

Abstract
Monocytes and macrophages are critical for the effectiveness of monoclonal antibody therapy. Responses to antibody-coated tumor cells are largely mediated by Fcγ receptors (FcγRs), which become activated upon binding to immune complexes. FcγRIIb is an inhibitory FcγR that negatively regulates these responses, and it is expressed on monocytes and macrophages. Therefore, deletion or down-regulation of this receptor may substantially enhance therapeutic outcomes. Here we screened a panel of Toll-like receptor (TLR) agonists and found that those selective for TLR4 and TLR8 could significantly down-regulate the expression of FcγRIIb. Upon further examination, we found that treatment of monocytes with TLR4 agonists could lead to the ubiquitination of FcγRIIb protein. A search of our earlier microarray database of monocytes activated with the TLR7/8 agonist R-848 (in which FcγRIIb was down-regulated) revealed an up-regulation of membrane-associated ring finger (C3HC4) 3 (MARCH3), an E3 ubiquitin ligase. Therefore, we tested whether LPS treatment could up-regulate MARCH3 in monocytes and whether this E3 ligase was involved with LPS-mediated FcγRIIb down-regulation. The results showed that LPS activation of TLR4 significantly increased MARCH3 expression and that siRNA against MARCH3 prevented the decrease in FcγRIIb following LPS treatment. These data suggest that activation of TLR4 on monocytes can induce a rapid down-regulation of FcγRIIb protein and that this involves ubiquitination.
Keywords: cell biology, Fc receptor, immunology, monocyte, signal transduction, Toll-like receptor 4 (TLR4), ubiquitylation (ubiquitination)
Introduction
Monocytes and macrophages play an important role in the innate immune response by phagocytosing IgG-opsonized infectious particles (1) and are major mediators in the destruction of tumor cells (2–5). Indeed, the importance of monocytes in clearing antibody-targeted tumor cells following the administration of therapeutic mAbs used in oncology indications has been well established (6–8). However, despite showing statistically significant effects, the low rates of complete remission combined with the relatively high relapse rate suggest strongly that there is much room for improvement (9–11). Therapeutic mAbs themselves are being improved, with the goal of increasing affinity toward Fcγ receptors in some cases (12, 13). Along with this, immune modulators such as interferons (14, 15), interleukins (16–20), synthetic compounds (21–23), and CpG oligonucleotides (16) are being explored as potential enhancers of antibody therapy.
Antibody-dependent destruction of target cells is largely mediated by Fcγ receptors (FcγRs)4 (5, 24, 25). Human monocytes and macrophages express at least four different functional FcγRs: FcγRI, FcγRIIa, FcγRIIb, and FcγRIIIa (26). Of these, FcγRI, FcγRIIa, and FcγRIIIa are activating receptors that drive cellular responses to antibodies. These receptors either contain, within their cytoplasmic tails, an immune receptor tyrosine-based activation motif (ITAM), as in the case of FcγRIIa (27), or are associated with the γ-chain homodimer that has an ITAM (28). The association of the γ-chain is critical not only for surface expression of FcγRI and FcγRIIIa but also for signaling from these receptors. In mice that do not express the ITAM-containing FcγRIIa, deficiencies in γ-chain expression abrogate the surface expression and function of activating FcγR (29).
In contrast, FcγRIIb is an inhibitory receptor that has an immune receptor tyrosine-based inhibitory motif in its cytoplasmic tail (30, 31). Co-clustering of FcγRIIb with ITAM-FcγR results in phosphorylation of the immune receptor tyrosine-based inhibitory motif tyrosine and association of Src homology 2 domain-containing inositol 5′-phosphatase with FcγRIIb (31–34). This clustering of FcγRIIb and its association with Src homology 2 domain-containing inositol phosphatase serves to inhibit FcγR-mediated responses (35). Without FcγRIIb (or Src homology 2 domain-containing inositol phosphatase), FcγR activity is increased. For example, bone marrow-derived macrophages from FcγRII-deficient mice display enhanced phagocytic ability compared with wild-type bone marrow-derived macrophages (36), and Src homology 2 domain-containing inositol phosphatase-deficient bone marrow-derived macrophages can more effectively phagocytose IgG-coated particles than wild-type bone marrow-derived macrophages (37). Therefore, the effectiveness of FcγR-mediated function is dictated by the ratio of activating to inhibitory FcγR on effector cells (38). Indeed, this has been demonstrated by Clynes et al. (25), who showed that antibody-mediated clearance of B16 melanoma cells was enhanced markedly in mice that had a genetic deletion of FcγRIIb.
The expression of FcγR is malleable. It has been shown that proinflammatory cytokines such as IFNγ up-regulate the expression of ITAM-FcγR, thereby enhancing monocyte/macrophage responses (39–41). In contrast, IL-13 has been shown to down-regulate these activating FcγRs (42), and IL-4 can up-regulate expression of the immune receptor tyrosine-based inhibitory motif-bearing FcγRIIb, with the combination of IL-4 and IL-10 leading to synergistic increases in this receptor (39, 43–45). Toll-like receptor (TLR) agonists can also influence FcγR expression. For example, previous work in our laboratory has shown that the TLR7/8 agonist R-848 could simultaneously increase the expression of activating FcγR and decrease expression of the inhibitory FcγRIIb (46). In this earlier study, we also found that up-regulation of activating FcγR depended on autocrine/paracrine signaling, whereas the down-regulation of FcγRIIb did not. However, the precise mechanisms involved in the TLR-mediated down-regulation of FcγRIIb are not fully understood.
Here we examined the down-regulation of FcγRIIb by TLR agonists in greater detail in an attempt to uncover the underlying mechanism(s) of the modulation. We began by screening a battery of TLR agonists to identify those capable of decreasing FcγRIIb and found that agonists for TLR4 and TLR8 caused a rapid and simultaneous decrease in transcript and protein levels. We interrogated the mechanisms behind the rapid reduction in FcγRIIb protein using the TLR4 agonist LPS and found that it involved the ubiquitination of FcγRIIb and that it depended on the E3 ubiquitin ligase MARCH3. Therefore, these results identify a novel mechanism by which TLR agonists can modulate expression of the inhibitory FcγR and, thereby, alter the ratio of activating to inhibitory Fcγ receptors.
Experimental Procedures
Antibodies and Reagents
LPS, used at 1 to 1000 ng/ml) was purchased from Sigma-Aldrich (St. Louis, MO). Agonists for TLR2 (Pam2CSK4, used at 100 ng/ml), TLR3 (polyI:C, used at 10 μg/ml), TLR5 (Flagellin, used at 100 ng/ml), TLR8 (CL075, used at 0.01–10 μm), and CpG (used at 10 μg/ml) were purchased from Invivogen (San Diego, CA). The TLR7-selective agonist 3M-055 (used at 1 μm) was provided by 3M Drug Delivery Systems (Minneapolis, MN). The TLR8-selective agonist motolimod, formerly known as VTX-2337 (used at 1 μm) was provided by VentiRx (Seattle, WA). Anti-FcγRIIb (CD32b) antibody for Western blotting was purchased from Abcam (Cambridge, MA). Anti-ubiquitin antibody was purchased from Cell Signaling Technology (Beverly, MA). Antibodies against actin and HRP-conjugated anti-goat and anti-mouse secondary antibodies were from Santa Cruz Biotechnology. Anti-rabbit HRP-conjugated secondary antibody was purchased from Cell Signaling Technology. TRIzol® was purchased from Invitrogen. Reverse transcriptase, random hexamers, and SYBR Green PCR mix were purchased from Applied Biosystems (Foster City, CA).
PCR primers were purchased from Invitrogen. Sequences for FcγRIIa, FcγRIIb, and GAPDH were as described previously (47). Primer sequences to detect MARCH transcripts were as follows: MARCH3 forward, GCGAGGACGATGGAAATCCT; MARCH3 reverse, CTTGCATGACATACTGCGGC; MARCH7forward, CAAGCACACGTGTCCGATTTA; MARCH7 reverse,TGGTCTCCGTCTTCTTCGGA; MARCH9 forward, AGAAGGTCCAGATTGCTGCC; and MARCH9 reverse, GATGAGGCCTATGCAGACGA.
Human and mouse whole-molecule IgG were from Jackson ImmunoResearch Laboratories (West Grove, PA). N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate liposomal transfection reagent was purchased from Roche Applied Science. Dharmacon control and MARCH3 siRNA constructs were purchased from GE Life Sciences (Lafayette, CO). Recombinant protein G-agarose beads were purchased from Invitrogen. Red blood cell lysis buffer was purchased from eBioscience (San Diego, CA). The ubiquitin E1 inhibitor UBEI-41 (PYR-41) was purchased from Biogenova (Potomac, MD).
Western Blotting and ELISAs
Western blotting was done as described previously (48). Cells were lysed in TN1 buffer (50 mm Tris (pH 8.0), 10 mm EDTA, 10 mm Na4P2O7, 10 mm NaF, 1% Triton X-100, 125 mm NaCl, 10 mm Na3VO4, and 10 μg/ml each aprotinin and leupeptin). Protein lysates were boiled in Laemmli sample buffer, separated by SDS-PAGE, transferred to nitrocellulose membranes, probed with the antibody of interest, and then developed by Pierce ECL 2 Western blotting substrate (Thermo Scientific, Rockford, IL) or SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific). Densitometry was performed using ImageJ software (National Institutes of Health, Bethesda, MD), and ratios between the indicated probes and their respective anti-actin reprobes were calculated. Cell supernatants were collected, centrifuged at full speed to clear cellular debris, and then assayed for cytokine via sandwich ELISA (R&D Systems, Minneapolis, MN) according to the protocol of the manufacturer.
Real-time RT-PCR (qPCR)
Cells were lysed in TRIzol® reagent (Invitrogen), and RNA isolation was completed according to the instructions of the manufacturer. Reverse transcription was done with 50–200 ng of total RNA. The cDNA was run in duplicate for each donor on an Applied Biosystems Step One Plus system with automatically calculated thresholds. Relative copy number was calculated as 2↽−ΔCt, with ΔCt calculated by subtracting the Ct of the housekeeping control (GAPDH) from the experimental sample Ct (49, 50).
Peripheral Blood Monocyte Isolation
Peripheral blood monocytes (PBM) were isolated from Red Cross Leukopaks via Ficoll separation (Mediatech, Manassas, VA), followed by CD14-positive selection using MACS® (Miltenyi Biotec, Inc., Cambridge, MA) as described previously (46). PBM were resuspended in RPMI 1640 containing 10% heat-inactivated FBS (Hyclone, Logan, UT), penicillin/streptomycin, and l-glutamate (Invitrogen). The purity of the monocytes obtained was > 97%, as determined by flow cytometry with CD14 antibody.
Phagocytosis
Phagocytosis assays were performed as described previously (47). Briefly, IgG-coated, PKH26-labeled sheep red blood cells (SRBC) were added to the PBM. Cells were pelleted briefly by slow centrifugation, followed by 30 min of incubation at 37 °C. Non-ingested SRBC were subjected to hypotonic lysis with RBC lysis buffer and PBS wash prior to fixation with 1% paraformaldehyde. Samples were analyzed by fluorescence microscopy in a blinded fashion. The phagocytic index was defined as the total number of SRBC ingested by 100 phagocytes.
Immunoprecipitations
Immunoprecipitations were performed as described previously (51). PBM were treated with or without TLR agonists at different time points. Cells were lysed in 500 μl of TN1 buffer (50 mm Tris (pH 8.0), 10 mm EDTA, 10 mm Na4P2O7, 10 mm NaF, 1% Triton X-100, 125 mm NaCl, 10 mm Na3VO4, and 10 μg/ml each aprotinin and leupeptin), and protein lysates were incubated overnight with the specified antibody and protein G-agarose beads. A negative control was included without the specified antibody or without protein G-agarose beads. After incubation, the beads were washed twice with 500 μl of TN1 buffer, boiled with 40 μl of 1× Laemmli sample buffer for 5 min, and subjected to SDS-PAGE. Proteins were transferred to nitrocellulose membranes, probed with the antibody of interest (anti-ubiquitin or anti-CD32B), and then developed by Pierce ECL 2 Western blotting substrate (Thermo Scientific).
Transfections
Transfections were performed using N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate liposomal transfection reagent (Roche Applied Science) in accordance with the instructions of the manufacturer. Briefly, N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate solution diluted in HBS buffer (20 mm HEPES (cell culture grade) and 150 mm NaCl (pH 7.4)) was incubated in a 2:1 ratio with either 1.5 μg of MARCH3 siRNA or 1.5 μg of control siRNA for 15 min at room temperature. After incubation, cells were treated with the N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate/nucleic acid mixture and incubated for 6 h at 37 °C. Cells were then stimulated with or without LPS and incubated for 18 h at 37 °C. Cells were either lysed in TN1 buffer for Western blotting or TRIzol® for real-time RT-PCR to verify transfection efficacy.
Statistical Analyses
For experiments that involved placing the cells of each donor across multiple conditions, data were analyzed by using analysis of variance with repeated measures. For experiments with only two groups involved, paired Student's t tests were used to test for statistically significant differences. Analyses of variance were performed using SAS statistical software (SAS, Inc., Cary, NC). p ≤ 0.05 was considered significant.
Results
TLR Ligands Down-regulate FcγRIIb
We have found previously that the TLR7/8 agonist R-848 was capable of down-regulating FcγRIIb in monocytes (46), which led us to ask whether other TLR agonists could do this. We treated human PBM overnight with selective agonists for TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9 and then measured the levels of FcγRIIb protein. The results (Fig. 1A) showed that agonists for TLR4 and TLR8 almost completely eliminated FcγRIIb, whereas agonists for TLR9 showed modest effects. TLR2 agonists led to variable results, occasionally leading to a slight decrease but, more often, to a modest increase in FcγRIIb, which is in agreement with our earlier findings (47). We repeated this and examined the transcript levels of FcγRIIb and found similar results (Fig. 1B), although only agonists for TLR4, TLR8, and TLR9 led to significant differences (Fig. 1C). There was a modest trend toward statistical significance for the TLR2 agonist, but the magnitude of change was not sufficient to achieve statistical significance despite the relatively low variability.
FIGURE 1.

Ligands for TLR4 and TLR8 down-regulate FcγRIIb. A, human PBM were isolated and incubated overnight (∼18 h) either without (UT, untreated) or with a panel of TLR agonists for TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9 (described under “Experimental Procedures”). Western blotting was done to measure FcγRIIb protein expression (n = 4, representative blot shown). IB, immunoblot. B and C, PBM were treated without or with the indicated TLR agonists as in A. Then mRNA was collected, and qPCR was done to measure FcγRIIb transcript (B). Statistical analyses were done to identify the agonists that down-regulated FcγRIIb transcript (n = 3) (C). RCN, relative copy number; CI, confidence interval. D and E, PBM were treated overnight without or with 500 ng/ml LPS, and then Western blotting and qPCR were done to measure protein levels of FcγRIIb (D) and FcγRIIa (E) (n = 3, representative blots shown). F, PBM were treated as in D and E, and then transcript of FcγRIIa was measured by qPCR (n = 3). For all blots, membranes were reprobed for actin to verify equivalent loading. *, p ≤ 0.05.
A protein Basic Local Alignment Search Tool alignment showed that the isoform of FcγRIIb expressed by monocytes was 90% identical to FcγRIIa with a 74% query coverage, so we next verified that the TLR agonist treatment was specific in down-regulating FcγRIIb and not FcγRIIa. We treated PBM overnight with the TLR4 agonist LPS and examined protein levels of FcγRIIb and FcγRIIa. The results showed that LPS significantly down-regulated FcγRIIb protein and significantly up-regulated FcγRIIa (Fig. 1, D and E, respectively). Similarly, qPCR analysis showed that LPS up-regulated the FcγRIIa transcript (Fig. 1F). Because of the robust response, we chose to focus on TLR4 activation by LPS.
Dose and Time Course Responses of FcγRIIb to LPS
Next we tested the concentration of LPS required to down-regulate FcγRIIb. We treated PBM overnight with LPS concentrations between 0–1000 ng/ml and found that as little as 1 ng/ml was sufficient to elicit at least a partial decrease in protein and transcript levels of the receptor (Fig. 2, A and B, respectively). We then examined the time course of FcγRIIb down-regulation. As shown in Fig. 2C, LPS treatment decreased the levels of FcγR protein in as little as 1 h, although some degree of donor-to-donor variability was seen. Transcript levels were also decreased as early as 1 h (Fig. 2D).
FIGURE 2.

Concentration and time course responses of FcγRIIb to LPS. A and B, human PBM were incubated overnight with LPS at increasing concentrations (0, 1, 10, 50, 100, 500, and 1000 ng/ml for protein analyses and 0, 1, 10, 50, 100, and 500 ng/ml for transcript analyses). Western blotting and qPCR were done to measure FcγRIIb protein (A) and mRNA expression (B) (n = 3). IB, immunoblot; RCN, relative copy number. C and D, PBM (n = 4) were treated with 500 ng/ml LPS for 0, 1, 3, 6, 18, or 24 h, and then Western blotting was done to measure FcγRIIb protein (C), and qPCR was done to measure transcript (D). For all blots, membranes were reprobed for actin to verify equivalent loading.
We next examined the activating receptor FcγRIIa as a control and found that 1 ng/ml was sufficient for up-regulation of protein and transcript (Fig. 3, A and B, respectively). However, in contrast to FcγRIIb, where changes were seen in as little as 1 h, changes in FcγRIIa protein were not seen until after the 6-h mark (Fig. 3C), with transcript increasing at 6 h (Fig. 3D). This is consistent with autocrine-paracrine signaling being required for up-regulation of FcγRIIa, as we have shown previously using the TLR7/8 agonist R-848 (46). These results show that LPS modulates FcγRIIb and the nearly identical activating receptor FcγRIIa through different means because it causes down-regulation of one and up-regulation of the other. Indeed, up-regulation of FcγRIIa has been shown to be caused by secreted factors such as IFNγ and IL-10 (39, 45, 53). Importantly, regarding FcγRIIb, the LPS-mediated decrease in transcript did not precede the decrease in protein, which suggested that LPS was driving the down-regulation of protein and transcript through separate mechanisms.
FIGURE 3.

Concentration and time course responses of FcγRIIa to LPS. A and B, human PBM were incubated overnight with LPS at increasing concentrations (0, 1, 10, 50, 100, 500, and 1000 ng/ml for protein analyses and 0, 1, 10, 50, 100, and 500 ng/ml for transcript analyses). Western blotting and qPCR were done to measure FcγRIIa protein (A) and mRNA expression (B). IB, immunoblot; RCN, relative copy number. C and D, PBM were treated with 500 ng/ml LPS for 0, 1, 3, 6, 18, or 24 h, and then Western blotting was done to measure FcγRIIa protein (C), and qPCR was done to measure transcript (D) (n = 3). For all blots, membranes were reprobed for actin to verify equivalent loading.
LPS Treatment Leads to FcγRIIb Ubiquitination
The apparent discrepancy between protein and transcript down-regulation of FcγRIIb was of interest. In particular, the N-terminal MG (54) and its lack of a PEST domain suggested that the normal half-life of FcγRIIb should be relatively long, meaning that the reduction in transcript would not account for the rapid decrease in protein levels. Indeed, in murine macrophages, the half-life of FcγRIIb has been measured at ∼10 h (55). Because the plasma membrane is recycled in its entirety two to three times per hour (56), it is likely that FcγRIIb is recycled back to the cell surface under normal conditions but that TLR4 activation somehow disrupts this. Previous reports have shown that binding of immune complexes in J774 cells can lead to lysosomal localization of FcγR within 1 h (57), and phagocytosis of IgG-coated particles in mouse peritoneal macrophages can result in over 50% FcγR degradation within 2 h (55).
In an effort to gain insights into the cause of this LPS-driven decrease in FcγRIIb protein, we examined the possibility that ubiquitin was involved. We used UbPred to predict possible ubiquitination sites in FcγRIIb and found one medium- and one high-confidence site at residues 101 and 263, respectively. Interestingly, FcγRIIa shared the medium-confidence site but contained a completely different high-confidence site. This distinct high-confidence ubiquitination site in FcγRIIb opened the possibility that this receptor, but not the activating receptor FcγRIIa, was ubiquitinated following LPS treatment.
We tested whether LPS led to the ubiquitination of FcγRIIb by treating PBM with LPS for time points from 15 min to 6 h and performing immunoprecipitations to detect receptor-ubiquitin associations. As shown in Fig. 4A, a strong ubiquitin association was found at 1 h. This decreased at later time points, likely because FcγRIIb levels themselves were decreasing. Reciprocal immunoprecipitation experiments in which ubiquitin was pulled down and blots done for FcγRIIb showed roughly similar results, with the strongest association occurring at 30 min (Fig. 4B). We also examined ubiquitination of FcγRIIa, which is up-regulated rather than degraded following LPS treatment. Results showed that, as expected, LPS did not increase its ubiquitination (data not shown).
FIGURE 4.
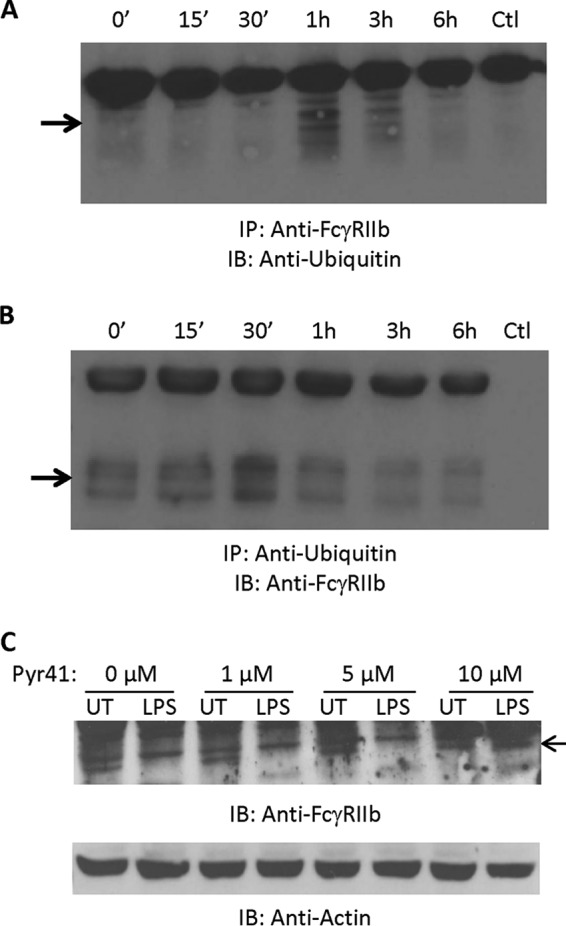
LPS treatment leads to FcγRIIb ubiquitination. A and B, human PBM were incubated with LPS (500 ng/ml) for 0, 15, or 30 min or 1, 3, or 6 h. For each time point, FcγRIIb was immunoprecipitated (IP), and Western blotting was done to detect ubiquitin (A). The reciprocal experiment was also done with immunoprecipitations of ubiquitin and Western blotting for FcγRIIb (B). Ctl, control; IB, immunoblot. C, human PBM were pretreated for 30 min with increasing doses of the E1 ubiquitin-activating enzyme inhibitor PYR-41 (0, 1, 5, or 10 μm) and then incubated overnight without (UT, untreated) or with 500 ng/ml LPS. Western blotting was done to analyze FcγRIIb expression (n = 3). C, membranes were reprobed for actin to verify equivalent loading.
We then examined the effect of blocking ubiquitination by using the E1-activating enzyme inhibitor Pyr-41. As shown in Fig. 4C, pretreatment of PBM with increasing concentrations of Pyr-41 led to attenuation of LPS-mediated FcγRIIb down-regulation. These results suggest that LPS treatment leads to ubiquitination of FcγRIIb and that this may mediate its degradation.
MARCH3 Is Required for LPS-induced FcγRIIb Down-regulation
A proteomics study of B cells identified the E3 ubiquitin ligase MARCH9 as a potential effector of FcγRIIb degradation (58). Because we have found previously that the TLR7/8 agonist R-848 could down-regulate FcγRIIb (46), we searched through our microarray datasets of monocytes treated with TLR7 or TLR8 agonists (50) to see whether MARCH9 had been up-regulated. We found a modest increase of MARCH9 but a very strong up-regulation of the related MARCH3 in monocytes treated with the TLR8 agonist. These results suggested that MARCH9 or MARCH3 might be involved in the down-regulation of FcγRIIb. To test this, we treated PBM overnight with LPS and measured the expression of MARCH9, MARCH3, and the related MARCH7 in response to TLR4 activation. The results showed that LPS significantly increased the expression of MARCH3 (Fig. 5A, left panel) but not of MARCH7 or MARCH9 (data not shown). Because the decrease in FcγRIIb levels occurred in as little as 1 h, we also repeated the LPS treatment and evaluated MARCH3 expression after 1 h. The results showed up-regulation of MARCH3 at this early time point as well (Fig. 5A, right panel).
FIGURE 5.

MARCH3 is required for LPS-induced FcγRIIb down-regulation. A, human PBM were incubated overnight without (UT, untreated) or with 500 ng/ml LPS, and qPCR was done to measure MARCH3 expression (left panel, n = 6). This was repeated, and transcript of MARCH3 was measured at the 1-h time point (right panel, n = 4). RCN, relative copy number. B, human PBM were transfected with either control (Ctl) siRNA or siRNA against MARCH3 and then incubated overnight without or with 500 ng/ml LPS. MARCH3 expression was measured by qPCR (n = 3). C–E, human PBM were transfected with control or MARCH3 siRNA and then LPS-treated as in B. qPCR was done to measure FcγRIIb expression (C), and Western blotting was to measure FcγRIIb protein (D). Densitometric analysis of the Western blotting results is shown in E (n = 3). *, p ≤ 0.05.
Next, to determine whether MARCH3 was required for the LPS-mediated decrease in FcγRIIb protein, we transfected PBM with siRNA against MARCH3 prior to treating them with LPS. To verify the efficacy of the knockdown, we performed qPCR to measure MARCH3 and found that LPS-treated PBM transfected with control siRNA showed significant up-regulation of MARCH3, whereas siRNA against MARCH3 prevented the increase in response to LPS treatment (Fig. 5B). We also verified that the siRNA treatments had no effect on the transcript levels of FcγRIIb, with qPCR showing that overnight LPS treatment reduced RNA levels of FcγRIIb in both control and MARCH3 siRNA treatments (Fig. 5C). Finally, we measured protein levels of FcγRIIb following siRNA and LPS treatments and found that knockdown of MARCH3 inhibited the LPS-mediated decrease in FcγRIIb (Fig. 5, D and E). Collectively, these results suggest that LPS up-regulates the expression of MARCH3 in monocytes, which then targets FcγRIIb for degradation.
LPS Treatment Enhances FcγR Function
To test whether LPS treatment led to a difference in FcγR function, we treated PBM overnight with LPS and then incubated them for 20 h with immobilized IgG to cluster the Fcγ receptors. Production of TNFα was then measured in the cleared supernatants, and the results showed a superadditive effect of LPS plus IgG (Fig. 6A). Next we treated PBM overnight with LPS and then subjected them to a phagocytosis assay using antibody-opsonized SRBC. As shown in Fig. 6B, LPS treatment significantly enhanced the number of ingested SRBC by the PBM. These results indicate that activation of TLR4 with LPS enhances FcγR function.
FIGURE 6.

LPS treatment enhances FcγR function. PBM were treated overnight without (UT, untreated) or with 500 ng/ml LPS. A, after treatment, cells were incubated for 20 h without (PBS) or with immobilized IgG, and then cleared supernatants were analyzed for TNFα by ELISA. B, after treatment, cells were incubated with fluoresceinated, antibody-coated SRBC for 30 min, and then the numbers of ingested SRBC were counted via fluorescence microscopy (n = 3). *, p ≤ 0.05.
Discussion
Modulation of FcγRIIb expression is important within the context of both tumor immunotherapy and autoimmune diseases. For example, genetic deletion of FcγRIIb can permit the development of collagen-induced arthritis in a typically non-susceptible mouse strain (59). In humans, levels of FcγRIIb on monocytes were found to be equivalent in rheumatoid arthritis patients and healthy donors, but the patient monocytes expressed more activating receptors. Treatment in vitro of both healthy donor and patient monocytes with IL-4 plus IL-10 led to increases in FcγRIIb expression and decreases in IgG-mediated cytokine production (45). It has also been shown that a FcγRIIb polymorphism that was less inhibitory could serve as a strong predictor of joint damage in rheumatoid arthritis patients (60).
Conversely, within a tumor setting, reductions rather than increases in FcγRIIb expression or function may be beneficial. In a mouse B16 melanoma model of antibody therapy, the genetic deletion of FcγRIIb led to an almost complete clearance of tumor cells in the lung following antibody treatment (25). It has been shown recently that the use of a blocking antibody against FcγRIIb led to enhanced antitumor effects from therapeutic antibody treatment, especially in stromal regions in which there is typically reduced effectiveness of antibodies (61).
In this study, we found that TLR agonists, most notably those for TLR4 and TLR8, led to a marked down-regulation of FcγRIIb transcript and, separately, to a down-regulationof FcγRIIb protein. Further examination showed that LPS caused FcγRIIb to become ubiquitinated and that the E3 ubiquitin ligase MARCH3 was required for the observed decrease in protein following LPS treatment. Of note, the ubiquitination of FcγRIIb preceded the strong LPS-driven up-regulation of MARCH3, suggesting that LPS had an earlier effect on MARCH3 activity prior to up-regulation of MARCH3 transcription. LPS treatment may have enhanced the ubiquitin system in general, perhaps affecting the E1-activating enzyme or MARCH3-binding E2-conjugating enzymes. This, in turn, could increase the activity of basally expressed MARCH3. Further studies are required to elucidate this.
Interestingly, treatment with the TLR8 agonist also led to rapid decreases in FcγRIIb protein as well as to receptor ubiquitination (data not shown). However, we were unable to see a reversal of TLR8-mediated FcγRIIb down-regulation after MARCH3 knockdown (data not shown). It is possible that our knockdown was not complete enough to have an effect or that other pathways are involved with TLR8-mediated FcγRIIb degradation. For example, TLR8 agonist treatment up-regulated MARCH9 and MARCH3, suggesting that, in contrast to TLR4 activation, both E3 ligases could be involved following TLR8 agonist treatment.
Ubiquitination can lead to degradation by either the proteasome or lysosome, depending on the type of ubiquitination and the cellular location (62). Because there is a natural recycling of FcγR from the cell surface to the endosome and back (with complete membrane recycling taking place as rapidly as two or three times per hour (56)), it is likely that TLR4 activation breaks this recycling pattern by triggering ubiquitination that shunts FcγRIIb to the lysosome. In partial support of ubiquitin-mediated lysosomal rather than proteasomal degradation, we found that the proteasomal inhibitor MG-132 did not block R-848-mediated degradation of FcγRIIb (data not shown).
The finding that some but not all TLR agonists led to FcγRIIb down-regulation is of interest because it not only brings attention to the qualitative and quantitative differences in signaling between the various TLR, but it also has implications for disease. For example, certain pathogens such as Gram-negative bacteria or RNA viruses might be predicted to exacerbate the symptoms of rheumatoid arthritis and other immune diseases involving autoantibodies. Conversely, it may also suggest that certain TLR ligands would be more suited than others as adjuvants for antibody therapy against tumors. Indeed, the TLR8-selective agonist motolimod has been tested in a phase 1 clinical trial in combination with cetuximab for the treatment of recurrent or metastatic squamous cell carcinomas of the head and neck (NCT01334177). A phase II trial to test this TLR8 agonist in combination with cisplatin or carboplatin, fluorouracil, and cetuximab is also underway (NCT01836029). The therapeutic hypothesis for the TLR8 agonist motolimod is increased antibody-dependent cellular cytotoxicity because of stronger activation of natural killer cells and monocytes, and enhancement of this activation via loss of the inhibitory FcγRIIb would be consistent with this.
Although the TLR4 pathway is commonly associated with excessive cytokine production during sepsis (63, 64), it is also being examined as a potential therapeutic target for the treatment of cancer. One preclinical study has shown that monophosphoryl lipid A significantly enhanced the effectiveness of antitumor antibody treatment in a mouse melanoma model (65). Similarly, the synthetic lipid A mimetic E6020 enhanced the survival of mice treated with trastuzumab in a model of HER2+ cancer (52). Interestingly, depletion of macrophages, but not of natural killer cells, led to a reduction in the efficacy of treatment (52). Although no clinical trials combining TLR4 agonists with antibody therapy are listed (https://clinicaltrials.gov), trials are being conducted to test several agonists as antitumor agents. The TLR4 agonist glucopyranosyl lipid A stable emulsion is being tested against sarcoma in combination with radiation therapy (NCT02180698). The same agonist is being tested as a vaccine adjuvant against melanoma (NCT02320305) and as a single agent against Merkel cell carcinoma (NCT02035657) and follicular non-Hodgkin lymphoma (NCT02501473).
In summary, we identified the mechanism by which TLR4 activation by LPS down-regulates the protein levels of the inhibitory FcγRIIb, namely up-regulation of the E3 ubiquitin ligase MARCH3. This uncovers a more specific potential therapeutic target that may be of value within certain disease settings in which FcγRs are involved. It is conceivable that selective inhibitors of MARCH3 might be beneficial within the context of rheumatoid arthritis or that the administration of agents that up-regulate MARCH3, in conjunction with antitumor antibodies, may lead to improved outcomes for cancer patients.
Author Contributions
K. F., L. R., S. E., and H. F. acquired, summarized, and plotted the data. X. M. analyzed the data and contributed to the experimental design. J. P. V., G. N. D., and R. M. H. participated in the experimental design and contributed reagents. S. T. and J. P. B. conceived the study, designed the experiments, and wrote the manuscript. All authors contributed to writing and proofreading of the manuscript, and all authors approved the final version.
This work was supported by National Institutes of Health Grants P01-CA095426 and R01 CA162411 (to S. T.) and K12 CA133250 (to J. P. B.), Ohio State University College of Medicine McWhinney Bridge Fund Grant 244749 (to J. P. B.), and American Cancer Society Institutional Research Grant IRG-67-003-47 (to J. P. B.). J. P. V. is an employee of 3M. G. N. D. and R. M. H. are employees of VentiRx Pharmaceuticals. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- FcγR
- Fcγ receptor
- ITAM
- immune receptor tyrosine-based activation motif
- TLR
- Toll-like receptor
- MARCH
- membrane-associated ring finger (C3HC4)
- qPCR
- quantitative PCR
- PBM
- peripheral blood monocyte(s)
- SRBC
- sheep red blood cell(s).
References
- 1. Aderem A., and Underhill D. M. (1999) Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593–623 [DOI] [PubMed] [Google Scholar]
- 2. Yazawa N., Hamaguchi Y., Poe J. C., and Tedder T. F. (2005) Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc. Natl. Acad. Sci. U.S.A. 102, 15178–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oflazoglu E., Stone I. J., Gordon K. A., Grewal I. S., van Rooijen N., Law C. L., and Gerber H. P. (2007) Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 110, 4370–4372 [DOI] [PubMed] [Google Scholar]
- 4. Oflazoglu E., Stone I. J., Brown L., Gordon K. A., van Rooijen N., Jonas M., Law C. L., Grewal I. S., and Gerber H. P. (2009) Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br. J. Cancer 100, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Uchida J., Hamaguchi Y., Oliver J. A., Ravetch J. V., Poe J. C., Haas K. M., and Tedder T. F. (2004) The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 199, 1659–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gong Q., Ou Q., Ye S., Lee W. P., Cornelius J., Diehl L., Lin W. Y., Hu Z., Lu Y., Chen Y., Wu Y., Meng Y. G., Gribling P., Lin Z., Nguyen K., Tran T., Zhang Y., Rosen H., Martin F., and Chan A. C. (2005) Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 174, 817–826 [DOI] [PubMed] [Google Scholar]
- 7. Minard-Colin V., Xiu Y., Poe J. C., Horikawa M., Magro C. M., Hamaguchi Y., Haas K. M., and Tedder T. F. (2008) Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcγRI, FcγRIII, and FcγRIV. Blood 112, 1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beers S. A., French R. R., Chan H. T., Lim S. H., Jarrett T. C., Vidal R. M., Wijayaweera S. S., Dixon S. V., Kim H., Cox K. L., Kerr J. P., Johnston D. A., Johnson P. W., Verbeek J. S., Glennie M. J., and Cragg M. S. (2010) Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191–5201 [DOI] [PubMed] [Google Scholar]
- 9. Byrd J. C., Murphy T., Howard R. S., Lucas M. S., Goodrich A., Park K., Pearson M., Waselenko J. K., Ling G., Grever M. R., Grillo-Lopez A. J., Rosenberg J., Kunkel L., and Flinn I. W. (2001) Rituximab using a thrice weekly dosing schedule in B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma demonstrates clinical activity and acceptable toxicity. J. Clin. Oncol. 19, 2153–2164 [DOI] [PubMed] [Google Scholar]
- 10. O'Brien S. M., Kantarjian H., Thomas D. A., Giles F. J., Freireich E. J., Cortes J., Lerner S., and Keating M. J. (2001) Rituximab dose-escalation trial in chronic lymphocytic leukemia. J. Clin. Oncol. 19, 2165–2170 [DOI] [PubMed] [Google Scholar]
- 11. Hainsworth J. D., Litchy S., Barton J. H., Houston G. A., Hermann R. C., Bradof J. E., Greco F. A., and Minnie Pearl Cancer Research Network (2003) Single-agent rituximab as first-line and maintenance treatment for patients with chronic lymphocytic leukemia or small lymphocytic lymphoma: a phase II trial of the Minnie Pearl Cancer Research Network. J. Clin. Oncol. 21, 1746–1751 [DOI] [PubMed] [Google Scholar]
- 12. Hutchins J. T., Kull F. C. Jr., Bynum J., Knick V. C., Thurmond L. M., and Ray P. (1995) Improved biodistribution, tumor targeting, and reduced immunogenicity in mice with a γ 4 variant of Campath-1H. Proc. Natl. Acad. Sci. U.S.A. 92, 11980–11984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lazar G. A., Dang W., Karki S., Vafa O., Peng J. S., Hyun L., Chan C., Chung H. S., Eivazi A., Yoder S. C., Vielmetter J., Carmichael D. F., Hayes R. J., and Dahiyat B. I. (2006) Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 103, 4005–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis T. A., Maloney D. G., Grillo-López A. J., White C. A., Williams M. E., Weiner G. J., Dowden S., and Levy R. (2000) Combination immunotherapy of relapsed or refractory low-grade or follicular non-Hodgkin's lymphoma with rituximab and interferon-α-2a. Clin. Cancer Res. 6, 2644–2652 [PubMed] [Google Scholar]
- 15. Bertè R., Vallisa D., Civardi G., Moroni C. F., Lazzaro A., and Cavanna L. (2001) Rituximab in combination with interferon-α in relapsed and refractory diffuse large B-cell non-Hodgkin's lymphoma. Acta Haematol. 106, 141–142 [DOI] [PubMed] [Google Scholar]
- 16. Moga E., Alvarez E., Cantó E., Vidal S., Rodríguez-Sánchez J. L., Sierra J., and Briones J. (2008) NK cells stimulated with IL-15 or CpG ODN enhance rituximab-dependent cellular cytotoxicity against B-cell lymphoma. Exp. Hematol. 36, 69–77 [DOI] [PubMed] [Google Scholar]
- 17. Ansell S. M., Witzig T. E., Kurtin P. J., Sloan J. A., Jelinek D. F., Howell K. G., Markovic S. N., Habermann T. M., Klee G. G., Atherton P. J., and Erlichman C. (2002) Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood 99, 67–74 [DOI] [PubMed] [Google Scholar]
- 18. Friedberg J. W., Neuberg D., Gribben J. G., Fisher D. C., Canning C., Koval M., Poor C. M., Green L. M., Daley J., Soiffer R., Ritz J., and Freedman A. S. (2002) Combination immunotherapy with rituximab and interleukin 2 in patients with relapsed or refractory follicular non-Hodgkin's lymphoma. Br. J. Haematol. 117, 828–834 [DOI] [PubMed] [Google Scholar]
- 19. Ansell S. M., Geyer S. M., Maurer M. J., Kurtin P. J., Micallef I. N., Stella P., Etzell P., Novak A. J., Erlichman C., and Witzig T. E. (2006) Randomized phase II study of interleukin-12 in combination with rituximab in previously treated non-Hodgkin's lymphoma patients. Clin. Cancer Res. 12, 6056–6063 [DOI] [PubMed] [Google Scholar]
- 20. Khan K. D., Emmanouilides C., Benson D. M. Jr., Hurst D., Garcia P., Michelson G., Milan S., Ferketich A. K., Piro L., Leonard J. P., Porcu P., Eisenbeis C. F., Banks A. L., Chen L., Byrd J. C., and Caligiuri M. A. (2006) A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin's lymphoma. Clin. Cancer Res. 12, 7046–7053 [DOI] [PubMed] [Google Scholar]
- 21. Tai Y. T., Li X. F., Catley L., Coffey R., Breitkreutz I., Bae J., Song W., Podar K., Hideshima T., Chauhan D., Schlossman R., Richardson P., Treon S. P., Grewal I. S., Munshi N. C., and Anderson K. C. (2005) Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer Res. 65, 11712–11720 [DOI] [PubMed] [Google Scholar]
- 22. Reddy N., Hernandez-Ilizaliturri F. J., Deeb G., Roth M., Vaughn M., Knight J., Wallace P., and Czuczman M. S. (2008) Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br. J. Haematol. 140, 36–45 [DOI] [PubMed] [Google Scholar]
- 23. Wu L., Adams M., Carter T., Chen R., Muller G., Stirling D., Schafer P., and Bartlett J. B. (2008) lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin. Cancer Res. 14, 4650–4657 [DOI] [PubMed] [Google Scholar]
- 24. Clynes R., Takechi Y., Moroi Y., Houghton A., and Ravetch J. V. (1998) Fc receptors are required in passive and active immunity to melanoma. Proc. Natl. Acad. Sci. U.S.A. 95, 652–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clynes R. A., Towers T. L., Presta L. G., and Ravetch J. V. (2000) Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446 [DOI] [PubMed] [Google Scholar]
- 26. Daëron M. (1997) Fc receptor biology. Annu. Rev. Immunol. 15, 203–234 [DOI] [PubMed] [Google Scholar]
- 27. Van den Herik-Oudijk I. E., Capel P. J., van der Bruggen T., and Van de Winkel J. G. (1995) Identification of signaling motifs within human Fc γ RIIa and Fc γ RIIb isoforms. Blood 85, 2202–2211 [PubMed] [Google Scholar]
- 28. Ernst L. K., Duchemin A. M., and Anderson C. L. (1993) Association of the high-affinity receptor for IgG (Fc γ RI) with the γ subunit of the IgE receptor. Proc. Natl. Acad. Sci. U.S.A. 90, 6023–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takai T., Li M., Sylvestre D., Clynes R., and Ravetch J. V. (1994) FcR γ chain deletion results in pleiotrophic effector cell defects. Cell 76, 519–529 [DOI] [PubMed] [Google Scholar]
- 30. Choquet D., Ku G., Cassard S., Malissen B., Korn H., Fridman W. H., and Bonnerot C. (1994) Different patterns of calcium signaling triggered through two components of the B lymphocyte antigen receptor. J. Biol. Chem. 269, 6491–6497 [PubMed] [Google Scholar]
- 31. Muta T., Kurosaki T., Misulovin Z., Sanchez M., Nussenzweig M. C., and Ravetch J. V. (1994) A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature 368, 70–73 [DOI] [PubMed] [Google Scholar]
- 32. Ono M., Okada H., Bolland S., Yanagi S., Kurosaki T., and Ravetch J. V. (1997) Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell 90, 293–301 [DOI] [PubMed] [Google Scholar]
- 33. Chacko G. W., Tridandapani S., Damen J. E., Liu L., Krystal G., and Coggeshall K. M. (1996) Negative signaling in B lymphocytes induces tyrosine phosphorylation of the 145-kDa inositol polyphosphate 5-phosphatase, SHIP. J. Immunol. 157, 2234–2238 [PubMed] [Google Scholar]
- 34. Tridandapani S., Pradhan M., LaDine J. R., Garber S., Anderson C. L., and Coggeshall K. M. (1999) Protein interactions of Src homology 2 (SH2) domain-containing inositol phosphatase (SHIP): association with Shc displaces SHIP from FcγRIIb in B cells. J. Immunol. 162, 1408–1414 [PubMed] [Google Scholar]
- 35. Hunter S., Indik Z. K., Kim M. K., Cauley M. D., Park J. G., and Schreiber A. D. (1998) Inhibition of Fcγ receptor-mediated phagocytosis by a nonphagocytic Fcγ receptor. Blood 91, 1762–1768 [PubMed] [Google Scholar]
- 36. Clynes R., Maizes J. S., Guinamard R., Ono M., Takai T., and Ravetch J. V. (1999) Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 189, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cox D., Dale B. M., Kashiwada M., Helgason C. D., and Greenberg S. (2001) A regulatory role for Src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fc γ receptors and complement receptor 3 (α(M)β(2); CD11b/CD18). J. Exp. Med. 193, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clynes R., and Ravetch J. V. (1995) Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity 3, 21–26 [DOI] [PubMed] [Google Scholar]
- 39. Pricop L., Redecha P., Teillaud J. L., Frey J., Fridman W. H., Sautès-Fridman C., and Salmon J. E. (2001) Differential modulation of stimulatory and inhibitory Fc γ receptors on human monocytes by Th1 and Th2 cytokines. J. Immunol. 166, 531–537 [DOI] [PubMed] [Google Scholar]
- 40. Vogel S. N., Finbloom D. S., English K. E., Rosenstreich D. L., and Langreth S. G. (1983) Interferon-induced enhancement of macrophage Fc receptor expression: β-interferon treatment of C3H/HeJ macrophages results in increased numbers and density of Fc receptors. J. Immunol. 130, 1210–1214 [PubMed] [Google Scholar]
- 41. Guyre P. M., Morganelli P. M., and Miller R. (1983) Recombinant immune interferon increases immunoglobulin G Fc receptors on cultured human mononuclear phagocytes. J. Clin. Invest. 72, 393–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Waal Malefyt R., Figdor C. G., Huijbens R., Mohan-Peterson S., Bennett B., Culpepper J., Dang W., Zurawski G., and de Vries J. E. (1993) Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes: comparison with IL-4 and modulation by IFN-γ or IL-10. J. Immunol. 151, 6370–6381 [PubMed] [Google Scholar]
- 43. Tridandapani S., Siefker K., Teillaud J. L., Carter J. E., Wewers M. D., and Anderson C. L. (2002) Regulated expression and inhibitory function of Fcγ RIIb in human monocytic cells. J. Biol. Chem. 277, 5082–5089 [DOI] [PubMed] [Google Scholar]
- 44. Joshi T., Ganesan L. P., Cao X., and Tridandapani S. (2006) Molecular analysis of expression and function of hFcγRIIbl and b2 isoforms in myeloid cells. Mol. Immunol. 43, 839–850 [DOI] [PubMed] [Google Scholar]
- 45. Wijngaarden S., van de Winkel J. G., Jacobs K. M., Bijlsma J. W., Lafeber F. P., and van Roon J. A. (2004) A shift in the balance of inhibitory and activating Fcγ receptors on monocytes toward the inhibitory Fcγ receptor IIb is associated with prevention of monocyte activation in rheumatoid arthritis. Arthritis Rheum. 50, 3878–3887 [DOI] [PubMed] [Google Scholar]
- 46. Butchar J. P., Mehta P., Justiniano S. E., Guenterberg K. D., Kondadasula S. V., Mo X., Chemudupati M., Kanneganti T. D., Amer A., Muthusamy N., Jarjoura D., Marsh C. B., Carson W. E. 3rd, Byrd J. C., and Tridandapani S. (2010) Reciprocal regulation of activating and inhibitory Fcγ receptors by TLR7/8 activation: implications for tumor immunotherapy. Clin. Cancer Res. 16, 2065–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shah P., Fatehchand K., Patel H., Fang H., Justiniano S. E., Mo X., Jarjoura D., Tridandapani S., and Butchar J. P. (2013) Toll-like receptor 2 ligands regulate monocyte Fcγ receptor expression and function. J. Biol. Chem. 288, 12345–12352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tridandapani S., Wang Y., Marsh C. B., and Anderson C. L. (2002) Src homology 2 domain-containing inositol polyphosphate phosphatase regulates NF-κ B-mediated gene transcription by phagocytic Fc γ Rs in human myeloid cells. J. Immunol. 169, 4370–4378 [DOI] [PubMed] [Google Scholar]
- 49. Gavrilin M. A., Bouakl I. J., Knatz N. L., Duncan M. D., Hall M. W., Gunn J. S., and Wewers M. D. (2006) Internalization and phagosome escape required for Francisella to induce human monocyte IL-1β processing and release. Proc. Natl. Acad. Sci. U.S.A. 103, 141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elavazhagan S., Fatehchand K., Santhanam V., Fang H., Ren L., Gautam S., Reader B., Mo X., Cheney C., Briercheck E., Vasilakos J. P., Dietsch G. N., Hershberg R. M., Caligiuri M., Byrd J. C., Butchar J. P., and Tridandapani S. (2015) Granzyme B expression is enhanced in human monocytes by TLR8 agonists and contributes to antibody-dependent cellular cytotoxicity. J. Immunol. 194, 2786–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ganesan L. P., Fang H., Marsh C. B., and Tridandapani S. (2003) The protein-tyrosine phosphatase SHP-1 associates with the phosphorylated immunoreceptor tyrosine-based activation motif of Fc γ RIIa to modulate signaling events in myeloid cells. J. Biol. Chem. 278, 35710–35717 [DOI] [PubMed] [Google Scholar]
- 52. Wang S., Astsaturov I. A., Bingham C. A., McCarthy K. M., von Mehren M., Xu W., Alpaugh R. K., Tang Y., Littlefield B. A., Hawkins L. D., Ishizaka S. T., and Weiner L. M. (2012) Effective antibody therapy induces host-protective antitumor immunity that is augmented by TLR4 agonist treatment. Cancer Immunol. Immunother. 61, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Roon J., Wijngaarden S., Lafeber F. P., Damen C., van de Winkel J., and Bijlsma J. W. (2003) Interleukin 10 treatment of patients with rheumatoid arthritis enhances Fc γ receptor expression on monocytes and responsiveness to immune complex stimulation. J. Rheumatol. 30, 648–651 [PubMed] [Google Scholar]
- 54. Bachmair A., Finley D., and Varshavsky A. (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 [DOI] [PubMed] [Google Scholar]
- 55. Mellman I. S., Plutner H., Steinman R. M., Unkeless J. C., and Cohn Z. A. (1983) Internalization and degradation of macrophage Fc receptors during receptor-mediated phagocytosis. J. Cell Biol. 96, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steinman R. M., Brodie S. E., and Cohn Z. A. (1976) Membrane flow during pinocytosis: a stereologic analysis. J. Cell Biol. 68, 665–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ukkonen P., Lewis V., Marsh M., Helenius A., and Mellman I. (1986) Transport of macrophage Fc receptors and Fc receptor-bound ligands to lysosomes. J. Exp. Med. 163, 952–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hör S., Ziv T., Admon A., and Lehner P. J. (2009) Stable isotope labeling by amino acids in cell culture and differential plasma membrane proteome quantitation identify new substrates for the MARCH9 transmembrane E3 ligase. Mol. Cell Proteomics 8, 1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuasa T., Kubo S., Yoshino T., Ujike A., Matsumura K., Ono M., Ravetch J. V., and Takai T. (1999) Deletion of fcγ receptor IIB renders H-2 (b) mice susceptible to collagen-induced arthritis. J. Exp. Med. 189, 187–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Radstake T. R., Franke B., Wenink M. H., Nabbe K. C., Coenen M. J., Welsing P., Bonvini E., Koenig S., van den Berg W. B., Barrera P., and van Riel P. L. (2006) The functional variant of the inhibitory Fcγ receptor IIb (CD32B) is associated with the rate of radiologic joint damage and dendritic cell function in rheumatoid arthritis. Arthritis Rheum. 54, 3828–3837 [DOI] [PubMed] [Google Scholar]
- 61. Roghanian A., Teige I., Mårtensson L., Cox K. L., Kovacek M., Ljungars A., Mattson J., Sundberg A., Vaughan A. T., Shah V., Smyth N. R., Sheth B., Chan H. T., Li Z. C., Williams E. L., Manfredi G., Oldham R. J., Mockridge C. I., James S. A., Dahal L. N., Hussain K., Nilsson B., Verbeek J. S., Juliusson G., Hansson M., Jerkeman M., Johnson P. W., Davies A., Beers S. A., Glennie M. J., Frendéus B., and Cragg M. S. (2015) Antagonistic human FcγRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell 27, 473–488 [DOI] [PubMed] [Google Scholar]
- 62. Clague M. J., and Urbé S. (2010) Ubiquitin: same molecule, different degradation pathways. Cell 143, 682–685 [DOI] [PubMed] [Google Scholar]
- 63. Savva A., and Roger T. (2013) Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front. Immunol. 4, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Duan G., Zhu J., Xu J., and Liu Y. (2014) Targeting myeloid differentiation 2 for treatment of sepsis. Front. Biosci. 19, 904–915 [DOI] [PubMed] [Google Scholar]
- 65. Bevaart L., Jansen M. J., van Vugt M. J., Verbeek J. S., van de Winkel J. G., and Leusen J. H. (2006) The high-affinity IgG receptor, FcγRI, plays a central role in antibody therapy of experimental melanoma. Cancer Res. 66, 1261–1264 [DOI] [PubMed] [Google Scholar]