

Abstract
The non-structural protein 5A (NS5A) is a hepatitis C virus (HCV) protein indispensable for the viral life cycle. Many prior papers have pinpointed several serine residues in the low complexity sequence I region of NS5A responsible for NS5A phosphorylation; however, the functions of specific phosphorylation sites remained obscure. Using phosphoproteomics, we identified three phosphorylation sites (serines 222, 235, and 238) in the NS5A low complexity sequence I region. Reporter virus and replicon assays using phosphorylation-ablated alanine mutants of these sites showed that Ser-235 dominated over Ser-222 and Ser-238 in HCV replication. Immunoblotting using an Ser-235 phosphorylation-specific antibody showed a time-dependent increase in Ser-235 phosphorylation that correlated with the viral replication activity. Ser-235 phosphorylated NS5A co-localized with double-stranded RNA, consistent with its role in HCV replication. Mechanistically, Ser-235 phosphorylation probably promotes the replication complex formation via increasing NS5A interaction with the human homologue of the 33-kDa vesicle-associated membrane protein-associated protein. Casein kinase Iα (CKIα) directly phosphorylated Ser-235 in vitro. Inhibition of CKIα reduced Ser-235 phosphorylation and the HCV RNA levels in the infected cells. We concluded that NS5A Ser-235 phosphorylated by CKIα probably promotes HCV replication via increasing NS5A interaction with the 33-kDa vesicle-associated membrane protein-associated protein.
Keywords: antibody, Hepatitis C virus (HCV), phosphoproteomics, phosphorylation, proteomics, casein kinase, serine 235
Introduction
Chronic HCV2 infection affects 130–170 million people worldwide (1). The infection is often asymptomatic until development of severe liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma, making chronic HCV infection the most common cause of liver transplant (2). HCV is an enveloped virus with a positive, single-stranded RNA genome encoding three structural (core, E1, and E2) and seven non-structural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (1). The structural proteins together with the host membranes make up the viral particles, whereas the non-structural proteins are required for a complete life cycle. Already, there are several approved highly efficient HCV antivirals targeting non-structural proteins, including NS3/4A protease inhibitors (boceprevir, telaprevir, and simeprevir) and an NS5B RNA-dependent RNA polymerase inhibitor (sofosbuvir) (3). However, their high costs prohibit their accessibility to most patients (4). New competitive alternatives are desirable.
NS5A is a multitasking protein required for the HCV life cycle and thus a good antiviral target (5). It is a phosphoprotein that appears as two bands at 56 and 58 kDa on immunoblots, respectively, referred to as hypophosphorylated (p56) and hyperphosphorylated (p58) NS5A (6). NS5A interacts with many viral and host proteins and participates in various aspects of the viral life cycle (7). For example, NS5A was reported to interact with the hVAP-A protein that takes part in the replication protein complex formation (8–10). NS5A mutations that disrupted the interaction with hVAP-A strongly reduced HCV RNA replication (8). A subset of the genotype 1 HCV with mutations that confer replication fitness shows enhanced NS5A interaction with hVAP-A and suppressed NS5A hyperphosphorylation (8). Thus, NS5A hyperphosphorylation was concluded to reduce genotype 1 HCV replication via reducing NS5A interaction with hVAP-A.
A lot of effort has been devoted to identifying NS5A phosphorylation sites: by mutating potential sites followed by immunoblotting (11–13), by overexpressing NS5A in non-liver cells followed by Edman degradation or mass spectrometry (14, 15), by transfecting the HCV replicon into liver cells followed by mass spectrometry (16–18), and by screening kinases that interact with NS5A in the liver cells (19). Most of the identifications centered around eight highly conserved serine residues in the LCS I region of NS5A. Among them, phosphorylation-ablated alanine mutation at Ser-229 or Ser-235 resulted in a profound reduction in the HCV genotype 2a activity (13, 19); however, the alanine mutations at these two sites seemed to have different effects on the levels of NS5A hyperphosphorylation. Potentially, the above observations are due to the lack of phosphorylation site-specific antibodies that could distinguish the so-called hyperphosphorylated band (p58) of NS5A. Recently, an antibody against Ser-222 phosphorylation was developed (18), but the functions of Ser-222 phosphorylation remain unclear because alanine mutation at Ser-222 does not have an apparent phenotype (11, 13, 19). Moreover, whereas alanine mutation at Ser-225, Ser-229, Ser-232, and Ser-235 reduced HCV genotype 2a activity, the same mutations enhanced genotype 1b activity (11), adding another layer of complexity to the functions of NS5A phosphorylation (7).
To discover HCV phosphoproteins in the conditions that resemble viral infection, we took advantage of the cell culture-derived infectious HCV system (20) and identified three serine phosphorylation sites (Ser-222, Ser-235, and Ser-238) in the LCS I region of NS5A in the HCV (J6/JFH-1)-infected Huh7.5.1 liver cells using LC-MS/MS-based phosphoproteomics. Subsequent study using molecular virology and a phosphorylation site-specific antibody showed that Ser-235 is a CKIα phosphorylation site of NS5A responsible for enhancing genotype 2a HCV replication, probably via enhancing interaction with hVAP-A.
Experimental Procedures
Cells, Reagents, and Constructs
Human hepatocarcinoma 7.5.1 cell line (Huh7.5.1) originated in Francis V. Chisari's laboratory in the Scripps Research Institute was used in most experiments (21). The cells were cultured in DMEM (Invitrogen, catalogue no. 12100-046) with 10% fetal bovine serum (Biological Industries, catalogue no. 040011B) without antibiotics.
A rabbit antibody specific to Ser-235-phosphorylated NS5A was custom-made by GeneTex Corp. using a synthetic peptide (SQLpSAPSLKATC, where pS indicates phosphorylated serine). Antibodies against HCV NS5A (7B5 and 2F6) and NS3 (2E3) were obtained from BioFront Technologies. The double-stranded RNA antibody (J2-1402) was from English and Scientific Consulting Kft., the casein kinase Iα antibody (sc-6477) was from Santa Cruz Biotechnology, Inc., and the β-actin antibody (A5316) was from Sigma-Aldrich. The hVAP-A antibody (15275-1-AP) was from the Proteintech Group. DTT (catalogue no. 3483-12-3) and iodoacetamide (catalogue no. 144-48-9) were obtained from Thermo. Casein kinase inhibitor (D4476, catalogue no. D1944) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (catalogue no. M2003) were obtained from Sigma-Aldrich. The lipid droplet was stained with BODIPY 493/503 (catalogue no. D-3922) from Life Technologies.
The full HCV genome (J6/JFH-1, 5′C19Rluc2AUbi) construct and the subgenome replicon (JFH-1/SG-Neo) were kindly provided by Charles M. Rice (Rockefeller University). The replication defect replicon construct (pSGR-JFH-1) was a gift from Timothy Tellinghuisen (Scripps Research Institute). The cytomegalovirus (CMV) promoter-driven NS3-NS5A expression construct was made via PCR amplification of an NS3-NS5A fragment from the full HCV genome (5′C19Rluc2AUbi) construct with the following primers: forward, 5′-AAGCTTATGGCTCCCA-3′ with a HindIII site at the 5′ end; reverse, 5′-TCTAGATCAGCAGCAC-3′ containing a stop codon and an XbaI site at the 3′ end. The NS3-NS5A fragment was first inserted into the pSTBlue-1 vector (Novagen, catalogue no. 70596). The NS3-NS5A fragment was then excised from the vector via HindIII and XbaI double digestion and ligated into the expression vector pcDNA3.1 (+) (Invitrogen, catalogue no. V790-20). The CMV-driven NS5A expression vectors were made using the Gateway system (Invitrogen). Briefly, the NS5A fragment was amplified with PCR and ligated into the entry vector using the pENTR directional TOPO cloning kits (catalog no. 2400-20). After a sequencing check, the NS5A insert was moved from the entry vector to the destination vector pcDNA-DEST40 (catalog no. 12274-015) using the Gateway LR Clonase II enzyme mix (catalog no. 11791-020). After transformation using the One Shot TOP10 chemically competent cells (catalog no. C4040-52, Invitrogen), the correct vectors were verified with DNA sequencing. Single or combinatory alanine mutations at Ser-222, Ser-235, and Ser-238 were made via site-directed mutagenesis using PCR (KOD hot start polymerase, Merck-Millipore, catalogue no. 71086-4) followed by DpnI (New England Biolabs, catalogue no. R0176L) digestion and transformation into DH5α competent cells. All plasmids were verified with DNA sequencing. Casein kinase Iα small hairpin RNA (shCSNK1A1) plasmid was obtained from the RNAi core facility at the Academia Sinica (Taipei, Taiwan). The target sequence is GCCACAGTTGTGATGGTTGTT. The control non-targeting shRNA sequence is CAAATCACAGAATCGTCGTAT.
Phosphoproteomics
The procedures were described previously (22). Briefly, the Huh7.5.1 cells were infected with HCV (J6/JFH-1) for 72 h before being harvested in a lysis buffer containing 8 m urea, 75 mm NaCl, and 50 mm Tris (pH 8.0). The proteins were reduced with 10 mm DTT, alkylated with 55 mm iodoacetamide, and digested into peptides with trypsin. The tryptic peptides were then desalted using an OASIS HLB column (Waters, catalogue no. WAT094225). To enrich for phosphopeptides, strong cation exchange high performance liquid chromatography (polysulfoethyl A column, 200 × 4.6 mm, 5 μm, 200 Å, capacity: 4 mg of peptides, PolyLC) and immobilized metal affinity chromatography (Pierce IMAC spin column) were used. The conditions used for strong cation exchange were 100% solvent A (5 mm KH2PO4 in 25% acetonitrile, pH 2.6), 2 min; 86% solvent A, 14% solvent B (5 mm KH2PO4 and 500 mm KCl in 25% acetonitrile, pH 2.6), 15 min; 30% solvent A, 70% solvent B, 16 min; 100% solvent B, 11 min; 100% solvent A, 11 min. Phosphopeptide identification was performed on an LTQ-Orbitrap mass spectrometer. Spectra were collected in a data-dependent mode with one full precursor MS1 scan in the Orbitrap followed by six-fragment MS2 scans in the LTQ. Collision-induced dissociation was done at 35% normalized energy level. Peptide peak lists were generated with the Bioworks software package (version 3.3.1, SP1 (23)) using the following criteria: precursor mass range between 600 and 3500 atomic mass units, precursor tolerance 1.4 atomic mass units for grouping spectra, a total ion current of >1000 (arbitrary units)/spectrum, and a minimum of 15 peaks/spectrum. Peptide identifications were done using the Sequest search algorithm against a database containing protein sequences of Homo sapiens (34,942 entries, NCBI RefSeq) and HCV JFH1 isolate (10 entries, Uniprot Q99IB8) plus common protein contaminants: porcine (P00761) and bovine (P00760) trypsin and human keratins (P35908, Q01546, P04264, P12035, P08729, and P35527). The search parameters were as follows: precursor mass tolerance 20 ppm, product mass tolerance 1.0 atomic mass unit, 2 missed cleavages, fixed cysteine carboxyiodomethylation, variable modifications: methionine oxidation plus serine, tyrosine, and threonine phosphorylation. The target-decoy strategy based on reversed protein sequences was used to set a false discovery rate of <1% for identified peptides (24). Common contaminants were excluded. Phosphorylation site-specific identification confidence was based on the Ascore algorithm (25).
Immunoblotting
Cells were washed with ice-cold phosphate-buffered saline (PBS) and resuspended in a lysis buffer (50 mm HEPES, 150 mm NaCl, 5 mm EDTA, 0.2% Nonidet P-40, pH 7.6) containing protease inhibitor (Calbiochem, catalogue no. 539134) and phosphatase inhibitor (Calbiochem, catalogue no. 524625). After centrifugation at 10,000 × g at 4 °C, the supernatant was collected and quantified using a BCA assay kit (BIOTOOLS, catalogue no. TAAR-ZBE6). 20 μg of total protein were separated on a 7.5% SDS-polyacrylamide gel before being transferred to a nitrocellulose membrane (Bio-Rad, catalogue no. 162-0112). The membrane was blocked with TBS-T (150 mm NaCl, 50 mm Tris plus 0.05% Tween 20 and 0.1% bovine serum albumin (BSA), pH 7.6) and incubated with primary antibodies followed by infrared dye-conjugated secondary antibodies (LI-COR, IRDye 680 or 800). The proteins of interest were quantified using the LI-COR Odyssey scanner and software.
Confocal Immunofluorescence Microscopy
Monolayer cells were seeded at a subconfluent density on a coverglass inside a Petri dish. Before staining, the cells were washed with ice-cold PBS and fixed with 4% paraformaldehyde in PBS for 20 min. The cells were treated with a permeabilization buffer (0.3% Triton X-100 and 0.1% BSA in PBS) for 30 min, followed by a blocking buffer (1% BSA, 0.05% saponin, and 0.2% gelatin) for 10 min three times. The cells were probed with primary antibody and then a fluorescence (Alexa 488, 568, or 594)-labeled secondary antibody (Invitrogen). The cell nuclei were counterstained with DAPI (Sigma-Aldrich, catalogue no. D9542). Immunofluorescence images were taken using a Leica SP5C spectral confocal laser-scanning microscope and software.
In Vitro Transcription and Electroporation of HCV RNA
HCV constructs were linearized with XbaI (New England Biolabs, catalogue no. R0145) digestion before being transcribed into viral RNAs using the Ambion MEGAscript T7 in vitro transcription kit (Invitrogen, catalogue no. AM1334). The viral RNA was purified using phenol/chloroform extraction and isopropyl alcohol precipitation. The quality and quantity of the RNA were assessed by gel electrophoresis and a Thermo NanoDrop spectrophotometer.
Electroporation was performed using the Amaxa 4D-Nucleofector system and the Amaxa SF 4D-Nucleofector X solution kit from Lonza. Briefly, the Huh7.5.1 cells were subcultured 1–2 days before electroporation so that they reached between 80 and 90% confluence on the day of electroporation. Prior to electroporation, the cells were trypsinized, washed in PBS, and suspended in the SF solution at a density of 1 × 106 cells/ml. A 100-μl aliquot of the cell suspension was mixed with 5 μg of the viral RNA and then transferred to a 100-μl cuvette for electroporation using the built-in program CM-137. The transfected cells were allowed to recover at room temperature for 10 min and then cultured in the DMEM supplemented with 10% FBS.
Viral RNA Transfection and Luciferase Assay
One day before the experiment, the Huh7.5.1 cells were seeded at a density so that they reached 80% confluence on the next day for transfection. The viral RNA was mixed with DMRIE-C (Invitrogen, catalogue no. 10459-014) at a ratio of 1 μg/3 μl and used to transfect the Huh7.5.1 cells. At various time points (4, 12, 24, 48, and 72 h) after transfection, the cells were lysed and subjected to luciferase assay using the luciferase assay kit (Promega, catalogue no. E1500).
In Vitro Kinase Assay
Active casein kinase Iα was purchased from SignalChem (catalogue no. C64-10G-10). Biotinylated synthetic peptides mapped to amino acids 216–243 of NS5A (101 peptide, biotin-RRLARGpSPPSEASSSVSQLSAPpSLRATC; 111 peptide, biotin-RRLARGpSPPSEASSSVSQLpSAPpSLRATC) were synthesized by GeneTex Corp. The active kinase was mixed with the 101 peptide on ice. ATP solution was then added to the mixture and incubated at 30 °C for 15 min prior to detection of phosphorylation using dot blotting.
Kinase Inhibition
The Huh7.5.1 cells were infected with HCV virus (J6/JFH-1) for 72 h at a multiplicity of infection of 0.001. The infected cells were then seeded in a 6-well plate at a density of 5 × 106 cells/well for 24 h before kinase inhibition with D4476. One μl of D4476 was dissolved in 3 μl of lipid-based transfection reagent (Mirus Bio, TransIT-LT1, catalogue no. MIR 2304) plus 6 μl of serum-free DMEM before being added to the cells. The cells were incubated with the inhibitor for 24 h before immunoblotting. For small hairpin RNA-based kinase silencing, shRNA harboring lentivirus was prepared and used to infect the cells before they were harvested for RNA and protein measurements.
Quantification of HCV RNA
The cells were lysed with the TRIzol LS reagent (Ambion, catalogue no. 10296-010), and the total RNA was isolated with the direct-zol RNA Mini Prep kit (ZYMO Research, catalogue no. R2052). The RNA concentrations were determined by the NanoDrop spectrophotometer. For absolute quantification, 10 ng of RNA were subjected to one-step quantitative analysis using the One-Step qRT-PCR kit (KAPA Biosystems, catalogue no. KK4660) and the StepOnePlus Real-Time PCR system (Applied Biosystem). For relative quantification, 500 ng of RNA were subjected to reverse transcription using SuperScript III reverse transcriptase (Invitrogen, catalogue no. 18080-044) and 2 pm HCV gene-specific primer (5′-CACTCGCAAGCACCCTATCA-3′). qPCR was done using HCV-specific primers: sense, 5′-TCTGCGGAACCGGTGAGTA-3′; antisense, 5′-TCAGGCAGTACCACAAGGC-3′. Abundance of the 18S rRNA was used as an internal control. The primers were 5′-AAACGGCTACCACATCCAAG-3′ (sense) and 5′-CCTCCAATGGATCCTCGTTA-3′ (antisense).
Results
Phosphoproteomics Identified Three NS5A Phosphorylation Sites
To identify phosphorylation sites of the HCV proteins, we analyzed the phosphoproteome of HCV (genotype 2a, J6/JFH-1)-infected Huh7.5.1 cells. 1,087 proteins with 1,773 phosphorylation sites were identified with high confidence (Ascore > 20; i.e. FDR < 0.01) (25). Fig. 1 classifies the phosphorylation sites identified. Serine phosphorylation sites were the largest group, which was dominated by proline-directed sites, followed by acidophilic sites and then basophilic sites. The Phosphoproteome Database of HCV-infected Human Hepatocellular Carcinoma 7.5.1 Cells was established.
FIGURE 1.

Motif logo representation of phosphopeptides identified in the HCV (J6/JFH-1)-infected Huh7.5.1 cells. A total of 1,773 unique phosphorylation sites were classified into phosphoserine, phosphothreonine, and phosphotyrosine groups. The phosphorylated residues were centered at position 0 with 6 amino acids flanking them on the right (positive numbers) and left (negative numbers). Phosphoserine peptides were further classified based on the frequencies of the neighboring amino acids. The bit score represents similarity in amino acids in each position and frequency of each amino acid in each position.
NS5A was the only HCV phosphoprotein identified with three high confidence serine phosphorylation sites (Table 1, Ser-222, Ser-235, and Ser-238) in the LCS I region and conserved across major HCV strains (Fig. 2A). All three sites were identified individually in singly phosphorylated peptides (Table 1). Ser-235 and Ser-238 were identified in a doubly phosphorylated peptide. Three additional phosphorylation sites identified with lower confidence were Ser-225 (Ascore = 16.8), Ser-229 (8.6), and Ser-232 (5.1). Raw mass spectrometry data were deposited in the ProteomeXchange Consortium with the identifier PXD000988.
TABLE 1.
Phosphopeptides identified for the HCV non-structural protein NS5A
The Ascore is an estimate for false discovery rate of a phosphorylation site (boldface and underlined letters). An Ascore of >20 indicates a false discovery rate of <1%. Numbering was based on NS5A (genotype 2a, JFH-1 isolate, Uniprot accession number Q99IB8). MH+, peptide mass; Xcorr, cross-correlation score; ΔCn, Xcorr difference from the next hit; Ions, number of peptides matched.
| Peptide sequence | MH+ | Charge | Xcorr | ΔCn | Ions | Ascore |
||
|---|---|---|---|---|---|---|---|---|
| Ser-222 | Ser-235 | Ser-238 | ||||||
| GSPPSEASSSVSQLSAPSLR | 2,023.9 | 3 | 4.96 | 0.09 | 41/112 | 24.75 | ||
| GSPPSEASSSVSQLSAPSLR | 2,023.9 | 2 | 4.98 | 0.17 | 32/56 | 44.73 | ||
| GSPPSEASSSVSQLSAPSLR | 2,023.9 | 2 | 2.99 | 0.31 | 22/56 | 27.99 | ||
| GSPPSEASSSVSQLSAPSLR | 2,103.9 | 3 | 3.81 | 0.26 | 45/148 | 39.63 | 45.69 | |
FIGURE 2.

Predominant roles of NS5A Ser-235 in HCV replication. A, schematic structure of NS5A with three serine phosphorylation sites identified by LC-MS/MS. Shown are NS5A sequences from six major HCV genotypes: 1a, H77 (Uniprot accession number, P27958); 1b, Con 1 (Q9WMX2); 2a, JFH1 (Q99IB8); 3a, K3a (Q81495); 4a, ED43 (O39929); 5a, EUH1480 (O39928); 6a, 6a33 (Q5I2N3). Amino acid numbering was based on the NS5A sequence. Numbers in parentheses indicate the amino acid position of the HCV genotype 2a polyprotein. LCS, low complex sequence. B and C, time course luciferase reporter virus activity assay in the Huh7.5.1 cells transfected with in vitro transcripts of the WT full-length genome (Rluc-J6/JFH-1) or alanine mutants at single or combined serine residues 222 (S222A), 235 (S235A), or 238 (S238A). Values are mean ± S.E. (error bars) standardized against those at 4 h post-transfection (three independent experiments). *, Student's t test against values of the WT at the same time point (p < 0.05). D and E, time course quantitative RT-PCR profile of the HCV RNA levels in the Huh7.5.1 cells transfected with in vitro transcripts of the wild type subgenome (pSGR-JFH-1) or the alanine mutant replicons. The GND mutant replicon with a mutation at the catalytic site of the RNA-dependent RNA polymerase NS5B served as a replication defect control. F, time course reporter virus activity in the Huh7.5.1 cells infected with the conditioned medium collected from the cells transfected with the wild type or the phosphorylation-ablated S235A mutant full-length viral transcript. G, time course reporter activity in the Huh7.5.1 cells transfected with the wild type or the phosphorylation mimetic S235D mutant full-length HCV transcript.
Ser-235 Dominated over Ser-222 and Ser-238 in Viral Replication
To investigate roles of Ser-222, Ser-235, and Ser-238 phosphorylation sites, we made single, double, and triple mutations of the three sites to phosphorylation-ablated alanine in a full-length HCV reporter construct (Rluc-J6/JFH-1). Fig. 2B summarizes the reporter activity at various time points after the in vitro transcripts of the viral constructs were transfected into the Huh7.5.1 cells. As seen, the reporter activities of the serine-to-alanine mutants, either at Ser-222 (S222A) or Ser-238 (S238A), did not differ from that of the wild type. In contrast, the reporter activity of the S235A mutant was significantly suppressed, suggesting a crucial role of Ser-235 phosphorylation in the HCV activity. In fact, as long as Ser-235 was mutated in double or triple alanine mutation, the reporter activity remained significantly lower than that of the wild type virus (Fig. 2C), consistent with a predominant role of Ser-235 phosphorylation over Ser-222 and Ser-238 phosphorylation in the HCV activity. Note that although single mutation in Ser-222 or Ser-238 alone did not have apparent effects on the reporter activity, double S222A/S238A mutation showed significantly reduced reporter activity (Fig. 2C).
To examine whether Ser-235 phosphorylation participates in HCV replication, a series of single or combinatory alanine mutations of Ser-222, Ser-235, and Ser-238 were made in the HCV replicon (pSG-JFH-1). The HCV replicon lacks the structural proteins and hence cannot produce infectious virus, permitting assessment of viral replication without complications from reinfection. As seen in Fig. 2D, the HCV RNA levels of the S222A and S238A mutant replicons showed a similar trend of increase with time as that of the wild type replicon. In contrast, the S235A mutant replicon failed to increase its RNA levels, which kept decreasing with time as those of the replication defect mutant (Fig. 2D, GND), indicating a replication defect of the S235A mutant. In fact, all mutant replicons failed to produce RNA once Ser-235 was mutated in double or triple alanine mutation (Fig. 2E). Note that single mutation in Ser-222 or Ser-238 alone did not have apparent effects on the replicon RNA levels. Double Ser-222/Ser-238 mutation showed reduced replicon RNA levels (Fig. 2E).
No infectivity was found in the conditioned medium of the Huh7.5.1 cells transfected with the S235A mutant full-length viral RNA (Fig. 2F), consistent with the lack of viral replication activity of the S235A mutant. Phosphorylation mimetic aspartate S235D mutant viral RNA had about 51% of the reporter activity of the wild type viral RNA (Fig. 2G), supporting a role of Ser-235 phosphorylation in HCV replication.
Ser-235 Phosphorylation Level Correlated with HCV Replication Activity
In order to directly measure NS5A Ser-235 phosphorylation, we generated a specific antibody against Ser-235 phosphorylation. On the dot blots (Fig. 3A), the antibody detected a synthetic NS5A peptide only when Ser-235 is phosphorylated. It did not detect other peptides regardless of their phosphorylation at Ser-222, Ser-229, Ser-232, or Ser-238. The detection of Ser-235 phosphorylation by the antibody was not interfered with when Ser-222 and Ser-238 were both phosphorylated on the same peptide, after standardizing the signals with the peptide loading in moles. On the immunoblots (Fig. 3B), the antibody detected a single NS5A band corresponding to the hyperphosphorylated NS5A (p58) band in the HCV (J6/JFH-1)-infected Huh7.5.1 cells. The antibody did not detect phosphorylation-ablated S235A or phosphorylation mimetic S235D mutant NS5A (Fig. 3C). On the confocal immunofluorescence micrographs (Fig. 3D), about 82% of the antibody staining overlapped with the total NS5A antibody staining in the HCV (J6/JFH-1)-infected Huh7.5.1 cells, confirming the specificity of the phospho-specific antibody to NS5A Ser-235 phosphorylation.
FIGURE 3.

Characterization of a phospho-specific antibody against Ser-235 phosphorylation. A, dot blot assessing specificity of the phospho-specific antibody. Serial diluted peptides with or without phosphorylation at Ser-222, Ser-229, Ser-232, Ser-235, or Ser-238 (red type) were spotted on a nitrocellulose membrane and detected with the phospho-specific antibody. B, immunoblotting for NS5A in naive (N) or HCV (genotype 2a, J6/JFH-1) virus-infected Huh7.5.1 cells. pS235, serine 235-phosphorylated NS5A; p58, hyperphosphorylated NS5A; p56, hypophosphorylated NS5A. C, immunoblotting for NS5A in the HEK293T cells transfected with the S235A or the S235D mutant NS5A expression vector using the phospho-specific antibody. D, immunofluorescence staining for NS5A (red) and Ser(P)-235 (green) in the HCV (J6/JFH-1)-infected Huh7.5.1 cells. The nuclei were counterstained with DAPI (blue).
Using the Ser-235 phosphorylation-specific antibody, immunoblotting showed that the levels of Ser-235 phosphorylation (Ser(P)-235) increased in a time-dependent manner in the HCV (J6/JFH-1)-infected Huh7.5.1 cells (Fig. 4, A and B). The ratios of Ser(P)-235 over total NS5A positively and significantly correlated with the wild type reporter virus activity at various time points post-transfection (Fig. 4C), consistent with a role of Ser-235 phosphorylation in HCV replication. Similar increases in the Ser-235 phosphorylation levels were observed in the Huh7.5.1 cells transfected with the wild type replicon (Fig. 4D).
FIGURE 4.

Correlation between Ser-235 phosphorylation and HCV replication. Shown are representative (A) and summary (B) results of time course immunoblotting for NS5A in the HCV (J6/JFH-1)-infected Huh7.5.1 cells. C, a scatter plot of the wild type HCV reporter virus activity (Fig. 2, B and C) against Ser(P)-235/NS5A ratio (Fig. 4B) at the indicated hours after HCV infection. r, Pearson's correlation coefficient with p value in parentheses. D, time course immunoblotting for NS5A in the Huh7.5.1 cells transfected with the wild type subgenomic replicon (pSGR-JFH-1). E, immunoblotting for NS5A in the Huh7.5.1 cells transfected with the WT full-length (J6/JFH-1) and various serine-to-alanine mutant transcripts. F, immunoblotting for NS5A in the Huh7.5.1 cells transfected with the wild type subgenome (pSG-JFH-1) and various serine-to-alanine mutant transcripts. G and H, representative and summary results of the NS5A protein levels in the Huh7.5.1 cells transfected with the wild type and various alanine mutant NS5A constructs. Error bars, S.E.
In the Huh7.5.1 cells transfected with the wild type or the alanine mutant full-length in vitro transcripts, the levels of the total NS5A abundance and the levels of Ser-235 phosphorylation varied greatly among the wild type and the alanine mutants (Fig. 4E), probably due to the effects of the mutations on the viral activity. Interestingly, the standardized Ser(P)-235/NS5A ratios showed correlation with the reporter virus activity (Fig. 2, B and C). Similarly, in the Huh7.5.1 cells transfected with the wild type or the alanine mutant replicons (Fig. 4F), the Ser(P)-235/NS5A ratios also showed correlation with the replicon RNA levels (Fig. 2, D and E).
Note that the alanine mutation per se did not affect the steady state NS5A protein levels in the Huh7.5.1 cells transfected with the wild type and various alanine mutant NS5A expression constructs (Fig. 4, G and H). Because all of the NS5A expression constructs were driven by the same CMV promoter for protein production, no change in the steady state NS5A protein levels suggests that the alanine mutation did not affect NS5A protein stability or degradation. Similar observations were made in the HEK293T cells transfected with the CMV-driven NS3-NS5A expression constructs (see Fig. 5). Thus, the observed changes in the viral replication activity of the various alanine mutants (Fig. 2, D and E) can be attributed to the lack of protein phosphorylation.
FIGURE 5.

Alanine mutation at Ser-235 reduced NS5A hyperphosphorylation. The HCV non-permissive HEK293T cells were transfected with the NS3-NS5A expression constructs, WT or various combinations of alanine mutations, at the indicated serine residues of NS5A. The amounts of total NS5A and Ser-235-phosphorylated NS5A (pS235) were detected with immunoblotting and quantified using the LI-COR system. The bar diagrams summarize mean ± S.E. (error bars) from at ≥3 independent experiments. The numbers inside the red bars indicate the percentage of non-p58 among total NS5A. *, significantly different from WT, Student's t test, p < 0.05.
Alanine Mutation at Ser-235 Reduced NS5A Hyperphosphorylation with a Concomitant Increase in NS5A Hypophosphorylation
To examine the effects of alanine mutations on NS5A phosphorylation without the interference from the HCV life cycle, single and combinatory alanine mutant constructs were made in the CMV-driven NS3-NS5A expression vectors and used to transfect the HEK293T cells that do not support HCV replication (26). Without the interference of the HCV life cycle in the HEK293T cells, the NS5A protein was expressed from all NS3-NS5A constructs at a similar level (Fig. 5, top and bottom). The wild type NS5A showed similar levels of hypo- (p56) and hyperphosphorylation (p58) as those observed in the HCV-infected Huh7.5.1 cells (Fig. 5, bottom). The percentage of the hyperphosphorylated NS5A (p58) among all (p56 + p58) NS5A was similar (∼34%) in the NS3-NS5A-expressing HEK293T and the HCV-infected Huh7.5.1 cells, suggesting that the HEK293T cells are a proper model for examining NS5A phosphorylation.
Alanine mutation at Ser-222 or Ser-238 did not significantly reduce the levels of Ser-235 phosphorylation (Fig. 5, top and middle). Alanine mutation at Ser-235 eliminated Ser-235 phosphorylation. The disappearance of Ser-235 phosphorylation was accompanied by a significant reduction in the hyperphosphorylated NS5A (p58) to about 8% and a concomitant increase in the hypophosphorylated NS5A (non-p58) to about 92% (Fig. 5, bottom). Similar changes were observed in double or triple alanine mutations involving Ser-235.
It is interesting to note that double alanine mutations in Ser-222 and Ser-238 led to a reduced level of Ser-235 phosphorylation (Fig. 5, middle) that correlated with the reduced reporter virus activity and RNA levels (Fig. 2, C and E). It is also interesting to note that there were additional NS5A protein bands between the hyper- and the hypophosphorylated NS5A bands in S235A single and S235A/S238A double mutant vector-transfected cells (Fig. 5, top). These additional NS5A protein bands decreased when Ser-222 was also mutated to alanine in S222A/S235A double and S222A/S235A/S238A triple mutant vector-transfected cells.
Ser-235-phosphorylated NS5A Co-localized with Double-stranded RNA
Consistent with a role of Ser-235 phosphorylation in HCV replication, confocal immunofluorescence staining showed co-localization of Ser-235-phosphorylated NS5A with double-stranded RNA and NS3, markers for HCV replication (Fig. 6, A and B). About 69.8 and 75.7% of the Ser-235-phosphorylated NS5A overlapped with double-stranded RNA and NS3, respectively. About 31.4% of the Ser-235-phosphorylated NS5A showed co-localization with lipid droplets, markers for HCV assembly (Fig. 6C).
FIGURE 6.
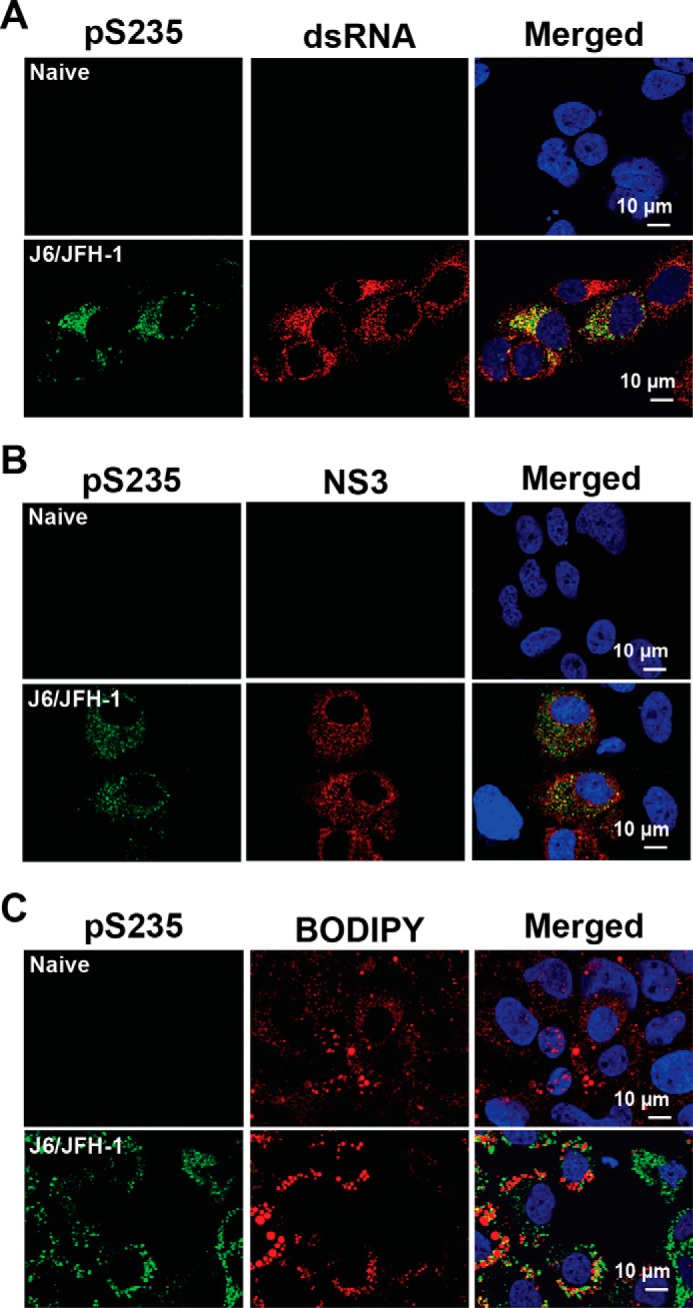
Intracellular localization of Ser-235-phosphorylated NS5A. Naive or HCV (J6/JFH-1)-infected Huh7.5.1 cells were stained for Ser-235-phosphorylated NS5A plus dsRNA (a replication marker), NS3 (a replication complex component), and lipid droplet (BODIPY, or an assembly marker). The nuclei were stained with DAPI.
NS5A Ser-235 Phosphorylation Enhanced Its Interaction with hVAP-A
The hVAP-A protein is a reported component of the HCV replication complex (8–10). We used synthetic peptides to test whether Ser-235 phosphorylation enhances its interaction with hVAP-A to facilitate HCV replication. As seen in Fig. 7, A and B, the synthetic peptide with Ser-235 phosphorylated was able to pull down 49% more hVAP-A than the Ser-235 non-phosphorylated peptide. In line with the enhanced interaction between Ser-235-phosphorylated NS5A and hVAP-A, twice as much (43%) of the S235D NS5A, compared with that (20%) of the S235A NS5A, co-localized with hVAP-A in the HEK293T cells transfected with the CMV-driven NS3-NS5B expression construct (Fig. 7C), supporting the above hypothesis.
FIGURE 7.

Ser-235 phosphorylation enhanced NS5A interaction with hVAP-A. A and B, representative and summary protein pull-down results showing enhanced interaction between the host protein hVAP-A with the synthetic Ser-235-phosphorylated versus non-phosphorylated NS5A peptide. The NS5A-111 peptide is phosphorylated at Ser-222, Ser-235, and Ser-238; the NS5A-101 peptide is phosphorylated at Ser-222 and Ser-238. These peptides were incubated with the Huh7.5.1 cell lysate before the bound proteins were eluted and detected for hVAP-A. C, immunofluorescence staining for NS5A and hVAP-A in the HEK293T cells transfected with WT, S235A, and S235D NS3-NS5B expression vectors. The percentages of the NS5A overlapping with hVAP-A are indicated. p < 0.05. Error bar, S.E.
Casein Kinase Iα Participated in NS5A Ser-235 Phosphorylation and HCV Replication
CKIα was known to be responsible for NS5A hyperphosphorylation (27). In the present study, we found that the CKIα protein levels were elevated in the HCV (J6/JFH-1)-infected Huh7.5.1 cells by 58% (Fig. 8A) and that almost all (99.3%) Ser-235-phosphorylated NS5A co-localized with CKIα in the infected cells (Fig. 8B), suggesting a role of CKIα in NS5A Ser-235 phosphorylation. An in vitro kinase assay followed by dot blotting analysis showed that CKIα directly phosphorylated Ser-235 of the synthetic NS5A-101 peptide that is phosphorylated at Ser-222 and Ser-238 except Ser-235 (Fig. 8C).
FIGURE 8.
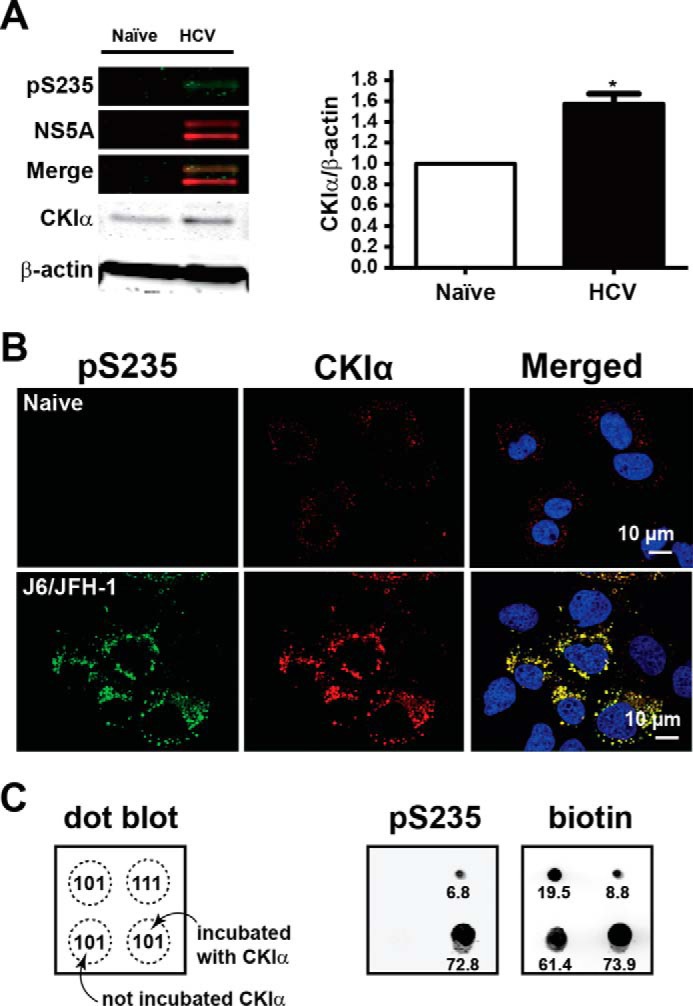
Phosphorylation of NS5A Ser-235 by casein kinase Iα. A and B, immunoblotting and confocal immunofluorescence staining for CKIα and NS5A in the HCV (J6/JFH-1)-infected Huh7.5.1 cells. C, in vitro kinase assay. The biotin-labeled synthetic NS5A-101 peptide was incubated with or without CKIα before Ser-235 phosphorylation (pS235) was assessed with dot blotting. The fluorescence-conjugated streptavidin was used to detect biotin for peptide loading control. The numbers indicate signal intensity. p < 0.05. Error bar, S.E.
In the HCV (J6/JFH-1)-infected Huh7.5.1 cells, the CKI-selective inhibitor D4476 significantly reduced the levels of Ser-235 phosphorylation in a dose-dependent manner (Fig. 9, A and B). CKI inhibition by D4476 also reduced the levels of total NS5A protein (Fig. 9C) and the HCV RNA levels (Fig. 9D) in the infected cells without significant effects on the cell viability (Fig. 9D). The above results were not general effects of CKI inhibition on the HCV life cycle, because in the NS3-NS5A-expressing HEK293T cells that do not support the HCV life cycle (Fig. 9, E and F), D4476 reduced NS5A Ser-235 phosphorylation levels without affecting the total NS5A protein levels. Thus, the reduced HCV RNA and NS5A protein levels in the Huh7.5.1 cells can be attributed to the reduced NS5A Ser-235 phosphorylation levels upon CKI inhibition. Similarly in the HCV (J6/JFH-1)-infected Huh7.5.1 cells, shRNA-mediated CKIα knockdown significantly reduced the levels of Ser-235 phosphorylation, total NS5A and HCV RNA (Fig. 10, A–C). However, when CKIα was knocked down in the Huh7.5.1 cells prior to transfection of the reporter viral RNA, both the wild type and the S235D mutant viral RNA failed to produce significant levels of luciferase activity (Fig. 10D), suggesting additional roles of CKIα next to Ser-235 phosphorylation.
FIGURE 9.

Reduced Ser-235 phosphorylation and HCV RNA levels upon casein kinase Iα inhibition. A, immunoblotting for NS5A in the HCV (J6/JFH-1)-infected Huh7.5.1 cells after they were treated with various doses of the casein kinase I inhibitor D4476 for 1 day. B and C, bar diagrams of three independent immunoblotting experiments. Values are mean ± S.E. (error bars). *, Student's t test against vehicle control (0 μm) at p < 0.05. D, quantitative RT-PCR measurements of HCV RNA levels and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay of Huh7.5.1 cells in response to D4476. The experiments were done in parallel to those in A. E and F, representative and summary results of immunoblotting for NS5A in the NS3-NS5A expressing HEK293T cells treated with D4476 for 2 days.
FIGURE 10.

Reduced Ser-235 phosphorylation and HCV RNA levels upon casein kinase Iα knockdown. A and B, representative and summary results of immunoblotting for CKIα (CSNK1A1) and NS5A in the non-targeting control (shControl) and CKIα knockdown (shCSNK1A1) Huh7.5.1 cells. The Huh7.5.1 cells were infected with HCV (J6/JFH-1) for 1 day prior, followed by CKIα knockdown for 5 days before immunoblotting. C, quantitative RT-PCR measurements of the HCV RNA (HCV) and CKIα RNA levels. The experiments were done in parallel to those in A and B. D, reporter virus activity in the CKIα knockdown Huh7.5.1 cells transfected with the WT, S235A, and S235D mutant full-length HCV RNA. CKIα in the Huh7.5.1 cells was knocked down for 4 days before the cells were transfected with the full-length HCV RNA. *, p < 0.05. Error bars, S.E.
Discussion
The phosphorylation status of NS5A regulates various stages of the HCV life cycle (6). For medical and virological interests, numerous efforts have been devoted to identifying and studying functions of specific NS5A phosphorylation sites (11–14, 17, 18, 28–30). For more than 20 years since its first description by Kaneko et al. in 1994 (31), genetic mutations that either ablate or mimic phosphorylation have been very instrumental in elucidating the functions of putative NS5A phosphorylation sites; however, direct observations or measurements of NS5A phosphorylation at specific sites have not been done until recently when the Harris group (18) produced the first phospho-specific antibody against Ser-222 phosphorylation and showed that Ser-222 phosphorylation corresponds to NS5A hyperphosphorylation. Recently, there are two major leaps that demonstrated critical roles of Ser-225 phosphorylation in the assembly of an active genome replication complex (32) as well as virion production (19); however, the functions of NS5A phosphorylation remain enigmatic and promiscuous (7). To dissect the functions of NS5A phosphorylation, we employed LC-MS/MS-based phosphoproteomics in the HCV (J6/JFH-1, genotype 2a)-infected Huh7.5.1 cells (33) and identified three high confidence NS5A phosphorylation sites conserved in the LCS I region across major genotypes (Table 1 and Fig. 2A, Ser-222, Ser-235, and Ser-238). Using site-directed mutagenesis, we showed that Ser-235 dominated over Ser-222 and Ser-238 in HCV replication because the viral replicon RNA levels were essentially blunted to the levels of the replication defect replicon once Ser-235 was mutated to alanine (Fig. 2, D and E).
We generated a high quality phospho-specific antibody against NS5A Ser-235 phosphorylation (Fig. 3A) and showed for the first time that the Ser-235 phosphorylation levels correlated with the viral activity in general (Fig. 4C) and replication in particular (Fig. 4D). Ser-235-phosphorylated NS5A corresponded to the hyperphosphorylated band of NS5A (Fig. 3B). Ser-235-phosphorylated NS5A co-localized with double-stranded RNA and NS3 in the HCV-infected cells (Fig. 6, A and B), consistent with a role of NS5A Ser-235 phosphorylation in genotype 2a HCV replication (13, 19). The importance of Ser-235 phosphorylation in genotype 2a HCV replication is further supported by the phosphorylation mimetic aspartate or glutamate mutation that maintained a similar level of the replication activity as the wild type virus (Fig. 2G) (13, 19). Mechanistically, we found that Ser-235 phosphorylation probably enhances NS5A interaction with the host replication factor hVAP-A to facilitate HCV replication (Fig. 7, A–C). Taken together, by combining tools of phosphoproteomics, molecular virology, and immunology, we established a coherent site-specific functional description of NS5A Ser-235 phosphorylation in genotype 2a HCV replication.
Although single mutation at either Ser-222 or Ser-238 did not show significant effects on genotype 2a viral activity (Fig. 2, B and D) (13, 16, 18), double alanine mutations in Ser-222 and Ser-238 reduced HCV replication, whereas Ser-235 remained intact (Fig. 2, C and E). The reduction in the replication activity was accompanied with the reduction in the Ser-235 phosphorylation levels in the HCV-infected Huh7.5.1 cells (Fig. 4, E and F). These results suggest that either Ser-222 or Ser-238 needs to remain intact for a full level of Ser-235 phosphorylation and a complete NS5A function. These results also suggest interdependence among Ser-222, Ser-235, and Ser-238 phosphorylation sites. In this regard, it is interesting to note that Ser-235 and Ser-238 were co-identified on the same phosphopeptide (Table 1).
One interesting observation was the additional NS5A bands between the hypo- and hyperphosphorylated NS5A bands in the HEK293T cells transfected with the S235A single and S235A/S238A double mutant constructs (Fig. 5). These additional NS5A bands decreased when Ser-222 was also mutated to alanine in S222A/S235A double and S222A/S235A/S238A triple mutant construct-transfected cells. Perhaps these additional NS5A bands represent phosphorylation at Ser-222 and/or other sites that depend on Ser-222 phosphorylation; however, the functions of these additional NS5A phosphorylation bands remain unknown because alanine mutation at Ser-222 did not seem to affect viral activity (Fig. 2, B and D (13, 16, 19)). It must be noted that the above observations were made in NS3-NS5A-expressing kidney-derived HEK293T cells, instead of Huh7.5.1 liver cells, due to technical issues in the transfection efficiency and expressing T7 polymerase in the Huh7.5.1 cells to drive NS5A expression. Nevertheless, the NS3-NS5A-expressing HEK293T cells permit NS5A hypo- and hyperphosphorylation at ratios not different from those in the HCV-infected Huh7.5.1 cells (Figs. 4A and 5), suggesting that both cells host a similar repertoire of kinases and phosphatases for NS5A phosphorylation.
Many efforts have also been devoted to identifying kinases responsible for NS5A phosphorylation using in vitro kinase assay, library of interference RNA, library of kinase inhibitors, chemical proteomics, and protein-kinase interaction (19, 27, 30, 34–40). CKIα was reported to be responsible for NS5A hyperphosphorylation and down-regulation of CKIα-attenuated HCV RNA replication (27). Recently, CKIα was shown to be responsible for NS5A hyperphosphorylation probably at Ser-225 and Ser-232 involved in viral replication and production (19, 32). CKII was reported to phosphorylate NS5A at Ser-457 at the COOH terminus involved in HCV virion production (30, 34). In addition, Polo-like kinase 1 (Plk1) was also reported responsible for NS5A hyperphosphorylation (40). In this study, we showed for the first time that CKIα can directly phosphorylate Ser-235 (Fig. 8C). Ser-235-phosphorylated NS5A co-localizes with CKIα in the HCV-infected cells (Fig. 8B). Moreover, CKIα inhibition reduced the Ser-235 phosphorylation and HCV RNA levels in the infected cells (Figs. 9 (A–D) and 10 (A–C)), consistent with a critical role of CKIα-mediated NS5A Ser-235 phosphorylation in HCV replication. However, NS5A Ser-235 phosphorylation is probably not sufficient to support HCV replication because the phosphorylation mimetic S235D mutant viral RNA could not maintain the reporter activity in the CKIα knockdown cells (Fig. 10D), compared with the apparent reporter activity in the control cells, where CKIα is present (Fig. 2G). The fact that even the wild type viral RNA could not maintain the reporter activity in the CKIα knockdown cells (Fig. 2G) suggests that CKIα probably has additional roles besides phosphorylating Ser-235. One of the roles could be phosphorylating other serine residues upstream from Ser-235 (e.g. Ser-232, another CKIα site important for the HCV life cycle) (39).
CKIα prefers to phosphorylate serine or threonine residue when the upstream −3 position is phosphorylated (41). This substrate preference makes CKIα particularly interesting in the event of NS5A phosphorylation and signifies the identification of the four NS5A phosphorylation sites (i.e. Ser-229, Ser-232, Ser-235, and Ser-238, separated by two amino acids) (Fig. 2A). To completely resolve the NS5A phosphorylation event by CKIα requires additional phospho-specific antibodies and phosphopeptides because an aspartate or a glutamate residue at the −3 position does not fulfill the substrate preference of CKIα (39, 41).
Author Contributions
M.-J. Y. and W. M. C. conceived and coordinated the study and wrote the paper. W. M. C. was responsible for the experiments shown in Table 1 and Figs. 1, 2, 3, 4, 9, and 10. S.-C. H. was responsible for the experiments shown in Figs. 3 (A and C), 4 (G and H), and 5. W.-T. K. was responsible for the experiments shown in Figs. 2 (B, C, and F) and 4E. C.-W. L. was responsible for the experiments shown in Figs. 6 and 7. K.-Y. L. was responsible for the experiments shown in Figs. 9 (E and F) and 10D. J.-S. S. was responsible for the experiments shown in Figs. 2 (D, E, G), 8B, and 10 (A–C). Y.-H. C. was responsible for the experiments shown in Fig. 8C. J. C. was responsible for the experiments shown in Fig. 10D. S. S.-L. C. critically revised the paper for important intellectual content. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Albert Wang at (Montgomery Blair High School, Silver Spring, MD) and Meredith Zhou at the American Taipei School (Taipei, Taiwan) for generating experiment reagents and proofreading the manuscript. We thank the First Core Laboratory, National Taiwan University College of Medicine for making the NS3-NS5A expression construct. We thank the National Taiwan University College of Medicine for hosting the Phosphoproteome Database of HCV-Infected Human Hepatocellular Carcinoma 7.5.1 Cells and the ProteomeXchange Consortium for hosting the raw mass spectrometry data.
This work was supported by National Health Research Institutes Grant NHRI-EX104-10213BI (to M. J. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
- HCV
- hepatitis C virus
- CKI and CKII
- casein kinase I and II, respectively.
References
- 1. Scheel T. K., and Rice C. M. (2013) Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas D. L. (2013) Global control of hepatitis C: where challenge meets opportunity. Nat. Med. 19, 850–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pawlotsky J. M. (2014) New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146, 1176–1192 [DOI] [PubMed] [Google Scholar]
- 4. Hill A., and Cooke G. (2014) Medicine: hepatitis C can be cured globally, but at what cost? Science 345, 141–142 [DOI] [PubMed] [Google Scholar]
- 5. Reghellin V., Donnici L., Fenu S., Berno V., Calabrese V., Pagani M., Abrignani S., Peri F., De Francesco R., and Neddermann P. (2014) NS5A inhibitors impair NS5A-PI4KIIIα complex formation and cause a decrease of PI4P and cholesterol levels in HCV-associated membranes. Antimicrob. Agents Chemother. 58, 7128–7140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang Y., Staschke K., De Francesco R., and Tan S. L. (2007) Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology 364, 1–9 [DOI] [PubMed] [Google Scholar]
- 7. Ross-Thriepland D., and Harris M. (2015) Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on!. J. Gen. Virol. 96, 727–738 [DOI] [PubMed] [Google Scholar]
- 8. Evans M. J., Rice C. M., and Goff S. P. (2004) Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U.S.A. 101, 13038–13043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tu H., Gao L., Shi S. T., Taylor D. R., Yang T., Mircheff A. K., Wen Y., Gorbalenya A. E., Hwang S. B., and Lai M. M. (1999) Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 263, 30–41 [DOI] [PubMed] [Google Scholar]
- 10. Gao L., Aizaki H., He J. W., and Lai M. M. (2004) Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78, 3480–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Appel N., Pietschmann T., and Bartenschlager R. (2005) Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79, 3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blight K. J., Kolykhalov A. A., and Rice C. M. (2000) Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 [DOI] [PubMed] [Google Scholar]
- 13. Fridell R. A., Valera L., Qiu D., Kirk M. J., Wang C., and Gao M. (2013) Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J. Virol. 87, 2320–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Katze M. G., Kwieciszewski B., Goodlett D. R., Blakely C. M., Neddermann P., Tan S. L., and Aebersold R. (2000) Ser(2194) is a highly conserved major phosphorylation site of the hepatitis C virus nonstructural protein NS5A. Virology 278, 501–513 [DOI] [PubMed] [Google Scholar]
- 15. Reed K. E., and Rice C. M. (1999) Identification of the major phosphorylation site of the hepatitis C virus H strain NS5A protein as serine 2321. J. Biol. Chem. 274, 28011–28018 [DOI] [PubMed] [Google Scholar]
- 16. Lemay K. L., Treadaway J., Angulo I., and Tellinghuisen T. L. (2013) A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J. Virol. 87, 1255–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nordle Gilliver A., Griffin S., and Harris M. (2010) Identification of a novel phosphorylation site in hepatitis C virus NS5A. J. Gen. Virol. 91, 2428–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ross-Thriepland D., and Harris M. (2014) Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J. Virol. 88, 1421–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Masaki T., Matsunaga S., Takahashi H., Nakashima K., Kimura Y., Ito M., Matsuda M., Murayama A., Kato T., Hirano H., Endo Y., Lemon S. M., Wakita T., Sawasaki T., and Suzuki T. (2014) Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-α in infectious virus production. J. Virol. 88, 7541–7555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., and Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhong J., Gastaminza P., Cheng G., Kapadia S., Kato T., Burton D. R., Wieland S. F., Uprichard S. L., Wakita T., and Chisari F. V. (2005) Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U.S.A. 102, 9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rinschen M. M., Yu M. J., Wang G., Boja E. S., Hoffert J. D., Pisitkun T., and Knepper M. A. (2010) Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc. Natl. Acad. Sci. U.S.A. 107, 3882–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 24. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 25. Beausoleil S. A., Villen J., Gerber S. A., Rush J., and Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 26. Kambara H., Fukuhara T., Shiokawa M., Ono C., Ohara Y., Kamitani W., and Matsuura Y. (2012) Establishment of a novel permissive cell line for the propagation of hepatitis C virus by expression of microRNA miR122. J. Virol. 86, 1382–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quintavalle M., Sambucini S., Di Pietro C., De Francesco R., and Neddermann P. (2006) The α isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J. Virol. 80, 11305–11312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanji Y., Kaneko T., Satoh S., and Shimotohno K. (1995) Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 69, 3980–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masaki T., Suzuki R., Murakami K., Aizaki H., Ishii K., Murayama A., Date T., Matsuura Y., Miyamura T., Wakita T., and Suzuki T. (2008) Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82, 7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tellinghuisen T. L., Foss K. L., and Treadaway J. (2008) Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4, e1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaneko T., Tanji Y., Satoh S., Hijikata M., Asabe S., Kimura K., and Shimotohno K. (1994) Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem. Biophys. Res. Commun. 205, 320–326 [DOI] [PubMed] [Google Scholar]
- 32. Ross-Thriepland D., Mankouri J., and Harris M. (2015) Serine phosphorylation of the hepatitis C virus NS5A protein controls the establishment of replication complexes. J. Virol. 89, 3123–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lindenbach B. D., Evans M. J., Syder A. J., Wölk B., Tellinghuisen T. L., Liu C. C., Maruyama T., Hynes R. O., Burton D. R., McKeating J. A., and Rice C. M. (2005) Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 [DOI] [PubMed] [Google Scholar]
- 34. Kim J., Lee D., and Choe J. (1999) Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem. Biophys. Res. Commun. 257, 777–781 [DOI] [PubMed] [Google Scholar]
- 35. Supekova L., Supek F., Lee J., Chen S., Gray N., Pezacki J. P., Schlapbach A., and Schultz P. G. (2008) Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J. Biol. Chem. 283, 29–36 [DOI] [PubMed] [Google Scholar]
- 36. Coito C., Diamond D. L., Neddermann P., Korth M. J., and Katze M. G. (2004) High-throughput screening of the yeast kinome: identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J. Virol. 78, 3502–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reed K. E., Xu J., and Rice C. M. (1997) Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J. Virol. 71, 7187–7197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neddermann P., Quintavalle M., Di Pietro C., Clementi A., Cerretani M., Altamura S., Bartholomew L., and De Francesco R. (2004) Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 78, 13306–13314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quintavalle M., Sambucini S., Summa V., Orsatti L., Talamo F., De Francesco R., and Neddermann P. (2007) Hepatitis C virus NS5A is a direct substrate of casein kinase I-α, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J. Biol. Chem. 282, 5536–5544 [DOI] [PubMed] [Google Scholar]
- 40. Chen Y. C., Su W. C., Huang J. Y., Chao T. C., Jeng K. S., Machida K., and Lai M. M. (2010) Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J. Virol. 84, 7983–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flotow H., Graves P. R., Wang A. Q., Fiol C. J., Roeske R. W., and Roach P. J. (1990) Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem. 265, 14264–14269 [PubMed] [Google Scholar]