

Abstract
The protozoan parasite Toxoplasma gondii develops within a parasitophorous vacuole (PV) in mammalian cells, where it scavenges cholesterol. When cholesterol is present in excess in its environment, the parasite expulses this lipid into the PV or esterifies it for storage in lipid bodies. Here, we characterized a unique T. gondii homologue of mammalian lecithin:cholesterol acyltransferase (LCAT), a key enzyme that produces cholesteryl esters via transfer of acyl groups from phospholipids to the 3-OH of free cholesterol, leading to the removal of excess cholesterol from tissues. TgLCAT contains a motif characteristic of serine lipases “AHSLG” and the catalytic triad consisting of serine, aspartate, and histidine (SDH) from LCAT enzymes. TgLCAT is secreted by the parasite, but unlike other LCAT enzymes it is cleaved into two proteolytic fragments that share the residues of the catalytic triad and need to be reassembled to reconstitute enzymatic activity. TgLCAT uses phosphatidylcholine as substrate to form lysophosphatidylcholine that has the potential to disrupt membranes. The released fatty acid is transferred to cholesterol, but with a lower transesterification activity than mammalian LCAT. TgLCAT is stored in a subpopulation of dense granule secretory organelles, and following secretion, it localizes to the PV and parasite plasma membrane. LCAT-null parasites have impaired growth in vitro, reduced virulence in animals, and exhibit delays in egress from host cells. Parasites overexpressing LCAT show increased virulence and faster egress. These observations demonstrate that TgLCAT influences the outcome of an infection, presumably by facilitating replication and egress depending on the developmental stage of the parasite.
Keywords: cell surface enzyme, host-pathogen interaction, parasitology, Phospholipase A, Toxoplasma gondii
Introduction
The phospholipase A2 (PLA2)2 family of serine lipases comprises more than 30 enzymes in mammals. The PLA2 members hydrolyze the sn-2 ester of phospholipids, yielding lysophospholipid and releasing a fatty acid (1). Approximately one-third of the PLA2 family members are secreted from cells and display various localizations post-secretion, reflecting their specialized biological functions (2). Among the secretory PLA2, lecithin:cholesterol acyltransferase (LCAT; EC 2.3.1.43) is characterized by dual activity, PLA2 and acyltransferase. In mammals, this enzyme catalyzes the transacylation of the sn-2 fatty acid liberated from various phospholipids (e.g. phosphatidylcholine or phosphatidylethanolamine) to the 3-β-hydroxyl group on the A-ring of cholesterol, thereby forming cholesteryl esters (3–5). The primary sequence of LCAT is well conserved between mammalian species (6). A structural model for LCAT predicts the conformation of a catalytic triad formed by Ser-Asp-His residues involved in the phospholipase reaction (7). Mammalian LCATs are primarily expressed in the liver and secreted to the plasma where they circulate in association with HDL (8). These enzymes are components of the reverse cholesterol transport pathway by which cholesterol from peripheral cells is delivered to the liver for excretion (9). LCAT deficiency syndromes (e.g. familial LCAT deficiency or fish-eye disease) result in low plasma concentrations of HDL and reduced plasma cholesteryl esters, which leads to cellular dysfunctions due to alterations in cell and membrane lipid composition (10).
Intriguingly, some organisms lacking a reverse cholesterol transport pathway also possess an LCAT homologue, suggesting functions for this enzyme other than cholesterol clearance. For example, Saccharomyces cerevisiae expresses a gene named LRO1 (LCAT-related open reading frame) that codes for a protein whose predicted sequence harbors the conserved catalytic triad SDH and shares 27% overall identity with human LCAT. In contrast to mammalian LCAT that esterifies cholesterol, yeast LRO1 mediates the esterification of diacylglycerol using phosphatidylcholine as the acyl donor (11, 12); LRO1 has thus been renamed phospholipid:diacylglycerol acyltransferase. In plants, Arabidopsis contains one gene product homologue to human LCAT and five genes with similarities to yeast phospholipid:diacylglycerol acyltransferase (13). The function of the human LCAT homologue in plants has not been studied.
The protozoan parasite Toxoplasma gondii multiplies in a parasitophorous vacuole (PV) within the cytoplasm of mammalian cells. T. gondii is a member of the phylum Apicomplexa, which includes several human and animal pathogens, e.g. the causative agents of malaria and cryptosporidiosis. Approximately 30% of the United States population is infected with T. gondii, a leading opportunistic parasite in immunosuppressive conditions (14, 15). Previous studies demonstrated that T. gondii has an unusual lipid metabolism, and interference with lipid transport pathways, e.g. for phospholipid, cholesterol, or sphingolipids, has been proven to be detrimental for the parasite (16, 17). We previously characterized in Toxoplasma two acyl-CoA:cholesterol acyltransferase enzymes that are responsible for cholesterol esterification and storage in lipid bodies (18, 19) and four ATP-binding cassette G family transporters that promote cholesterol and phospholipid efflux (20), reflecting the importance of the regulation and exportation of lipids for the parasite.
In this study, we identified in the Toxoplasma genome database (www.ToxoDB.org) a single gene product that contains the conserved motifs characteristic of PLA2 serine lipases and that is most similar to mammalian LCAT. To gain a wider perspective into the biological significance of conserved LCAT proteins during evolution and to add information to lipid regulatory mechanisms in T. gondii, we examined the activity and function of the T. gondii LCAT homologue (herein named TgLCAT) during the developmental stages of the parasite. Our results demonstrate that TgLCAT is secreted by T. gondii and has a dual PLA2 and cholesteryl esterase activity. Compared with wild-type parasites, T. gondii lacking TgLCAT has reduced replication rate, impaired egress from host cells, and decreased virulence, whereas Toxoplasma overexpressing TgLCAT escapes faster from cells and is more virulent, suggesting a contribution of TgLCAT to the parasite intracellular development and pathogenicity.
Experimental Procedures
Chemicals and Antibodies
All chemicals were obtained from either Sigma or Fisher unless indicated otherwise. Solvents and standards for chromatography were of the highest analytical grade (Avanti Polar Lipids, Alabaster, AL). Silica gel 60 TLC plates were from EM Science (Gibbstown, NJ). Radiolabeled reagents included 1-palmitoyl-2[2-palmitoyl-9,10-3H]-sn-glycero-3-phosphocholine ([3H]DPPC), [4-14C]cholesterol, and [5,6-3H]uracil purchased from PerkinElmer Life Sciences. Primary antibodies were from the following: rat anti-TgLCAT (this study); mouse anti-SAG1, rabbit anti-GRA6, and mouse anti-GRA4 provided by J. F. Dubremetz (University of Montpellier, France); rabbit anti-GRA7 (21); mouse α-tubulin, and mouse, or rat anti-HA (Covance, Berkeley, CA). Secondary antibodies used for immunofluorescence were conjugated to Alexa488, Alexa594, or Alexa350 (Invitrogen).
Mammalian Cell Line, Culture Conditions, and Parasite Propagation
Primary human foreskin fibroblasts (HFF; ATCC CRL-1635) were used in this study and cultivated in Dulbecco's modified Eagle's medium supplemented with 10% FCS, 2 mm glutamine, and penicillin/streptomycin (100 units/ml per 100 μg/ml). The strains of T. gondii (tachyzoite forms) used throughout this study were propagated in vitro by serial passages in monolayers of HFF (22).
LCAT Sequence Analyses
Nucleotide and amino acid sequences were searched against the T. gondii database, EuPathDB, and the NCBI database using the BLAST algorithm (23). Multiple sequence alignment was created using ClustalW, and the resulting similarities were then visualized by subjecting the alignment to Boxshade. Percent identity and similarity were calculated using standard tools for sequence analysis from NCBI (ncbi.nlm.nih.gov). I-TASSER was used for ab initio structural prediction of the LCAT inserted element queried with TgLCAT amino acids 434–606. The highest scoring models encompassed amino acids 465–605. Phyre2 was used for template-dependent modeling of TgLCAT queried with amino acids 40–763, allowing the program to identify the best candidate template for modeling. Human lysosomal phospholipase A2 (LPLA2; Protein Data Bank code 4X91) was the highest scoring template upon which the structural model was built. Models were visualized with PyMOL. For the phylogenetic analysis, LCAT orthologues were identified via BLAST search using the TGME49_272420 sequence from the EuPathDB. Representative eukaryotic sequences were chosen from major groups via BLAST of the non-redundant protein database (blast.ncbi.nlm.nih.gov). To assess the phylogenetic relationship between the homologues, a multiple sequence alignment of LCAT proteins was performed using ClustalX, from which a bootstrap neighbor-joining phylogenetic tree using p-distance method and 1,000 bootstrap support were generated (MEGA version 6.0 software). A schematic tree was predicted by neighbor-joining using the LCAT sequences from Hammondia hammondi (HHA_272420), Neospora caninum (NCLIV_034880), Sarcocystis neurona (SN3_00202440), Eimeria tenella (ETH_00005915), Vitrella brassicaformis (Vbra_21178), Gregarina niphandrodes (GNI_089950), Plasmodium berghei (PBANKA_112810), Plasmodium falciparum (PF3D7_0629300), Plasmodium knowlesi (PKH_111960), Plasmodium vivax (PVX_114565), Trichuris trichiura (GI:669226237), Arabidopsis thaliana (GI:30689946), Xenopus laevis (GI:148229443), Alligator mississipiensis (GI:944443680), Homo sapiens (AAB34898.1), and Mus musculus (AAL11035). Yeast LRO1 was not included in the unrooted analyses because the bootstrap value was below 40%.
Rapid Amplification of cDNA Ends of lcat
The FirstChoice RLM-RACE kit from Ambion (Austin, TX) was used to perform 5′-rapid amplification of cDNA ends (5′-RACE) and 3′-RACE. Total RNA was first purified from freshly harvested tachyzoites (RNeasy, Qiagen). Briefly, total RNA from the RH strain of T. gondii was treated with calf intestinal phosphatase and subsequently with tobacco acid pyrophosphatase. An RNA adapter oligonucleotide (supplied with the kit) was ligated to the decapped RNA population using T4 RNA ligase. The ligated mRNA was then reverse-transcribed using Moloney murine leukemia virus reverse transcriptase. For the 5′-RLM-RACE, the obtained cDNA was subjected to two rounds of nested PCR. The first PCR was performed using a 5′-RACE outer primer (5′-GCTGATGGCGATGAATGAACACTG-3′) provided with the kit and a 5′-RACE gene-specific outer primer (5′-GTTCTTCTGCCTTCGCCAGTTCTT-3′). The second nested PCR was performed using 5′-RACE inner primer (5′-CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG-3′) and 5′-RACE gene-specific inner primer (5′-CCGTGTATCGGTCCTCGATGTCTGTCTTGA-3′). For the 3′-RLM-RACE also two nested PCRs were performed using 3′-RACE outer primer (5′-GCGAGCACAGAATTAATACGACT-3′) and 3′-RACE outer gene-specific primer (5′-CTCACCCTATTCGCATCTTCGATT-3′) for the first PCR and 3′-RACE inner primer (5′-CGCGGATCCGAATTAATACGACTCACTATAGG-3′) and 3′-RACE gene-specific inner primer (5′-CTGATGCTAACCCGTTCTTGTCCTCCCT-3′) for the second PCR). The thermal cycling conditions were as follows: initial denaturation at 94 °C for 3 min; 35 cycles of 94 °C for 30 s, 62 °C for 30 s, and 72 °C for 1 min 30 s; and final elongation of 7 min at 72 °C. PCR products of lcat were analyzed using agarose gel electrophoresis and cloned into pCR4-TOPO vector for sequencing.
Cloning of Full-length cDNA Encoding TgLCAT and Stable Transfection of TgLCAT-HA or TgLCAT-YFP in Toxoplasma
To engineer transgenic RH parasites expressing YFP or HA in fusion with TgLCAT, the 2,292-kb ORF of TgLCAT plus 463 nucleotides upstream of the initiation codon were amplified from a T. gondii cDNA library3 using primers 5′-GATCAGATCTGGAGCTCATCCAGCGTTTACTAAC-3′ (forward, BglII site underlined) and 5′-GATCCCTAGGCGTGCTGTCTGCCATAATC-3′ (reverse, AvrII site underlined) and cloned into plasmids tubYFP or tubHA provided by D.S. Roos (24), which harbor a sagCATsag cassette. Extracellular parasites (107) were transfected with 50 μg of the plasmid pTub-TgLCAT-YFP or pTub-TgLCAT-HA, selected with chloramphenicol, and cloned by limiting dilution as described previously (25).
Recombinant Peptide Expression in E. coli and Affinity Purification
To generate anti-TgLCAT antibodies, the region of TgLCAT encompassing nucleotides 311–606 was PCR-amplified using the primers 5′-CAGGCATGCGAAAATGTTCCCGCCTTTCCCTCGCGTGACCCTTCATT-3′ and 5′-ATAAAGCTTACGAAAGGGGTTGGGATAACTAATTGGGGATTTGCCG-3′ and cloned into the SphI and HindIII sites of the pQE-30 vector (Qiagen) to generate N-terminal His6-tagged fusion protein. The recombinant peptide TgLCAT(106–202) was expressed in Escherichia coli M15 strain, purified on Ni2+-nitrilotriacetic acid resin according to the Qiagen protocol, and injected into a rat to produce polyclonal anti-LCAT antibodies (Covance). Before use, the sera containing antibodies were affinity-purified against the recombinant TgLCAT(106–202) according to the protocol by AminoLink Kit (Pierce).
Immunoblot Analysis of Parasites
For immunodetection, RH and transgenic parasites were lysed by suspension in SDS gel-loading buffer (50 mm Tris-HCl, 50 mm 2-mercaptoethanol, 2% SDS, 0.1% bromphenol blue, 10% glycerol, pH 6.8) followed by boiling in a water bath. The samples were subjected to SDS-PAGE, and the proteins were then electrophoretically transferred to a membrane (Immobilon Transfer Membranes, Millipore, Bedford, MA). The membrane was immersed in blocking buffer (PBST containing 3% skim milk) for 60 min, and then incubated with antibodies against TgLCAT(1:300), HA (1:5,000), GRA7 (1:3,000), α-tubulin (1:2,500), or SAG1 (1:4,000) in the blocking buffer for 60 min. Unbound antibody was removed by washing the membrane six times with blocking buffer. Next, the membrane was incubated with horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Amersham Biosciences) at 1:10,000 in PBST for an additional hour, before detection by chemiluminescence using ECL-Plus. Quantification of scanned ECL films was performed using NIH Image J software (rsb.info.nih.gov/ij). For TgLCAT cross-linking assays, HFF were infected with transgenic parasites expressing TgLCAT-HA at a multiplicity of infection of 3. After 24 h, cells were washed and incubated with the membrane-permeable cross-linker dithiobis(succinimidyl propionate)) at 1 mm (diluted from a 100 mm stock in DMSO) in PBS or with DMSO (control) at room temperature for the intracellular conjugation of TgLCAT fragments. After 30 min, the cross-linking reaction was quenched by washing cells with 20 mm Tris-HCl, pH 7.6, in PBS, and samples were subjected to SDS-PAGE in the absence of reducing agent and Western blotting using anti-HA antibodies (1:2,000).
Recombinant LCAT Cloning and Expression in the Baculovirus System
TgLCAT coding sequence was amplified from the T. gondii cDNA library with the primers 5′-GTACGAATTCTTCAAGACGGACGTTGCGTTGTCG-3′ (forward, EcoRI site underlined) and 5′-GATCTCTAGACTACGTGCTGTCTGCCATAATCGC-3′ (reverse, XbaI site underlined) using Expand High Fidelity PCR system (Roche Applied Science). The PCR was cloned into TOPO 2.1 vector and sequenced. For the expression of the recombinant TgLCAT in insect cells, the Bac-to-Bac Baculovirus Expression System (Invitrogen) was used. The pFastBac HT A donor vector was used in a first cloning step. For cloning reactions, both vector and amplified cDNA cloned in TOPO 2.1 vector were digested with EcoRI and XbaI restriction enzymes. The ligation reaction was performed using Quick ligation kit (New England Biolabs, Ipswich, MA). After generation of the pFastBacHT A donor construct (with TgLCAT cDNA), the purified plasmid was transformed into DH10BacTM E. coli for transposition into the bacmid. Extraction of bacmids was performed according to the manufacturer's protocol (Bac-to-Bac Baculovirus Expression System). To verify the presence of the lcat gene after transposition, PCR with M13 primers was performed. The obtained amplicons were further sequenced using TgLCAT(EcoRI)f primers and TgLCAT(XbaI)r primers for confirmation of the presence of the lcat gene in the bacmid after transposition. Transfection of insect cells was performed according to the manufacturer's protocol (Bac-to-Bac Baculovirus Expression System). SF9 insect cells were co-transfected with the TgLCAT transfer vector and linearized baculovirus genomic DNA. After initial passages in SF21 cells, further infection was carried out in High Five cells. The culture medium containing recombinant TgLCAT protein (rTgLCAT) was harvested 96 h p.i. Expression of rTgLCAT and secretion in the culture medium was confirmed by Western blotting using anti-LCAT antibodies.
Immunoprecipitation of Recombinant TgLCAT
One hundred ml of the culture serum-free medium of insect cells expressing rTgLCAT was centrifuged at 10,000 × g, and the supernatant was concentrated ∼15–20 times using the Amicon ultrafiltration system. DTT and PMSF were added to the concentrated supernatant at final concentrations of 10 μg/ml and 0.5 mm, respectively. rTgLCAT was immunoprecipitated by mixing the concentrated supernatant with 50 μl of anti-TgLCAT antiserum (or prebleed serum as negative control) overnight at 4 °C. Antigen-antibody complexes were then incubated for 2 h with 500 μl of washed 25% protein A-Sepharose beads at 4 °C. Beads were collected by low speed centrifugation, washed three times with 5 ml of cold washing buffer (0.1 m sodium phosphate, 1 mm EDTA, 0.1% Triton X-100, 0.5% Nonidet P-40, and 150 mm NaC1; pH 7.4), and then three times with cold PBS. Purified rTgLCAT was quantified by BCA protein assay (Thermo Fisher Scientific Inc., Rockford, IL) and stored at −80 °C in an aliquot at a concentration of 100 ng of rTgLCAT/μl until use. The purity of rTgLCAT in our preparation was examined by SDS-PAGE after elution of absorbed material by heating the beads at 90 °C for 10 min in electrophoresis sample buffer (0.1 m Tris-HCl, 1% SDS and 40% glycerol, pH 6.8) and silver-staining of the gel.
Hemolysis Assay with TgLCAT
Hemolysis was measured using sheep red blood cells as described (26). Briefly, red blood cells were washed in PBS, diluted at 2% (v/v) suspension, and incubated at 37 °C in the Hemolysis Assay Buffer (HAB: 0.125 m NaCl, 0.035 m Na2HPO4, 0.1 mg/ml BSA; pH 7.4) with rTgLCAT, at different volumes and time points. Hemolysis was monitored by measuring the absorbance at 550 nm, using 1% Triton X-100 to induce total lysis of the red blood cells.
Enzymatic Assays for TgLCAT
TgLCAT activity was measured using single monolayer liposomes containing radioactive lipid substrates as described (27). For detecting phospholipase activity of TgLCAT, liposomes were made from DPPC, phosphatidylcholine (PC), and phosphatidylglycerol in molar ratio 10:5:2 with 0.3 μmol of [3H]DPPC as described (28). [3H]DPPC-containing liposomes were resuspended in an assay medium containing 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.01% NaN3, and various concentrations of rTgLCAT for different times at 37 °C. The reaction was stopped by addition of chloroform/methanol (1:2, v/v), and the lipid mixture was incubated for 2 h at room temperature to extract lipids. The phases were separated with chloroform and the same volume of 150 mm NaCl, and samples of the lower phase were collected for radiochemical analysis of the products of labeled DPPC catabolism by thin layer chromatography (TLC). The radiolabeled DPPC, lyso-PC, and free fatty acids were separated on silica gel using chloroform/ethanol/triethylamine/water at 30:35:35:7 as solvent, identified by lipid standards, and quantified by scintillation counting. LCAT lipase activity was expressed in nanomoles of fatty acid released per μg of LCAT. For detecting esterase activity of TgLCAT, liposomes were made from DPPC, PC, phosphatidylglycerol, and cholesterol in molar ratio 10:5:2:3 with 0.025 μmol of [14C]cholesterol as described (28). [14C]Cholesterol-containing liposomes were resuspended in the same assay medium as described above with various concentrations of rTgLCAT for 1 h at 37 °C. The reaction was stopped by addition of chloroform/methanol, and TLC using a neutral solvent system of hexane/ether/acetic acid at 70:30:2 was used to monitor the amount of radioactive cholesteryl esters by scintillation counting. LCAT transferase activity was expressed in nanomoles of cholesteryl esters formed per μg of LCAT.
Light and Electron Microscopy Studies
Light and epifluorescence microscopy were performed on infected cells seeded on sterile coverslips in 24-well culture dishes. IFA on parasites or mammalian cells was performed using primary antibodies against TgLCAT (1:100), SAG1 (1:500), GRA7 (1:200), GRA6 (1:250), GRA4 (1:50), or HA (1:1,000) and fluorescently labeled secondary antibodies diluted at 1:2,000. Cells were viewed with either 1) a Nikon Eclipse E800 microscope (Nikon, Melville, NY) equipped with an oil-immersion plan Apo ×100 NA 1.4 objective. Images were acquired with a Spot RT CCD camera (Diagnostic Instruments, Sterling Heights, MI) and Image-Pro-Plus software (Media Cybernetics, Silver Spring, MD). Photoshop (Adobe, San Jose, CA) was used to adjust levels and crop and resize images, or 2) a Nikon Eclipse 90i equipped with an oil-immersion plan Apo ×100 NA 1.4 objective and a Hamamatsu GRCA-ER camera (Hamamatsu Photonics, Hamamatsu, Japan). Optical z-sections with 0.2-μm spacing were acquired using Volocity software (PerkinElmer Life Sciences). The images were deconvolved using an iterative restoration algorithm, and the registry was corrected using Volocity software. Photoshop (Adobe) was used to adjust levels and crop and resize images. For ultrastructural observation of the Δlcat strain by thin section transmission electron microscopy (EM), infected cells were fixed in 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) and processed as described (29). Ultrathin sections of infected cells were stained by lead citrate and uranyl acetate before examination with a Philips CM120 EM (Eindhoven, The Netherlands) under 80 kV. For immunoelectron microscopy (immuno-EM), Toxoplasma-infected cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in 0.25 m HEPES, pH 7.4, for 1 h at room temperature and then in 8% paraformaldehyde in the same buffer overnight at 4 °C. They were infiltrated, frozen, and sectioned as described previously (21). The sections were immunolabeled with anti-TgLCAT antibodies (1:20) in PBS, 1% fish skin gelatin, then with anti-IgG antibodies, followed directly by 10-nm protein A-gold particles to detect TgLCAT. For the double immunogold staining, parasite sections were labeled with anti-TgLCAT (1:20) and anti-GRA7 antibodies (1:100) in PBS, 1% fish skin gelatin, incubated with anti-IgG antibodies, and then 5- or 10-nm protein A-gold particles to detect TgLCAT and GRA7, respectively.
Genetic Disruption of the lcat Locus
The lcat targeting construct was engineered by fusion PCR amplifying the 5′- and 3′-flanking sequences of lcat from RH genomic DNA and the dhfrHXGPRTdhfr selectable marker cassette from pminiHXGPRT provided by D. R. Roos (30). Primers used to amplify the 5′-flanking sequence (3.8 kb) were Pf1, 5′-GATTCATGCCTCCGTTGACTCGCAATC-3′ (forward), and Pr1, 5′-gaatgcaaggtttcgtgcttCTGGAAGGAGCGGAACAC-3′ (reverse); primers to amplify the selectable marker (1.8 kb) were Pf2, 5′-GTGTTCCGCTCCTTCCAGAagcacgaaaccttgcattc-3′ (forward), and Pr2, 5′-GTCCCGACTCAGTTCCCTcactgactggcatgaatggc-3′ (reverse); primers to amplify the 3′-flanking sequence (3.3 kb) were Pf3, 5′-gccattcatgccagtcagtgAGGGAACTGAGTCGGGAC-3′, and Pr3, 5′-CCACCCTGTATCCTAGAGCGTAG-3′. Primers Pf2 and Pr2 and primers Pr3 and Pf3 were designed to have overlapping complementary ends (lowercase letters) to facilitate further fusion PCR. Expand High Fidelity system was used in this step. The three PCR products were gel-purified and joined in a second step fusion PCR using Expand Long Template PCR and primers Pf1 and Pr3 resulting in an lcat targeting construct of 8.9 kb. RHΔHXGPRT parasites (107) were transfected with 30 μg of the 8.1-kb fusion PCR and selected in mycophenolic acid/xanthine as described previously (30). Surviving populations were screened for the following: (a) the loss of the lcat locus using primers 5′-GCATTCGTCACACTGCTGCATCGTC-3′ (forward) and primer 5′-GATCCCTAGGcgtgctgtctgccataatc-3′ (reverse, AvrII underlined); (b) the presence of the HXGPRT-selectable marker using primers Pf1 and primer Pr5, 5′-CATCCTGCAAGTGCATAGAAGGAAAG-3′ (reverse); and (c) homologous recombination of HXGPRT into the lcat locus using primers Pf5, 5′-ATCGTGGCGTGTCTACTGCGTTAAG-3′ and Pr5. Positive populations were cloned by serial dilution in 96-well plates. For functional complementation, 50 μg of the plasmid Tub-TgLCAT-HA harboring a sagCATsag cassette were transfected into the Δlcat strain, selected with chloramphenicol, and cloned by limiting dilution.
Transcriptional Profiles of lcat by qRT-PCR
2 μg of total RNA isolated from tachyzoites of RH WT, RH Δlcat, and Δlcat::LCAT strains with an RNeasy minikit (Qiagen, Valencia, CA) was reverse-transcribed using random primers and SuperScript II (Invitrogen). Gene expression was measured by TaqMan qRT-PCR using an Applied Biosystems 7900HT real time PCR system. The cycling program included 2 min at 50 °C and 10 min of incubation at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The Toxoplasma GAPDH gene was used as a reference to normalize the quantity of transcripts. For lcat, the primers were 5′-AAGTGGCCTCTTCGCTACAG-3′ (forward) and 5′-AGTGGAGCGACATGAGAGTG-3′ (reverse), and the dual-labeled TaqMan probe was 5′-(6-carboxyfluorescein)-TCATGCGGCAAATCCCCAATTAGT-(Black Hole Quencher 1)-3′. For GAPDH, the primer was 5′-TATTAACGGTTTTGGCCGTA-3′ (forward) and the dual-labeled TaqMan probe was 5′-(6-carboxyfluorescein)-CGTTGGTCAAGTGAGCCTGAGCCT-(Black Hole Quencher 1)-3′. Transcript levels were represented as 2ΔCT to show absolute level of transcripts relative to every strain examined.
Parasite Assays in Vitro
For invasion assays, 24-well plates were infected with 107 parasites per well and incubated for 20 min at 37 °C before fixation. Slides were differentially stained with anti-SAG1 antibodies for as per the red-green invasion assay (31). To visualize parasites invading cells by fluorescence microscopy, we used the high potassium-based Toxoplasma synchronous invasion assays as described previously (32). Briefly, freshly egressed parasites were added to host cells in high K+ Endo buffer at a multiplicity of infection of 10:1, followed by incubation for 20 min at 37 °C to allow parasites to adhere to the host cells. The buffer was then removed and replaced with pre-warmed invasion medium (DMEM plus 10% FBS). Temperature shift-based synchronized invasion assays were performed by adding parasites to host cells at room temperature and then immediately centrifuging the cells at 250 × g for 3 min, followed by incubation at room temperature for 5 min. The plates were then placed on a 37 °C heat block. Cells were fixed with 0.1% glutaraldehyde, 4% formaldehyde for 15 min, and incubated with anti-SAG1, permeabilized, and then exposed to anti-LCAT antibodies. For induced-egress assay, parasites were inoculated into 24-well plates and allowed to replicate for 28 h. After washing in Hanks' balanced salt solution supplemented with 1 mm CaCl2 and 1 mm MgCl2 and placed in a 37 °C water bath, cells were incubated for 4 min with 0.5 mm zaprinast (or DMSO as control), followed by the addition of 8% formaldehyde in PBS, and immunostaining with anti-SAG1 and anti-GRA7 antibodies to identify parasites and vacuoles, respectively. The detection of lactate dehydrogenase (LDH) in the culture medium released from infected cells was used as an indicator of host cell membrane rupture to quantify Toxoplasma egress. HFF were seeded in 96-well plates and infected with 5 × 105 parasites per well. After 24 h, monolayers were washed with Ringer's buffer supplemented with 1% FBS and then incubated for 20 min in 50 μl of zaprinast from 30 to 1,000 μm concentration or DMSO control. After treatment, culture supernatants (40 μl) were collected into another 96-well plate and were incubated for 20 min with equal amounts of reaction mixture used to determine the LDH concentration using a commercially available kit (cytotoxicity detection kit LDH, Roche Applied Science, Switzerland). The enzyme reaction was stopped by 0.1 m HCl. Absorbance was measured at a wavelength of 492 nm with a multiwall plate reader (Fluostar Optima, BMG Labtechnologies, Germany). Cytotoxicity was calculated as the percentage of total LDH released from cells treated by 1% Triton X-100 detergent. No significant release of LDH was monitored with zaprinast up to 1 mm in uninfected cells. Data were analyzed using Prism (GraphPad) to determine IC50 values by plotting normalized log-transformed data, using non-linear regression analysis as a sigmoidal dose-response curve with variable slope. To investigate the role of PLA2 activity on egress, we monitored the time of parasite egress in the presence of 4-bromophenacyl bromide (4-BPB). HFF cells were seeded in 96-well plates and infected with 5 × 105 RH parasites per well. After 30 h, monolayers were washed, incubated for 15 min with 10 μm 4-BPB (or DMSO control) in culture medium without serum, then washed, and exposed to 250 μm zaprinast before monitoring parasite egress over time using live microscopy. For growth assays, we used plaque assays to monitor parasite development over several days. Two hundred parasites were added to 24-well plates and incubated at 37 °C for 6 days. The cells were fixed and stained as described previously (33). The plates were scanned (ScanWizard 5, Microtek), and the area of each plaque was measured using Volocity software (PerkinElmer Life Sciences) by tracing each plaque using the region of interest tool. To examine parasite replication, we infected 24-well plates with 104 or 105 parasites for 4 h and washed to remove extracellular parasites, and replication was assessed at 16 and 36 h p.i. after fixation, immunostaining for SAG1 and GRA7, and scoring the number of intracellular parasites in randomly selected PV. In another replication assay, we monitored the incorporation of radioactive uracil into the parasite. HFF cells were grown until confluent in 24-well plates, infected with 5 × 104 parasites for 4 h at 37 °C, washed with PBS, and incubated for 24 and 48 h. Cells were then incubated with 1 μCi of [3H]uracil for 2 h, and the samples were processed as described previously to monitor the radioactivity associated with the parasites (22).
Ethics Statement for Animals and Parasite Assays in Vivo
All animal procedures were approved by the Institutional Animal Care and Use Committee of The Johns Hopkins University following the National Institutes of Health guidelines for animal housing and care. The protocol was approved by The Johns Hopkins University Animal Care and Use Committee (protocol MO12H448), which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All efforts were made to minimize suffering. Virulence assays were performed by infecting intradermally 6-week-old female Swiss Webster mice with 10 or 50 parasites (dissolved in 200 μl of PBS) from the parental RH, Δlcat, and Δlcat::LCAT strains (10 mice per strain) and monitoring mouse survival daily.
Statistical Analysis
For comparison of means, p values were determined by analysis of variance against control (one-way analysis of variance). For comparison of medians on dot plot graphs, p values were determined by the Mann Whitney test.
Results
T. gondii Has a Unique Gene Homologue to Lecithin:Cholesterol Acyltransferase Enzymes
Our BLAST homology searches for PLA2 and LCAT motifs retrieved one genomic sequence from the T. gondii database. The gene sequence (TGME49_272420) located on chromosome VIII was annotated as “putative phosphatidylcholine:sterol acyltransferase/acylceramide synthase.” We amplified the ORF of this gene by PCR from a cDNA library of the tachyzoite stage of T. gondii. The ORF is 2.3-kb nucleotides long, consists of seven exons, and encodes a polypeptide of 763 amino acids, predicting a protein of ∼84 kDa (Fig. 1A). The protein sequence reveals 22% identity and 36% similarity with human LCAT (EScore: e-22), and 32–35% identity and 39–43% similarity with LCAT from different Plasmodium species (EScores ranging from e-47 to e-37). Acylceramide synthase is a lysosomal enzyme that has also PLA2 and transacylase activities but, unlike LCAT, acylceramide synthase catalyzes the esterification of ceramide (34). The parasite sequence shows 16% identity and 25% similarity with human acylceramide synthase, but 1-O-acylceramide, the metabolite of acylceramide synthase, has not been detected in T. gondii (35), making the parasite protein more likely an LCAT homologue. Deduced amino acid sequence of the Toxoplasma protein, which we named TgLCAT, shares sequence motifs with the LCAT family members (6) as follows: AHSLG as the pentapeptide motif (G/A)XSXG with the serine residue being an acylated center characteristic of the serine lipase family; the catalytic triad of serine, aspartate, and histidine (SDH); two potential disulfide bonds predicted to partially cover the active site of the enzyme. TgLCAT also contains three potential N-linked glycosylation sites and a predicted N-terminal hydrophobic signal peptide, suggesting that TgLCAT is a secretory protein. TgLCAT contains a 141-amino acid “inserted element” (465–605) not present in any LCAT orthologs. To further analyze the relationship of TgLCAT with orthologues present in other Apicomplexa, we constructed a phylogenetic consensus tree for LCAT-like proteins using the neighbor-joining method (Fig. 1B). The LCAT-like sequences included in the tree were selected for the presence of a PLA2/LCAT motif (G/A)HSLG. The closest orthologue of TgLCAT has been observed for the LCAT-like enzyme present in the non-human pathogen H. hammondi, a tissue-dwelling Coccidia previously characterized as the Toxoplasma's closest extant relative. Indeed, H. hammondi shares many biological features, a high degree of genomic conservation, and synteny with T. gondii but is highly attenuated in mice (36). TgLCAT is similarly well conserved in another tissue-encysting coccidian parasite, N. caninum, which has same genome size, gene content, and synteny as Toxoplasma, but it has a more restrictive host range than either H. hammondi or T. gondii. No LCAT orthologues could be retrieved among Cryptosporidia species or in Chromera velia from the Chromerid branch. TgLCAT has a closer relationship with the LCAT sequences of the hematozoan cluster than with mammalian LCAT sequences, suggesting divergence of function between primitive and higher eukaryotic cells. However, this shared phylogeny between the hematozoan and coccidian LCAT is unsurprising given their common phylogeny as apicomplexan parasites with similar inheritance of the apicoplast red algal organelle that is unique to their eukaryotic decent (37).
FIGURE 1.

Comparison of sequences of the predicted ORF of LCAT from T. gondii and other organisms and molecular phylogeny of LCAT proteins. A, alignment of the primary sequences of TgLCAT, P. berghei LCAT (PbLCAT; PBANKA_112810), and human LCAT (HsLCAT; AAB34898.1). Identical amino acids are indicated by asterisks. The conserved (G/A)XSXG pentapeptide sequence of the serine lipase family is boxed; the carboxyesterase active-site serine motifs and cysteine residues for potential disulfide linkages are highlighted in yellow and green, respectively; the lid region is shown in blue; potential hydrophobic regions are indicated in brown; N-linked glycosylation sites are in red; and N-myristoylation site is in green. B, sequence relationships among LCAT family proteins. Unrooted phylogenetic tree of the LCAT family proteins constructed using neighbor joining analysis. Numbers at the branch nodes represent bootstrap values (as percentages) obtained in 1,000 replications. Branch lengths indicate the number of amino acid differences.
We screened cDNA libraries from different parasite stages for transcriptional analyses and all stages of T. gondii, e.g. the intracellular asexual stages, both tachyzoites (proliferative forms) and bradyzoites (tissue cyst forms); and the extracellular sexual stages, both oocysts (zygotes) and sporozoites (environmental cyst forms), expressed the lcat transcript (Fig. 2A). A 2.2-kb band was present in all samples and corresponded to the mature cDNA of lcat, although a larger transcript of 2.4-kb was detected in the tachyzoite and oocyst stages and may represent an alternately spliced transcript of lcat. Although the potential presence of splice variants for lcat remains to be established experimentally, a recent article (38) identified ∼2,000 genes, including lcat, that were predicted to be alternatively spliced based on informatics analyses.
FIGURE 2.

TgLCAT expression and secretion by T. gondii. A, transcriptional profiles of lcat in tachyzoites (Tz), bradyzoites (Bz), partially sporulated oocysts (Ooc), and sporozoites (Spz). Expression of lcat transcripts from all parasite stages was assayed by RT-PCR. To verify the absence of genomic DNA contamination, RT-PCRs were set up in duplicate with (+) and without (−) reverse transcriptase (RT). B, expression of TgLCAT. Immunoblots on parasite lysates separated by SDS-PAGE and probed with anti-TgLCAT antibodies reveal two bands, one corresponding to the predicted size of TgLCAT (double arrow) and the other one corresponding to cleaved product of the protein. No signal on immunoblots was observed on these lysates probed with preimmune serum. C, secretion of TgLCAT. Panel a, immunoblots on lysates of transgenic parasites expressing LCAT-HA separated by SDS-PAGE and probed with either anti-TgLCAT or anti-HA antibodies show two forms of TgLCAT products with different sizes of peptides (single arrows) and TgLCAT full-length (double arrow). Panel b, immunoblots of lysates of transgenic parasites expressing LCAT-HA after intracellular cross-linking of TgLCAT fragments using DSP, separation by SDS-PAGE, and probing with anti-HA antibodies show full-length TgLCAT (double arrow) and an upper band at 105 kDa absent from lysates under control conditions (DMSO). Panel c, immunoblots of material secreted by transgenic parasites expressing LCAT-HA separated by SDS-PAGE and probed with anti-TgLCAT and anti-HA antibodies show stronger bands corresponding to LCAT fragments compared with a weaker band for full-length protein (double arrow). Parasite-secreted material was also probed with antibodies against GRA7 (positive control) and against SAG1 (control for parasite cell integrity). D, schematic illustration of TgLCAT. Predicted domain structure of TgLCAT is shown with putative signal peptide (SP), membrane-binding domain (MBD) based on homology with human LPLA2, N-terminal catalytic fragment (N-term cat. frag.), cap domain (Cap), inserted element (IE), and C-terminal catalytic fragment (C-term). Numbers above the illustration indicate amino acid position at the beginning of select domains. Position of the recombinant polypeptide used to immunize rats for anti-TgLCAT (α-TgLCAT) is indicated. Positions of catalytic residues SDH are indicated with red spheres. A putative coiled-coil domain within the inserted element is shown based on the highest scoring ab initio model from I-TASSER. The precursor TgLCAT species and putative proteolytic fragments are indicated as black lines below the schematic, with dashed lines indicating the estimated region of proteolysis. E, structural model of TgLCAT. Model was constructed with Phyre2, which identified human LPLA2 (Protein Data Bank code 4X91) as the highest scoring template for threading. Domains are colored according to the published structure of human LPLA2 (34). The position of the inserted element is indicated with an arrow, with the two flanking amino acid backbone residues shown in cyan. Catalytic residues, including their side changes, are shown as spheres and labeled with the single letter designations for the corresponding amino acids. The image on the right is viewed from the perspective of the membrane, with the catalytic pocket shown centrally. The left image is rotated 90° from the right image.
TgLCAT Is Expressed and Cleaved by T. gondii
The expression of TgLCAT was analyzed by Western blotting using anti-TgLCAT antibodies, which were generated against a recombinant fragment of TgLCAT (amino acids 106–202). Two bands were detected in immunoblots of lysates from RH parasites (tachyzoite forms) as follows: an ∼83-kDa band, approximating the predicted size of TgLCAT, and ∼62 kDa, potentially representing a cleaved form of TgLCAT although no other LCAT enzymes have been reported to undergo proteolytic cleavage (Fig. 2B). To further probe the cleavage of TgLCAT, we engineered a parasite strain expressing LCAT tagged at its C terminus with an HA epitope (LCAT-HA). Lysates from LCAT-HA-expressing parasites were analyzed by immunoblotting using both anti-TgLCAT and anti-HA antibodies. For both conditions, the ∼83-kDa band of full-length LCAT-HA was detected. Additional bands at ∼62 and ∼33 kDa were observed on blots probed with anti-TgLCAT (N-terminal) and anti-HA (C-terminal) antibodies, respectively (Fig. 2C, panel a). The endogenous untagged LCAT bands were difficult to resolve from the predominant tagged LCAT species. The sum of these two fragments exceeds that of the precursor species, suggesting that cleavage results in aberrant migration or is followed by additional post-translational modification(s).
We next examined whether the two proteolytic fragments of TgLCAT remain together in a non-covalent complex. Treatment of intracellular TgLCAT-HA-expressing parasites with a cell-permeable cross-linker (DSP) followed by immunoblotting suggests that the two proteolytic fragments are associated in an ∼105-kDa cross-linked species, potentially representing a secondary protein complex (Fig. 2C, panel b).
The TgLCAT sequence contains a signal peptide, indicating that this enzyme is associated with secretory organelles and/or secreted by T. gondii. To determine whether TgLCAT is released from Toxoplasma, we examined its presence among proteins secreted by the parasite by Western blotting using anti-TgLCAT antibodies. Parasites expressing LCAT-HA were maintained under axenic conditions, and secretions were collected by centrifugation and subjected to immunoblotting using anti-TgLCAT and anti-HA antibodies. The two cleaved products of LCAT-HA were detected as strong bands, whereas the band corresponding to the full-length protein was weaker, suggesting that T. gondii predominantly secretes the cleaved forms of the protein (Fig. 2C, panel c). As controls, we probed the same blots with antibodies against GRA7, a secreted dense granule protein, and against SAG1, a protein residing in the plasma membrane. Our data show the presence of GRA7 and the absence of SAG1 in the material secreted by the parasite.
To visualize how enzyme activity may potentially be maintained despite the observed proteolytic processing, we performed structural modeling of TgLCAT based on the recently solved crystal structure of human LPLA2 (39). This analysis suggested that the active site residues remain in close proximity due to the folding of the core catalytic domain, its associated cap domain, and putative membrane-binding domain. The inserted element maps to a site within the cap domain where it is unlikely to interfere with access of TgLCAT to its substrates. Because the inserted element lacks strong homology to proteins of known structure, we performed ab initio structural modeling to predict its features. This analysis suggested that the inserted element mainly consists of two to three long α-helices predicted to form a two-stranded coiled-coil (Fig. 2D). Together, these structural predictions indicate that TgLCAT has the potential to retain catalytic activity despite proteolytic processing.
Recombinant TgLCAT Displays Membrane Lytic Activity
The presence of the lipase motif AHSLG and catalytic triad SDH in the TgLCAT sequence suggests that the parasite protein has phospholipase activity. PLA2 activity is associated with the release of fatty acids from phospholipid membranes, leading to the production of lysophospholipids that can destabilize membranes. In particular, the product of phosphatidylcholine lysis, lysophosphatidylcholine, possesses a strong membrane lytic activity (40, 41). To investigate the potential effect of TgLCAT on cell membranes, we generated a recombinant form of TgLCAT (rTgLCAT) using the baculovirus expression system (42), as we previously exploited to engineer recombinant Plasmodium LCAT (43). The culture medium of SF21 cells infected with baculoviruses containing the Toxoplasma lcat gene was analyzed by Western blot using anti-TgLCAT antibodies (Fig. 3A, lanes a and b). We detected a major band at ∼92 kDa secreted by insect cells and a minor band at ∼65 kDa. We used the culture medium of the SF21 cells containing rTgLCAT to assess the potential membranolytic activity of TgLCAT. rTgLCAT was exposed to red blood cells, and the release of hemoglobin was monitored by fluorimetry. rTgLCAT induced hemolysis in a concentration- and time-dependent manner, with maximal lytic activity at 24 h (Fig. 3B, panels a and b). Addition of anti-TgLCAT antibodies to the assay medium significantly decreased the lysis of red blood cells induced by TgLCAT, confirming the specific activity of the parasite protein for hemolysis.
FIGURE 3.

Hemolytic and enzymatic activities of recombinant TgLCAT. A, detection and purification of rTgLCAT in the medium from insect cells infected with TgLCAT-expressing baculovirus. Immunoblots of culture medium using anti-TgLCAT antibodies show a major band at ∼92 kDa (lane a, arrow). Silver-stained SDS-PAGE of the culture medium is shown in lane b. Silver-stained SDS-PAGE of purified rTgLCAT by immunoprecipitation using anti-TgLCAT antibodies is shown (lane c). No protein was detected on silver-stained gel using preimmune serum in the immunoprecipitation assay (lane d). B, hemolytic activity of rTgLCAT. Panel a, red blood cells were incubated for 30 min with different concentrations of rTgLCAT before measuring hemolysis by absorbance at 550 nm (closed circles). Control includes hemolytic assay performed in the presence of preimmune serum (triangles, dashed line) or antiserum with TgLCAT antibodies (open square). Data are means ± S.D. (n = 3 independent assays), and values in % are relative to absorbance values calculated for Triton X-100 (100%). Panel b, time dependence of the hemolytic reaction determined with 5 μg of rTgLCAT (closed circles) or Hemolysis Assay Buffer alone (open circles). Data are means ± S.D. (n ≥ 3 separate experiments), and values in % are relative to absorbance values calculated for Triton X-100 (100%). C and D, phospholipase activity of rTgLCAT. C, TLC analysis of PC used as substrate incubated with rTgLCAT at different concentrations for 3 h at 37 °C, showing liberation of free fatty acid (FA) from PC and production of lysophosphatidylcholine (lyso-PC). D, quantification of fatty acid released from PC by rTgLCAT (panel a). 25 μg of radiolabeled PC incorporated into liposomes were incubated for 1 h at 37 °C with 5 μg of rTgLCAT in the presence of anti-LCAT antibodies or an irrelevant antibody (rat immunoglobulin G, rIgG). PC, fatty acid, and lyso-PC were separated on TLC plates and measured by liquid scintillation counting. Fractional fatty acid hydrolysis in % was measured as follows: cpm of lyso-PC/(cpm of lyso-PC + cpm of PC). Data are means ± S.D. (n = 3 independent assays). *, p < 0.01. Panel b, concentration dependence of rTgLCAT for fatty acid hydrolysis. Same assay as described in panel a using different concentrations of rTgLCAT incubated with radiolabeled PC for 1 h is shown. Data are means ± S.D. (n = 3 independent assays). Panel c, time dependence of fatty acid hydrolysis using 4 μg of rTgLCAT. Data are means ± S.D. (n = 3 independent assays). Panel d, effect of calcium and cysteine modifications on phospholipase activity mediated by rTgLCAT. Same assay as described in panel a using 5 μg of rTgLCAT for 1 h in the presence of 20 mm EDTA, 50 mm Ca2+, 0.5 mm DTNB, 0.5 mm diisopropyl fluorophosphate, (DFP) 0.5 mm diethylpyrocarbonate (DEPC), or no addition (control). Data in % normalized to control (100%) are means ± S.D. (n = 3 independent experiments). *, p < 0.05. E, cholesterol transferase activity of rTgLCAT. 1.5 μg of radiolabeled cholesterol incorporated into PC-containing liposomes were incubated for 4 h at 37 °C in the presence of the indicated rTgLCAT concentrations in the absence (closed circles) or presence (open circles) of DTNB. Cholesterol and cholesterol esters (CE) were separated on TLC plates and measured by liquid scintillation counting. Data are means ± S.D. (n = 3 independent assays).
Recombinant TgLCAT Has a Dual Enzymatic Activity as a PLA2 and a Cholesterol Transacyltransferase
To determine whether TgLCAT has phospholipase and/or acyltransferase activities, we performed a series of enzymatic assays with various lipid substrates using purified rTgLCAT. To purify rTgLCAT proteins for these assays, we immunoprecipitated the recombinant protein from the culture medium of SF21 cells infected with lcat-containing baculoviruses with affinity-purified antibodies against TgLCAT, and we detected the ∼92-kDa protein as major band (Fig. 3A, lane c). As a negative control, preimmune serum was used in the purification protocol, and as expected, no protein band was detected (Fig. 3A, lane d).
To determine whether rTgLCAT has a PLA2 activity, we incubated rTgLCAT with phosphatidylcholine as a substrate and monitored the production of lysophosphatidylcholine and free fatty acid by TLC. We detected spots corresponding to lysophosphatidylcholine and free fatty acid on TLC plates in the presence of rTgLCAT (Fig. 3C). With the addition of increasing amounts of rTgLCAT, we detected proportionally larger amounts of lysophosphatidylcholine and free fatty acid. To quantify the phospholipase reaction mediated by rTgLCAT, we incubated the enzyme with liposomes composed of phosphatidylcholine and containing radioactive 1-palmitoyl-2[2-palmitoyl-9,10-3H]sn-glycero-3-phosphocholine ([3H]DPPC, with tritium on the palmitate at the sn-2 position) and monitored the release of radioactive fatty acid from DPPC. In this assay, rTgLCAT hydrolyzed about 10% of fatty acid from DPPC in 1 h (Fig. 3D, panel a). To assess the specificity of the reaction, anti-LCAT antibodies were added to the incubation medium, resulting in a significant decrease in the amount of fatty acid released, although an irrelevant antibody had no effect. Hydrolysis of fatty acid was proportional to both rTgLCAT concentration and incubation time, with a specific activity of 0.95 ± 0.025 nmol of free fatty acid hydrolyzed per h/μg of rLCAT (Fig. 3D, panels b and c). Because human LCAT has a calcium-independent PLA2 activity, differentiating it from calcium-dependent PLA2 enzymes, we examined whether TgLCAT activity required calcium by chelating this cation or adding it in excess to the phospholipase activity assay. The activity of rTgLCAT was unaffected by EDTA and excess calcium (Fig. 3D, panel d). Finally, we added amino acid-specific inhibitors to the reaction assay to assess the role of the conserved sequence motifs on TgLCAT activity. Human LCAT activity is sensitive to sulfhydryl-reactive agents (44) because disulfide-linked cysteine residues form part of a lid region that protects the catalytic triad SDH. To assess the importance of the conserved cysteine residues surrounding the active site and the residues of the catalytic triad on TgLCAT phospholipase activity, we added into the reaction mixture the following: sulfhydryl-modifying reagent for cysteine residues, dithiobis(2-nitrobenzoic acid) (DTNB); the serine anti-esterase inhibitor, diisopropyl fluorophosphate; and the histidine anti-esterase inhibitor, diethylpyrocarbonate (Fig. 3D, panel d). A dramatic reduction in TgLCAT activity was observed for each inhibitor. This suggests that the enzymatic PLA2 activity of TgLCAT is independent of calcium and requires functional cysteine, serine, and histidine residues.
To examine whether rTgLCAT has a cholesterol acyltransferase activity, we incubated the enzyme with phosphatidylcholine-cholesterol liposomes containing [14C]cholesterol and monitored the production of esters of cholesterol by TLC. We show spots corresponding to cholesteryl esters on TLC plates. The synthesis of cholesteryl esters increased with rTgLCAT concentrations, reaching a plateau at 1 μg of rTgLCAT (Fig. 3E). At this concentration, the specific acyltransferase activity is 0.047 ± 0.009 nmol of cholesteryl esters/h/μg of rTgLCAT, with a Vmax of 12.8 ± 3.4 pmol/h/mg and a Km of 26 ± 4 nm. Altogether, these data indicate that LCAT from T. gondii, like mammalian LCAT, has a dual phospholipase A2 and cholesterol acyltransferase activity.
TgLCAT Localizes at the Plasma Membrane of Extracellular Parasites
Next, we examined the localization of TgLCAT in parasites by indirect IFA and immuno-EM using anti-TgLCAT antibodies. On extracellular parasites, TgLCAT was distributed to the parasite surface and co-localized with SAG1, a plasma membrane protein (Fig. 4A, panel a). Immunogold staining of parasite sections for ultrastructural examinations detected gold particles on the parasite plasma membrane and in organelles inside the parasite (Fig. 4A, panel b).
FIGURE 4.

TgLCAT localization in extracellular and intracellular Toxoplasma. A, LCAT staining on extracellular parasites. Panel a, IFA of extracellular parasites using anti-SAG1 antibodies, followed by permeabilization and immunolabeling for TgLCAT, showing co-staining of the parasite surface with anti-TgLCAT and anti-SAG1 antibodies. Panel b, immuno-EM on extracellular parasites using anti-TgLCAT, confirming surface staining of the enzyme (arrows) as well as the presence of gold particles in dense granule-like organelles. Scale bar, 500 nm. B, LCAT staining on invading parasites by IFA. A synchronized population of invading parasites for 15 min was fixed, stained with anti-SAG1 antibodies, then permeabilized, and immunolabeled for TgLCAT, showing co-localization for TgLCAT and SAG1. C, LCAT staining on intracellular parasites. Live fluorescence microscopy of intracellular parasites expressing LCAT-YFP viewed at the indicated times p.i., illustrating the fluorescent signal associated with intracellular organelles and the PV lumen increasing with time post-invasion.
We also inspected the localization of TgLCAT during the process of invasion. T. gondii invasion is a two-step process in which parasites attach to and then penetrate into mammalian host cells (45). These two steps can be dissociated from one another by adding parasites to cells in a buffer with high levels of potassium, which permits adhesion of parasites to cells but blocks penetration (32). Penetration is then triggered by replacing the medium with a buffer poor in potassium that is permissive for invasion. Freshly egressed parasites were added to fibroblasts in high potassium medium for 20 min, and invasion was stimulated by the addition of invasion buffer for 5 min. Cells were fixed, stained, with anti-SAG1 antibodies to detect the surface-exposed SAG1 protein, and then permeabilized and immunolabeled for TgLCAT. The staining of SAG1 under non-permeabilization allows the visualization of the gradual penetration of the parasite into cells. As expected, the SAG1 labeling was progressively reduced as the parasite forges ahead into the host cell and remained confined to the extracellular portion of the parasite (Fig. 4B). Under permeabilization conditions, the TgLCAT staining was progressively reduced as the parasite penetrates into the host cell, suggesting that most of TgLCAT was exposed at the cell surface of the parasite during invasion and then removed from the parasite surface upon invasion.
Replicating Toxoplasma Secretes TgLCAT from a Subset of Dense Granules into the PV
To analyze TgLCAT localization after invasion, we engineered a parasite line expressing TgLCAT-YFP. We infected fibroblasts with parasites expressing TgLCAT-YFP and visualized the fluorescence staining of live parasites from 2 min to 31 h post-invasion (p.i.) (Fig. 4C). Shortly after invasion and until 90 min, no TgLCAT-YFP was observed on parasites, consistent with our observations that TgLCAT is stripped off invading parasites (Fig. 4B). At 3 h p.i., a very weak punctate signal was visible within parasites. The punctate TgLCAT-YFP signal in the parasite became stronger at the onset of parasite replication ∼7 h p.i. and a staining appeared inside the PV, indicating that TgLCAT was secreted by the parasite. At later time points (>16 h p.i.), TgLCAT-YFP remained localized to intraparasitic organelles and between parasites in the PV lumen.
The intraparasitic punctate pattern observed in Fig. 4C was reminiscent of that of dense granules, organelles that contain proteins secreted into the PV and/or exported into the host cell during the intracellular life cycle of T. gondii (46). To examine whether TgLCAT localizes to dense granules, we compared the localization of TgLCAT and various GRA proteins in wild-type or TgLCAT-HA parasites (Fig. 5, A and B). In PV containing one parasite, TgLCAT localized to some organelles containing GRA7, GRA6, or GRA4. Some puncta contained TgLCAT, and in both of the GRA proteins assayed, some contained LCAT-HA and one GRA protein, whereas others contained only LCAT-HA. Other examples of partial co-localization between TgLCAT and GRA proteins are illustrated in Fig. 5B. Organelles containing exclusively TgLCAT may correspond to either a subset of dense granules or organelles unrelated to dense granules. In PV with more than one parasite, TgLCAT staining was present in both the PV lumen and within the parasite, although the fluorescence signal for GRA7, GRA6, or GRA4 proteins was predominantly detected in the PV lumen (Fig. 5C).
FIGURE 5.

Intraparasitic localization of TgLCAT in intracellular Toxoplasma. A–C, IFA for TgLCAT localization relative to dense granule proteins in PV with one (A and B) or two parasites (C). A, triple IFA of TgLCAT-HA-expressing parasites using anti-HA, anti-GRA7, and anti-GRA6 antibodies, illustrating partial or no co-localization between the three proteins. Yellow squares, granules positively labeled for HA, GRA6, and GRA7. White squares, granules labeled for HA and GRA7; orange squares, granules labeled for HA and GRA6; green squares, granule solely labeled for HA. B, IFA of TgLCAT-HA-expressing parasites using anti-HA, anti-GRA6, and anti-GRA4 antibodies, confirming observations in A using a different set of dense granule proteins shown as insets. C, IFA of wild-type parasites using anti-TgLCAT, anti-GRA6, and anti-GRA4 antibodies, showing persistent signal for TgLCAT within the parasites, as compared with GRA4 and GRA6 staining mainly in the PV lumen. Dotted lines delineate the PV contour.
We performed immunogold EM labeling to examine to what extent TgLCAT localizes to dense granules by EM. These organelles are characterized by a spherical shape, a diameter of 250 nm, and a uniformly electron dense matrix surrounded by a unit membrane. Our observations reveal an accumulation of gold particles in organelles presenting the ultrastructural hallmarks of dense granules; no other secretory organelles of the parasites, e.g. micronemes or rhoptries, contained gold particles (Fig. 6A, panel a). We observed gold-labeled dense granules docked onto the plasma membrane, suggestive a process of exocytosis of TgLCAT (Fig. 6A, panel b). TgLCAT also localized to some areas of the parasite plasma membrane (Fig. 6A, panel c). Examinations of the immunostaining associated with the dense granules show TgLCAT present in the matrix of the organelles or at the limiting membrane of dense granules. In addition to dense granules, few electron-lucent structures were labeled for TgLCAT, as exemplified in panel e of Fig. 6A. These variable locations of TgLCAT may explain the partial overlap observed between TgLCAT and other GRA proteins as seen by IFA in Fig. 5. Double immuno-EM using anti-TgLCAT and anti-GRA7 antibodies demonstrated the localization of both proteins within dense granules at a variable extent (Fig. 6B).
FIGURE 6.

Ultrastructural localization of TgLCAT in the parasite and PV. A, immuno-EM to detect TgLCAT using anti-TgLCAT antibodies (coupled to 10 nm gold particles) on wild-type parasites 24 h p.i. Panel a is a large view of PV showing the presence of gold particles in dense granules (DG) and the PV lumen. P, parasite; PVM, parasitophorous vacuole membrane; n, nucleus; mt, mitochondrion; mi, micronemes; rh, rhoptry; hcell, host cell. Panel b illustrates the docking of a dense granule containing TgLCAT onto the parasite plasma membrane (PM) prior to exocytosis. Panel c shows the distribution of TgLCAT at the parasite plasma membrane. Panel d shows the distribution of TgLCAT in the matrix of dense granules (panel i), at the limiting membrane (panels ii–iv), or between dense granules (arrow in panel v). Panel e illustrates gold particles associated with an electron-lucent organelle (arrow). Scale bars are 500 nm for panels a–c and 100 nm for panels d and e. B, double immuno-EM using anti-TgLCAT and anti-GRA7 antibodies (coupled to 5 and 10 nm gold particles, respectively), showing the localization of the two proteins in dense granules. The relative distribution of the 5 and 10 nm gold particles in dense granules was quantified from 80 sections of parasites, totaling ∼300 dense granules, and data were expressed in percent of total viewed sections of dense granules for each category of dense granules classified as follows: panel i, dense granules with neither GRA7 nor TgLCAT; panel ii, with GRA7 only; panel iii, with [GRA7] > [TgLCAT] (circles); panels iv and v, [with GRA7] = [TgLCAT] at low and high abundance, respectively; panel vi, with [TgLCAT] > [GRA7]; and panel vii, with GRA7 alone in lesser dense organelle (arrow). Scale bar 100 nm. C, triple IFA of TgLCAT-HA-expressing parasites using anti-HA, anti-GRA7, and anti-GRA6 antibodies, illustrating the accumulation of TgLCAT into the PV lumen in co-localization with GRA7 and GRA6 but no association with the PV membrane (PVM) in contrast to GRA7 and GRA6 (white rectangles). D, double immuno-EM using anti-TgLCAT and anti-GRA7-antibodies, (coupled to 5 and 10 nm gold particles, respectively), revealing the presence of GRA7 and TgLCAT in the PV lumen and their association with vesicular structures of the IVN (inset). Scale bar, 500 nm.
We next inspected the distribution of TgLCAT following its secretion by immunostaining infected cells. At 24 h p.i., TgLCAT accumulated in the PV lumen between parasites as observed by IFA for GRA7 and GRA6 (Fig. 6C). Unlike GRA6 and GRA7, no staining of TgLCAT was detected on the PV membrane. Even in large PVs, the punctate staining pattern of TgLCAT within individual parasites remained substantial, in contrast to GRA6 and GRA7, suggesting that TgLCAT has different secretion dynamics than these GRA proteins. To visualize the fine distribution of secreted TgLCAT, we performed immuno-EM. We observed TgLCAT on membranous structures that form the intravacuolar network or IVN (Fig. 6D), a membranous interface derived from multilamellar vesicles secreted from the parasite (47). No gold particles were observed in the host cell.
Toxoplasma Lacking LCAT Show Delayed Egress whereas Parasites Overexpressing LCAT Exit Faster from Host Cells than Parental Parasites
The localization of TgLCAT to the surface of axenic parasites suggests that this enzyme may be involved in a process occurring during the extracellular stage of the parasite, e.g. invasion and/or exit from host cells. To test this hypothesis, we generated a parasite strain lacking LCAT and examined the ability of the mutant parasite to invade and egress, in comparison with the parental strain. To genetically disrupt the lcat gene, the entire locus of lcat was replaced by homologous recombination with the selectable marker HXGPRT in the parental RHΔHXGPRT strain (referred as RH). The successful generation of knock-out parasites (Δlcat strain) was confirmed by PCR (Fig. 7, A, B, and D); the absence of TgLCAT expression was further verified by Western blotting analysis and IFA using anti-TgLCAT antibodies, showing no signal for TgLCAT in the knock-outs (Fig. 8A, panels a and b). We also examined the ultrastructure of Δlcat parasites by EM. The mutant parasites had all the hallmarks of tachyzoites, however, they accumulated amylopectin granules in their cytoplasm (Fig. 8A, panel c), which is a characteristic of tissue cyst forms (bradyzoites) that grow very slowly and rely on sugar stores for glycolysis and gluconeogenesis (48). The presence of these glucose storage granules suggests that the Δlcat parasites exhibited signs of stress.
FIGURE 7.

Targeted deletion of the lcat gene and complementation strategies. A, top, scale diagram of the genomic locus of lcat. The boxes represent the seven exons of lcat (open, untranslated; filled, coding regions). Middle, plasmid pminiHXGPRT used to PCR-amplify the selectable marker cassette dhfrHXGPRTdhfr. Bottom, schematic representation of the fusion PCR to generate the targeting construct. The 5′- and 3′-flanking sequences of lcat were amplified using the primers Pf1/Pr1 and Pf3/Pr3, respectively, and fused to the 5′dhfr-HXGPRT3′dhfr cassette PCR-amplified with primers Pf2/Pr2. B, strategy for disrupting lcat in the parental RH parasites. C, schematic of the plasmid Tub-lcatcDNA-HA used to complement Δlcat parasites. Arrows in B and C indicate primers for PCR screening. D, PCR of RHΔHXGPRT, Δlcat, and complement. The P1/P2 primers were used to verify the absence of lcat in the knock-out strain (lane 2) and its presence in both the parental and complemented strains (lanes 1 and 3). Primers P9/Pcr were used to confirm the insertion of the targeting construct by homologous recombination (lanes 5 and 6) and the absence of this construct in the parental line (lane 4). The expected sizes of the fragments are shown on the right side of the gels.
FIGURE 8.

Characterization of the Δlcat and Δlcat::LCAT strains. A, LCAT deficiency. Immunoblots on lysates (panel a) or IFA (panel b) of parental RH and Δlcat parasites using anti-TgLCAT antibodies, confirming the absence of LCAT protein in the knock-out strain. α-Tubulin was used as a loading control for the Western blot. Panel c, ultrastructure of TgLCAT-deficient Toxoplasma 24 h p.i., characterized by the accumulation of amylopectin granules (arrow). Scale bar, 500 nm. B, LCAT complementation and overexpression. Panel a, double IFA of Δlcat::LCAT parasites using anti-TgLCAT and anti-HA antibodies, illustrating the presence of LCAT protein in dense granules and PV lumen. Panel b, gene expression measured by qRT-PCR. The GAPDH gene was used as a reference to normalize the quantity of transcripts. Transcript levels were represented as 2ΔCT to show absolute level of transcripts relative to the three strains examined. Data are means ± S.D. (n = 4 independent parasite preparations). Transcript levels of lcat were statistically significant between RH and Δlcat::LCAT parasites (p < 0.001). Panel c, immunoblots of RH and Δlcat::LCAT parasite lysates separated by SDS-PAGE and probed with anti-LCAT antibodies, showing the bands of full-length and cleaved form of LCAT. α-Tubulin was used as a loading control for the Western blot. Panel d, quantification of band intensities of TgLCAT normalized to α-tubulin (α-Tub) on the scanned ECL films revealed ∼3.5 times higher expression levels of TgLCAT in Δlcat::LCAT, as compared with WT. Data are means ± S.D. from three independent lysates of RH and Δlcat::LCAT strains. Expression levels of TgLCAT, either uncleaved or cleaved, were statistically significant between RH and Δlcat::LCAT parasites (p < 0.05).
In parallel, we complemented the deletion by transfecting Δlcat parasites with a plasmid containing, under the control of the strong tubulin promoter, lcat fused to the HA epitope (Δlcat::LCAT strain; Fig. 7, C and D). In the complemented (acting like overexpressor) strain, we performed double IFA using anti-TgLCAT and anti-HA antibodies to confirm the localization of TgLCAT-HA to dense granules and PV (Fig. 8B, panel a). The levels of lcat transcripts in the Δlcat::LCAT strain were ∼70 times higher than in parental RH parasites (Fig. 8B, panel b), resulting in an ∼3 times increase in TgLCAT protein expression, both of the intact and cleaved forms (Fig. 8B, panels c and d).
We compared the ability of Δlcat, Δlcat::LCAT, and parental parasites to invade host cells. Extracellular (surface-attached) and intracellular (invaded) parasites were enumerated 20 min p.i., and all three strains were equally proficient in invading mammalian cells (Fig. 9A). Next, we examined the capability of the strains to egress from host cells upon chemical induction. Zaprinast (2-O-propoxyphenyl-8-dazapurin-6-one), a cGMP-specific phosphodiesterase inhibitor that modulates intracellular Ca2+ transiently (49), is used to induce synchronized egress of T. gondii from fibroblasts. Compared with most egress inducers, e.g. the calcium ionophore A21387, zaprinast releases parasites from cells over a longer period of time and therefore closely approximates natural egress conditions of the parasite (50). The morphology of the PV at 180 and 360 s post-addition of 0.5 mm zaprinast is illustrated in panel a in Fig. 9B. By 180 s, some of the parental parasites had egressed while the Δlcat parasites were still in their PV. Interestingly, the vast majority of Δlcat::LCAT parasites were extracellular at that time. At 360 s, Δlcat parasites started to egress, although ∼85% of parental PV were already lysed and all of the Δlcat::LCAT PV were disrupted. We examined the localization of TgLCAT on parasites egressing upon zaprinast addition, and we observed TgLCAT at the parasite surface (Fig. 9B, panel b), consistent with our observations in Fig. 4 (panels a and b). To monitor the dynamics of egress, we used phase-contrast microscopy to determine the time of parasite egress after the addition of zaprinast for each strain. After 1-min of zaprinast induction, the three strains of T. gondii began to move within their PV, as marked by the disassembly of the rosette structure of intracellular parasites. Δlcat::LCAT parasites first egressed from their PV at 88 ± 7 s while parental parasites escaped at 152 ± 12 s, and the Δlcat parasites remained encased in their PV until 325 ± 16 s.
FIGURE 9.

Invasion and egress of Δlcat and Δlcat::LCAT parasites. A, quantification of invasion using the red/green assay for Δlcat, parental, and Δlcat::LCAT parasites stained with antibodies against SAG1. Data are means ± S.D. (n = 3 separate assays), counting 20 randomly selected fields for each sample at ×20 objective. B, zaprinast-induced egress from RH, Δlcat, and Δlcat::LCAT PV. Intracellular parasites for 24 h were incubated 4 min with 0.5 mm zaprinast or DSMO vehicle before fixation for IFA. Panel a, microscopic observations on intracellular parasites incubated with 0.5 mm zaprinast to monitor time until egress. Representative images are shown at 180 and 360 s post-drug induction. Panel b, double IFA using anti-TgLCAT and anti-SAG1 antibodies, showing TgLCAT at the parasite surface stained for SAG1. Panel c, scoring of the number of intact versus ruptured PV on parasites immunostained for GRA7 and SAG1 following addition of zaprinast at 0.5 mm at the indicated times post-drug addition. Data are means ± S.D. (n = 3 independent assays), counting 29 randomly selected fields for each sample at ×20 objective. Egress values for eitherΔlcat or Δlcat::LCAT parasites were statistically significant for each time point as compared with parental parasites. **, p < 0.01; *, p < 0.05. Panel d, dose-response for egress following a 20-min exposure with zaprinast at the indicated concentrations. Egress was measured as a function of LDH released from host cells and normalized to the levels resulting from Triton X-100-treated cells. Data are means ± S.D. (n = 3 independent experiments). Panel e, effect of 4-BPB on parasite egress. HFF infected with RH parasites for 30 h were incubated with 10 μm 4-BPB (or DMSO) before egress induction with 250 μm zaprinast, and time of parasite egress was measured. Data shown in dot plots are medians from four independent infections with four or five replicates. *, p < 0.01.
To further quantify the egress rate of the three strains, we incubated infected cells with 0.5 mm zaprinast for 5–30 min, followed by immunostaining for SAG1 and GRA7 and counted the number of PVs that have been ruptured by escaping parasites for each treatment time (Fig. 9B, panel c). At all times post-induction with zaprinast, Δlcat::LCAT parasites egressed faster, and Δlcat parasites exited significantly slower than parental parasites (Fig. 9B, panel c). At 20 min post-induction, ∼50% of Δlcat PV were still occupied while the PV of the two other strains had already released their parasites. In addition, we examined the dose response of each strain to concentrations of zaprinast ranging from 30 μm to 1 mm. To assess egress efficiency, we measured the release of cytosolic LDH from infected cells. The IC50 values, calculated from sigmoidal dose-response curves, were 140 μm for Δlcat::LCAT parasites, 574 μm for Δlcat parasites, as compared with 248 μm for the parental parasites. Finally, to determine the physiological relevance of PLA2 activities for T. gondii egress, we treated infected cells with the irreversible inhibitor of PLA2 enzymes, 4-BPB, prior to zaprinast exposure. Although no studies have reported the influence of host cell lipase activities on T. gondii egress, our experiments were conducted on cells terminally infected, hence metabolically impaired, to minimize the potential contribution of the host cell to this event. Pretreatment of infected cells with 4-BPB significantly increased the time of parasite release from cells (median values of 241 and 198 s post-zaprinast induction, in 4-BPB-treated cells and control cells, respectively; Fig. 9B, panel e).
Jointly, these results suggest that the loss of TgLCAT expression leads to an egress defect, and its overexpression confers an egress advantage.
TgLCAT-null Parasites Replicate Slower than Parental Parasites in Vitro
Because TgLCAT is secreted into the PV during the intracellular growth of Toxoplasma, we hypothesized that expression of TgLCAT may proffer a growth advantage in cultured cells. First, we performed plaque-based assays to follow the intracellular development of the parasite over time. Fibroblasts were infected with the Δlcat, Δlcat::LCAT, and parental strains for 7 days to allow for multiple rounds of invasion, replication, and egress. We measured the size of the lysed area in infected monolayers for each strain and observed that Δlcat parasites developed ∼1.5 times slower and Δlcat::LCAT parasites ∼1.3 times faster than the parental strain (Fig. 10A). These significant differences in plaque sizes may be a consequence of the parasite's ability to egress and/or to replicate. To more directly monitor the replication rate of each strain, we counted the number of parasites per PV (16 h and 36 h p.i.). At both time points, the PV of Δlcat parasites contained significantly fewer parasites than the PV of the parental strain; no difference in replication rate was observed between Δlcat::LCAT and parental parasites (Fig. 10B). We confirmed these observations by measuring the incorporation of radiolabeled uracil into the parasites grown for 24 and 48 h, and Δlcat parasites displayed a significant growth delay after 2 days in culture, although Δlcat::LCAT parasites showed no defect (Fig. 10C).
FIGURE 10.
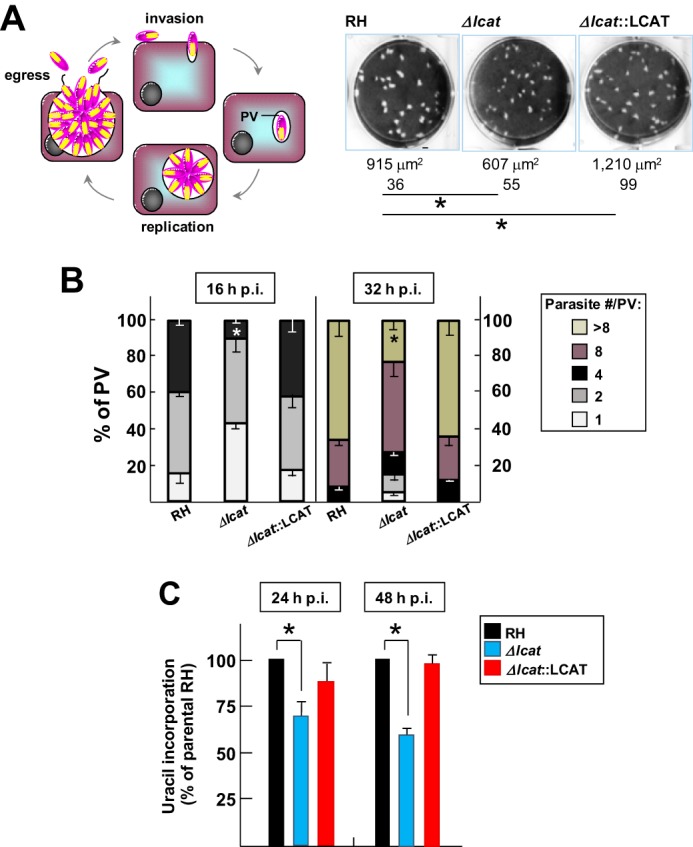
Growth rate of Δlcat parasites in cultured cells. A, comparison of growth rate of Δlcat with parental and LCAT-complemented strains using plaque assays. Left, schematic representation of the parasite lytic cycle. Right, representative images of area of destroyed cells by the parasites. The mean area of the plaques in μm2 ± S.D. was calculated from three independent experiments. *, p < 0.05. B, quantification of replication rate of Δlcat, Δlcat::LCAT, and parental parasites. The distribution of PV sizes expressed in % was determined at 16 and 36 h after infection of fibroblasts with these three strains. Data represent the means ± S.D. for at least 75 randomly selected vacuoles in three different experiments. *, p < 0.05. C, uracil incorporation assays. Fibroblasts were infected for 24 or 48 h with 5 × 104 Δlcat, RH, and Δlcat::LCAT parasites. Data in % are expressed relative to RH parasites taken as 100% ± S.D. from three separate experiments. Differences between values of RH and Δlcat parasites were statistically significant (*, p < 0.01).
TgLCAT Is Associated with Virulence in Vivo
Finally, to assess the importance of TgLCAT for in vivo infections, we inoculated outbred mice intradermally with freshly harvested Δlcat, Δlcat::LCAT, and parental parasites, and we monitored mouse viability daily. Mice infected with 10 or 50 wild-type parasites generally died within 22 days. In three independent assays, deletion of lcat revealed an attenuation of virulence: mice infected with 50 Δlcat parasites died 10 days later than the parental parasites, and mouse inoculation with 10 Δlcat parasites resulted in animal survival to the end point (Fig. 11). Interestingly, Δlcat::LCAT overexpressors were more virulent than parental parasites, as mice died systematically 1 week earlier.
FIGURE 11.
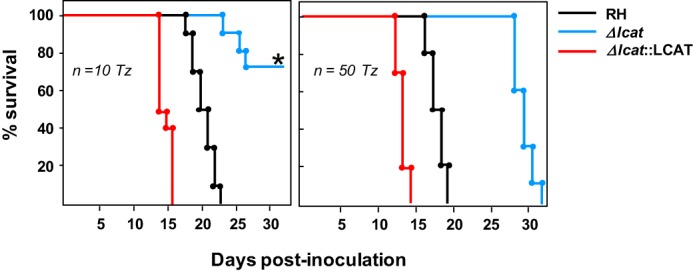
Virulence of Δlcat and Δlcat::LCAT parasites in vivo. Virulence in outbred mice following inoculation of Δlcat, RH, and Δlcat::LCAT parasites. Female BALB/c mice were infected subcutaneously with 10 or 50 tachyzoites (Tz) for each strain, and the viability of the mice was monitored daily. Sero-conversion of all surviving mice was confirmed by ELISA 3 weeks p.i. Shown are mean values from four independent infections using 10 mice per strain for each assay. The asterisk denotes a statistically significant time until moribundity of the Δlcat strain compared with the parental strain as determined using the Kaplan-Meier estimator (p < 0.01). 70% of Δlcat strain-infected mice survived to the end point.
Discussion
Microbial PLA2 enzymes have attracted increasing interest as drug targets due to their participation in diverse pathophysiological settings of infection. In this study, we have characterized the single protein of T. gondii that harbors a PLA2 motif and is a close homologue to LCAT enzymes. TgLCAT has the dual enzymatic activity of a PLA2 and a cholesteryl esterase, and it localizes to both the IVN of the PV and the parasite plasma membrane, depending on the parasite stage. LCAT-deficient parasites exhibit both replication and egress delays, which suggests the participation of TgLCAT in distinct functions to sustain the infectivity of the parasite throughout its life cycle. The involvement of TgLCAT in lipid membrane remodeling could benefit T. gondii during egress, by destabilizing membranes to facilitate parasite escape from host cells and during replication by reshaping the IVN to possibly supply the parasite with nutrients, as the IVN has been supposedly involved in nutrient uptake.
Role of TgLCAT for Egress
The event of egress, which liberates infectious progeny from a nutritionally exhausted habitat, is a crucial step in the propagation of intracellular pathogens within a host. The exit of T. gondii from mammalian cells is an active process regulated by the parasite itself, and not a breakdown of host cell membranes due to parasite overload (51). We observed loss of TgLCAT delays egress, whereas overexpression of this enzyme hastens this process, suggesting that TgLCAT may act as a facilitator for parasite exit from host cells. In fact, exposure of infected cells with a PLA2 inhibitor extends the time of egress. Comparable findings have been reported for the malaria parasite P. berghei, which expresses an LCAT-like enzyme PbP, at the surface of sporozoites (the insect form that infects mammalian hosts) and on the PV membrane of intrahepatic parasites (the mammalian form that multiplies in the liver (43, 52)). PbPL-null sporozoites are impaired in their ability to pass through cell monolayers in vitro and to migrate through mammalian tissues to reach the liver, whereas PbLP-deficient liver forms have delayed release from liver cells, presumably due to a defect in PV membrane disruption. More broadly, several intracellular bacteria also secrete phospholipases to break down host membranes and establish successful infections in mammalian hosts (53, 54).
We demonstrated that TgLCAT is an active phospholipase, able to attack phospholipids both of cellular (red blood cells) and non-cellular (liposome) sources. The PLA2 activity of recombinant TgLCAT (average of 1.2 nmol of fatty acid/h/μg of LCAT) is similar to human LCAT, which hydrolyzes about 40% of free fatty acid in 1 h (average of 1.9 nmol of fatty acid/h/μg of LCAT) (55). Lysophosphatidylcholine is a very potent membrane-perturbing agent, causing cytolysis (56–59). In contrast to phospholipids that possess a cylindrical molecular shape with a bulky hydrophilic headgroup similar in size to fatty acid chains, lysophospholipids consist of a large polar head and a thin hydrophobic tail yielding a cone shape. Migrating cone-shaped lysophospholipids into an outer leaflet of the bilayer enhances the intrinsic curvature and surface area of the outer leaflet of the cell membrane, leading to modifications of the mechanical properties of the bilayer and potentially facilitating the egress of T. gondii from the host cell.
The process of egress mediated by T. gondii seems to be multifactorial. It involves the activity of a parasite-expressed perforin-like protein, TgPLP1 (26, 60), and of host enzymes activated by the parasite for rupturing membranes from the inside out (51). The mechanism of action of TgPLP1 may be linked to the protein capability to induce pores (∼10 nm in size) in the PV membrane, which may permit passage of effectors across this membrane to aid in egress. Toxoplasma egress is a calcium-dependent process as exposure to calcium ionophores results in the rapid escape of parasites from cells. An increase in calcium induces the exocytosis of micronemes containing TgPLP1 and subsequent host cell lysis. The enzymatic activity of TgLCAT may contribute to the calcium-dependent events that lead to parasite egress. Indeed, by disrupting the integrity and permeability of the cell membrane, lysophospholipids induce changes in the conformation and activity of numerous membrane proteins, e.g. ion channels (57). If TgLCAT forms lysophospholipids within the PV membrane, the concentration of these lipids in this membrane may result in local membranous injuries and increases in PV membrane permeability, allowing the passage of molecules/ions that may subsequently activate downstream effectors required for egress, such as TgPLP1. The calcium-independent activity of TgLCAT and the calcium-dependent pathway of TgPLP1 may have synergic roles in the egress of Toxoplasma from cells, suggesting multiple layers of regulation for this event. Any potential functional redundancy between TgPLP1 and TgLCAT could be verified by deleting both genes from Toxoplasma genome and examining the ability of ΔΔlcat/plp1 parasites to egress.
Involvement of TgLCAT during Replication
At the onset of division, T. gondii secretes TgLCAT into the PV, specifically onto the IVN. The IVN, a network of tubules and vesicles, is generated from proteins and lipids secreted by the parasite as well as lipids scavenged from the host cell, including phosphatidylcholine, cholesterol from host low density lipoproteins (LDL), and sphingolipids from the host Golgi (21, 62–65). The distribution of TgLCAT to the IVN suggests a role for this enzyme in the remodeling of the IVN membranes by producing lysophosphatidylcholine that can shape membranes and change their curvature. A precedent for potential activity of lipid-modifying enzymes in the PV is illustrated by a phosphatidylserine decarboxylase, TgPSD, that is secreted by the parasite into the PV lumen (66). Phosphatidylethanolamine produced by TgPSD is a potent inducer of changes in protein conformation, which influences the lateral movement and activity of membrane-bound proteins (67). Thus, intravacuolar TgPSD1 and TgLCAT may have the potential to regulate the sequestration or processing of host-derived vesicles and lipids by providing an optimal bilayer environment for PV protein functions or liberating host lipids present in vesicles for the parasite (62, 64). To this point, our preliminary observations illustrate the distribution of TgLCAT on membranes surrounding host Rab vesicles sequestered into the PV.4
In addition to processing phospholipids, TgLCAT is an esterase for cholesterol, suggesting that cholesteryl esters may be synthesized in the PV. The transesterification capacity of TgLCAT in our assays is lower than that reported for human LCAT (average 2.1 nmol of cholesteryl esters/h/μg of LCAT) (55) as recombinant TgLCAT produces 50 times fewer cholesteryl esters, although TgLCAT has a high affinity for cholesterol as substrate. T. gondii scavenges cholesterol from LDL in host endocytic compartments (62). Incubating infected cells with excess LDL leads to a significant increase in cholesterol levels in the parasite as a result of its uncontrolled uptake of cholesterol. To circumvent toxic accumulation of free cholesterol, the parasite activates two acyl-CoA:cholesterol acyltransferase enzymes that are dedicated to the synthesis of cholesteryl esters for storage in lipid bodies (18, 19). Inhibition of T. gondii acyl-CoA:cholesterol acyltransferase activities is detrimental for growth, demonstrating the importance of maintaining proper levels of cholesterol in the parasite interior. As T. gondii acyl-CoA:cholesterol acyltransferase enzymes adjust the levels of free cholesterol within the parasite, TgLCAT may function in preventing the accumulation of free cholesterol inside the PV by promoting the esterification of this lipid scavenged in excess from the host cell.
Our EM observations illustrate the presence of TgLCAT in dense granules prior to secretion. These organelles secrete their contents, GRA proteins, into the PV or beyond the PV membrane. There is a burgeoning concept that distinct dense granule subpopulations exist in the parasite, with different GRA protein contents and secretion dynamics. Conventional GRA proteins, e.g. GRA1 to GRA9, GRA12, GRA14, GRA21, GRA23, and GRA25, play a role in transforming the PV into a metabolically active compartment and its maturation into a tissue cyst (46). They are typified by several common characteristics as follows: (i) lack of homology with one another or any known proteins; (ii) a monomeric structure with molecular masses ranging from 20 to 75 kDa; (iii) an N-terminal hydrophobic signal peptide; (iv) high transcription during the tachyzoite and/or bradyzoite stage; (v) peak of secretion at 10–30 min p.i. (tachyzoite stage); (vi) partial solubility and membrane association via a single hydrophobic α-helical domain; (vii) dispersion in the PV lumen, anchorage to the PV membrane, and/or association with the IVN; and (viii) a lack of lethality upon their gene deletion. Recent studies, however, have identified GRA-like proteins stored in dense granules that have unrelated functions and different physical properties or transcriptional profiles to the “conventional” GRA proteins. For example, GRA15, GRA16, and GRA24 are “unconventional” GRA proteins as they secreted beyond the PV membrane (∼16 to 32 h p.i.) and targeted to the host nucleus to reprogram host cell activities (68). GRA22 expression peaks at the sporozoite stage, and this protein also functions as a regulator of T. gondii egress (69), which together with TgLCAT establishes an unanticipated role for dense granules in the control of Toxoplasma exit from host cells. As with other unconventional GRA proteins, TgLCAT diverges from conventional GRA proteins based on its homology with LCAT enzymes, higher molecular mass, localization to the parasite plasma membrane, involvement in egress, and secretion into the PV only from 7 h p.i. until late in infection.
The mechanism used by TgLCAT to tether to membranes, e.g. parasite plasma membrane or the IVN, remains to be elucidated. However, the sequence of TgLCAT has an overall hydrophobic character and contains potential interfacial binding sites for lipids, such as several extended linear sequences of hydrophobic amino acids at the N terminus, which may mediate TgLCAT interactions with a membrane bilayer. TgLCAT contains two sets of disulfide bridges meditated by cysteines that are present in human LCAT. One of these disulfide bridges (Cys-74–Cys-98) was reported to be essential for the binding of human LCAT to lipoprotein surfaces (8), and by analogy, this function may be conserved in TgLCAT. TgLCAT is synthesized as an ∼83-kDa protein that is further cleaved into ∼62- and ∼33-kDa fragments. The cleaved products are predominantly secreted by the parasite. Interestingly, the catalytic triad of LCAT is split between the two fragments, suggesting that neither fragment is enzymatically active by itself. These two fragments need then to be restored for enzymatic activity. The TgLCAT sequence contains a structural coiled-coil motif, located in both fragments, which could allow their dimerization by interhelical contact and the restoration of the catalytic site. In support of this hypothesis, we detected higher molecular species for TgLCAT in the presence of a cell-permeable cross-linker. The cleavage of TgLCAT into two fragments following their reassembly may represent a post-translational regulatory mechanism in the parasite.
TgLCAT influences the pathogenicity of T. gondii as TgLCAT-null parasites display reduced virulence in vivo. Likewise, mice infected with parasites overexpressing TgLCAT succumbed to infection before animals infected with parental parasites. Secreted PLA2 enzymes have been implicated in virulence in many organisms. In bacteria, these enzymes are involved in host cell invasion, phagosomal escape, and initiation of a pro-inflammatory response (70, 71). The localization of TgLCAT on the surface of extracellular parasites may confer an advantage for rapid egress and dissemination within the host and explain the hypervirulence of overexpressors. The TgLCAT product lysophosphatidylcholine, aside from its detergent-like action, is also a bioactive lipid mediator that acts as an immune suppressor (72). It has been documented that this lipid is involved in the blockade of nitric oxide production, stimulation of reactive oxygen species, modulation of the Toll-like receptor-mediated signaling pathway, or attenuation of pathogen-induced immune responses by macrophages and dendritic cells. As TgLCAT is surface-exposed, it raises the possibility that phospholipids on the parasite plasma membrane may be substrates, thus allowing the local production of lysophosphatidylcholine from phosphatidylcholine and impacting the outcome of infection. Another example emphasizing the influence of phospholipid composition at the T. gondii plasma membrane on immune evasion is illustrated in studies showing that T. gondii survival in activated macrophages depends on phosphatidylserine exposure in the outer leaflet of the plasma membrane, which results in inhibition of nitric oxide production (61, 73). More detailed characterization on the host immune responses activated upon infection with parasite strains either deficient in TgLCAT or overexpressing TgLCAT will be required to assess the mechanisms of virulence of TgLCAT in the mouse infection model.
Author Contributions
I. C. conceived and coordinated the study, did the experiments related to enzymatic properties and EM studies, and wrote the paper. J. D. R. performed the fluorescence microscopy analyses and edited the manuscript. V. P., K. E., and D. S. R. conceived and/or did the molecular constructs and analyses. V. P., I. C., and M. E. G. performed the virulence assays. A. S. and A. K. performed the phylogenetic analyses. A. S. and V. B. C. contributed to the egress assays and modeling of TgLCAT. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to all members of the Coppens laboratory for their helpful discussions during the course of this work. We are thankful to Kimberly Zichichi at the Yale Center for Cell and Molecular Imaging for excellent assistance with electron microscopy.
This work was supported by National Institutes of Health Grant AI060767 from NIAID (to I. C.) and in part by the Intramural Research Program Grant AI001018 from NIAID (to M. E. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
V. B. Carruthers, unpublished data.
J. D. Romano, S. J. Nolan, R. Hsia, and I. Coppens, manuscript in preparation.
- PLA2
- phospholipase A2
- PV
- parasitophorous vacuole
- LCAT
- lecithin:cholesterol acyltransferase
- RACE
- rapid amplification of cDNA end
- 4-BPB
- 4-bromophenacyl bromide
- IVN
- intravacuolar network
- DPPC
- 1-palmitoyl-2[2-palmitoyl-9,10-3H]sn-glycero-3-phosphocholine
- HFF
- human foreskin fibroblast
- PC
- phosphatidylcholine
- qRT
- quantitative RT
- LDH
- lactate dehydrogenase
- DSP
- dithiobis(succinimidylpropionate)
- DTNB
- dithiobis(2-nitrobenzoic acid)
- IFA
- immunofluorescence assay
- rTgLCAT
- recombinant TgLCAT
- LPLA2
- lysosomal phospholipase A2.
References
- 1. Murakami M., Taketomi Y., Sato H., and Yamamoto K. (2011) Secreted phospholipase A2 revisited. J. Biochem. 150, 233–255 [DOI] [PubMed] [Google Scholar]
- 2. Lambeau G., and Gelb M. H. (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77, 495–520 [DOI] [PubMed] [Google Scholar]
- 3. Glomset J. A. (1968) The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 9, 155–167 [PubMed] [Google Scholar]
- 4. Pownall H. J., Pao Q., and Massey J. B. (1985) Acyl chain and headgroup specificity of human plasma lecithin:cholesterol acyltransferase. Separation of matrix and molecular specificities. J. Biol. Chem. 260, 2146–2152 [PubMed] [Google Scholar]
- 5. Christiaens B., Vanloo B., Gouyette C., Van Vynckt I., Caster H., Taveirne J., Verhee A., Labeur C., Peelman F., Vandekerckhove J., Tavernier J., and Rosseneu M. (2000) Headgroup specificity of lecithin cholesterol acyltransferase for monomeric and vesicular phospholipids. Biochim. Biophys. Acta 1486, 321–327 [DOI] [PubMed] [Google Scholar]
- 6. Rousset X., Vaisman B., Amar M., Sethi A. A., and Remaley A. T. (2009) Lecithin:cholesterol acyltransferase–from biochemistry to role in cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 16, 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peelman F., Vinaimont N., Verhee A., Vanloo B., Verschelde J. L., Labeur C., Seguret-Mace S., Duverger N., Hutchinson G., Vandekerckhove J., Tavernier J., and Rosseneu M. (1998) A proposed architecture for lecithin cholesterol acyl transferase (LCAT): identification of the catalytic triad and molecular modelling. Protein Sci. 7, 587–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sorci-Thomas M., Kearns M. W., and Lee J. P. (1993) Apolipoprotein A-I domains involved in lecithin-cholesterol acyltransferase activation. Structure:function relationships. J. Biol. Chem. 268, 21403–21409 [PubMed] [Google Scholar]
- 9. Schaefer E. J., Anthanont P., and Asztalos B. F. (2014) High-density lipoprotein metabolism, composition, function, and deficiency. Curr. Opin. Lipidol. 25, 194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saeedi R., Li M., and Frohlich J. (2015) A review on lecithin:cholesterol acyltransferase deficiency. Clin. Biochem. 48, 472–475 [DOI] [PubMed] [Google Scholar]
- 11. Oelkers P., Tinkelenberg A., Erdeniz N., Cromley D., Billheimer J. T., and Sturley S. L. (2000) A lecithin cholesterol acyltransferase-like gene mediates diacylglycerol esterification in yeast. J. Biol. Chem. 275, 15609–15612 [DOI] [PubMed] [Google Scholar]
- 12. Dahlqvist A., Stahl U., Lenman M., Banas A., Lee M., Sandager L., Ronne H., and Stymne S. (2000) Phospholipid:diacylglycerol acyltransferase: an enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants. Proc. Natl. Acad. Sci. U.S.A. 97, 6487–6492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ståhl U., Carlsson A. S., Lenman M., Dahlqvist A., Huang B., Banas W., Banas A., and Stymne S. (2004) Cloning and functional characterization of a phospholipid:diacylglycerol acyltransferase from Arabidopsis. Plant Physiol. 135, 1324–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luft B. J., and Remington J. S. (1992) Toxoplasma encephalatis in AIDS. Clin. Infect. Dis. 15, 211–222 [DOI] [PubMed] [Google Scholar]
- 15. Nissapatorn V., Lee C., Quek K. F., Leong C. L., Mahmud R., and Abdullah K. A. (2004) Toxoplasmosis in HIV/AIDS patients: a current situation. Jpn. J. Infect. Dis. 57, 160–165 [PubMed] [Google Scholar]
- 16. Ramakrishnan S., Serricchio M., Striepen B., and Bütikofer P. (2013) Lipid synthesis in protozoan parasites: a comparison between kinetoplastids and apicomplexans. Prog. Lipid Res. 52, 488–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coppens I. (2013) Targeting lipid biosynthesis and salvage in apicomplexan parasites for improved chemotherapies. Nat. Rev. Microbiol. 11, 823–835 [DOI] [PubMed] [Google Scholar]
- 18. Nishikawa Y., Quittnat F., Stedman T. T., Voelker D. R., Choi J. Y., Zahn M., Yang M., Pypaert M., Joiner K. A., and Coppens I. (2005) Host cell lipids control cholesteryl ester synthesis and storage in intracellular Toxoplasma. Cell Microbiol. 7, 849–867 [DOI] [PubMed] [Google Scholar]
- 19. Lige B., Sampels V., and Coppens I. (2013) Characterization of a second sterol-esterifying enzyme in Toxoplasma highlights the importance of cholesterol storage pathways for the parasite. Mol. Microbiol. 87, 951–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ehrenman K., Sehgal A., Lige B., Stedman T. T., Joiner K. A., and Coppens I. (2010) Novel roles for ATP-binding cassette G transporters in lipid redistribution in Toxoplasma. Mol. Microbiol. 76, 1232–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coppens I., Dunn J. D., Romano J. D., Pypaert M., Zhang H., Boothroyd J. C., and Joiner K. A. (2006) Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125, 261–274 [DOI] [PubMed] [Google Scholar]
- 22. Roos D. S., Donald R. G., Morrissette N. S., and Moulton A. L. (1994) Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 45, 27–63 [DOI] [PubMed] [Google Scholar]
- 23. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nishi M., Hu K., Murray J. M., and Roos D. S. (2008) Organellar dynamics during the cell cycle of Toxoplasma gondii. J. Cell Sci. 121, 1559–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soldati D., Kim K., Kampmeier J., Dubremetz J. F., and Boothroyd J. C. (1995) Complementation of a Toxoplasma gondii ROP1 knock-out mutant using phleomycin selection. Mol. Biochem. Parasitol. 74, 87–97 [DOI] [PubMed] [Google Scholar]
- 26. Roiko M. S., and Carruthers V. B. (2013) Functional dissection of Toxoplasma gondii perforin-like protein 1 reveals a dual domain mode of membrane binding for cytolysis and parasite egress. J. Biol. Chem. 288, 8712–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aron L., Jones S., and Fielding C. J. (1978) Human plasma lecithin-cholesterol acyltransferase. Characterization of cofactor-dependent phospholipase activity. J. Biol. Chem. 253, 7220–7226 [PubMed] [Google Scholar]
- 28. Fisher A. B., and Dodia C. (2001) Lysosomal-type PLA2 and turnover of alveolar DPPC. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L748–L754 [DOI] [PubMed] [Google Scholar]
- 29. Coppens I., and Joiner K. A. (2003) Host but not parasite cholesterol controls Toxoplasma cell entry by modulating organelle discharge. Mol. Biol. Cell 14, 3804–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Donald R. G., Carter D., Ullman B., and Roos D. S. (1996) Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J. Biol. Chem. 271, 14010–14019 [DOI] [PubMed] [Google Scholar]
- 31. Huynh M. H., Rabenau K. E., Harper J. M., Beatty W. L., Sibley L. D., and Carruthers V. B. (2003) Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 22, 2082–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kafsack B. F., Beckers C., and Carruthers V. B. (2004) Synchronous invasion of host cells by Toxoplasma gondii. Mol. Biochem. Parasitol. 136, 309–311 [DOI] [PubMed] [Google Scholar]
- 33. Striepen B., and Soldati-Favre D. (2013) in Toxoplasma gondii The Model Apicomplexan: Perspectives and Methods (Weiss L. M., and Kim K., eds) pp. 408–409, Academic Press, London, UK [Google Scholar]
- 34. Hiraoka M., Abe A., and Shayman J. A. (2002) Cloning and characterization of a lysosomal phospholipase A2, 1-O-acylceramide synthase. J. Biol. Chem. 277, 10090–10099 [DOI] [PubMed] [Google Scholar]
- 35. Welti R., Mui E., Sparks A., Wernimont S., Isaac G., Kirisits M., Roth M., Roberts C. W., Botté C., Maréchal E., and McLeod R. (2007) Lipidomic analysis of Toxoplasma gondii reveals unusual polar lipids. Biochemistry 46, 13882–13890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walzer K. A., Adomako-Ankomah Y., Dam R. A., Herrmann D. C., Schares G., Dubey J. P., and Boyle J. P. (2013) Hammondia hammondi, an avirulent relative of Toxoplasma gondii, has functional orthologs of known T. gondii virulence genes. Proc. Natl. Acad. Sci. U.S.A. 110, 7446–7451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arisue N., and Hashimoto T. (2015) Phylogeny and evolution of apicoplasts and apicomplexan parasites. Parasitol. Int. 64, 254–259 [DOI] [PubMed] [Google Scholar]
- 38. Yeoh L. M., Goodman C. D., Hall N. E., van Dooren G. G., McFadden G. I., and Ralph S. A. (2015) A serine-arginine-rich (SR) splicing factor modulates alternative splicing of over a thousand genes in Toxoplasma gondii. Nucleic Acids Res. 43, 4661–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Glukhova A., Hinkovska-Galcheva V., Kelly R., Abe A., Shayman J. A., and Tesmer J. J. (2015) Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 6, 6250–6262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bierbaum T. J., Bouma S. R., and Huestis W. H. (1979) A mechanism of erythrocyte lysis by lysophosphatidylcholine. Biochim. Biophys. Acta 555, 102–110 [DOI] [PubMed] [Google Scholar]
- 41. Silverman B. A., Weller P. F., and Shin M. L. (1984) Effect of erythrocyte membrane modulation by lysolecithin on complement-mediated lysis. J. Immunol. 132, 386–391 [PubMed] [Google Scholar]
- 42. Chawla D., and Owen J. S. (1995) Secretion of active human lecithin-cholesterol acyltransferase by insect cells infected with a recombinant baculovirus. Biochem. J. 309, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhanot P., Schauer K., Coppens I., and Nussenzweig V. (2005) A surface phospholipase is involved in the migration of plasmodium sporozoites through cells. J. Biol. Chem. 280, 6752–6760 [DOI] [PubMed] [Google Scholar]
- 44. Francone O. L., and Fielding C. J. (1991) Effects of site-directed mutagenesis at residues cysteine-31 and cysteine-184 on lecithin-cholesterol acyltransferase activity. Proc. Natl. Acad. Sci. U.S.A. 88, 1716–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carruthers V., and Boothroyd J. C. (2007) Pulling together: an integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 10, 83–89 [DOI] [PubMed] [Google Scholar]
- 46. Mercier C., and Cesbron-Delauw M. F. (2015) Toxoplasma secretory granules: one population or more? Trends Parasitol. 31, 60–71 [DOI] [PubMed] [Google Scholar]
- 47. Mercier C., Dubremetz J. F., Rauscher B., Lecordier L., Sibley L. D., and Cesbron-Delauw M. F. (2002) Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol. Biol. Cell 13, 2397–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guérardel Y., Leleu D., Coppin A., Liénard L., Slomianny C., Strecker G., Ball S., and Tomavo S. (2005) Amylopectin biogenesis and characterization in the protozoan parasite Toxoplasma gondii, the intracellular development of which is restricted in the HepG2 cell line. Microbes Infect. 7, 41–48 [DOI] [PubMed] [Google Scholar]
- 49. Corbin J. D., and Francis S. H. (2002) Pharmacology of phosphodiesterase-5 inhibitors. Int. J. Clin. Pract. 56, 453–459 [PubMed] [Google Scholar]
- 50. Lourido S., Tang K., and Sibley L. D. (2012) Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J. 31, 4524–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blackman M. J., and Carruthers V. B. (2013) Recent insights into apicomplexan parasite egress provide new views to a kill. Curr. Opin. Microbiol. 16, 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burda P. C., Roelli M. A., Schaffner M., Khan S. M., Janse C. J., and Heussler V. T. (2015) A Plasmodium phospholipase is involved in disruption of the liver stage parasitophorous vacuole membrane. PLoS Pathog. 11, e1004760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith G. A., Marquis H., Jones S., Johnston N. C., Portnoy D. A., and Goldfine H. (1995) The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 63, 4231–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heffernan B. J., Thomason B., Herring-Palmer A., Shaughnessy L., McDonald R., Fisher N., Huffnagle G. B., and Hanna P. (2006) Bacillus anthracis phospholipases C facilitate macrophage-associated growth and contribute to virulence in a murine model of inhalation anthrax. Infect. Immun. 74, 3756–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang J., Gebre A. K., Anderson R. A., and Parks J. S. (1997) Amino acid residue 149 of lecithin:cholesterol acyltransferase determines phospholipase A2 and transacylase fatty acyl specificity. J. Biol. Chem. 272, 280–286 [DOI] [PubMed] [Google Scholar]
- 56. Weltzien H. U. (1979) Cytolytic and membrane-perturbing properties of lysophosphatidylcholine. Biochim. Biophys. Acta 559, 259–287 [DOI] [PubMed] [Google Scholar]
- 57. Martin I., Dubois M. C., Saermark T., Epand R. M., and Ruysschaert J. M. (1993) Lysophosphatidylcholine mediates the mode of insertion of the NH2-terminal SIV fusion peptide into the lipid bilayer. FEBS Lett. 333, 325–330 [DOI] [PubMed] [Google Scholar]
- 58. Martin G. P., el-Hariri L. M., and Marriott C. (1992) Bile salt- and lysophosphatidylcholine-induced membrane damage in human erythrocytes. J. Pharm. Pharmacol. 44, 646–650 [DOI] [PubMed] [Google Scholar]
- 59. Safeukui I., Buffet P. A., Deplaine G., Perrot S., Brousse V., Ndour A., Nguyen M., Mercereau-Puijalon O., David P. H., Milon G., and Mohandas N. (2012) Quantitative assessment of sensing and sequestration of spherocytic erythrocytes by the human spleen. Blood 120, 424–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kafsack B. F., Pena J. D., Coppens I., Ravindran S., Boothroyd J. C., and Carruthers V. B. (2009) Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 323, 530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Santos T. A., Portes Jde A., Damasceno-Sá J. C., Caldas L. A., Souza Wd, Damatta R. A., and Seabra S. H. (2011) Phosphatidylserine exposure by Toxoplasma gondii is fundamental to balance the immune response granting survival of the parasite and of the host. PLoS ONE 6, e27867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coppens I., Sinai A. P., and Joiner K. A. (2000) Toxoplasma gondii exploits host low-density lipoprotein receptor-mediated endocytosis for cholesterol acquisition. J. Cell Biol. 149, 167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Caffaro C. E., and Boothroyd J. C. (2011) Evidence for host cells as the major contributor of lipids in the intravacuolar network of Toxoplasma-infected cells. Eukaryot. Cell 10, 1095–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Romano J. D., Sonda S., Bergbower E., Smith M. E., and Coppens I. (2013) Toxoplasma gondii salvages sphingolipids from the host Golgi through the rerouting of selected Rab vesicles to the parasitophorous vacuole. Mol. Biol. Cell 24, 1974–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. de Melo E. J., and De Souza W. (1996) Pathway of C6-NBD-ceramide on the host cell infected with Toxoplasma gondii. Cell Struct. Funct. 21, 47–52 [DOI] [PubMed] [Google Scholar]
- 66. Gupta N., Hartmann A., Lucius R., and Voelker D. R. (2012) The obligate intracellular parasite Toxoplasma gondii secretes a soluble phosphatidylserine decarboxylase. J. Biol. Chem. 287, 22938–22947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grzelczyk A., and Gendaszewska-Darmach E. (2013) Novel bioactive glycerol-based lysophospholipids: new data–new insight into their function. Biochimie. (Paris) 95, 667–679 [DOI] [PubMed] [Google Scholar]
- 68. Bougdour A., Tardieux I., and Hakimi M. A. (2014) Toxoplasma exports dense granule proteins beyond the vacuole to the host cell nucleus and rewires the host genome expression. Cell Microbiol. 16, 334–343 [DOI] [PubMed] [Google Scholar]
- 69. Okada T., Marmansari D., Li Z. M., Adilbish A., Canko S., Ueno A., Shono H., Furuoka H., and Igarashi M. (2013) A novel dense granule protein, GRA22, is involved in regulating parasite egress in Toxoplasma gondii. Mol. Biochem. Parasitol. 189, 5–13 [DOI] [PubMed] [Google Scholar]
- 70. Istivan T. S., and Coloe P. J. (2006) Phospholipase A in Gram-negative bacteria and its role in pathogenesis. Microbiology 152, 1263–1274 [DOI] [PubMed] [Google Scholar]
- 71. Sitkiewicz I., Stockbauer K. E., and Musser J. M. (2007) Secreted bacterial phospholipase A2 enzymes: better living through phospholipolysis. Trends Microbiol. 15, 63–69 [DOI] [PubMed] [Google Scholar]
- 72. Kabarowski J. H., Xu Y., and Witte O. N. (2002) Lysophosphatidylcholine as a ligand for immunoregulation. Biochem. Pharmacol. 64, 161–167 [DOI] [PubMed] [Google Scholar]
- 73. Seabra S. H., de Souza W., and Damatta R. A. (2004) Toxoplasma gondii exposes phosphatidylserine inducing a TGF-β1 autocrine effect orchestrating macrophage evasion. Biochem. Biophys. Res. Commun. 324, 744–752 [DOI] [PubMed] [Google Scholar]