

Abstract
The NMDA receptor antagonist ketamine can improve major depressive disorder (MDD) within hours. To evaluate the putative role of glutamatergic and GABAergic systems in ketamine’s antidepressant action, medial prefrontal cortical (mPFC) levels of glutamate + glutamine (Glx) and γ-aminobutyric acid (GABA) were measured before, during, and after ketamine administration using proton magnetic resonance spectroscopy. Ketamine (0.5 mg/kg i.v.) was administered to eleven depressed patients with MDD. Glx and GABA mPFC responses were measured as ratios relative to unsuppressed voxel tissue water (W) successfully in 8/11 patients. Ten of 11 patients remitted (50% reduction in 24-item Hamilton Depression Rating Scale and total ≤ 10) within 230 minutes of commencing ketamine. mPFC Glx/W and GABA/W peaked at 37.8%±7.5% and 38.0%±9.1% above baseline in ~26 minutes. Mean areas under the curve (AUC) for Glx/W (p = 0.025) and GABA/W (p = 0.005) increased and correlated (r = 0.796; p=0.018). Clinical improvement correlated with 90-minute norketamine concentration (df=6, r=−0.78, p=0.023), but no other measures.
Rapid increases in Glx and GABA in MDD following ketamine administration support the postulated antidepressant role of glutamate and for the first time raises the question of GABA’s role in the antidepressant action of ketamine. These data support the hypothesis1 that ketamine administration may cause an initial increase in glutamate that potentially activates mammalian target of rapamycin (mTOR) pathway via AMPA receptors, since ketamine blocks NMDA receptors. The role of the contemporaneous surge in GABA remains to be determined.2
Keywords: proton magnetic resonance spectroscopy, glutamate/glutamine (Glx), Major Depressive Disorder
Introduction
Major depressive disorder (MDD) affects approximately 14.8 million American adults. Contributing to the disease burden is the multi-week lag in onset of antidepressant effect and the fact that only about one third of patients remit after 6–8 weeks. A single subanesthetic dose of ketamine, a glutamate N-methyl-D-aspartate (NMDA) receptor antagonist, based on two meta-analyses3, 4 of ketamine’s antidepressant effect in randomized placebo-controlled trials (10 trials and 246 patients total; 6 trials and 163 patients overlapped; 34 patients had bipolar disorder), produces an antidepressant effect in hours to days with standardized mean differences of −0.91 and 0.9. These findings held true even in previously treatment-resistant depression. Although studies have examined the duration of antidepressant response to a single ketamine dose5 and following repeated ketamine infusions,6, 7 the number of completed short and long-term controlled and dose-dependent studies is insufficient to establish ketamine as a treatment for general clinical use. However, given the promise of ketamine’s efficacy as a rapid-acting antidepressant, identification of ketamine’s mechanism of action may permit development of a new class of fast-acting antidepressants.
To date, most studies of ketamine’s antidepressant action have been conducted in animals. Ketamine activates the mTOR pathway8, 9 via glutamatergic α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors. mTOR activation increases synaptic proteins and sprouting of new synaptic spines in prefrontal cortex (PFC) within hours, consistent with the time-course of its antidepressant effect.1 The mTOR signaling pathway in hippocampal neurons10 activated by glutamatergic synaptic activity11, 12 is required for long-term potentiation (LTP),10, 12, 13 long-term depression (LTD)14 and memory consolidation.15 Deficits in mTOR expression and in several mTOR-dependent translation initiation factors are reported in prefrontal cortex (PFC) in MDD postmortem, suggesting that this ketamine target may also be part of the pathogenesis of MDD.16 In vivo brain proton magnetic resonance spectroscopy (1H MRS) studies in healthy volunteers report increased glutamine17 and unchanged18 or increased glutamate19 levels in response to ketamine administration. A study in depressed patients20 found no effect of ketamine on glutamatergic compounds. Thus, it remains unclear how ketamine enhances glutamatergic signaling in MDD in vivo.
This pilot study sought to test the hypothesis that ketamine administration in depressed patients produces a rapid, robust surge in glutamatergic compounds in medial PFC (mPFC), as observed in rodent studies.21, 22 1H MRS was used to dynamically measure the time-course of brain glutamatergic response to ketamine (via the combined resonance of glutamate + glutamine, or Glx) from baseline through 40 minutes of infusion to approximately 30 minutes after infusion in depressed DSM IV-defined MDD patients. An exploratory objective was to synchronously measure ketamine’s effect on brain γ-aminobutyric acid (GABA), reported to be low in severe MDD.23–25 GABAergic abnormalities associated with MDD include low GABA in cerebrospinal fluid26 and plasma27, 28 and are consistent with magnetic resonance spectroscopy (MRS) findings of low brain GABA levels.23, 24, 29–31
Materials and Methods
Patients
All subjects provided written informed consent as approved by the Institutional Review Board prior to participation. Eleven outpatients (eight female) with mean age of 38.8 ± 12.8 years participated and met DSM-IV criteria for major depressive episode (MDE) and MDD, scoring at least 16 on the 17-item Hamilton Depression Rating Scale (HDRS-17) (mean score= 20.7± 3.7). All participants were free of psychotropic medications for at least 14 days prior to scanning, off fluoxetine for 6 weeks and off serotonin depleting drugs for 3 months.
Patients were excluded for: lack of capacity; history of other major Axis I disorders; suicidal ideation with a plan or intent or attempted suicide within the preceding 6 months; current or past drug or alcohol dependence; prior use of ketamine; electroconvulsive therapy in the preceding three months; having a first-degree relative with a psychotic disorder (for subjects under age 33); any significant active physical illness; previous loss of consciousness for more than a few minutes that required medical evaluation; pregnancy or intent to conceive during study participation or having ferromagnetic implants or other magnetic resonance imaging (MRI) contraindications. Medical history, physical examination and standard blood tests (including urinalysis and toxicology) confirmed the absence of active physical illness, pregnancy and drug use. Prior treatment for depression was a requirement for inclusion; treatment resistance was not.
Study Design
Subjects fasted for approximately 8 hours prior to scanning. Baseline ratings were administered within 24-hours of scanning [24-item Hamilton Depression Rating Scale (HDRS-24), Beck Depression Inventory (BDI), Profile of Mood States (POMS), Young Mania Rating Scale (YMRS) and the Brief Psychiatric Rating Scale (BPRS)].
Prior to ketamine administration, patients were positioned in the MRI scanner. After structural MRI and baseline 1H MRS scans, 0.5 mg/kg of ketamine hydrochloride in saline was administered intravenously over approximately 40 minutes. Six 1H MRS data frames were acquired, each of ~13-minute duration: one pre-ketamine, four during the 40-minute ketamine infusion, and one after the ketamine infusion. The POMS was completed 80 and 110 minutes after initiation of the ketamine infusion. Blood samples were obtained from a second venous line at 90 minutes and 120 minutes post-ketamine for levels of ketamine, norketamine and dehydronorketamine.
Baseline psychiatric ratings were repeated 230 minutes post-ketamine infusion, excluding items not expected to change (eg: sleep). HDRS-24 was also administered 24 hours post-infusion. Remission was defined as ≥50% improvement from baseline and a total score of ≤10. Response was defined as ≥50% improvement.5 The HDRS-24 was the primary outcome measure, as in most other ketamine studies.5 The BPRS was administered at baseline and at 230 minutes post-infusion to monitor potential adverse effects of ketamine. The POMS was used to measure clinical state during the first 230 minutes post-infusion because it is better suited for short-term (hours) re-administration.32, 33
MRI and MRS Data Acquisition
Neuroimaging data were acquired on a General Electric Signa EXCITE 3.0T MR scanner using commercial 8-channel phased-array head coil. A three-plane localizer imaging series was obtained, followed by a volumetric T1 weighed spoiled gradient-recalled (SPGR) echo acquisition (TE=2.86ms, TR=7.12 ms, flip angle = 9°, field of view = 256×256 mm2, image matrix size = 256×256, slice thickness 1 mm; voxel size 1×1×1 mm3). Next, in vivo brain spectra of the GABA and combined resonance of glutamate and glutamine (Glx) were recorded from a 3.0×2.5×.2.5-cm3 mPFC voxel (Figure 1A, B) using the standard J-edited spin echo difference method.34, 35 A pair of frequency-selective inversion pulses was inserted into the standard point-resolved spectroscopy (PRESS) method and then applied to the GABA C-3 resonance at 1.9 ppm on alternate scans using TE/TR 68/1500ms. This resulted in two subspectra (Figure 1C, traces [a] and [b]) in which the GABA C-4 resonance at 3.03 ppm and Glx C-2 at 3.71 ppm were alternately inverted. Subtracting these two subspectra yielded a spectrum consisting of only the edited GABA C-4 and Glx C-2 resonances, with all overlapping resonances eliminated (Figure 1B). Data were acquired in 13-minute frames using 256 interleaved excitations (512 total) with the editing pulse alternatingly on or off. The resultant raw 8-channel phased-array coil data were combined into a single regular free-induction decay signal using the coil sensitivity factors derived from the unsuppressed water signal acquired with each receiver coil. The magnetic field homogeneity for all acquisitions was required to be less than ≤20 Hz, as assessed by the full width at half of the unsuppressed water resonance.
Figure 1.

(A) Axial and (B) sagittal localizer images showing the size and location of the mPFC voxel of interest. (C) Demonstration of in vivo human brain GABA and Glx detection by 1H MRS: (a) and (b), single-voxel subspectra acquired in 13.4 minutes with the editing pulse on and off and 256 (512 total) interleaved averages; spectrum (c), difference between spectra (a) and (b) showing the edited brain GABA and Glx resonances; spectrum (d), model fitting of spectrum (c) to obtain the GABA and Glx peak areas; spectrum (e), individual components of the fits; spectrum (f), residual of the difference between spectra (c) and (d). Abbreviations: GABA, γ-aminobutyric acid; Glx, glutamate + glutamine; NAA, N-acetyl-aspartate; tCho, total choline; tCr, total creatine; MPFC, medial prefrontal cortex.
Areas under the Glx and GABA peaks, which are proportional to their concentrations, were obtained as illustrated in Figure 1C (traces [a-f]) by fitting each resonance to a Gauss-Lorentz (i.e., pseudo-Voigt) function in the frequency-domain using a Levenberg-Marquardt nonlinear least-squares minimization routine written in IDL (ITT EXELIS, McLean, VA). The levels of Glx and GABA in the edited spectra were then expressed as ratios of peak areas relative to the simultaneously acquired and similarly fitted unsuppressed voxel water signal (W)—a commonly used36, 37, 38–40 method with high test-retest reliability.41
Plasma Ketamine, Norketamine and Dehydronorketamine
Plasma ketamine, norketamine and dehydronorketamine were assayed by liquid chromatographic (LC) procedure with UV detection. Within-day coefficient of variation of ketamine, norketamine and dehydronorketamine did not exceed 12.8% (range 2000–5ng/mL), (n=12 for each of seven concentrations). Day-to-day variation of ketamine and norketamine quality controls at 1250, 250, and 50ng/mL did not exceed 4.3 and 3.4%, respectively (n=11 days). For dehydronorketamine, day-to-day variation at 500, 100 and 20 ng/mL did not exceed 8.8% (n=11 days). The minimum quantifiable limits were set at 10ng/mL for both ketamine and norketamine, and 5ng/mL for dehydronorketamine (such low levels were not seen in this study).
Statistical Analysis
Linear mixed-effects models tested for: (1) effect of time point on POMS score with time point as a (categorical) fixed effect and subject as a random effect; (2) ketamine’s effect on log-transformed Glx/W and GABA/W levels, with subject as a random effect and each 13-minute MRS frames as a fixed effect. Correlations were calculated using Pearson product moment. MRS outcome measures were log-transformed to ensure normality of distribution, sphericity and compound symmetry. A first-order autoregressive covariance structure was applied to account for time dependency within subjects.
Results
Effect of Ketamine on MDD Symptoms
Ten of 11 subjects met criteria for remission by 230 minutes post-infusion, 9/11 at 24 hrs post-infusion and 7/10 at three days. HDRS-24 and BDI scores declined dramatically 24 hours post-ketamine (F=52.5; df=1,10; p<0.0001 and F=21.7; df=1,10; p=0.0009, respectively) (Figure 2B–C).
Figure 2.
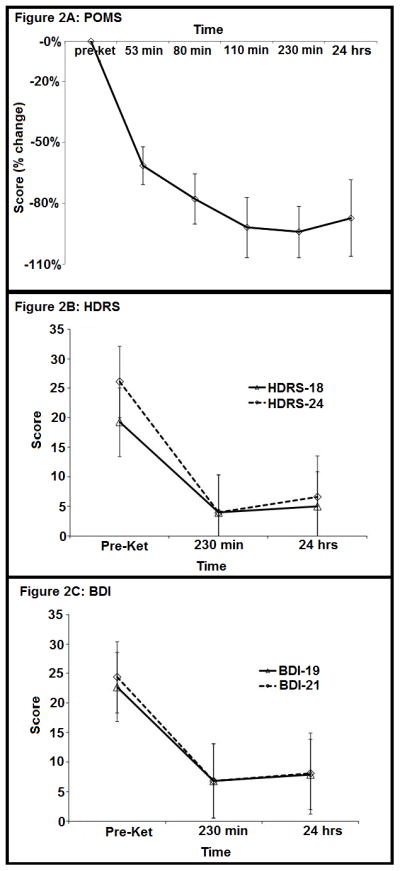
Figure 2A. Profile of Mood States (POMS) total score, 24 hours pre-ketamine infusion, 80, 110 and 230 minutes after ketamine infusion and 24 hours after ketamine infusion. Error bars denote standard error of the mean.
Figure 2B. 18 Item Hamilton Depression Rating Scale (HDRS-18) total score, 24 hours pre-ketamine infusion, 230 minutes after ketamine infusion and 24 hours after ketamine infusion. Error bars denote standard error of the mean.
Figure 2C. 19 Item Beck Depression Inventory (BDI) total score, 24 hours pre-ketamine infusion, 230 minutes after ketamine infusion and 24 hours after ketamine infusion. Error bars denote standard error of the mean.
POMS total score (Figure 2A) declined rapidly (F=18.0; df=4,35; p<0.0001), reached a nadir at 230 minutes, and remained low 24 hours later. Notably, POMS sub-score vigor gradually increased in contrast to total score and confusion decreased comparably to the total score (Table 3).
Table 3.
Change over time in BPRS and POMS sub-scores.
| Scale | Sub-Score | F-Value | DF | P-Value |
|---|---|---|---|---|
| POMS | Tension | 16.1 | 3, 38 | <0.0001 |
| Depression | 16.5 | 3, 38 | <0.0001 | |
| Anger | 9.5 | 3, 38 | 0.0001 | |
| Fatigue | 18.0 | 3, 38 | <0.0001 | |
| Confusion | 16.1 | 3, 38 | <0.0001 | |
| Vigor | 7.1 | 3, 38 | 0.0007 | |
| BPRS | Anxiety-Depression | 33.5 | 1, 10 | 0.0002 |
| Anergia | 9.8 | 1, 10 | 0.01 | |
| Thought Disturbance | p=1, all values are equal. | |||
| Activation | 1.9 | 1, 10 | 0.20 | |
| Hostile-Suspiciousness | 3.2 | 1, 10 | 0.10 | |
Abbreviations: POMS, Profile of Mood States; BPRS; Brief Psychiatric Rating Scale.
BPRS scores (Figure 3) declined from baseline to 230 minutes post-ketamine infusion (F= 26.2; df=1,10; p=.0004). There were decreases in BPRS sub-scores 42 anxiety-depression (F=33.5; df=1,10; p=.0002) and anergia (F=9.8; df=1,10; p=.01),. Psychotic symptom sub-scores remained stable or declined after infusion. No patient’s thought disturbance score changed from baseline after ketamine infusion. Subjects experienced mild or no adverse cognitive or dissociative effects (Table 3). There was no correlation between change in BPRS score and change in Glx or GABA levels.
Figure 3.
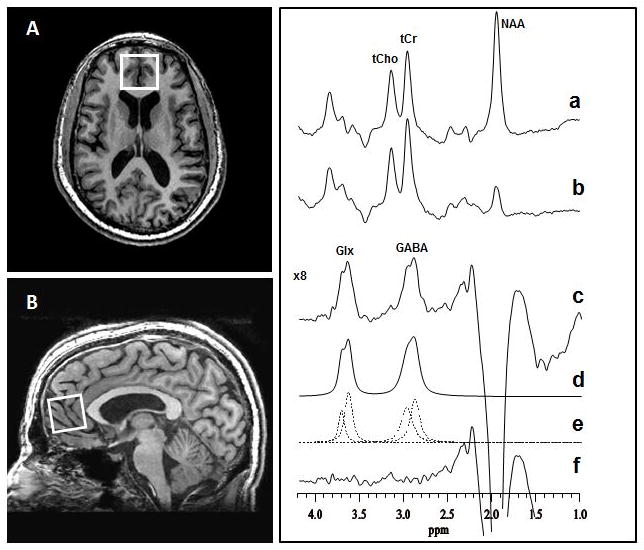
Figure 3A. Magnetic Resonance Spectroscopy Measurement of GABA/water and Glx/water concentrations in Medial Prefrontal Cortex in Major Depressive Disorder before (baseline frame), during (frame 1–4-) and after (frame 5–6) an intravenous ketamine infusion (40 minutes duration). Frame duration was 13:20 minutes. Asterisks denote statistically significant group increases in GABA and Glx concentrations relative to pre-ketamine baseline levels.
Abbreviations: GABA/W, mean of water-corrected γ-aminobutyric acid level; Glx/W, mean of water-corrected glutamate + glutamine level. Error bars denote standard deviation from the mean.
Figure 3B. Individual subjects’ Glx/W and GABA/W responses to ketamine as measured by the area under the curve. Abbreviations: AUC, area under the curve; GABA/W, water-corrected γ-aminobutyric acid level; Glx/W, sum of water-corrected glutamate + glutamine level.
Response of mPFC Glx and GABA to Ketamine
MRS data for three subjects were excluded from all analyses due to head motion, manifested as distorted peak phases and large residual water resonance in difference spectra, which degraded spectral quality. For the remaining subjects, there was a main effect of ketamine infusion on Glx/W (F=6.16; df=5,31; p=0.0004), that post-hoc analysis attributed to higher Glx/W in MRS frame 1 (p=0.037), frame 2 (p=0.001), and frame 3 (p=0.027) compared with the baseline frame (Figure 3A). For GABA/W there was a main effect of ketamine infusion (F=2.90; df=5,31; p=0.029) and post hoc analysis indicated higher GABA/W in MRS frame 1 (p=0.036) and frame 2 (p=0.031) compared with the baseline frame (Figure 3A).
The peak frame for each subject revealed a 38% ± 8% (mean ± SD) increase in Glx/W from baseline and a 38% ± 9% increase in GABA/W from baseline. The time points for these peak values, obtained by averaging the mid-time point of the frame of each peak, were 28 ± 8 and 26 ± 21 minutes from the start of infusion for Glx/W and GABA/W, respectively. When responses to ketamine were quantified as area under the curve (AUC), both Glx/W and GABA/W increases were statistically significant: Glx/W AUC was 0.013 ± 0.01 (SD) (one sample t-test: t = 2.8479, df = 7, p = 0.025); and GABA/W AUC was 0.016 ± 0.01 (t = 4.0841, df = 7, p = 0.005) (Figure 3B). The correlation between AUCs of Glx/W and GABA/W was 0.796 (p=0.018).
Vital Signs and Clinical/Imaging/Ketamine Level Correlations
Neither Glx/W nor GABA/W changes correlated with clinical response to ketamine. Of ketamine and its two active metabolites, norketamine and dehydronorketamine, measured at two time points (see Table 3), only norketamine concentration at 90 minutes correlated with clinical outcome (df= 6; r= −0.78; p= 0.023; uncorrected) expressed as a percent change in HDRS-24 scores. No effects were observed on blood pressure, heart rate, oxygen saturation, or conscious state during and after the ketamine infusion.
Discussion
This is the first study to report rapid and robust in vivo increases in both mPFC Glx and GABA in response to intravenous administration of a single subanesthetic dose of ketamine for treatment of MDD. A prior study failed to detect an effect of ketamine in depressed subjects.20 While differences in MRS methods cannot be ruled out as an explanation of this discrepancy, MRS data in the prior study were acquired after completion of the ketamine infusion, and comport with our study, which found that most of the Glx and GABA responses to be dissipated by the end of infusion. Consistent with our findings, two studies in healthy volunteers also reported increases in glutamine17 and glutamate19 levels, and one study did not.18 The infusion methods used in the studies with positive findings may have produced higher ketamine blood levels than those achieved in the negative study and caused a more robust glutamatergic response in healthy volunteers. This highlights the need for future studies of the glutamatergic response.
A rapid increase in Glx in response to ketamine is consistent with microdialysis rodent studies21,43 that found a 250% increase in extracellular glutamate levels in response to ketamine administration at 40 minutes compared to baseline, and using 13-C labeled glucose found that 13-C enrichment of Glu-C4 increased roughly 18% from baseline, significantly more than in saline-control rats. The relatively modest increase of de novo glutamate synthesis measured by incorporation of 13-C labeled glucose into the carbon backbone of glutamate belies the substantial increase in extracellular glutamate by microdialysis. These findings support a role for the glutamatergic system in the rapid antidepressant action of ketamine.
We also observed a parallel increase in total tissue GABA (as measured by 1H MRS) in MDD in response to ketamine in depressed subjects that is consistent with animal studies.22 The potential role of GABA in the antidepressant action of ketamine is an area for future research.
We also confirm previous reports of rapid remission of MDD in response to ketamine.44 POMS scores declined more than 50% improvement within one hour of initiating the ketamine infusion (Figure 2A). Remission rates observed in this open study are comparable with those in previous studies (e.g.5), or perhaps better because this study’s subjects were younger, had shorter duration of illness, fewer previous MDEs, and were less treatment-resistant.
Norketamine levels, which showed substantial variance at 90 minutes (in contrast to ketamine), correlated negatively with HDRS-24 scores 24 hours after ketamine infusion. Plasma ketamine and dehydronorketamine did not correlate with clinical outcome or with Glx/W or GABA/W changes. Since norketamine levels were relatively high, their correlation with antidepressant effect suggests that norketamine may be a long-lived and active metabolite due to slow conversion to dehydronorketamine, and thus may have a more pronounced role in blocking NMDA receptors. The absence of a correlation of ketamine level with clinical response or the Glx or GABA response may be due, in part, to the standardized dose given to all subjects, which minimized variance in ketamine blood levels, and to the generally robust clinical response exhibited by most subjects in the study. A dose-finding study, using a wider range of doses, may clarify how ketamine and its active metabolite levels relate to glutamate and GABA responses and to antidepressant action.
The nearly 40% increase in the concentration of both Glx and GABA may be explained by changes in brain glucose utilization. Following ketamine administration in patients with bipolar disorder, an increase in regional metabolic rate of glucose (rMRGlu) correlated with improvement in depressive symptoms in the right ventral striatum.45 The cerebral metabolic rate for glucose in human brain is approximately 0.4 μmol/min/g tissue and turnover of glutamate is approximately 0.8 μmol/min/g tissue.46 Virtually all the glucose that enters the brain is metabolized through glutamate since one molecule of glucose gives rise to two molecules of acetyl-CoA, which enter the tricarboxylic acid cycle (TCA) to become α-ketoglutarate and then glutamate. In most cells of the body, glutamate is in equilibrium with a-ketoglutarate (a-KG) as it is continuously reconverted and then metabolized through the TCA cycle. However, in glutamatergic neurons, the enzyme aspartate aminotransferase (which aminates a-KG to glutamate) has much higher activity than the enzyme a-KG dehydrogenase, which can lead to an accumulation of intracellular glutamate.47 In GABAergic neurons, this same process feeds GABA synthesis because glutamate is the precursor of GABA. The robust correlation between Glx and GABA increases supports the hypothesis that glucose utilization drives the increase in both neurotransmitters.
Preclinical studies indicate that ketamine’s antidepressant action may depend on activation of glutamatergic AMPA receptors8 and the downstream mTOR pathway.1 Our findings support the model1 positing that ketamine causes a rapid increase in cortical glutamate through an unknown mechanism that, in combination with blockade of NMDA receptors by ketamine, diverts glutamate signaling to AMPA receptors. AMPA activation leads to the downstream activation of the mTOR pathway, leading to increased BDNF release,48–51 dendritic protein synthesis,1 mushroom spine formation and associated downstream effects.52–54 Other NMDA receptor antagonists exhibit antidepressant effects,55–64 and monoaminergic antidepressants and modulators of metabotropic glutamatergic receptors with antidepressant effects also reduce NMDA signal transduction.54, 65–71 The glutamatergic system may also be a target for treatment of depression because it is abnormal in major depression.72, 73 A 1H-MRS study reported glutamate deficits in anterior cingulate cortex (ACC) in MDD.74 Conversely, higher glutamate in cerebrospinal fluid (CSF) has been reported and a cytotoxic role for glutamate transmission via NMDA receptors has been implicated in the loss of mature granule cells in dentate gyrus and glia in hippocampus and in amygdala.75
Our finding of a parallel GABA elevation following ketamine administration is a novel observation. Though the mechanism is uncertain, the synchronous surge in this inhibitory neurotransmitter could limit ketamine-mediated glutamate release and reduce excessive spread of glutamatergic excitation. CSF,26 plasma27, 28 and in vivo brain GABA levels23–25, 30 are reported in MDD during depression; but not when not depressed.78 GABA(B) receptor agonists and positive modulators have antidepressant-like effects in rodent depression models79, 80 and antidepressant treatment (SSRI or ECT) normalizes GABAergic deficits in MDD patients.81–84 Postmortem studies report fewer GABA neurons in MDD and bipolar disorder and a GABAergic deficit may be a part of the pathophysiology of major depressive episodes.75,85, 86 GABAergic deficits are found postmortem with decreased density of calbindin immunoreactive GABA neurons in MDD compared with controls.87 GABA(A) receptor deficits have also been demonstrated postmortem in the brains of MDD suicides.88 Olfactory bulbectomy and learned helplessness models in rodents have shown deficits in GABAergic function.76,77 GABAergic system involvement in the pathophysiology of MDD also may involve a complex interplay between GABA and Glu that is more abnormal in TRD depression.
This pilot proof of concept study has a small sample size, but all subjects were medication-free and the measured clinical effects of ketamine were robust. While overall neurochemical effect of ketamine was statistically significant, lack of effect on Glx or GABA in some patients (Figure 3B) calls for further investigation via larger randomized controlled trials. 1H MRS measures total tissue levels, including intracellular, synaptic, and vesicular levels, but not transmission or shifts from one compartment to another, which limits interpretation. The tissue concentration of GABA requires relatively large voxels for reliable quantification; consequently, associated partial volume effects could make measurement more difficult, but since this is a within-subject design, partial volume effects were stable throughout the infusion. The Glx peak consists of the combined resonances of glutamate and glutamine; however, we recently reported that the Glx measured by the J-editing technique contains mainly glutamate, and little or no glutamine.92 The contribution of macromolecules known to co-edit with GABA was not taken into account and is a potential confound.
Our findings require replication in a larger sample using a wider range of ketamine doses, which will generate a more complete dose response curve and more effectively reveal correlations, including determining if Glx or GABA responses are predictors of antidepressant response. Such a multi-dose study with a larger sample size would also allow for further analyses of covariates in antidepressant response to ketamine such as sex differences, as a preclinical study93 found gonadal hormones enhanced antidepressant-like effects of ketamine in female rats. The effect on Glx and GABA observed in this study is an early effect that may or may not be the initial step in antidepressant effect, though animal data with mTOR suggest that AMPA effects of glutamate initiate the antidepressant cascade. Lack of a relationship between a proximal drug effect and clinical response is common in the study of the action of psychotropic medications. For example, SSRIs require occupancy of at least 80% of transporter sites and MAOIs need to block at least 80% of MAO in order to work.94,95 Greater occupancy does not correlate with antidepressant response but there is little doubt that this initial pharmacological effect is needed for an antidepressant effect.
Conclusion
This pilot study found rapid and comparably robust increases in glutamatergic compounds and GABA in most MDD patients in response to ketamine antidepressant treatment of MDD, which supports the potential involvement of these amino acid neurotransmitters in the antidepressant action of ketamine. Further studies are needed to clarify the relationship between clinical response, glutamate and GABA levels and ketamine dose.
Table 1A.
Demographic and clinical characteristics of subjects with Major Depressive Disorder.
| (N=11) | Table 1A: Demographic and Clinical Characteristics | |||||||
|---|---|---|---|---|---|---|---|---|
| Gender | Ethnicity | Race | Responder Status | |||||
| Male | Female | Hispanic | Non-Hispanic | White | African American | At 230 minutes | At 24 hrs | |
| N | 3 | 8 | 0 | 11 | 10 | 1 | 10 | 9 |
| (Mean ± SD): | Age | Age at MDD onset (yrs) | Duration of Current MDE (yrs) | Duration of MDD (yrs) | Number of Previous MDEs* | |||
| 38.8 ± 12.8 | 22.1 ± 14.0 | 8.9 ± 10.7 | 16.6 ± 9.4 | 1.1 ± 0.9 | ||||
Abbreviations: MDD, Major Depressive Disorder; MDE, Major Depressive Episode.
2 patients were excluded from this analysis because we were unable to ascertain the number of previous MDEs.
Table 1B.
Clinical severity measures of current Major Depressive Episode.
| (N=11) | Table 1B: Clinical Severity Measures of Current MDE | ||
|---|---|---|---|
| (Mean ± SD): | Baseline | 230 mins | 24 hrs |
| HDRS-18 | 19.3 ± 5.8 | 4.0 ± 6.3 | 5.0 ± 5.9 |
| HDRS-24 | 26.1 ± 6.1 | 6.6 ± 6.9 | |
| BDI-19 | 22.7 ± 7.6 | 6.8 ± 5.8 | 7.9 ± 8.0 |
| BDI-21 | 24.4 ± 8.3 | 8.1 ± 8.0 | |
| BPRS | 33.1 ± 6.6 | 21.0 ± 4.5 | |
| POMS | 85.3 ± 42.2 | 14.6 ± 30.7 | 20.2 ± 38.8 |
Abbreviations: Baseline, 24 hours pre-ketamine infusion; 230 minutes, 230 minutes post-ketamine infusion; HDRS-17, 17 Item Hamilton Depression Rating Scale; HDRS-18, 18 Item Hamilton Depression Rating Scale; HDRS-24, 24 Item Hamilton Depression Rating Scale; BDI-21, 21 Item Beck Depression Inventory; BDI-18, 18 Item Beck Depression Inventory; POMS, Profile of Mood States; BPRS, Brief Psychiatric Rating Scale.
Table 2A.
Correlation analyses between change in Glx over water and GABA over water, frame 1 to frame 4, and clinical outcome (HDRS and BDI, pre-ketamine to Day 1; POMS, pre-ketamine to 230 minutes post-ketamine).
| Table 2A: Glx/W and GABA/W and Clinical Outcome | ||||||
|---|---|---|---|---|---|---|
| df = 6 | Change in HDRS-24 | Change in BDI-21 | Change in POMS | |||
| r | p | r | p | r | p | |
| Glx/W | −0.21 | 0.60 | −0.24 | 0.57 | 0.34 | 0.41 |
| GABA/W | −0.05 | 0.90 | −0.13 | 0.75 | 0.20 | 0.63 |
Abbreviations: GABA, γ-aminobutyric acid; Glx, glutamate + glutamine; HDRS-24, 24 Item Hamilton Depression Rating Scale; BDI-21, 21 Item Beck Depression Inventory; POMS, Profile of Mood States.
Table 2B.
Correlation analyses between ketamine and its metabolites norketamine and dehydronorketamine and clinical outcome.
| Table 2B: Ketamine and Its Metabolites and Clinical Outcome | ||||||
|---|---|---|---|---|---|---|
| df=6 | % Change in HDRS-24 | % Change in BDI-21 | % Change in POMS | |||
| r | p | r | p | r | p | |
| Ket 90 min | −0.17 | 0.68 | 0.24 | 0.56 | −0.19 | 0.68 |
| Ket 120 min | 0.01 | 0.98 | −0.02 | 0.97 | −0.42 | 0.34 |
| Norket 90 min | −0.78 | 0.023* | −0.09 | 0.83 | −0.06 | 0.90 |
| Norket 120 min | −0.66 | 0.07 | −0.36 | 0.38 | −0.35 | 0.44 |
| Deh 90 min | −0.10 | 0.81 | 0.07 | 0.87 | 0.09 | 0.85 |
| Deh 120 min | −0.02 | 0.97 | 0.02 | 0.97 | 0.02 | 0.96 |
Abbreviations: ket, ketamine; norket, norketamine; deh, dehydronorketamine; HDRS-24, 24 Item Hamilton Depression Rating Scale; BDI-21, 21 Item Beck Depression Inventory; POMS, Profile of Mood States.
Acknowledgments
This work was supported by a Brain and Behavior Research Foundation NARSAD Distinguished Investigator Award to Dr. Mann and NIMH grants R01 MH-075895 to Dr. Shungu and R01 MH-093637 to Dr. Milak.
Footnotes
Financial Disclosures
Dr. Milak, Ms. Proper, Ms. Mulhern, Ms. Parter, Dr. Ogden, Dr. Keilp, Ms. Mao, Mr. Cooper, Dr. Shungu, Dr. Rodriguez and Dr. Suckow reported no biomedical financial interests or potential conflicts of interest. Dr. Kegeles has received research grants from Pfizer and Amgen. Dr. Oquendo receives royalties for use of the Columbia Suicide Severity Rating Scale and received financial compensation from Pfizer for the safety evaluation of a clinical facility, unrelated to this study. She has received unrestricted educational grants and/or lecture fees from Astra-Zeneca, Bristol Myers Squibb, Eli Lilly, Janssen, Otsuko, Pfizer, Sanofi-Aventis, and Shire. Her family owns stock in Bristol Myers Squibb. Dr. Mann received prior unrelated grants from Novartis and GSK and receives royalties for commercial use of the C-SSRS from the Research Foundation for Mental Health.
References
- 1.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychological medicine. 2014:1–12. doi: 10.1017/S0033291714001603. [DOI] [PubMed] [Google Scholar]
- 4.Fond G, Loundou A, Rabu C, Macgregor A, Lancon C, Brittner M, et al. Ketamine administration in depressive disorders: a systematic review and meta-analysis. Psychopharmacology. 2014;231(18):3663–3676. doi: 10.1007/s00213-014-3664-5. [DOI] [PubMed] [Google Scholar]
- 5.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of general psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 6.Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biological psychiatry. 2013;74(4):250–256. doi: 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiroma PR, Johns B, Kuskowski M, Wels J, Thuras P, Albott CS, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. Journal of affective disorders. 2014;155:123–129. doi: 10.1016/j.jad.2013.10.036. [DOI] [PubMed] [Google Scholar]
- 8.Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biological psychiatry. 2008;63(4):349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 9.Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224(1):107–111. doi: 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 10.Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99(1):467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenz G, Avruch J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J Biol Chem. 2005;280(46):38121–38124. doi: 10.1074/jbc.C500363200. [DOI] [PubMed] [Google Scholar]
- 12.Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, et al. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A. 2003;100(24):14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cracco JB, Serrano P, Moskowitz SI, Bergold PJ, Sacktor TC. Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus. 2005;15(5):551–556. doi: 10.1002/hipo.20078. [DOI] [PubMed] [Google Scholar]
- 14.Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24(28):6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tischmeyer W, Schicknick H, Kraus M, Seidenbecher CI, Staak S, Scheich H, et al. Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur J Neurosci. 2003;18(4):942–950. doi: 10.1046/j.1460-9568.2003.02820.x. [DOI] [PubMed] [Google Scholar]
- 16.Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011 doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. The American journal of psychiatry. 2005;162(2):394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- 18.Taylor MJ, Tiangga ER, Mhuircheartaigh RN, Cowen PJ. Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. Journal of psychopharmacology. 2012;26(5):733–737. doi: 10.1177/0269881111405359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17(7):664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry research. 2011;191(2):122–127. doi: 10.1016/j.pscychresns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. The Journal of neuroscience. 1997;17(8):2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNair DM, Lorr M, Droppleman LF. Profile of Mood States. 1971. [Google Scholar]
- 23.Price RB, Shungu DC, Mao X, Nestadt P, Kelly C, Collins KA, et al. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: relationship to treatment resistance in major depressive disorder. Biological psychiatry. 2009;65(9):792–800. doi: 10.1016/j.biopsych.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Archives of general psychiatry. 2007;64(2):193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- 25.Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61(7):705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 26.Gold BI, Bowers MB, Jr, Roth RH, Sweeney DW. GABA levels in CSF of patients with psychiatric disorders. Am J Psychiatry. 1980;137(3):362–364. doi: 10.1176/ajp.137.3.362. [DOI] [PubMed] [Google Scholar]
- 27.Sanacora G. Cortical inhibition, gamma-aminobutyric acid, and major depression: there is plenty of smoke but is there fire? Biological psychiatry. 2010;67(5):397–398. doi: 10.1016/j.biopsych.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Sanacora G, Saricicek A. GABAergic contributions to the pathophysiology of depression and the mechanism of antidepressant action. CNS Neurol Disord Drug Targets. 2007;6(2):127–140. doi: 10.2174/187152707780363294. [DOI] [PubMed] [Google Scholar]
- 29.Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Archives of general psychiatry. 2004;61(7):705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 30.Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Archives of general psychiatry. 1999;56(11):1043–1047. doi: 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- 31.Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biological psychiatry. 2010;67(5):458–464. doi: 10.1016/j.biopsych.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 32.Biersack HJ. Letter to the Editor. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2012 [Google Scholar]
- 33.Graham MM, Menda Y. Letter to the Editor. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2012 [Google Scholar]
- 34.Rothman DL, Petroff OA, Behar KL, Mattson RH. Localized 1H NMR measurements of gamma-aminobutyric acid in human brain in vivo. Proc Natl Acad Sci U S A. 1993;90(12):5662–5666. doi: 10.1073/pnas.90.12.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sailasuta N, LeRoux P, Hurd R, Wang P, Sachs N, Ketter T. Detection of cerebral gamma-aminobutyric acid (GABA) in bipolar disorder patients and healthy volunteers at 3 T. Proc Intl Soc Magn Reson Med. 2001;9:1011. [Google Scholar]
- 36.Derom ML, Martinez-Gonzalez MA, Sayon-Orea Mdel C, Bes-Rastrollo M, Beunza JJ, Sanchez-Villegas A. Magnesium intake is not related to depression risk in Spanish university graduates. The Journal of nutrition. 2012;142(6):1053–1059. doi: 10.3945/jn.111.155572. [DOI] [PubMed] [Google Scholar]
- 37.Derom ML, Sayon-Orea C, Martinez-Ortega JM, Martinez-Gonzalez MA. Magnesium and depression: a systematic review. Nutritional neuroscience. 2013;16(5):191–206. doi: 10.1179/1476830512Y.0000000044. [DOI] [PubMed] [Google Scholar]
- 38.Wilson K, Brakoulias V. Magnesium intake and depression. The Australian and New Zealand journal of psychiatry. 2009;43(6):580. [PubMed] [Google Scholar]
- 39.Douglas BG, Dagirmanjian R. The effects of magnesium deficiency of ketamine sleeping times in the rat. British journal of anaesthesia. 1975;47(3):336–340. doi: 10.1093/bja/47.3.336. [DOI] [PubMed] [Google Scholar]
- 40.Murck H. Ketamine, magnesium and major depression--from pharmacology to pathophysiology and back. Journal of psychiatric research. 2013;47(7):955–965. doi: 10.1016/j.jpsychires.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 41.Kegeles LS, Mao X, Dyke J, Gonzales R, Soones T, Shungu DC. Test-retest reliability of dorsolateral prefrontal cortical GABA measurement using an 8-channel phased-array head coil with the J-editing technique at 3T. Proc Intl Soc Mag Reson Med. 2006;14:489. [Google Scholar]
- 42.Guy W National Institute of Mental Health (U.S.). Psychopharmacology Research Branch., Early Clinical Drug Evaluation Program. ECDEU assessment manual for psychopharmacology. U. S. Dept. of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; Rockville, Md: 1976. p. 603. Rev. edn. [Google Scholar]
- 43.Chowdhury GM, Behar KL, Cho W, Thomas MA, Rothman DL, Sanacora G. (1)H-[(1)(3)C]-nuclear magnetic resonance spectroscopy measures of ketamine’s effect on amino acid neurotransmitter metabolism. Biological psychiatry. 2012;71(11):1022–1025. doi: 10.1016/j.biopsych.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lapidus KAB, Mathew SJ. Ketamine in treatment-resistant depression. In: Mann JJ, Roose SP, McGrath PJ, editors. Clinical handbook for the management of mood disorders. 1. Cambridge University Press; Cambridge: 2013. pp. 345–357. [Google Scholar]
- 45.Nugent AC, Diazgranados N, Carlson PJ, Ibrahim L, Luckenbaugh DA, Brutsche N, et al. Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar disorders. 2014;16(2):119–128. doi: 10.1111/bdi.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith DH, Okiyama K, Gennarelli TA, McIntosh TK. Magnesium and ketamine attenuate cognitive dysfunction following experimental brain injury. Neuroscience letters. 1993;157(2):211–214. doi: 10.1016/0304-3940(93)90739-8. [DOI] [PubMed] [Google Scholar]
- 47.Brady ST, Siegel GJ, Albers RW, Price DL, Benjamins J. Basic neurochemistry: principles of molecular, cellular, and medical neurobiology. 8. xxiv. Elsevier/Academic Press; Amsterdam; Boston: 2012. p. 1096. [Google Scholar]
- 48.Bernard J, Ohayon M, Massicotte G. Modulation of the AMPA receptor by phospholipase A2: effect of the antidepressant trimipramine. Psychiatry research. 1994;51(2):107–114. doi: 10.1016/0165-1781(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 49.Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. The Journal of neuroscience. 2000;20(1):8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Legutko B, Li X, Skolnick P. Regulation of BDNF expression in primary neuron culture by LY392098, a novel AMPA receptor potentiator. Neuropharmacology. 2001;40(8):1019–1027. doi: 10.1016/s0028-3908(01)00006-5. [DOI] [PubMed] [Google Scholar]
- 51.Mackowiak M, O’Neill MJ, Hicks CA, Bleakman D, Skolnick P. An AMPA receptor potentiator modulates hippocampal expression of BDNF: an in vivo study. Neuropharmacology. 2002;43(1):1–10. doi: 10.1016/s0028-3908(02)00066-7. [DOI] [PubMed] [Google Scholar]
- 52.Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098) Neuropharmacology. 2001;40(8):1028–1033. doi: 10.1016/s0028-3908(00)00194-5. [DOI] [PubMed] [Google Scholar]
- 53.Bai F, Bergeron M, Nelson DL. Chronic AMPA receptor potentiator (LY451646) treatment increases cell proliferation in adult rat hippocampus. Neuropharmacology. 2003;44(8):1013–1021. doi: 10.1016/s0028-3908(03)00104-7. [DOI] [PubMed] [Google Scholar]
- 54.Skolnick P. Antidepressants for the new millennium. European journal of pharmacology. 1999;375(1–3):31–40. doi: 10.1016/s0014-2999(99)00330-1. [DOI] [PubMed] [Google Scholar]
- 55.Kroczka B, Branski P, Palucha A, Pilc A, Nowak G. Antidepressant-like properties of zinc in rodent forced swim test. Brain Res Bull. 2001;55(2):297–300. doi: 10.1016/s0361-9230(01)00473-7. [DOI] [PubMed] [Google Scholar]
- 56.Layer RT, Popik P, Olds T, Skolnick P. Antidepressant-like actions of the polyamine site NMDA antagonist, eliprodil (SL-82. 0715) Pharmacol Biochem Behav. 1995;52(3):621–627. doi: 10.1016/0091-3057(95)00155-p. [DOI] [PubMed] [Google Scholar]
- 57.Papp M, Moryl E. Antidepressant-like effects of 1-aminocyclopropanecarboxylic acid and D-cycloserine in an animal model of depression. European journal of pharmacology. 1996;316(2–3):145–151. doi: 10.1016/s0014-2999(96)00675-9. [DOI] [PubMed] [Google Scholar]
- 58.Przegalinski E, Tatarczynska E, Deren-Wesolek A, Chojnacka-Wojcik E. Antidepressant-like effects of a partial agonist at strychnine-insensitive glycine receptors and a competitive NMDA receptor antagonist. Neuropharmacology. 1997;36(1):31–37. doi: 10.1016/s0028-3908(96)00157-8. [DOI] [PubMed] [Google Scholar]
- 59.Ossowska G, Klenk-Majewska B, Szymczyk G. The effect of NMDA antagonists on footshock-induced fighting behavior in chronically stressed rats. J Physiol Pharmacol. 1997;48(1):127–135. [PubMed] [Google Scholar]
- 60.Panconi E, Roux J, Altenbaumer M, Hampe S, Porsolt RD. MK-801 and enantiomers: potential antidepressants or false positives in classical screening models? Pharmacol Biochem Behav. 1993;46(1):15–20. doi: 10.1016/0091-3057(93)90310-p. [DOI] [PubMed] [Google Scholar]
- 61.Maj J, Rogoz Z, Skuza G, Sowinska H. The effect of CGP 37849 and CGP 39551, competitive NMDA receptor antagonists, in the forced swimming test. Polish journal of pharmacology and pharmacy. 1992;44(4):337–346. [PubMed] [Google Scholar]
- 62.Maj J, Rogoz Z, Skuza G, Sowinska H. Effects of MK-801 and antidepressant drugs in the forced swimming test in rats. Eur Neuropsychopharmacol. 1992;2(1):37–41. doi: 10.1016/0924-977x(92)90034-6. [DOI] [PubMed] [Google Scholar]
- 63.Papp M, Moryl E. Antidepressant activity of non-competitive and competitive NMDA receptor antagonists in a chronic mild stress model of depression. European journal of pharmacology. 1994;263(1–2):1–7. doi: 10.1016/0014-2999(94)90516-9. [DOI] [PubMed] [Google Scholar]
- 64.Papp M, Moryl E, Willner P. Pharmacological validation of the chronic mild stress model of depression. European journal of pharmacology. 1996;296(2):129–136. doi: 10.1016/0014-2999(95)00697-4. [DOI] [PubMed] [Google Scholar]
- 65.Nowak G, Legutko B, Skolnick P, Popik P. Adaptation of cortical NMDA receptors by chronic treatment with specific serotonin reuptake inhibitors. European journal of pharmacology. 1998;342(2–3):367–370. doi: 10.1016/s0014-2999(97)01589-6. [DOI] [PubMed] [Google Scholar]
- 66.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. The Journal of neuroscience. 1995;15(11):7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brandoli C, Sanna A, De Bernardi MA, Follesa P, Brooker G, Mocchetti I. Brain-derived neurotrophic factor and basic fibroblast growth factor downregulate NMDA receptor function in cerebellar granule cells. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18(19):7953–7961. doi: 10.1523/JNEUROSCI.18-19-07953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palucha A, Branski P, Szewczyk B, Wieronska JM, Klak K, Pilc A. Potential antidepressant-like effect of MTEP, a potent and highly selective mGluR5 antagonist. Pharmacol Biochem Behav. 2005;81(4):901–906. doi: 10.1016/j.pbb.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 69.Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20(21):7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zahorodna A, Bijak M. An antidepressant-induced decrease in the responsiveness of hippocampal neurons to group I metabotropic glutamate receptor activation. European journal of pharmacology. 1999;386(2–3):173–179. doi: 10.1016/s0014-2999(99)00757-8. [DOI] [PubMed] [Google Scholar]
- 71.Pilc A, Branski P, Palucha A, Tokarski K, Bijak M. Antidepressant treatment influences group I of glutamate metabotropic receptors in slices from hippocampal CA1 region. European journal of pharmacology. 1998;349(1):83–87. doi: 10.1016/s0014-2999(98)00169-1. [DOI] [PubMed] [Google Scholar]
- 72.Zarate CA, Quiroz J, Payne J, Manji HK. Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacology bulletin. 2002;36(4):35–83. [PubMed] [Google Scholar]
- 73.Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Molecular psychiatry. 2002;7 (Suppl 1):S71–80. doi: 10.1038/sj.mp.4001021. [DOI] [PubMed] [Google Scholar]
- 74.Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biological psychiatry. 2000;47(4):305–313. doi: 10.1016/s0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- 75.Boldrini M, Santiago AN, Hen R, Dwork AJ, Rosoklija GB, Tamir H, et al. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38(6):1068–1077. doi: 10.1038/npp.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petty F. GABA and mood disorders: a brief review and hypothesis. Journal of affective disorders. 1995;34(4):275–281. doi: 10.1016/0165-0327(95)00025-i. [DOI] [PubMed] [Google Scholar]
- 77.Lloyd KG, Morselli PL, Bartholini G. GABA and affective disorders. Medical biology. 1987;65(2–3):159–165. [PubMed] [Google Scholar]
- 78.Hasler G, Neumeister A, van der Veen JW, Tumonis T, Bain EE, Shen J, et al. Normal prefrontal gamma-aminobutyric acid levels in remitted depressed subjects determined by proton magnetic resonance spectroscopy. Biological psychiatry. 2005;58(12):969–973. doi: 10.1016/j.biopsych.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 79.Cryan JF, Kaupmann K. Don’t worry ‘B’ happy!: a role for GABA(B) receptors in anxiety and depression. Trends Pharmacol Sci. 2005;26(1):36–43. doi: 10.1016/j.tips.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 80.Pilc A, Nowak G. GABAergic hypotheses of anxiety and depression: focus on GABA-B receptors. Drugs Today (Barc) 2005;41(11):755–766. doi: 10.1358/dot.2005.41.11.904728. [DOI] [PubMed] [Google Scholar]
- 81.Streeter CC, Hennen J, Ke Y, Jensen JE, Sarid-Segal O, Nassar LE, et al. Prefrontal GABA levels in cocaine-dependent subjects increase with pramipexole and venlafaxine treatment. Psychopharmacology. 2005;182(4):516–526. doi: 10.1007/s00213-005-0121-5. [DOI] [PubMed] [Google Scholar]
- 82.Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159(4):663–665. doi: 10.1176/appi.ajp.159.4.663. [DOI] [PubMed] [Google Scholar]
- 83.Bhagwagar Z, Wylezinska M, Taylor M, Jezzard P, Matthews PM, Cowen PJ. Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. Am J Psychiatry. 2004;161(2):368–370. doi: 10.1176/appi.ajp.161.2.368. [DOI] [PubMed] [Google Scholar]
- 84.Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160(3):577–579. doi: 10.1176/appi.ajp.160.3.577. [DOI] [PubMed] [Google Scholar]
- 85.Underwood MD, Kassir SA, Bakalian MJ, Galfalvy H, Mann JJ, Arango V. Neuron density and serotonin receptor binding in prefrontal cortex in suicide. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. 2012;15(4):435–447. doi: 10.1017/S1461145711000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Benes FM, Majocha R, Bird ED, Marotta CA. Increased vertical axon numbers in cingulate cortex of schizophrenics. Archives of general psychiatry. 1987;44(11):1017–1021. doi: 10.1001/archpsyc.1987.01800230097015. [DOI] [PubMed] [Google Scholar]
- 87.Maciag D, Hughes J, O’Dwyer G, Pride Y, Stockmeier CA, Sanacora G, et al. Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biological psychiatry. 2010;67(5):465–470. doi: 10.1016/j.biopsych.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One. 2009;4(8):e6585. doi: 10.1371/journal.pone.0006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Unschuld PG, Ising M, Specht M, Erhardt A, Ripke S, Heck A, et al. Polymorphisms in the GAD2 gene-region are associated with susceptibility for unipolar depression and with a risk factor for anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(8):1100–1109. doi: 10.1002/ajmg.b.30938. [DOI] [PubMed] [Google Scholar]
- 90.Hettema JM, An SS, Neale MC, Bukszar J, van den Oord EJ, Kendler KS, et al. Association between glutamic acid decarboxylase genes and anxiety disorders, major depression, and neuroticism. Mol Psychiatry. 2006;11(8):752–762. doi: 10.1038/sj.mp.4001845. [DOI] [PubMed] [Google Scholar]
- 91.Alcaro A, Panksepp J, Witczak J, Hayes DJ, Northoff G. Is subcortical-cortical midline activity in depression mediated by glutamate and GABA? A cross-species translational approach. Neurosci Biobehav Rev. 2010;34(4):592–605. doi: 10.1016/j.neubiorev.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 92.Shungu DC, Mao X, Gu M, Milak MS, Weiduscha N, Mayer D, et al. ‘Glx’ measured by J-editing/MEGA-PRESS is primarily ‘pure’ glutamate…or is it? Proc Intl Soc Magn Reson Med. 2013;21:1. [Google Scholar]
- 93.Carrier N, Kabbaj M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology. 2013;70:27–34. doi: 10.1016/j.neuropharm.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 94.Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry. 2004;161(5):826–835. doi: 10.1176/appi.ajp.161.5.826. [DOI] [PubMed] [Google Scholar]
- 95.Nair NP, Ahmed SK, Kin NM. Biochemistry and pharmacology of reversible inhibitors of MAO-A agents: focus on moclobemide. Journal of psychiatry & neuroscience: JPN. 1993;18(5):214–225. [PMC free article] [PubMed] [Google Scholar]