonary TB. This effort revealed a small number of variable epitopes, indicating that, even though epitope sequence conservation dominates in M. tuberculosis, there are exceptions, and these had not been identified by standard approaches. Together, the results reveal that M. tuberculosis employs epitope sequence variation only rarely as an evolutionary strategy to evade recognition by human T cells. Since epitope peptides are the sole molecular interface of the pathogen with T cells, and since T cells are the most important component of protective adaptive immunity in TB, our results indicate that these bacteria have evolved to use mechanisms other than antigenic variation to evade T cell immunity.
The findings we report here are consistent with the conclusions of our recent analysis of the pe_pgrs gene family in M. tuberculosis that had been proposed to be involved in antigenic variation (Cole et al., 1998). That analysis of 27 pe_pgrs genes in 94 phylogenetically diverse strains of M. tuberculosis revealed that, although certain of the pe_pgrs genes are highly polymorphic, their sequence polymorphisms and indels are concentrated in the C-terminal PGRS domains, while their T cell epitopes are concentrated in the conserved N-terminal PE domain (Copin et al., 2014). Therefore, in the products of the pe_pgrs genes, despite their high frequency of sequence and structural variants, the T cell epitopes are conserved.
The evidence that M. tuberculosis does not employ antigenic variation as a major mechanism of adaptation and immune evasion is unexpected, since M. tuberculosis causes chronic infection that can persist for the life of the host. This indicates that the bacteria are highly successful in using other mechanisms to evade elimination by adaptive immune responses. Since other pathogens that cause chronic infections, including HIV (Liu et al., 2013), hepatitis C virus (Farci, 2011), Trypanosoma cruzei (Mugnier et al., 2015), and Treponema pallidum (Reid et al., 2014) employ antigenic variation to cause persistent infection, M. tuberculosis stands as a prominent exception to an increasingly widely accepted rule.
The unexpected finding of antigen and epitope conservation in M. tuberculosis compels consideration of an explanation for the results. One potential explanation is that during coevolution with humans, M. tuberculosis has derived a net evolutionary benefit from T cell recognition, despite the within-host cost that T cell responses impose on the bacteria in the majority of infected individuals. As noted previously, one possible mechanism of an evolutionary benefit to the bacteria in the context of epitope conservation is through the inflammatory lung tissue damage characteristic of human cavitary tuberculosis, whose incidence is directly related to the number of circulating CD4 T cells at the time of TB diagnosis in HIV coinfected individuals (Kwan and Ernst, 2011) and which is associated with high transmission of infection (Reichler et al., 2002). A second potential explanation for the dominance of conserved epitopes in M. tuberculosis is that the epitopes are derived from domains of proteins that serve an essential function for the bacteria, and are therefore constrained in their inherent mutational tolerance. Although this is a plausible explanation, 76% of the epitopes we analyzed here are encoded by nonessential genes, and our efforts to date have not detected evidence that human T cell epitopes in M. tuberculosis have common structural motifs, or that they are preferentially derived from active sites in proteins with known enzymatic functions (R. Copin and J.D. Ernst, unpublished). However, in support of this latter model is the recent observation that certain of the known T cell epitopes of M. tuberculosis are also conserved in nonpathogenic mycobacteria, which are not under selection pressure in a mammalian host (Lindestam Arlehamn et al., 2014). Similarly, T cell epitopes are also conserved in Mycobacterium canettii, which is a member of the MTBC that otherwise shows much genomic diversity (Supply et al., 2013).
In addition to being unexpected, the finding that antigenic variation is the exception, rather than the rule in M. tuberculosis, has implications for understanding the immunopathogenesis of TB. If antigenic variation does not contribute to the persistence of M. tuberculosis in immunocompetent individuals with measurable T cell responses, then other potent mechanisms for evading elimination of the bacteria by immune responses must account for bacterial persistence and chronic infection. Since M. tuberculosis occupies professional antigen-presenting cells such as dendritic cells and macrophages (Ernst, 2012; Philips and Ernst, 2012), it is not surprising that there is evidence for the bacteria manipulating antigen presentation to T cells. For example, multiple studies provide evidence that M. tuberculosis interferes with MHC class II antigen presentation to CD4 T cells (reviewed in (Baena and Porcelli, 2009)), and M. tuberculosis inhibits apoptosis in diverse subsets of antigen presenting cells in vivo (Blomgran et al., 2012; Velmurugan et al., 2007), which decreases cross presentation to CD8 T cells (Divangahi et al., 2010). These and other mechanisms may allow the bacteria to survive and cause progressive disease and transmission of infection without requiring antigenic variation as a mechanism of immune evasion.
Apart from the importance for understanding TB pathogenesis, the significance of antigen and epitope conservation should be considered in developing TB vaccines directed at inducing T cell responses. If bacterial persistence and survival in TB is predominantly accomplished by manipulating infected antigen presenting cells to minimize their recognition by antigen-specific CD4 or CD8 T cells, then these mechanisms may also limit the efficacy of vaccine-induced T cells. The modest impact of existing (e.g., BCG) and experimental TB vaccines to date (Andersen and Woodworth, 2014) suggests that there are potent mechanisms that restrict the efficacy of antigen-specific T cells at the site of infection; our results that human T cell recognition does not impose a measurable selection pressure on M. tuberculosis to evade these responses is consistent with this possibility. The finding of T cell epitopes that exhibit sequence variation implies that these epitopes are not subject to the same evasion mechanisms employed by those in the hyperconserved immunodominant antigens of M. tuberculosis. This suggests that vaccines that include such variable epitopes may be more efficacious than those containing conserved, immunodominant epitopes. In this sense, our search for variable epitopes under diversifying selection can be considered to be a variation of 'reverse vaccinology' (Sette and Rappuoli, 2010).
Finally, our finding that M. tuberculosis does not adhere to the canonical model of a host-pathogen arms race should prompt reconsideration of the generalizability of that model. Although it is clear that antigenic variation is employed by diverse, highly successful pathogens, our results indicate that in other pathogens, especially those with unique and narrow host ranges, antigen conservation may prevail, and this should be considered in understanding their pathogenesis and in devising methods to prevent them.
Experimental procedures
Computational analysis
Methods and associated references referring to the genetic diversity analysis, HLA allele selection, epitope predictions and statistical analysis are available in the Supplemental Experimental Procedures.
Subject recruitment and patient information
The subjects studied were recruited and studied at the MRC Unit - Gambia. They were HIV-seronegative adults with newly-diagnosed pulmonary TB who gave informed consent to a prospective study reviewed and approved by the New York University Institutional Review Board and by the Gambia Government/Medical Research Council (MRC) Joint Ethics Committee. After written informed consent, 30 ml of heparin-anticoagulated blood was obtained by venipuncture for the diluted whole blood assay with individual epitope peptides as the antigenic stimuli (Black et al., 2001; Black et al., 2009).
Human T cell responses to candidate epitope peptides
Peptides were synthesized by EZ Biolabs and reconstituted in water or DMSO. Stocks were diluted to 1 mg/ml and stored in aliquots. In a 96-well plate, each peptide was added to a final concentration of 10 µg/mL in an individual well containing whole blood diluted with 9 volumes of RPMI 1640. Samples without stimulus were used as negative controls to allow calculation of the magnitude of the induced responses. Phytohemagglutinin (PHA; 5 µg/mL), purified protein derivative (PPD; Staten Serum Institute, Denmark) and a pool of 35 overlapping 15 mer peptides (2.5 µg/mL) derived from the 6 kDa Early Secreted Antigenic Target and the Culture Filtrate Protein-10 protein (ESAT-6/CFP-10) were used as positive controls (Tientcheu et al., 2014). Antigens were stimulated in triplicate and after seven days incubation at 37°C in a humidified CO2 incubator, supernatants were removed for assay of IFN-γ.
ELISA of whole blood stimulated supernatants
Supernatants were analysed for IFN-γ by ELISA as previously described (Black et al., 2009). In Table S7, the values shown represent the average of duplicate wells for each antigen. The stimulation index (SI) was calculated for each response by dividing the concentration of IFN-γ in the stimulated wells by the unstimulated negative control for each subject. A positive response was defined as a SI ≥2.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 AI090928 and HHSN266200700022C) and the Swiss National Science Foundation (PP00P3_150750). R.C. was supported by the Potts Memorial Foundation and the Belgian American Educational Foundation. Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing core facility at University of Basel.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional Title Page Footnotes
MC and RC are co-first authors; JDE and SG are co-senior authors
Author contributions
JDE and SG designed the project, supervised the research, and wrote the paper. MC and RC acquired and analyzed genome diversity, evolutionary selection parameters, and predicted T cell epitopes. OO enrolled subjects, generated and organized clinical data and sample collection; GM generated and organized laboratory data, both in the Gambia. JS, FGe, BdJ, and RC organized, supervised, and contributed to analysis of human subjects data, including experimental responses to candidate epitope peptides. FG contributed to statistical analyses. RC and MC also contributed to writing the paper.
References
- Andersen P, Woodworth JS. Tuberculosis vaccines--rethinking the current paradigm. Trends Immunol. 2014;35:387–395. doi: 10.1016/j.it.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Arlehamn CSL, Gerasimova A, Mele F, Henderson R, Swann J, Greenbaum JA, Kim Y, Sidney J, James EA, Taplitz R, et al. Memory T Cells in Latent Mycobacterium tuberculosis Infection Are Directed against Three Antigenic Islands and Largely Contained in a CXCR3(+)CCR6(+) Th1 Subset. Plos Pathog. 2013;9 doi: 10.1371/journal.ppat.1003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena A, Porcelli SA. Evasion and subversion of antigen presentation by Mycobacterium tuberculosis. Tissue Antigens. 2009;74:189–204. doi: 10.1111/j.1399-0039.2009.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black GF, Fine PEM, Warndorff DK, Floyd S, Weir RE, Blackwell JM, Bliss L, Sichali L, Mwaungulu L, Chaguluka S, et al. Relationship between IFN-gamma and skin test responsiveness to Mycobacterium tuberculosis PPD in healthy, non-BCG-vaccinated young adults in Northern Malawi. Int J Tuberc Lung Dis. 2001;5:664–672. [PubMed] [Google Scholar]
- Black GF, Thiel BA, Ota MO, Parida SK, Adegbola R, Boom WH, Dockrell HM, Franken KLMC, Friggen AH, Hill PC, et al. Immunogenicity of Novel DosR Regulon-Encoded Candidate Antigens of Mycobacterium tuberculosis in Three High-Burden Populations in Africa. Clin Vaccine Immunol. 2009;16:1203–1212. doi: 10.1128/CVI.00111-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgran R, Desvignes L, Briken V, Ernst JD. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe. 2012;11:81–90. doi: 10.1016/j.chom.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell S, Gagneux S. Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin Microbiol Infect. 2011;17:815–820. doi: 10.1111/j.1469-0691.2011.03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos KI, Harkins KM, Herbig A, Coscolla M, Weber N, Comas I, Forrest SA, Bryant JM, Harris SR, Schuenemann VJ, et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature. 2014;514:494–497. doi: 10.1038/nature13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brites D, Gagneux S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev. 2015;264:6–24. doi: 10.1111/imr.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–U1311. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copin R, Coscolla M, Seiffert SN, Bothamley G, Sutherland J, Mbayo G, Gagneux S, Ernst JD. Sequence diversity in the pe_pgrs genes of Mycobacterium tuberculosis is independent of human T cell recognition. mBio. 2014;5:e00960–00913. doi: 10.1128/mBio.00960-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin G, Chiller JM. Lymphocyte Specificity to Protein Antigens .2. Fine Specificity of T-Cell Activation with Cytochrome-C and Derived Peptides As Antigenic Probes. J Exp Med. 1979;149:436–447. doi: 10.1084/jem.149.2.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26:431–444. doi: 10.1016/j.smim.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawkins R, Krebs JR. Arms races between and within species. Proc R Soc Lond B Biol Sci. 1979;205:489–511. doi: 10.1098/rspb.1979.0081. [DOI] [PubMed] [Google Scholar]
- de Souza GA, Leversen NA, Malen H, Wiker HG. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J Proteomics. 2011;75:502–510. doi: 10.1016/j.jprot.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Deitsch KW, Lukehart SA, Stringer JR. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat Rev Microbiol. 2009;7:493–503. doi: 10.1038/nrmicro2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin LB, Rice CM. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- Ernst JD, Lewinsohn DM, Behar S, Blythe M, Schlesinger LS, Kornfeld H, Sette A. Meeting Report: NIH Workshop on the Tuberculosis Immune Epitope Database. Tuberculosis (Edinb) 2008;88:366–370. doi: 10.1016/j.tube.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P. New insights into the HCV quasispecies and compartmentalization. Semin Liver Dis. 2011;31:356–374. doi: 10.1055/s-0031-1297925. [DOI] [PubMed] [Google Scholar]
- Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, Strazzera A, Chien DY, Munoz SJ, Balestrieri A, et al. The Outcome of Acute Hepatitis C Predicted by the Evolution of the Viral Quasispecies. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;149:143–151. doi: 10.1016/S1473-3099(07)70108-1. [DOI] [PubMed] [Google Scholar]
- Goldberg DE, Siliciano RF, Jacobs WR., Jr. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell. 2012;148:1271–1283. doi: 10.1016/j.cell.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulder PJ, Brander C, Tang Y, Tremblay C, Colbert RA, Addo MM, Rosenberg ES, Nguyen T, Allen R, Trocha A, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–338. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- Heaton NS, Sachs D, Chen C-J, Hai R, Palese P. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc Natl Acad Sci USA. 2013;110:20248–20253. doi: 10.1073/pnas.1320524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson FW, Collier AM, Clyde WA, Jr., Denny FW. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N Engl J Med. 1979;300:530–534. doi: 10.1056/NEJM197903083001004. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, Roach JC, Kremer K, Petrov DA, Feldman MW, et al. High Functional Diversity in Mycobacterium tuberculosis Driven by Genetic Drift and Human Demography. PLoS Biol. 2008;6:e311. doi: 10.1371/journal.pbio.0060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature. 2009;460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan CK, Ernst JD. HIV and Tuberculosis: a Deadly Human Syndemic. Clin Microbiol Rev. 2011;24:351–376. doi: 10.1128/CMR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancioni C, Nyendak M, Kiguli S, Zalwango S, Mori T, Mayanja-Kizza H, Balyejusa S, Null M, Baseke J, Mulindwa D, et al. CD8+ T Cells Provide an Immunologic Signature of Tuberculosis in Young Children. Am J Respir Crit Care Med. 2012;185:206–212. doi: 10.1164/rccm.201107-1355OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList-10 years after. Tuberculosis. 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Lin PL, Rodgers M, Smith L.k., Bigbee M, Myers A, Bigbee C, Chiosea I, Capuano SV, Fuhrman C, Klein E, et al. Quantitative Comparison of Active and Latent Tuberculosis in the Cynomolgus Macaque Model. Infect Immun. 2009;77:4631–4642. doi: 10.1128/IAI.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindestam Arlehamn CS, Paul S, Mele F, Huang C, Greenbaum JA, Vita R, Sidney J, Peters B, Sallusto F, Sette A. Immunological consequences of intra-genus conservation of Mycobacterium tuberculosis T cell epitopes. Proc Nat Acad Sci USA. 2014 doi: 10.1073/pnas.1416537112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MK, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, Li H, Pavlicek JW, Cai F, Rose-Abrahams M, et al. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest. 2013;123:380–393. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Forst CV, Dobos KM, Belisle JT, Chen J, Bradbury EM, Bradbury ARM, Chen X. Mycobacterium tuberculosis Functional Network Analysis by Global Subcellular Protein Profiling. Mol Biol Cell. 2005;16:396–404. doi: 10.1091/mbc.E04-04-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnier MR, Cross GA, Papavasiliou FN. The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science. 2015;347:1470–1473. doi: 10.1126/science.aaa4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B, Borrell S, Rose G, Gagneux S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013;29:160–169. doi: 10.1016/j.tig.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S. NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. ImmunomeRes2010Nov13. 2006:9–6. doi: 10.1186/1745-7580-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ, Jung YJ. Immunity to Tuberculosis. Annu Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- Palmer GH, Bankhead T, Lukehart SA. 'Nothing is permanent but change'- antigenic variation in persistent bacterial pathogens. Cell Microbiol. 2009;11:1697–1705. doi: 10.1111/j.1462-5822.2009.01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annual review of pathology. 2012;7:353–384. doi: 10.1146/annurev-pathol-011811-132458. [DOI] [PubMed] [Google Scholar]
- Reichler MR, Reves R, Bur S, Thompson V, Mangura BT, Ford J, Valway SE, Onorato IM. Evaluation of investigations conducted to detect and prevent transmission of tuberculosis. JAMA. 2002;287:991–995. doi: 10.1001/jama.287.8.991. [DOI] [PubMed] [Google Scholar]
- Reid TB, Molini BJ, Fernandez MC, Lukehart SA. Antigenic variation of TprK facilitates development of secondary syphilis. Infect Immun. 2014;82:4959–4967. doi: 10.1128/IAI.02236-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkrands I, King A, Weldingh K, Moniatte M, Moertz E, Andersen P. Towards the proteome of Mycobacterium tuberculosis. Electrophoresis. 2000;21:3740–3756. doi: 10.1002/1522-2683(200011)21:17<3740::AID-ELPS3740>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Schubert O, Mouritsen J, Ludwig C, Rîst H-L, Rosenberger G, Arthur PK, Claassen M, Campbell D-S, Sun Z, Farrah T, et al. The Mtb Proteome Library: A Resource of Assays to Quantify the Complete Proteome of Mycobacterium tuberculosis. Cell Host Microbe. 2013;13:602–612. doi: 10.1016/j.chom.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A, Rappuoli R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity. 2010;33:530–541. doi: 10.1016/j.immuni.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, Majlessi L, Criscuolo A, Tap J, Pawlik A, et al. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. 2013;45:172–179. doi: 10.1038/ng.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tientcheu LD, Sutherland JS, de Jong BC, Kampmann B, Jafali J, Adetifa IM, Antonio M, Dockrell HM, Ota MO. Differences in T- cell responses between Mycobacterium tuberculosis and Mycobacterium africanum- infected patients. Eur J Immunol. 2014;44:1387–1398. doi: 10.1002/eji.201343956. [DOI] [PubMed] [Google Scholar]
- Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR, Jr., Porcelli SA, Briken V. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. Plos Pathog. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B. The Immune Epitope Database 2.0. Nucleic Acids Res. 2010;38:D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BD, Korber BT. Immune control of HIV: the obstacles of HLA and viral diversity. Nat Immunol. 2001;2:473–475. doi: 10.1038/88656. [DOI] [PubMed] [Google Scholar]
- WHO . Global tuberculosis report. World Health Organization; Geneva: 2014. [Google Scholar]
- Woolhouse ME, Webster JP, Domingo E, Charlesworth B, Levin BR. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat Genet. 2002;32:569–577. doi: 10.1038/ng1202-569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.