

Abstract
As an innate defense mechanism, macrophages produce reactive oxygen species that weaken pathogens and serve as secondary messengers involved in immune function. The Gram-negative bacterium Francisella tularensis utilizes its antioxidant armature to limit the host immune response, but the mechanism behind this suppression is not defined. Here we establish that F. tularensis limits Ca2+ entry in macrophages, thereby limiting actin reorganization and IL-6 production in a redox-dependent fashion. Wild type (live vaccine strain) or catalase-deficient F. tularensis (ΔkatG) show distinct profiles in their H2O2 scavenging rates, 1 and 0.015 pm/s, respectively. Murine alveolar macrophages infected with ΔkatG display abnormally high basal intracellular Ca2+ concentration that did not increase further in response to H2O2. Additionally, ΔkatG-infected macrophages displayed limited Ca2+ influx in response to ionomycin, as a result of ionophore H2O2 sensitivity. Exogenously added H2O2 or H2O2 generated by ΔkatG likely oxidizes ionomycin and alters its ability to transport Ca2+. Basal increases in cytosolic Ca2+ and insensitivity to H2O2-mediated Ca2+ entry in ΔkatG-infected cells are reversed by the Ca2+ channel inhibitors 2-aminoethyl diphenylborinate and SKF-96365. 2-Aminoethyl diphenylborinate but not SKF-96365 abrogated ΔkatG-dependent increases in macrophage actin remodeling and IL-6 secretion, suggesting a role for H2O2-mediated Ca2+ entry through the transient receptor potential melastatin 2 (TRPM2) channel in macrophages. Indeed, increases in basal Ca2+, actin polymerization, and IL-6 production are reversed in TRPM2-null macrophages infected with ΔkatG. Together, our findings provide compelling evidence that F. tularensis catalase restricts reactive oxygen species to temper macrophage TRPM2-mediated Ca2+ signaling and limit host immune function.
Keywords: antioxidant, bacterial pathogenesis, calcium channel, catalase, cellular immune response, hydrogen peroxide, infection, macrophage, Francisella
Introduction
Francisella tularensis is a Gram-negative intracellular bacterium currently classified as a category A biological warfare agent. F. tularensis is transmissible through virtually all routes (airborne, contact, and ingestion), and inhalation of the bacteria is associated with a 50% lethality rate in the absence of antibiotic intervention (1, 2). These attributes of F. tularensis necessitate research into the mechanisms of bacterial pathogenicity and/or host response to make any advances in prevention or treatment of infection.
Upon infection, host macrophages take up Francisella via phagocytosis. Francisella has been shown to prevent the formation of the NADPH oxidase complex at the phagosomal membrane, which in turn allows its initial survival within the phagosome (3). Francisella subsequently escapes from the phagosome and proliferates within the cytoplasm prior to exiting the macrophage (4–6). As a defense mechanism, macrophages produce ROS including, but not limited to, superoxide (O2˙̄) and H2O2. All F. tularensis subspecies contain multiple mechanisms for ROS detoxification. Francisella superoxide dismutases (SodB and SodC) are responsible for converting superoxide to H2O2 and oxygen. H2O2 is further detoxified by catalase (KatG) and/or alkyl hydroperoxide reductase family members (AhpC) (7, 8). Although all subspecies of F. tularensis contain a similar repertoire of antioxidant proteins, their ability to evade destruction by host ROS is variable. We and other groups have previously demonstrated that the highly virulent SchuS4 strain is extremely resistant to ROS when compared with less virulent subspecies (7, 9, 10).
Francisella has been shown to subvert numerous host immune processes following infection to enhance survival. A pathway that is central to Francisella manipulation is the PI3K/Akt signal cascade (9, 11–14). Signal propagation through this path is highly reliant on phosphatase and kinase signaling, which have been shown to be regulated in part by the redox state of the cell (15). Our group has previously shown that there is a relationship between the scavenging capacity of Francisella and its ability to restrict redox-mediated protein modifications, thereby altering downstream signaling events. Specifically, the increased ability of Francisella to remove H2O2 from its environment allows for the preservation of host PTEN phosphatase activity, an important negative regulator of AKT (9). The rapid replication of this bacterium coupled with its ability to manipulate the host immune response allows Francisella to remain virtually undetected early after infection, contributing to its ability to widely disseminate.
Parallel to the ability of H2O2 to act on numerous pathways, calcium is also a small molecule mediator that facilitates cross-talk throughout the cell (15–19). Immune cell function is highly impacted by intracellular Ca2+ concentrations because many processes rely on local alterations of Ca2+. Subsequent to a rise in intracellular Ca2+ necessary for phagocytosis, Ca2+ signaling is also required for later fusion events, as well as the secretion of inflammatory cytokines (20–25). Several microarray studies have shown altered regulation of host genes important for Ca2+ homeostasis after Francisella infection, but no further studies into the contribution of Ca2+ signaling to the immune response have been conducted (26, 27). Several other intracellular bacteria including Mycobacterium spp., Salmonella spp., and Listeria spp. all have been shown to manipulate host Ca2+ signaling through various mechanisms, suggesting that manipulation by Francisella is also possible (17, 28–32).
Cytosolic Ca2+ homeostasis is maintained through mobilization of Ca2+ from both intracellular stores and the extracellular space. The interplay between intracellular and extracellular Ca2+ is regulated by diverse signals that control cell function and fate. Receptor-mediated Ca2+ signaling involves release of internal stores mainly from the endoplasmic reticulum and subsequent activation of Ca2+ influx channels at the plasma membrane (33). Other Ca2+ entry pathways exist that are not dependent on the state of internal Ca2+ stores. These pathways are mediated in part by members of the transient receptor potential (TRP)2 channel family, of which there are 28 channel members (34). Although most members of the TRP family are permeable to Ca2+, the selectivity varies greatly among members because of differences in both pore structure and regulation (35). Ca2+ mobilization is also intimately associated with the production of ROS. The transient receptor potential melastatin 2 (TRPM2) channels have recently been shown not only to be involved in regulation of immune function as it relates to bacterial infection but also to be gated by H2O2 through cysteine oxidation (36–38). We have identified a H2O2-sensitive mobilization of Ca2+ through TRPM2 in Francisella-infected macrophages that appears to be integral for the increased host inflammatory response upon infection with catalase-deficient F. tularensis, suggesting that H2O2 is responsible for TRPM2 activation in response to live vaccine strain (LVS) infection. This study identifies the critical components controlling redox-dependent Ca2+ mobilization that are crucial to the regulation of the inflammatory response to F. tularensis infection.
We propose that in addition to being a means of protection for itself, antioxidants of Francisella, and likely those of other intracellular pathogens, function to modulate Ca2+-dependent host cell signaling in a redox-dependent manner. We establish that Francisella is capable of scavenging host-derived ROS and that this scavenging leads to early modulation of immune Ca2+ signaling.
Experimental Procedures
Bacterial Strains, Cell Lines, and Media
F. tularensis subsp. Holarctica LVS (ATCC 29684; American Type Culture Collection, Manassas, VA) was used in this study. The catalase (katG) mutants were kindly provided by Dr. Anders Sjostedt. Dr. Karsten R. O. Hazlett generously provided wbtA, fopA, mglA, and iglC mutants. All bacteria were grown on chocolate agar plates supplemented with IsoVitaleX (Becton Dickinson) at 37 °C with 5% CO2. Bacteria were grown in Mueller Hinton broth supplemented with IsoVitaleX in a shaking incubator (160 rpm) at 37 °C where indicated. GFP-expressing LVS were created by transforming electrocompetent Francisella with the plasmid pkk::GFP. Selection of GFP-positive colonies was achieved with kanamycin-containing chocolate agar plates and confirmation via microscopy.
Mouse alveolar macrophage cells (ATCC® CRL-2019) and MH-S cells were cultured in RPMI medium (Cellgro) supplemented with 10% fetal bovine serum (Biowest) in a 37 °C humidified incubator that maintained 5% CO2 levels.
Generation of Bone Marrow-derived Macrophages
Femurs from adult WT C57BL/6 and TRPM2−/− mice (39) were used for generation of bone marrow-derived macrophages (BMDM).BMDM were isolated and differentiated as previously described (40).
Bacterial Scavenging Assay
Bacteria were grown in MHB broth to an A600 of 0.200 four times consecutively prior to being spun and washed with PBS. The bacteria were resuspended to an A600 of 0.200, and 1 × 109 bacteria were incubated with 50 μm H2O2 at 37 °C. At the indicated time points, the aliquots were removed, and H2O2 concentration was determined via the Amplex Red method (41).
ImageStream Analysis
MHS cells were grown to 90% confluence prior to infection. Cells were infected with an MOI of 100 for time indicated. 20,000 events were collected for each sample after gating out debris. Sample data were collected utilizing an ImageStream imaging flow cytometer from Amnis Corp. Data files were analyzed using IDEAS statistical analysis software (Amnis).
Phalloidin
Prior to analysis, cells were thoroughly washed with PBS and lifted with trypsin EDTA at the indicated time point. The cells were fixed and stained with Invitrogen Phalloidin Alexa 488 per the manufacturer's instructions. All cells were spun and resuspended at a concentration of 1 × 106 cells/60 μl for ImageStream analysis. Immediately prior to analysis, cells were vortexed to disrupt cell clumps. Utilizing a 488 laser, brightfield and GFP channels were collected.
Fura Red
Macrophages were loaded with Fura Red (Invitrogen) for 15 min at 4 °C and 30 min at 37 °C. The cells were then washed and infected with indicated bacteria for 1 h at an MOI of 100. The wells were washed three times with HBSS containing 100 mm CaCl2 and 1 mm dextrose and lifted with trypsin at the indicated time point. The cells were concentrated as described above. The samples were excited with a 488 laser, and the brightfield, GFP, and RFP channels were collected on the ImageStream.
An additional panel of LVS mutants was assayed for Fura Red fluorescence utilizing a high throughput plate reader method. Briefly, MHS cells were seeded in a 96-well plate 24 h prior to the experiment. The cells were then loaded and infected as described above. Following three HBSS washes, fluorescence was measured on a FlexStation 3 Molecular Devices plate reader with the software SoftMaxPro 5.4.1 at an excitation wavelength of 420 nm and emission at 650 nm.
Calcium Imaging
Macrophages were seeded on #30 glass coverslips 24 h prior to the experiment to a confluence of 80%. The cells were washed several times and loaded with 4 μm Fura-2-AM (Invitrogen) for 15 min at 4 °C and 30 min at 37 °C. The cells were then washed and infected with indicated bacteria for 1 h at an MOI of 100. Coverslips were washed three times with HBSS containing 2 mm CaCl2 and 1 mm dextrose. Fluorescence experiments were recorded and analyzed using a digital fluorescence imaging system (Intracellular Imaging). At the indicated times, the solutions on the coverslips were exchanged to include 300 μm hydrogen peroxide ± 2 mm calcium (42, 43).
Intracellular calcium levels were determined real time by ratiometric analysis of the calcium binding dye Fura-2-AM. This cell-permeable dye excites at 340 nm when bound to calcium and 380 nm when unbound. Fluorescence intensities were then measured at the emission wavelength of 510 nm. These intensities were represented as a ratio of 340/380.
Microplate-based Fura-2-AM Analysis
MHS cells were seeded on 96-well plates 24 h before performing assay. At the time of assay, the medium was removed, and the cells were loaded with 4 μm Fura-2-AM for 15 min at 4 °C followed by incubation at 37 °C for 30 min. Then the cells were washed with 1× HBSS with dextrose, three times. The 96-well plate was placed in a FlexStation 3 Molecular Devices plate reader, the treatments were added at specific time points using the Flex mode, and fluorescence measurements were obtained. For monitoring effects of H2O2 on ionomycin Ca2+ transport activity, the medium was removed, and cells were loaded with 4 μm Fura-2-AM for 15 min at 4 °C followed by incubation at 37 °C for 30 min. The cells were washed with 1× HBSS with dextrose and incubated with indicated doses of H2O2 at 37 °C for 10 min with no loss in cell viability. 10 μm ionomycin with 2 mm Ca2+ was added at 30 s during the read, and fluorescence traces were monitored for 240 s.
Cytokine Analysis
Secretion of proinflammatory cytokine IL-6 was measured 24 h postinfection. Mouse alveolar macrophage cells were plated in 6-well plates and incubated for 24 h at 37 °C. The cells were infected with MOI 100 or 50 ng of LPS and incubated for 24 h at 37 °C. Supernatants were collected and analyzed using a MSD Mouse Proinflammatory 7-plex ultrasensitive kit according to the manufacturer's instructions. Samples and standards were run in duplicate, and the data were collected utilizing a Sector 2400 imager (Meso Scale Discovery, Gaithersburg, MD).
Bone marrow-derived macrophages were assessed for increased transcription of IL-6 as follows. Total RNA was extracted from cells using an RNeasy isolation kit (Qiagen) per the manufacturer's instructions and stored at −80 °C until use. cDNA was synthesized utilizing a Maxima Universal First Strand cDNA synthesis kit (Fisher) with random hexamers. Real time PCR was performed with an Applied Biosystems 7500 real time thermocycler with SYBR green. Specific primers were used for each target: IL-6 sense, 5′-TGG AGT CAC AGA AGG AGT GGC TAA G-3′; and IL-6 antisense, 5′-TCT GAC CAC AGT GAG GAA TGT CCA C-3′. PCR conditions were as follows: 95 °C for 10 min, followed by 40 cycles of melting at 95 °C for 15 s, and annealing and elongation at 60 °C for 1 min. Product specificity was determined by melt curve CT values were normalized to a control housekeeping gene, GAPDH (sense, 5′-TTC ACC ACC ATG GAG AAG GC-3′; and antisense, 5′-GGC ATG GAC TGT GGT CAT GA-3′), and the fold change was found using the ΔΔCT method of analysis.
Statistical Analysis
All statistical analyses were performed with the GraphPad 5.04 statistical software package. Statistical significance was assessed using the two-tailed Student's t test or one-way analysis of variance with a Tukey-Kramer post-test. p values of <0.05 were considered significant.
Results
Infection with Catalase-deficient F. tularensis LVS Alters Macrophage Ca2+ Response
Previous studies from our lab and others have shown that F. tularensis antioxidants are capable of altering immune signaling postinfection through a disturbance of the AKT, MAPK, and NF-κB pathways (9, 44–46). Although ROS on their own can act as secondary messengers in the course of infection (15, 16, 47), the impact that ROS may have on the secondary messenger, Ca2+, has yet to be investigated in the context of Francisella. Other groups have shown that a number of Ca2+ channels are impacted by the intracellular redox state of a cell (48–51). Given our knowledge that infection with a catalase-deficient mutant of Francisella increases the intracellular steady state concentration of H2O2 within 30 min (1), we hypothesize that intracellular Ca2+ signaling may also be altered.
Several TRP channels have been implicated in the response to oxidative stress including members of the TRPC, TRPM, and TRPV families (52). Zou et al. (43) recently published a study illustrating the Ca2+ response of several different types of macrophages (both immortal cell lines and primary cells) to an exogenous H2O2 stimulus. Utilizing their experimental approach, we aimed to determine whether infection with Francisella will impact the ability of mouse alveolar macrophages to respond to a secondary stimulus shown to elicit a calcium response.
MHS cells were infected with wild type F. tularensis subspp. holartica LVS and catalase-deficient LVS ΔkatG), respectively, and the relative concentration of intracellular Ca2+ was assessed with the ratiometric dye Fura-2. One hour postinfection, baseline Ca2+ levels within ΔkatG-infected macrophages was significantly higher than LVS-infected cells (Fig. 1, A and B). Cells stimulated with 300 μm H2O2 in the absence of extracellular Ca2+ displayed no response when either strain was used for infection. However, when extracellular Ca2+ (2 mm) was reintroduced to the bath solution, both uninfected and LVS-infected MHS cells showed Ca2+ influx in response to H2O2, whereas the ΔkatG failed to respond (Fig. 1C). Additionally, macrophages infected with ΔkatG showed a reduced Ca2+ signal upon ionomycin treatment (Fig. 1A). Together, these data indicate that Francisella LVS catalase modulates macrophage Ca2+ homeostasis following infection.
FIGURE 1.

Antioxidant capacity of Francisella directly controls macrophage response to hydrogen peroxide stimulus. The calcium-sensitive ratiometric dye Fura-2-AM was utilized to obtain live cell measurements of intracellular calcium postinfection of the MHS mouse alveolar macrophage cell line. Calcium imaging was initiated 1 h postinfection, and mean traces with S.E. are plotted in A. At 300 s, medium was exchanged to include 300 μm H2O2; at 600 s, medium was exchanged to include 2 mm Ca2+; and at 2400 s, medium was exchanged to include 10 μm ionomycin. B depicts the baseline Fura ratio as measured from time 150–250. C illustrates the total change in intracellular calcium after stimulus. *, p < 0.01; **, p < 0.001; ***, p < 0.0001.
Francisella Catalase Is Necessary to Maintain Ca2+ Homeostasis Following Infection
To determine whether this disruption in Ca2+ homeostasis is exclusive to infected cells, relative intracellular Ca2+ levels were also measured with Fura Red, the fluorescence of which is inhibited by the presence of calcium (53). MHS cells were infected with GFP-expressing LVS, and imaging flow cytometry was used to monitor Fura Red intensity 1 h postinfection. Similar, to Fura-2, Fura Red intensity profiling revealed that intracellular Ca2+ is increased postinfection with ΔkatG as shown by the decrease in Fura Red fluorescence (Fig. 2A). Fig. 2B depicts images from the mean bins of each sample, illustrating the vast decrease in fluorescence after infection with ΔkatG.
FIGURE 2.
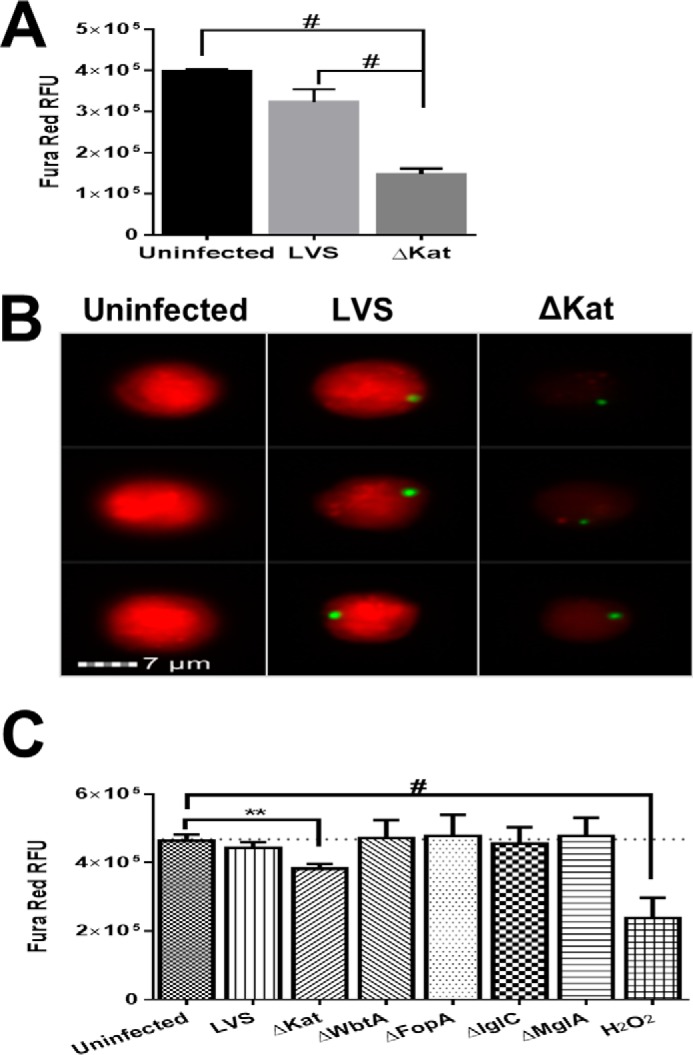
Francisella catalase is necessary to maintain calcium homeostasis following infection. Murine alveolar macrophages (MHS cell line) were infected with LVS or ΔkatG for 1 h. Macrophages were loaded with Fura Red, whose fluorescence is inhibited by calcium, lifted, and analyzed on ImageStream flow cytometer. A depicts the mean Fura Red fluorescence. B depicts representative images from the mean. C depicts screening of multiple LVS mutants was accomplished by the analysis of Fura Red fluorescence with a plate reader. 300 μm H2O2 was used as a control for calcium influx. The data represent the mean fluorescence intensity values ± S.E. of three independent experiments. **, p < 0.001; #, p < 0.00001.
To ensure that the alterations in intracellular Ca2+ are not attributed to the hypercytotoxicity that often accompanies engineered mutations in Francisella, we tested a panel of mutants that have been reported to display aberrant bacterial lysis and are inherently proinflammatory (54). Monack and co-workers (54) previously demonstrated that several Francisella mutants in the literature induce proinflammatory responses caused by decreased bacterial integrity and increased the release of pathogen-associated molecular patterns. The integrity of ΔkatG was assessed by scanning electron microscopy and vital staining and was found to be structurally sound (data not shown). To determine whether Ca2+ modulation was specific to the ΔkatG mutant, we chose a panel of four LVS mutants including ΔwbtA, ΔfopA, Δiglc, and ΔmglA (Table 1) to examine their impact on host cell Ca2+ modulation. Utilizing a high throughput assay, we monitored Fura Red fluorescence 1 h postinfection with the indicated bacterial strains in Fig. 2C. ΔkatG is the only mutant that exhibited a significant increase in intracellular Ca2+ as illustrated by a decrease in Fura Red fluorescence. Overall, these findings establish that Francisella catalase is likely the key participant in the regulation of host cell Ca2+ homeostasis.
TABLE 1.
List of Francisella mutant strains used in this study
| Mutant strain | Gene description | References |
|---|---|---|
| ΔkatG | Catalase | Ref. 7 |
| ΔwbtA | Involved in O-antigen synthesis | Ref. 76 |
| ΔfopA | Major outer membrane protein of the OmpA family | Ref. 76 |
| ΔiglC | Member of the Francisella intracellular growth locus; decreased KatG expression | Ref. 76 |
| ΔmglA | Transcriptional regulator; increased KatG expression | Ref. 77 |
Calcium Channel Inhibitors 2APB and SKF-96365 Prevent Increased Ca2+ in ΔkatG-infected MHS Cells
To determine whether a redox-sensitive plasma membrane ion channel controls the rise in intracellular Ca2+ postinfection, we employed two broad pharmacological inhibitors of Ca2+ influx channels: 2APB and SKF-96365. 2APB was first described as a membrane-permeable inositol 1,4,5-trisphosphate receptor antagonist that was shown later to inhibit store-operated Ca2+ entry channels mediated by STIM/Orai proteins, as well as several TRP channels (55–59). SKF-96365 was first introduced as a store-operated Ca2+ entry inhibitor but was later shown to inhibit several members of the TRPC channel family (60). To confirm that the rise in cytosolic Ca2+ is mediated by a plasma membrane channel, we treated MHS cells with the aforementioned modulators during infection and measured intracellular Ca2+.
Treatment with the broad spectrum inhibitor 2APB had no effect on the baseline level of intracellular Ca2+ of uninfected cells; however, it did significantly decrease the basal intracellular Ca2+ of cells infected with either LVS or ΔkatG (Fig. 3, A–C and G). The ability of uninfected and LVS-infected macrophages to mobilize Ca2+ in response to H2O2 was impaired by 2APB treatment, while rescuing the H2O2 response of ΔkatG-infected MHS cells (Fig. 3, B, C, and H). Together, these data suggests that Francisella catalase regulates Ca2+ influx through a channel sensitive to 2APB.
FIGURE 3.

Pharmacological calcium manipulation of calcium channels abrogates post-Francisella infection calcium increase. MHS cells (murine alveolar macrophage cell line) were treated with 50 μm 2APB or 10 μm SKF-96365 prior to 1 h of infection with Francisella, and calcium response to stimuli was measured by real time fluorescence traces. Raw ratiometric traces for 2APB-treated macrophages (A–C) and SKF-treated macrophages (d–F) are shown. G and I represent the baseline intracellular calcium levels 1 h postinfection. H and J illustrate the total calcium influx post-H2O2 addition to the cells. *, p < 0.01; **, p < 0.001; ***, p < 0.0001; #, p < 0.00001.
The Ca2+ channel modulator SKF-96365 has previously been used to illustrate the dependence of macrophage innate immune survival signaling on Ca2+ fluxes (61–63). Uninfected and LVS-infected cells exhibited no statistical difference in baseline intracellular Ca2+ following SKF-96365 treatment (Fig. 3, D, E, and I). However, cells pretreated with SKF-96365 and subsequently infected with ΔkatG display basal intracellular Ca2+ levels similar to those of LVS-infected cells (Fig. 3, E, F, and I). SKF-96365 treatment also rescues the ability of macrophages infected with ΔkatG to increase cytosolic Ca2+ in response to H2O2 (Fig. 3, F and J). Taken together, these data suggest that the failure of catalase-deficient Francisella to detoxify H2O2 dramatically alters the macrophages ability to mobilize Ca2+ in response to infection.
Francisella Requires Catalase in Order to Inhibit Macrophage Function
Ca2+ is required for many steps of phagocytosis, including the depolymerization of actin that encases phagosomes, phagolysosomal fusion, and the assembly and activation of the ROS producing complex NADPH oxidase (21). Other intracellular pathogens such as Mycobacteria and Listeria have been shown to abrogate the necessary rises in Ca2+ for proper phagosome maturation, therefore enhancing their survival (17). To determine whether the ability of Francisella to modulate Ca2+ also impacts the cytoskeleton, global cytoskeletal rearrangements after infection were analyzed with the use of imaging flow cytometry. Within 30 min postinfection, macrophages infected with catalase-deficient Francisella exhibit a significant rise in F-actin as monitored by mean phalloidin intensity (Fig. 4A). Fig. 4B depicts images from the mean bin of each data set represented in Fig. 4A, clearly illustrating the increased fluorescence following infection with GFP-expressing ΔkatG relative to control GFP-LVS. Restriction of Ca2+ influx with 2APB prevented the increase in actin polymerization following infection with ΔkatG (Fig. 4C). Overall, these data indicate that the H2O2 scavenging activity Francisella limits actin polymerization and may impair efficient phagosome formation in a Ca2+-dependent manner.
FIGURE 4.

Francisella catalase inhibits macrophage function in a calcium-dependent manner. Amnis ImageStream imaging flow cytometer was utilized to carry out high throughput analysis of actin polymerization postinfection at the indicated time points. A, infection of murine alveolar macrophages (MHS cell line) with catalase-deficient LVS leads to an increase in polymerized actin as measured by phalloidin staining. B, representative images from the mean fluorescent bin of each sample are shown. C, the increase in polymerized actin is mediated by calcium flux as it is inhibited by treatment with 2APB. Additionally, Francisella catalase mediated proinflammatory cytokine output in a calcium-dependent manner as measured by IL-6 secretion. D, macrophages infected for 24 h with LVSΔKat secrete increased amounts of IL-6, which can be abrogated by treatment with calcium modulator 2APB but not SKF. * = p < 0.01, ** = p < 0.001, # = p < 0.0001. ND, not detected.
Our lab has gathered compelling evidence that Francisella antioxidants contribute to virulence and actively participate in restricting host innate immune function (9, 64). We have previously published that infection with antioxidant-deficient Francisella potentiates macrophage proinflammatory cytokine production relative to LVS infection. The studies illustrated that loss of catalase impairs the cytokine-suppressing capacity of F. tularensis by enhancing NF-κB activation. Kinase signaling cascades upstream of NF-κB can also be influenced by the intracellular Ca2+ concentration (63). To elucidate whether modulation by Francisella of intracellular Ca2+ postinfection contributes to the cytokine suppression, MHS cells were treated with Ca2+ channel blockers prior to infection, and IL-6 secretion was measured. Pretreatment of alveolar macrophages with either 2APB or SKF-96365 reduced the secretion of IL-6 after wild type infection or following TLR engagement by LPS. However, IL-6 production was only significantly decreased with 2APB and not SKF-96365 following infection with ΔkatG, suggesting that the Ca2+ channel involved in this response is likely more efficiently blocked by 2APB than by SKF-96365 (Fig. 4D). Based on this pharmacology, we therefore reasoned that this Ca2+-dependent response is likely mediated by a TRP channel family member rather than the store-operated Ca2+ entry pathway mediated by Orai channels. Additionally, this pattern was only present in the absence of catalase, suggesting a redox-dependent mechanism in the Ca2+-dependent cytokine response.
Catalase-dependent Restriction of Intracellular Ca2+ Increase Appears to be TRPM2-dependent
We next sought to define the precise channel responsible for the increase in basal Ca2+ in response to infection with catalase-deficient Francisella. Among all receptor-activated Ca2+-conducting channels, TRPM2 stands out for being unambiguously characterized as redox-sensitive (48, 65–67). Recent work demonstrating that macrophage TRPM2 is highly redox-responsive (36, 43, 51) and the block of both actin polymerization and cytokine production by 2APB in ΔkatG-infected MHS cells led us to explore using molecular tools the contribution of TRPM2 to the Ca2+ entry pathway involved post-Francisella infection. BMDM from TRPM2 KO mice, which lack transmembrane domains 5 and 6, as well as the TRPM2 C terminus including the putative calcium pore, rendering the TRPM2 channel inactive (39), were infected with both LVS and ΔkatG, and intracellular Ca2+ was monitored. In the absence of TRPM2, ΔkatG no longer elicits a significant increase of intracellular Ca2+ 1 h postinfection, whereas WT BMDM behave similarly to MHS cells (Fig. 5D). WT and TRPM2 KO BMDM were also assessed for their ability to respond to H2O2 following Francisella infection. Similar to the MHS cell line (Fig. 1), WT BMDM that were either uninfected or infected with LVS increased cytosolic Ca2+ in response to 300 μm H2O2. WT BMDM infected with ΔkatG were in this case capable of responding to H2O2 although to a lesser extent than that of LVS-infected cells (Fig. 5E, dark bars). In accordance with other published studies, TRPM2 KO BMDM were defective in their response to H2O2 (Fig. 5E, light bars). Together, these data suggest that Francisella modulation of intracellular Ca2+ is mediated by TRPM2.
FIGURE 5.

Francisella catalase modulation of calcium homeostasis is mediated by TRPM2. WT and TRPM2−/− murine bone marrow-derived macrophages were examined to determine the contribution of TRPM2. Ratiometric calcium imaging 1 h postinfection illustrates that the increased intracellular calcium and ability to respond to redox stimulus are abrogated in TRPM2-null BMDM. A, B, and C represent the raw traces of uninfected, LVS-infected, and ΔkatG-infected macrophages, respectively. D depicts the baseline Fura ratio as measured from time 150–250. E illustrates the total change in intracellular calcium after stimulus. *, p < 0.01; **, p < 0.001; #, p < 0.0001.
We next determined whether ΔkatG-dependent increase in macrophage actin polymerization was TRPM2-dependent. Infection of WT BMDM with LVS caused a small decrease in actin polymerization, whereas infection with ΔkatG significantly increased actin polymerization as measured by phalloidin staining. This increase is completely abrogated in TRPM2-null BMDM (Fig. 6A), suggesting that Ca2+ influx through TRPM2 is responsible for increased actin polymerization following infection with ΔkatG infection.
FIGURE 6.

Francisella catalase modulation of macrophage function is mediated by TRPM2. WT and TRPM2−/− murine bone marrow-derived macrophages were examined to determine the contribution of TRPM2 to the immune response to Francisella. Macrophage function as measured by actin polymerization and IL-6 message were decreased in TRPM2-null BMDM following infection with LVSΔKat (A and B). **, p < 0.001; #, p < 0.0001.
We set out to determine whether TRPM2 plays a role in the cytokine response to Francisella infection. Following 24 h of infection, mRNA was collected from both WT and TRPM2 KO BMDM and assayed for IL-6 expression. Consistent with our previous findings (9), WT BMDM infected with ΔkatG displayed a significant elevation in IL-6 expression as compared with LVS infection. This pattern was not observed in the TRPM2 KO BMDM, further supporting the importance of TRPM2-mediated Ca2+ signaling in enhancing cytokine production in response to ΔkatG infection (Fig. 6B).
Finally, in an effort to determine the mechanistic basis for the impaired ionomycin response in response to ΔkatG infection (Figs. 1A, 3C, and 5C), we first evaluated whether thapsigargin induced changes in cytosolic calcium levels. Although treatment of uninfected, LVS- and ΔkatG-infected MHS cells with thapsigargin did not increase Fura-2 ratio in the absence of extracellular Ca2+, addition of Ca2+ caused an equivalent increase in the levels of cytosolic Ca2+ that was not affected by infection with either LVS or ΔkatG (Fig. 7, A and B). Thus, the ΔkatG-infected macrophages do not display an intrinsic defect in extracellular Ca2+ entry.
FIGURE 7.

Francisella catalase does not affect responses to thapsigargin but renders ionomycin H2O2-sensitive. A and B, total changes in intracellular calcium after addition of 2 μm thapsigargin in the absence or presence of 2 mm Ca2+ or 10 μm ionomycin are illustrated. C, the calcium-sensitive ratiometric dye Fura-2-AM was utilized to obtain live cell measurements of intracellular calcium 1 h postinfection in a microplate reader; mean traces with S.E. are plotted. D, effects of H2O2 (600 μm) pretreatment on ionomycin-mediated Ca2+ entry. E, fresh or frozen stocks of ionomycin display dose-dependent inhibition of Ca2+ transport activity in response to H2O2 treatment. *, p < 0.05; **, p < 0.01; ***, p < 0.001; #, p < 0.0001.
We have previously reported that upon infection with the ΔkatG, there occurs a more than 2-fold increase in the steady state levels of H2O2 relative to LVS-infected macrophages (9). Ionomycin has a complex chemical structure that coordinates Ca2+ hexavalently, and it is possible that its Ca2+ binding activity may be impaired by H2O2. We first validated the impairment in ionomycin-dependent Ca2+ entry by ΔkatG infection using a microplate-based assay (Fig. 7C). We next evaluated whether H2O2 alters the capacity of ionomycin to transport Ca2+ into the cytosol. H2O2 displayed a dose-dependent inhibition of ionomycin Ca2+ transport capacity that is exacerbated when the drug is not freshly prepared (Fig. 7, D and E). Together, these data suggest that Francisella catalase restricts Ca2+ mobilization by impairing TRPM2 activity and suppressing macrophage function (Fig. 8).
FIGURE 8.
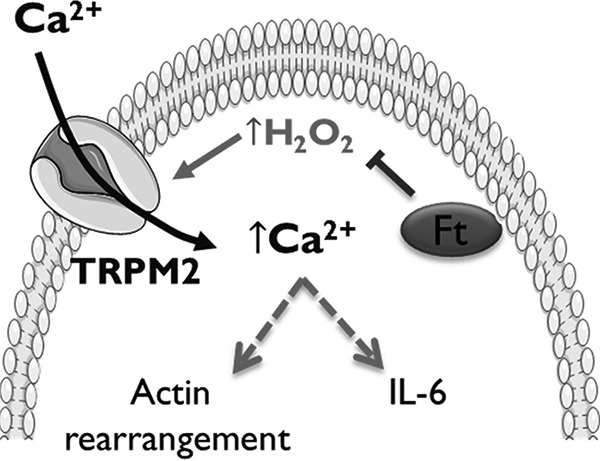
F. tularensis LVS catalase restricts immune function by impairing TRPM2 channel activity. Francisella scavenges H2O2 within the macrophage cytosol limiting TRPM2 activity. In the absence of catalase, intracellular H2O2 increases in response to infection activating the redox-sensitive TRPM2 channel facilitating macrophage Ca2+-dependent signaling, actin rearrangement, and IL-6 production.
Discussion
Overall, elucidating how Francisella survives in and restricts macrophage function is fundamental to understanding the pathogenic mechanisms of this highly virulent bacterium. To further our understanding of how Francisella antioxidants impede host immune signaling, we have begun investigating the role of intracellular Ca2+ in this process. Here, we establish that F. tularensis limits Ca2+ entry in a redox-dependent fashion. Utilizing two separate fluorescence detection methods, the Fura Red results (Fig. 2) that suggested an effect on basal Ca2+ were confirmed/supported by Fura-2 experiments (Fig. 3). Murine alveolar macrophages infected with LVSΔkatG display a high basal intracellular Ca2+ concentration and fail to show further increase of cytosolic Ca2+ in response to stimulation by H2O2. Given that TRPM2 has been firmly established to be activated by H2O2 (38, 49, 50, 52), we propose that LVS catalase restricts the activation of TRPM2 by limiting the amount of H2O2 within the cytoplasm. Therefore, in the absence of Francisella catalase, there is a chronic rise of H2O2 concentration upon infection (9) activating TRPM2 leading to an influx of Ca2+. We propose that TRPM2 is already “on” in the case of ΔkatG infection, and as such it can no longer respond to the addition of exogenous H2O2 as seen in Fig. 1A. Together, the dysregulation of intracellular Ca2+ can severely influence downstream kinase signaling, which in the case of Francisella infection is already impaired (9, 45, 68–71).
Additionally, macrophages infected with ΔkatG fail to respond to ionomycin treatment. We demonstrate that ionomycin Ca2+ transport capacity is sensitive to direct inhibition by H2O2. Thus, it is very likely that the increases in steady state H2O2 production resulting from the ΔkatG infection may limit the capacity of ionomycin to transport Ca2+. The chemistry of H2O2 is complex because it can work as a reductant, oxidant, and a radical initiator; thus, the exact mechanism behind the H2O2-dependent inhibition is unclear. We must emphasize that agents that alter the cellular redox state, such as infection with ΔkatG, may impair the Ca2+ binding capacity of ionomycin. Moreover, air exposure (oxidation) sensitizes ionomycin to H2O2-dependent inhibition (Fig. 8D). Although we are actively exploring the redox responsiveness of ionomycin, biochemically, this work is beyond the scope of the current report.
In conjunction with their Ca2+ profiles, macrophages infected with ΔkatG also displayed distinct actin polymerization patterns as compared with macrophages infected with LVS. We established that Francisella catalase restricts actin polymerization in a Ca2+-dependent manner. Actin polymerization is essential for several key steps in the clearance of infection including phagocytosis, as well as phagolysosome fusion. It has already been established that Francisella inhibits the phagolysosome fusion and goes on to escape and replicate within the cytoplasm (72, 73). Constraints on actin polymerization caused by the restriction of Ca2+ mobilization may represent a redundant pathway to achieve the same goal of bacterial survival.
Pharmacological inhibitors 2APB and SKF-96365 not only rescue the ability of LVSΔkatG-infected macrophages to respond to H2O2 but also differentially alter proinflammatory cytokine secretion, suggesting that there is interplay between intracellular Ca2+ release and the activation of membrane channels that are redox-regulated. The use of these inhibitors, as well as recent publications, led us to hypothesize that the channel involved was TRPM2.
We established that increases in intracellular Ca2+ following infection with ΔkatG, as well as the failure to respond to exogenous H2O2, is mediated by TRPM2. Additionally, we demonstrate that the catalase-dependent restriction of actin mobilization postinfection is mediated by Ca2+ influx through TRPM2. Finally, we present evidence that the increased cytokine output observed following infection with ΔkatG is dependent upon intracellular Ca2+ mediated by TRPM2. Recently several studies have illustrated the importance of TRPM2 for the clearance of the intracellular bacteria Listeria monocytogenes (36, 37, 74, 75). Knock-out of TRPM2 led to increased morbidity due in part to decreased cytokine output. A hallmark of Francisella infection is the delayed secretion of cytokines followed by a cytokine storm. A variety of mechanisms have been proposed for how Francisella accomplishes this feat (69). Intracellular bacteria are known to have redundant mechanisms to accomplish the same task, thereby increasing their odds of survival, and we propose that catalase restriction of Ca2+ signaling via TRPM2 represents an additional mechanism that Francisella utilizes to thwart the hosts immune response.
Together, our findings provide compelling evidence that bacterial antioxidants harness H2O2 to limit Ca2+ signaling and restrict host immune function. Continued work will further the understanding of how bacterial antioxidant enzymes impair macrophage antimicrobial mechanisms and will contribute to design and implementation of effective vaccination strategies against Francisella and potentially other infectious pathogens.
Author Contributions
N. L. S. and J. A. M. designed the study and wrote the paper. A. C. preformed and analyzed the experiments shown in Fig. 7. M. T. and B. A. M. provided technical assistance and contributed to the preparation of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Karsten R. O. Hazlett for providing the LVSΔwbtA, LVSΔfopA, LVSΔiglc, and LVSΔmglA mutants and Dr. Robert Brainard for helpful discussions with respect to H2O2 chemistry.
This study was supported by funded by NIAID, National Institutes of Health Grant PO1AI056320 (to J. A. M.). This study was also supported in part by National Institutes of Health Grants R01HL097111 and R01HL123364 and American Heart Association Grant 14GRNT18880008 (to M. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TRP
- transient receptor potential
- TRPM2
- TRP melastatin 2
- ROS
- reactive oxygen species
- LVS
- live vaccine strain
- 2APB
- 2-aminoethyl diphenylborinate
- BMDM
- bone marrow-derived macrophage
- MOI
- multiplicity of infection
- HBSS
- Hanks' buffered salt solution.
References
- 1. Oyston P. C., Sjostedt A., and Titball R. W. (2004) Tularaemia: bioterrorism defence renewa interest in Francisella tularensis. Nat. Rev. Microbiol. 2, 967–978 [DOI] [PubMed] [Google Scholar]
- 2. Oyston P. C. (2008) Francisella tularensis: unravelling the secrets of an intracellular pathogen. J. Med. Microbiol. 57, 921–930 [DOI] [PubMed] [Google Scholar]
- 3. Mohapatra N. P., Soni S., Rajaram M. V., Dang P. M., Reilly T. J., El-Benna J., Clay C. D., Schlesinger L. S., and Gunn J. S. (2010) Francisella acid phosphatases inactivate the NADPH oxidase in human phagocytes. J. Immunol. 184, 5141–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asare R., and Abu Kwaik Y. (2010) Molecular complexity orchestrates modulation of phagosome biogenesis and escape to the cytosol of macrophages by Francisella tularensis. Environ. Microbiol. 12, 2559–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clemens D. L., Lee B.-Y., and Horwitz M. A. (2004) Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72, 3204–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forestal C. A., Malik M., Catlett S. V., Savitt A. G., Benach J. L., Sellati T. J., and Furie M. B. (2007) Francisella tularensis has a significant extracellular phase in infected mice. J. Infect. Dis. 196, 134–137 [DOI] [PubMed] [Google Scholar]
- 7. Lindgren H., Shen H., Zingmark C., Golovliov I., Conlan W., and Sjöstedt A. (2007) Resistance of Francisella tularensis strains against reactive nitrogen and oxygen species with special reference to the role of KatG. Infect. Immun. 75, 1303–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parsonage D., Karplus P. A., and Poole L. B. (2008) Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc. Natl. Acad. Sci. U.S.A. 105, 8209–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melillo A. A., Bakshi C. S., and Melendez J. A. (2010) Francisella tularensis antioxidants harness reactive oxygen species to restrict macrophage signaling and cytokine production. J. Biol. Chem. 285, 27553–27560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindgren H., Honn M., Salomonsson E., Kuoppa K., Forsberg Å., and Sjöstedt A. (2011) Iron content differs between Francisella tularensis subspecies tularensis and subspecies holarctica strains and correlates to their susceptibility to H2O2-induced killing. Infect. Immun. 79, 1218–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cremer T. J., Butchar J. P., and Tridandapani S. (2011) Francisella subverts innate immune signaling: focus on PI3K/Akt. Front. Microbiol. 10.3389/fmicb.2011.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cremer T. J., Ravneberg D. H., Clay C. D., Piper-Hunter M. G., Marsh C. B., Elton T. S., Gunn J. S., Amer A., Kanneganti T.-D., Schlesinger L. S., Butchar J. P., and Tridandapani S. (2009) MiR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS One 4, e8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parsa K. V., Butchar J. P., Rajaram M. V., Cremer T. J., and Tridandapani S. (2008) The tyrosine kinase Syk promotes phagocytosis of Francisella through the activation of Erk. Mol. Immunol. 45, 3012–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Al-Khodor S., and Abu Kwaik Y. (2010) Triggering Ras signalling by intracellular Francisella tularensis through recruitment of PKCα and βI to the SOS2/GrB2 complex is essential for bacterial proliferation in the cytosol. Cell. Microbiol. 12, 1604–1621 [DOI] [PubMed] [Google Scholar]
- 15. Veal E. A., Day A. M., and Morgan B. A. (2007) Hydrogen peroxide sensing and signaling. Mol. Cell 26, 1–14 [DOI] [PubMed] [Google Scholar]
- 16. Dickinson B. C., and Chang C. J. (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 7, 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. TranVan Nhieu G., Clair C., Grompone G., and Sansonetti P. (2004) Calcium signalling during cell interactions with bacterial pathogens. Biol. Cell 96, 93–101 [DOI] [PubMed] [Google Scholar]
- 18. Hogan P. G., Lewis R. S., and Rao A. (2010) Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vig M., and Kinet J. (2009) Calcium signaling in immune cells. Nat. Immunol. 10, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raddassi K., Berthon B., Petit J. F., and Lemaire G. (1994) Role of calcium in the activation of mouse peritoneal macrophages: induction of NO synthase by calcium ionophores and thapsigargin. Cell. Immunol. 153, 443–455 [DOI] [PubMed] [Google Scholar]
- 21. Nunes P., and Demaurex N. (2010) The role of calcium signaling in phagocytosis. J. Leukoc. Biol. 88, 57–68 [DOI] [PubMed] [Google Scholar]
- 22. Nunes P., Demaurex N., and Dinauer M. C. (2013) Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 14, 1118–1131 [DOI] [PubMed] [Google Scholar]
- 23. Kim E., Enelow R. I., Sullivan G. W., and Mandell G. L. (1992) Regional and generalized changes in cytosolic free calcium in monocytes during phagocytosis. Infect. Immun. 60, 1244–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Massullo P., Sumoza-Toledo A., Bhagat H., and Partida-Sánchez S. (2006) TRPM channels, calcium and redox sensors during innate immune responses. Semin. Cell Dev. Biol. 17, 654–666 [DOI] [PubMed] [Google Scholar]
- 25. Vieira O. V., Botelho R. J., and Grinstein S. (2002) Phagosome maturation: aging gracefully. Biochem. J. 366, 689–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paranavitana C., Pittman P. R., Velauthapillai M., Zelazowska E., and Dasilva L. (2008) Transcriptional profiling of Francisella tularensis infected peripheral blood mononuclear cells: a predictive tool for tularemia. FEMS Immunol. Med. Microbiol. 54, 92–103 [DOI] [PubMed] [Google Scholar]
- 27. Andersson H., Hartmanová B., Kuolee R., Rydén P., Conlan W., Chen W., and Sjöstedt A. (2006) Transcriptional profiling of host responses in mouse lungs following aerosol infection with type A Francisella tularensis. J. Med. Microbiol. 55, 263–271 [DOI] [PubMed] [Google Scholar]
- 28. Ernst R. K., Guina T., and Miller S. I. (1999) How intracellular bacteria survive: surface modifications that promote resistance to host innate immune responses. J. Infect. Dis. 179, S326–S330 [DOI] [PubMed] [Google Scholar]
- 29. Yang R., Xi C., Sita D. R., Sakai S., Tsuchiya K., Hara H., Shen Y., Qu H., Fang R., Mitsuyama M., and Kawamura I. (2014) The RD1 locus in the Mycobacterium tuberculosis genome contributes to the maturation and secretion of IL-1α from infected macrophages through the elevation of cytoplasmic calcium levels and calpain activation. Pathog. Dis. 70, 51–60 [DOI] [PubMed] [Google Scholar]
- 30. Lischke T., Heesch K., Schumacher V., Schneider M., Haag F., Koch-Nolte F., and Mittrücker H.-W. (2013) CD38 controls the innate immune response against Listeria monocytogenes. Infect. Immun. 81, 4091–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malik Z. A., Denning G. M., and Kusner D. J. (2000) Inhibition of Ca2+ signaling by Mycobacterium tuberculosis is associated with reduced phagosome-lysosome fusion and increased survival within human macrophages. J. Exp. Med. 191, 287–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Azenabor A. A., Kennedy P., and York J. (2009) Free intracellular Ca2+ regulates bacterial lipopolysaccharide induction of iNOS in human macrophages. Immunobiology 214, 143–152 [DOI] [PubMed] [Google Scholar]
- 33. Potier M., and Trebak M. (2008) New developments in the signaling mechanisms of the store-operated calcium entry pathway. Pflugers Arch. 457, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pedersen S. F., Owsianik G., and Nilius B. (2005) TRP channels: an overview. Cell Calcium 38, 233–252 [DOI] [PubMed] [Google Scholar]
- 35. Gees M., Colsoul B., and Nilius B. (2010) The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2, a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knowles H., Li Y., and Perraud A. (2013) The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 55, 241–248 [DOI] [PubMed] [Google Scholar]
- 37. Knowles H., Heizer J. W., Li Y., Chapman K., Ogden C. A., Andreasen K., Shapland E., Kucera G., Mogan J., Humann J., Lenz L. L., Morrison A. D., and Perraud A.-L. (2011) Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc. Natl. Acad. Sci. U.S.A. 108, 11578–11583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Di A., Gao X. P., Qian F., Kawamura T., Han J., Hecquet C., Ye R. D., Vogel S. M., and Malik A. B. (2012) The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 13, 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller B. A., Wang J., Hirschler-Laszkiewicz I., Gao E., Song J., Zhang X.-Q., Koch W. J., Madesh M., Mallilankaraman K., Gu T., Chen S. J., Keefer K., Conrad K., Feldman A. M., and Cheung J. Y. (2013) The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 304, H1010–H1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilson J. E., Katkere B., and Drake J. R. (2009) Francisella tularensis induces ubiquitin-dependent major histocompatibility complex class II degradation in activated macrophages. Infect. Immun. 77, 4953–4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seaver L. C., and Imlay J. A. (2001) Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183, 7173–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Motiani R. K., Zhang X., Harmon K. E., Keller R. S., Matrougui K., Bennett J. A., and Trebak M. (2013) Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 27, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zou J., Ainscough J. F., Yang W., Sedo A., Yu S.-P., Mei Z.-Z., Sivaprasadarao A., Beech D. J., and Jiang L.-H. (2013) A differential role of macrophage TRPM2 channels in Ca2+ signalling and cell death in early responses to H2O2. Am. J. Physiol. Cell Physiol. 305, C61–C69 [DOI] [PubMed] [Google Scholar]
- 44. Mahawar M., Atianand M. K., Dotson R. J., Mora V., Rabadi S. M., Metzger D. W., Huntley J. F., Harton J. A., Malik M., and Bakshi C. S. (2012) Identification of a novel Francisella tularensis factor required for intramacrophage survival and subversion of innate immune response. J. Biol. Chem. 287, 25216–25229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Telepnev M., Golovliov I., and Sjöstedt A. (2005) Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb. Pathog. 38, 239–247 [DOI] [PubMed] [Google Scholar]
- 46. Jones C. L., Napier B. A., Sampson T. R., Llewellyn A. C., Schroeder M. R., and Weiss D. S. (2012) Subversion of host recognition and defense systems by Francisella spp. Microbiol. Mol. Biol. Rev. 76, 383–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Day A. M., Brown J. D., Taylor S. R., Rand J. D., Morgan B. A., and Veal E. A. (2012) Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 45, 398–408 [DOI] [PubMed] [Google Scholar]
- 48. Miller B. A., and Zhang W. (2011) TRP channels as mediators of oxidative stress. in Transient Receptor Potential Channels (Islam M. S., ed.), pp. 531–544, Springer, Netherlands: [DOI] [PubMed] [Google Scholar]
- 49. Kolisek M., Beck A., Fleig A., and Penner R. (2005) Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 18, 61–69 [DOI] [PubMed] [Google Scholar]
- 50. Kashio M., Sokabe T., Shintaku K., Uematsu T., Fukuta N., Kobayashi N., Mori Y., and Tominaga M. (2012) Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. U.S.A. 109, 6745–6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Song M. Y., Makino A., and Yuan J. X. (2011) Role of reactive oxygen species and redox in regulating the function of transient receptor potential channels. Antioxid. Redox Signal. 15, 1549–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trebak M., Ginnan R., Singer H. A., and Jourd'heuil D. (2010) Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 12, 657–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lipp P., and Niggli E. (1993) Ratiometric confocal Ca2+-measurements with visible wavelength indicators in isolated cardiac myocytes. Cell Calcium 14, 359–372 [DOI] [PubMed] [Google Scholar]
- 54. Peng K., Broz P., Jones J., Joubert L.-M., and Monack D. (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell. Microbiol. 13, 1586–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Togashi K., Inada H., and Tominaga M. (2008) Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2APB). Br. J. Pharmacol. 153, 1324–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maruyama T., Kanaji T., Nakade S., Kanno T., and Mikoshiba K. (1997) 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 122, 498–505 [DOI] [PubMed] [Google Scholar]
- 57. Trebak M., Bird G. S., McKay R. R., and Putney J. W. Jr. (2002) Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J. Biol. Chem. 277, 21617–21623 [DOI] [PubMed] [Google Scholar]
- 58. Braun F. J., Broad L. M., Armstrong D. L., and Putney J. W. (2001) Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J. Biol. Chem. 276, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 59. Peppiatt C. M., Collins T. J., Mackenzie L., Conway S. J., Holmes A. B., Bootman M. D., Berridge M. J., Seo J. T., and Roderick H. L. (2003) 2-Aminoethoxydiphenyl borate (2APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34, 97–108 [DOI] [PubMed] [Google Scholar]
- 60. Merritt J. E., Armstrong W. P., Benham C. D., Hallam T. J., Jacob R., Jaxa-Chamiec A., Leigh B. K., McCarthy S. A., Moores K. E., and Rink T. J. (1990) SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 271, 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ye Y., Huang X., Zhang Y., Lai X., Wu X., Zeng X., Tang X., and Zeng Y. (2012) Calcium influx blocked by SK&F 96365 modulates the LPS plus IFN-γ-induced inflammatory response in murine peritoneal macrophages. Int. Immunopharmacol. 12, 384–393 [DOI] [PubMed] [Google Scholar]
- 62. Evans J. H., and Falke J. J. (2007) Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 16176–16181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tano J.-Y., and Vazquez G. (2011) Requirement for non-regulated, constitutive calcium influx in macrophage survival signaling. Biochem. Biophys. Res. Commun. 407, 432–437 [DOI] [PubMed] [Google Scholar]
- 64. Melillo A. A., Mahawar M., Sellati T. J., Malik M., Metzger D. W., Melendez J. A., and Bakshi C. S. (2009) Identification of Francisella tularensis live vaccine strain CuZn superoxide dismutase as critical for resistance to extracellularly generated reactive oxygen species. J. Bacteriol. 191, 6447–6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen S. J., Zhang W., Tong Q., Conrad K., Hirschler-Laszkiewicz I., Bayerl M., Kim J. K., Cheung J. Y., and Miller B. A. (2013) Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am. J. Physiol. Cell Physiol. 304, C548–C560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gao G., Wang W., Tadagavadi R. K., Briley N. E., Love M. I., Miller B. A., and Reeves W. B. (2014) TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Invest. 124, 4989–5001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller B. A. (2006) The role of TRP channels in oxidative stress-induced cell death. J. Membr. Biol. 209, 31–41 [DOI] [PubMed] [Google Scholar]
- 68. Telepnev M., Golovliov I., Grundström T., Tärnvik A., and Sjöstedt A. (2003) Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-α and IL-1 from murine macrophages. Cell. Microbiol. 5, 41–51 [DOI] [PubMed] [Google Scholar]
- 69. Asare R., and Kwaik Y. A. (2010) Exploitation of host cell biology and evasion of immunity by Francisella tularensis. Front. Microbiol. 1, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bosio C. M. (2011) The subversion of the immune system by Francisella tularensis. Front. Microbiol. 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bosio C. M., Bielefeldt-Ohmann H., and Belisle J. T. (2007) Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J. Immunol. 178, 4538–4547 [DOI] [PubMed] [Google Scholar]
- 72. Clemens D. L., and Horwitz M. A. (2007) Uptake and intracellular fate of Francisella tularensis in human macrophages. Ann. N.Y. Acad. Sci. 1105, 160–186 [DOI] [PubMed] [Google Scholar]
- 73. Santic M., Asare R., Skrobonja I., Jones S., and Abu Kwaik Y. (2008) Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 76, 2671–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Perraud A.-L., Knowles H. M., and Schmitz C. (2004) Novel aspects of signaling and ion-homeostasis regulation in immunocytes. The TRPM ion channels and their potential role in modulating the immune response. Mol. Immunol. 41, 657–673 [DOI] [PubMed] [Google Scholar]
- 75. Knowles G. C., and McCulloch C. A. (1992) Simultaneous localization and quantification of relative G and F actin content: optimization of fluorescence labeling methods. J. Histochem. Cytochem. 40, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 76. Su J., Yang J., Zhao D., Kawula T. H., Banas J. A., and Zhang J. R. (2007) Genome-wide identification of Francisella tularensis virulence determinants. Infect. Immun. 75, 3089–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vonkavaara M., Telepnev M. V., Rydén P., Sjöstedt A., and Stöven S. (2008) Drosophila melanogaster as a model for elucidating the pathogenicity of Francisella tularensis. Cell. Microbiol. 10, 1327–1338 [DOI] [PubMed] [Google Scholar]