

Background: Little is known about cardiac function of myozap, a novel intercalated disc (ID) protein.
Results: Myozap deficiency leads to the junctional remodeling, cardiomyopathy, heart failure, and increased mortality in response to increased biomechanical stress.
Conclusion: Myozap is required for proper adaptation to increased biomechanical stress.
Significance: We provide an essential role of the ID in general and myozap in particular in the context of cardiac remodeling.
Keywords: animal model, cadherin, cardiac development, cardiac hypertrophy, cardiac muscle, cardiomyocyte, cardiomyopathy, cardiovascular disease, heart, heart failure
Abstract
The intercalated disc (ID) is a “hot spot” for heart disease, as several ID proteins have been found mutated in cardiomyopathy. Myozap is a recent addition to the list of ID proteins and has been implicated in serum-response factor signaling. To elucidate the cardiac consequences of targeted deletion of myozap in vivo, we generated myozap-null mutant (Mzp−/−) mice. Although Mzp−/− mice did not exhibit a baseline phenotype, increased biomechanical stress due to pressure overload led to accelerated cardiac hypertrophy, accompanied by “super”-induction of fetal genes, including natriuretic peptides A and B (Nppa/Nppb). Moreover, Mzp−/− mice manifested a severe reduction of contractile function, signs of heart failure, and increased mortality. Expression of other ID proteins like N-cadherin, desmoplakin, connexin-43, and ZO-1 was significantly perturbed upon pressure overload, underscored by disorganization of the IDs in Mzp−/− mice. Exploration of the molecular causes of enhanced cardiac hypertrophy revealed significant activation of β-catenin/GSK-3β signaling, whereas MAPK and MKL1/serum-response factor pathways were inhibited. In summary, myozap is required for proper adaptation to increased biomechanical stress. In broader terms, our data imply an essential function of the ID in cardiac remodeling beyond a mere structural role and emphasize the need for a better understanding of this molecular structure in the context of heart disease.
Introduction
The intercalated disc (ID)4 is an indispensable structural constituent of the heart that mechanically and electrically connects neighboring cardiomyocytes. IDs are composed of three distinct structural and functional subunits or junctions as follows: fascia adherens and desmosomes, both contributing to mechanical coupling, and gap junctions that form ion channels between cardiomyocytes providing electrical coupling and a synchronized heart rhythm (1). In recent times, the ID has emerged as a hot spot and the focus of research due to a direct association between mutations of several ID proteins and cardiac muscle diseases. Human mutations of plakophilin, plakoglobin, desmoplakin, and desmin may cause arrhythmogenic right ventricular cardiomyopathy (ARVC) (and/or dilated cardiomyopathy) (2–5). Moreover, loss-of-function studies in mice demonstrated that deficiency of ID proteins like plakoglobin, β-catenin, and N-cadherin is lethal (6–8). Considerable efforts have thus been made to better understand the structural and functional complexity of ID proteins and their contribution to the pathogenesis of heart muscle disease.
Cardiac remodeling is a pathological process of the heart that involves progressive changes in cellular size, structure, and function along with molecular alterations due to extrinsic or intrinsic stress (9–11). It can be triggered by various overlapping or independent disease conditions like hypertension, myocardial infarction, inflammation, cardiac ischemia, and/or genetic defects (12–15). Cardiac hypertrophy is one of the hallmarks of cardiac remodeling prompted by hemodynamic or metabolic stress. Although it has traditionally been considered a beneficial and adaptive process, it has also been associated with activation of maladaptive cellular processes and signaling pathways that eventually lead to cardiomyopathy (15–18).
The development of cardiac hypertrophy is a complex process that involves several distinct and interconnected signal transduction pathways involved in its induction and advancement. These pathways include multiple kinases, phosphatases, and transcription factor cascades like MAPKs, calcineurin/NFAT, PI3K/AKT, and Rho/SRF, etc. (19–28). MAPK signaling pathways consist of kinases that phosphorylate and activate p38, JNK1/2, ERK1/2, and ERK5, which on activation can promote the hypertrophic gene program via phosphorylation of multiple downstream targets (19, 21, 26, 27). Calcineurin, however, is a serine/threonine protein phosphatase that dephosphorylates transcription factors of the NFAT family resulting in its translocation to the nucleus and subsequent activation of pro-hypertrophic gene expression (22, 28). An important pathway directly involving the ID in the context of cardiac hypertrophy is Wnt/β-catenin signaling (29–33). It has been shown that constitutive overexpression of the ID protein β-catenin induces hypertrophic growth in vitro in cultured adult cardiomyocytes (34). To the contrary, its deletion in mouse heart attenuates the hypertrophic response to transverse aortic constriction (TAC) in vivo (30).
We have recently identified myozap as a new member of cardiac-enriched ID proteins (35). Our earlier in vitro and in vivo data revealed that overexpression of myozap activates Rho-dependent SRF signaling (35–37). Moreover, cardiac restricted overexpression of myozap in mice resulted in protein aggregate-associated cardiomyopathy, although the knockdown of its ortholog in zebrafish led to severe contractile dysfunction and cardiomyopathy (35, 36). We have generated myozap-null mutant mice (Mzp−/−) to further characterize its role in vivo and to study the cardiac consequences of a targeted deletion of myozap in a higher vertebrate. Here, we show that deletion of myozap did not disrupt the structural and functional integrity of the heart suggesting that myozap is dispensable for cardiac development and function under baseline conditions. However, in response to increased biomechanical stress, Mzp−/− mice displayed severe cardiac and cellular hypertrophy, cardiac remodeling, increased fibrosis, and alterations in hypertrophic signaling cascades, i.e. inhibition of MAPK/SRF signaling and activation of β-catenin/GSK-3β and calcineurin/NFAT signaling.
Experimental Procedures
Myozap Null Mutant Mice Generation
Myozap-deficient mice were generated using a classical constitutive germ line knock-out approach by deleting exon 1 that carries the translation initiation site. The 5′ arm contained 5.8 kb of the myozap promoter region upstream of exon 1, whereas the 3′ arm contained 2.7 kb of intronic region between exons 1 and 2. A lacZ reporter and a neomycin resistance cassette under the control of endogenous myozap promoter or phosphoglycerate kinase, respectively, were fused to the 5′-untranslated myozap sequences contained in the 5′ arm of the targeting vector. The resulting vector was linearized and electroporated into mouse embryonic stem cells. Positive stem cell clones, identified by Southern blot analyses, were injected into C57BL/6J mice blastocysts. Derived chimeric animals were bred with C57BL/6J mice to obtain heterozygous mice that carry the targeted myozap locus in their germ line. Subsequent genotyping was performed by PCR using primers, neoF, 5′-GAT GCA TCG CCA TGG GTC AC-3′, and neoR, 5′-ACC TGC CCA TTC GAC CAC CA-3′; and myozap_wtF, 5′-CTT GGC AGA GGC TTT CTC TC-3′, and 5′-CAG GAA ACA GAG CTA TAC GGA C-3′.
Induction of Hypertrophy in Cultured Cardiomyocytes
Neonatal mouse or rat cardiomyocytes were isolated with standard isolation methods as described elsewhere. After 24 h of isolation, cells were infected with respective viruses and stimulated either with the pharmacological agent, phenylephrine (PE, Sigma, Munich Germany), or by mechanical stretch, as reported earlier (37). Non-stimulated or un-stretched cardiomyocytes served as controls in respective experiments.
Transverse Aortic Constriction Isoproterenol Osmotic Pump Implantation and Echocardiography
TAC and echocardiography were performed in 8-week-old male Mzp−/− mice and their wild-type counterparts as described previously (38). Osmotic mini-pumps filled with isoproterenol (20 μg/kg body weight/min) prepared in PBS with 1 mg/ml l-ascorbate (Sigma) were implanted subcutaneously. Control group received vehicle l-ascorbate.
Antibodies
Antibodies were as follows: α-actinin, mouse monoclonal (1:200; Sigma); Akt, rabbit polyclonal (1:1,000; Cell Signaling Technology); p-Akt, mouse monoclonal (1:1,000; Cell Signaling Technology); connexin-43, rabbit polyclonal (1:1000, Sigma); catenin, mouse monoclonal (1:100; Zymed Laboratories Inc.); desmin, rabbit polyclonal (1:25; Cell Signaling Technology); desmoplakin, mouse monoclonal (1:10; Millipore); ERK1/2, mouse monoclonal (1:2,000; Cell Signaling Technology); p-ERK1/2, rabbit monoclonal (1:1,000; Cell Signaling Technology); MAPK p38, rabbit polyclonal (1:1,000; Cell Signaling Technology); MAPK p-p38, mouse monoclonal (1:2,000; Cell Signaling Technology); myozap, mouse monoclonal (1:50; Progen); N-cadherin, mouse monoclonal (1:250; BD Biosciences); TRITC-labeled phalloidin (1:500; Sigma); ZO-1, rabbit polyclonal (1:250; Millipore).
Histology
Standard routine protocols were applied for both H&E- and Sirius-red/fast green staining as detailed before (36). Briefly, sections were stained with H&E or Sirius red/fast green to analyze tissue architecture and degree of fibrosis, respectively. Several images were taken in “merge” mode to cover the entire heart section on BZ-9000-E HS all-in-one fluorescence microscope (Keyence) at ×4 magnification (CFI Plan Apochromatic λ ×4 objective, NA 0.20; Nikon), and all the images were merged to form a single image. Fibrotic areas stained in red were measured using Keyence's software module with the non-fluorescence intensity single-extraction mode. Mouse heart sections prepared as above were also used for lectin staining, carried out using FITC-conjugated lectin from Triticum vulgaris (wheat) (Sigma) according to the manufacturer's instructions. The cross-sectional area of cardiomyocytes differentially visible by lectin staining was determined using ImageJ software (version 1.46).
Electron Microscopy
Mice were weighed and anesthetized with an intraperitoneal injection consisting of ketamine (12 mg/ml) and xylazine (1.6 mg/ml) (10 μl/g body weight). The heart was pre-perfused with 1% procaine in 0.1 m PBS and fixed with 6% glutaraldehyde in 0.1 m PBS by transcardial vascular perfusion to harvest papillary muscles. Tissue blocks were post-fixed with 2% osmium tetroxide and embedded in araldite. Ultrathin sections were processed with uranyl acetate and lead citrate and viewed with Zeiss EM 900 microscope (Carl Zeiss).
Immunofluorescence Microscopy
Mouse hearts were harvested, molded into Tissue-Tek Cryomolder (Sakura Finetek), and frozen on dry ice. Sections were cut into 5–7-μm slices and plated on glass slides, fixed in 4% paraformaldehyde for 10 min, permeabilized with methanol at −20 °C, and blocked with 12% BSA in PBS for 1 h at room temperature. The sections were then incubated with primary antibodies diluted in 2.5% BSA in PBS at 4 °C overnight followed by incubation with respective AlexaFluor-conjugated secondary antibodies (Invitrogen) for 90 min at room temperature at a dilution of 1:200 in 2.5% BSA in PBS containing DAPI (4′,6′-diamidino-2-phenylindole). Fluor-mount medium (Biozol) was used for mounting. Images were captured on BZ-9000-E HS all-in-one fluorescence microscope (Keyence) at ×20 (CFI Plan Apochromatic λ ×20 objective, NA 0.75; Nikon) or ×40 (CFI Plan Apochromatic λ ×40 objective, NA 0.95; Nikon) magnifications.
Real Time PCR
Total RNA was isolated from left ventricles using QIAzol lysis reagent (Qiagen) and a Precellys homogenizer with plastic beads. Transcription into cDNA was carried out with 1 μg of DNA-free RNA using the Superscript III first strand cDNA synthesis kit (Life Technologies, Inc.). The EXPRESS SYBR GreenER reagent (Life Technologies, Inc.) was used for the qRT-PCR setup, and measurements were carried out in a CFX96 real time cycler (Bio-Rad). Cycling conditions used were as follows: 3 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 45 s at 60 °C, step for annealing and extension as well as for data collection. Rpl32 was used as a normalization control (37).
Immunoblotting
Left ventricles were homogenized with a Precellys homogenizer with plastic beads in RIPA buffer containing protease inhibitor mixture (Roche Applied Science) and phosphatase inhibitor mixtures 2 and 3 (Sigma). Protein concentration was determined with the DC assay according to the manufacturers' instructions (Bio-Rad). Protein samples were resolved by SDS-PAGE with a 10 or 12% polyacrylamide gel and transferred to nitrocellulose membrane. Immunoblotting was carried out with target-specific primary antibodies diluted in 5% milk prepared in 0.1% TBST overnight at 4 °C. Subsequently, the incubation in HRP-coupled secondary antibody was carried out for 1 h at room temperature. Proteins were visualized using a chemiluminescence kit (ECL-select; GE Healthcare) and detected by FluorChem Q imaging system (Biozym). Densitometric analyses were performed with the ImageJ software version 1.46.
Reporter Gene Assays
NRVCMs were infected with adenovirus encoding synthetic microRNA against myozap (50 m.o.i.) for its knockdown, and mir-NEG (50 m.o.i.), an unspecific synthetic microRNA, was used as a control. AdSRF-RE-luc (10 m.o.i.) or AdNFAT-RE-luc (20 m.o.i.) carrying a firefly luciferase, and AdRen-luc carrying Renilla luciferase (for normalization of the measurements) were co-infected. After 72 h of infections, cells were harvested in passive lysis buffer (Promega) and vortexed for 15 min, and luciferase assay was performed using a dual-luciferase reporter assay (Promega) according to the manufacturer's protocol on infinite m200 PRO system (Tecan). All the experiments were performed in quadruplicates or hexaplicates and repeated at least three times.
Cloning and Purification of myozap in Bacterial System
Full-length mouse myozap was cloned from mouse heart cDNA using the following primers: MZP forward (5′-GCT GGC ACC ATG CTG CGC TCC ACG TCC-3′) and MZP reverse (5′-GCT GGG TCG CCC TAA GTC AGC GTT TTC TT-3′) for a first PCR, and attB forward (5′-GGG GAC AAG TTT GTA CAA AAA AGC TGG CAC C-3′) and attB reverse (5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC GCC-3′) for a second step PCR. The PCR product was recombined into the pDonR221 plasmid (Invitrogen, Karlsruhe, Germany) using the Gateway technology and was subsequently recombined into expression plasmid pDest17 to obtain His-tagged expression construct. Plasmids were transformed into Escherichia coli BL21 cells (Invitrogen, Karlsruhe, Germany), grown in 500 ml of Luria-Bertani medium with 100 mg/liter carbenicillin until an A600 of 0.5–0.6 was reached, induced with 0.2% arabinose (Sigma), and grown further at 20 °C for 16 h. Overexpressed protein was purified using Ni2+ charged nickel-nitrilotriacetic acid (Qiagen) affinity column, and eluted fractions of high purity were pooled and concentrated by centrifugation with the Pierce Concentrator, PES (molecular mass cutoff of 30 kDa) (Thermo Scientific).
Actin Co-sedimentation Assay
Purified myozap from the bacterial system was used to perform actin co-sedimentation assays using a kit purchased from Cytoskeleton Inc. Briefly, BSA (as a negative control), actinin (as a positive control), or myozap were incubated with or without F-actin provided with the kit for 30 min. Samples were then ultracentrifuged (Beckman Coulter Inc.) at 150,000 × g. Supernatants were carefully dispensed in fresh tubes followed by addition of Laemmli buffer and SDS-PAGE.
Statistical Analyses
All the presented results are the means ± S.E., unless stated otherwise. Statistical analyses were performed using two-tailed Student's t test, one-way or two-way ANOVA (followed by Student-Newman-Keuls post-hoc tests when appropriate), respectively. p values < 0.05 were considered statistically significant.
Results
Myozap Mutant Mice Appear Normal and Develop Mild Cardiac Hypertrophy upon Aging
We previously reported that knockdown of the myozap ortholog in zebrafish leads to contractile dysfunction and cardiomyopathy (35). To determine whether myozap deficiency also alters cellular morphology, we knocked down myozap in neonatal rat cardiomyocytes and observed no baseline effect on cell surface area compared with control cells (Fig. 1, A and B). Phenylephrine treatment, however, provoked cellular hypertrophy in control cells, which was further exacerbated by myozap deficiency (Fig. 1, A and B). Moreover, the hypertrophic gene program (Nppa, Nppb) was strongly induced in cardiomyocytes upon down-regulation of myozap (Fig. 1, C and D).
FIGURE 1.

Knockdown of myozap in NRVCMs and basal characterization of Mzp−/− mice. A, primary NRVCMs were infected with adenovirus encoding synthetic microRNA to knock down myozap in the absence or presence of PE and immunostained against α-actinin. B, cell surface area was measured using Keyence's HybridCellCount software module in fluorescence intensity single-extraction mode. Statistical significance was determined using Shapiro-Wilk (for equal distribution) and Kruskal-Wallis (one-way ANOVA) tests. n > 500. Expression of fetal genes Nppa (C) and Nppb (D) was determined by quantitative real time PCR. Data represented are mean of two independent experiments performed in triplicate. E, strategic diagram showing the targeting vector and myozap genomic locus. Homologous recombination replaces exon 1 encoding the translation initiation site with a neomycin-lacZ reporter cassette. Deletion of myozap was confirmed by genotyping (F), qRT-PCR (G), immunoblotting (H), and immunofluorescence microscopy (I). J, electron micrographs showing the structural architecture of cardiac muscle. As to the intercalated discs, fasciae adherents (FA) are shown at higher magnification (see boxes). There are no ultrastructural differences between wild-type (WT) and myozap−/− (Mzp−/−) mice. GJ, gap junction; M, M-line; Mi, mitochondria; Z, Z-line. Statistical significance was calculated by two-way ANOVA. Error bars show mean ± S.E. *, p < 0.05; ***, p < 0.001.
To gain a deeper understanding of its cardiac function and to study the loss-of-function effects of myozap, we now generated a Mzp−/− mouse line with germ line deletion of the myozap allele by replacing exon 1 (which contains the translational start site) with a neomycin resistance cassette and a lacZ reporter gene (Fig. 1E). Genotyping was performed by PCR (Fig. 1F), and the absence of myozap was confirmed by qRT-PCR, immunoblotting, and immunofluorescence (Fig. 1, G–I). Mzp−/− mice were fertile, born with expected Mendelian ratios of offspring, and revealed a normal life span. As myozap is an ID protein, we assessed hearts from these mice at the ultrastructural level to find potential structural alterations of the ID. However, electron microscopy data revealed no obvious structural abnormalities at the ID (Fig. 1J).
Myozap-null Mice Develop Mild Cardiac Hypertrophy upon Aging and in Response to Isoproterenol Treatment
We next analyzed the cardiac phenotype of Mzp−/− mice at 8 weeks and after 52 weeks of age. In line with the in vitro data, young myozap-deficient mice did not exhibit any phenotypic, morphometric, and functional (echocardiography) differences from that of age- and gender-matched wild-type littermates (data not shown). Nevertheless, upon aging, 1-year-old mice developed mild cardiac hypertrophy (Fig. 2, A–H), as evident from increased heart weight/body weight ratios (Fig. 2C), significantly increased expression of natriuretic peptide A (Nppa, Fig. 2G), and a trend toward reduced fractional shortening (Fig. 2F). However, neither ventricular dilatation nor pulmonary congestion as a potential sign of heart failure was observed (Fig. 2, D and E). Electrocardiography measurements in 1-year-old mice provided no difference in QRS duration, QT interval, or PQ interval between genotypes (Table 1). Similarly, Langendorff heart model-based electrophysiology study revealed no difference in spontaneous or induced arrhythmias (neither ventricular nor atrial) nor in AV nodal conduction. Likewise, orciprenaline treatment did not cause more arrhythmias in either genotype (data not shown). However, significantly shorter ventricular action potential durations at paced cycle length of 100–200 ms were observed in myozap-KO mice (Table 2).
FIGURE 2.

Mild hypertrophy and cardiomyopathy upon aging and isoproterenol treatment in Mzp−/− mice. Comparative images of 1-year-old myozap-KO (Mzp−/−) and WT mouse hearts (A) and H&E-stained whole heart sections (B). Comparative phenotypic parameters from 1-year-old Mzp−/− and WT mice, heart weight/body weight (HW/BW, C), lung weight/body weight (Lung W/BW, D), diastolic left ventricular internal dimension (LVIDd, E), fractional shortening (FS, F), and the expression of fetal genes Nppa (G) and Nppb (H) show no severe abnormalities in Mzp−/− mice. n = 6. Similarly, comparative phenotypic parameters from isoproterenol (Iso)/PBS-treated Mzp−/− and WT mice, heart weight/body weight (HW/BW, I), lung weight/body weight (Lung W/BW, J), diastolic left ventricular internal dimension (LVIDd, K), fractional shortening (FS, L), and the expression of myozap (M) fetal genes Nppa (N) and Nppb (O) show no severe abnormalities in Mzp−/− mice. n = 8. Statistical significance was determined using two-tailed Student's t test. Error bars show mean ± S.E. *, p < 0.05; ***, p < 0.001. Cont, control.
TABLE 1.
Comparative ECG measurements of 1-year-old myozap-KO and wild-type littermate mice indicating no significant differences between the genotypes
Data are represented as mean ± S.E. Statistical significance was calculated by two-tailed Student's t test (n = 7).
| Parameter | Unit | Wild type | Myozap | p value |
|---|---|---|---|---|
| RR interval | ms | 114.11 ± 2.699 | 107.24 ± 2.264 | 0.097 |
| Heart rate | Beats/min | 527.96 ± 13.001 | 561.26 ± 12.028 | 0.107 |
| PQ duration | ms | 39.91 ± 1.756 | 45.04 ± 4.684 | 0.371 |
| Isoelectric line | mV | −0.03 ± 0.005 | −0.03 ± 0.005 | 0.985 |
| P duration | ms | 16.61 ± 0.459 | 15.73 ± 1.128 | 0.520 |
| QRS duration | ms | 14.01 ± 0.579 | 13.47 ± 0.185 | 0.435 |
| QT short | ms | 50.00 ± 2.285 | 44.46 ± 2.837 | 0.185 |
| QT long | ms | 54.14 ± 2.317 | 49.87 ± 2.072 | 0.228 |
| P amplitude | mV | 0.07 ± 0.016 | 0.06 ± 0.005 | 0.702 |
| Q amplitude | mV | 0.01 ± 0.006 | 0.01 ± 0.004 | 0.377 |
| R amplitude | mV | 0.50 ± 0.071 | 0.54 ± 0.045 | 0.671 |
| S amplitude | mV | −0.15 ± 0.045 | −0.27 ± 0.070 | 0.206 |
| T amplitude | mV | 0.00 ± 0.003 | 0.00 ± 0.002 | 0.884 |
| P wave area | mVX time unit label | 0.25 ± 0.059 | 0.31 ± 0.047 | 0.459 |
| QRS area | mVX time unit label | 2.23 ± 0.385 | 1.96 ± 0.421 | 0.666 |
| T wave area | mVX time unit label | −0.05 ± 0.120 | −0.13 ± 0.185 | 0.746 |
| P area (positive and negative) | mVX time unit label | 0.46 ± 0.093 | 0.43 ± 0.039 | 0.762 |
| QRS area (positive and negative) | mVX time unit label | 2.92 ± 0.333 | 3.30 ± 0.193 | 0.391 |
| T wave area (positive and negative) | mVX time unit label | 0.31 ± 0.052 | 0.41 ± 0.127 | 0.533 |
TABLE 2.
Electrophysiological findings in the isolated heart
APD denotes action potential duration; the number denotes the repolarization level (e.g. APD90 = action potential duration at 90% repolarization). AT denotes activation time. All APDs are mean over all three recording locations. Data represented as a mean ± S.E. Statistical significance was calculated by two-tailed Student's t-test (n = 7).
| Paced cycle length | Measured parameter | Wild type | Myozap-ko | p value |
|---|---|---|---|---|
| 100 | APD90 | 43.9 ± 3.4 | 33.1 ± 2.4 | 0.04 |
| APD70 | 23.3 ± 2.2 | 16.2 ± 2.4 | 0.07 | |
| APD50 | 12.7 ± 1.5 | 10.1 ± 1.6 | 0.30 | |
| 150 | APD90 | 50.0 ± 3.3 | 37.5 ± 2.6 | 0.03 |
| APD70 | 27.7 ± 2.3 | 18.6 ± 2.4 | 0.03 | |
| APD50 | 14.1 ± 2.0 | 10.0 ± 1.7 | 0.19 | |
| 200 | APD90 | 52.8 ± 5.1 | 41.5 ± 3.7 | 0.17 |
| APD70 | 29.8 ± 2.6 | 20.3 ± 3.2 | 0.08 | |
| APD50 | 15.8 ± 2.2 | 9.0 ± 2.3 | 0.10 |
Additionally, to translate the in vitro findings in vivo, we subjected Mzp−/− mice to treatment with the non-selective β-adrenergic agonist isoproterenol. But unlike in vitro, we observed only a mild cardiac phenotype (Fig. 2, I–O). The majority of the parameters like heart/body (Fig. 2I) or lung/body (Fig. 2J) weights, left ventricular inner diameter (Fig. 2K), or the expression of Nppa (Fig. 2N) in Mzp−/− mice (Fig. 2M) was not indifferent from the respective wild-type groups. However, Nppb was significantly up-regulated in knock-out compared with WT mice (Fig. 2O).
Severe Cardiomyopathy in Myozap-deficient Mice Subjected to Pressure Overload
In the absence of an obvious baseline phenotype, and insignificant differences with β-agonist treatment, we next asked whether myozap might be required for cardiac adaptation to increased biomechanical stress. Thus, we challenged Mzp−/− mice by pressure overload due to TAC. TAC resulted in significant cardiac hypertrophy in wild-type mice, which was further enhanced in myozap-null animals as shown by an increase in the heart size (Fig. 3A) and increased heart weight/body weight ratios, as well as left ventricular weight/body weight ratios (Fig. 3, B and C). Echocardiography assessments further substantiated the notion of a pathological growth of myozap-deficient mouse hearts after TAC as severe contractile dysfunction and a markedly reduced fractional shortening were observed (Fig. 3, D–F); nevertheless, posterior wall thickness was unchanged at diastole and rather reduced at systole compared with wild-type counterparts (Fig. 3, G and H). The presence of overt heart failure in these mice was corroborated by a pronounced increase in lung weight/body weight ratio (Fig. 3I). Finally, mortality post-TAC was found to be significantly increased in Mzp−/− mice compared with TAC-operated wild-type littermates (Fig. 3J).
FIGURE 3.

Biomechanical stress-induced severe cardiomyopathy in myozap-deficient mice. A, 8-week-old wild-type (WT) and myozap-KO (Mzp−/−) mice underwent TAC or SHAM operations. Four weeks post-operations (n = 9 (WT-SHAM), 15 (WT-TAC), 9 (KO-SHAM), 9 (KO-TAC)), phenotypic abnormalities were analyzed by measuring cross-sectional areas of the heart (A), heart weight/body weight (HW/BW, B), left ventricular weight/body weight (LVW/BW, C), diastolic left ventricular internal dimension (LVIDd, D), systolic left ventricular internal dimension (LVIDs, E), fractional shortening (FS, F), posterior wall thickness at diastole (LVPWd, G) and systole (LVPWs, H), lung weight/body weight (Lung W/BW, I), and mortality rate observed during post-operative 4-week housing (J). Mzp−/− mice displayed severe cardiac pathology, disturbed contractility, signs of heart failure and increased mortality under the influence of TAC. n = 17 (SHAM) or 24 (TAC). Statistical significance was calculated by two-way ANOVA. Error bars show mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Biomechanical Stress Accentuates Cellular Hypertrophy and Fibrosis in Myozap-deficient Mice
Consistently, cardiomyocytes in Mzp−/− mice displayed a significantly increased cell surface area compared with wild-type littermates after TAC despite no significant differences between genotypes in SHAM operated mice (Fig. 4, A and B). Furthermore, there was a highly “super” increase in the expression of the fetal genes, Nppa and Nppb, in Mzp−/− mice due to pressure overload compared with wild-type counterparts (Fig. 4C). Similarly, the expression of β-MHC (Myh7) was dramatically increased (Fig. 4, D–F), whereas levels of α-MHC (Myh6) (Fig. 4, G–I) and Serca2a (Fig. 4, J–L) were significantly reduced at both mRNA and protein levels in TAC Mzp−/− mice. Altogether, these findings reveal an exacerbation of cardiac and cellular hypertrophy in myozap-null mice upon biomechanical stress. Cardiac fibrosis is an important sign of pathological hypertrophy and typically an indicator of a functionally deteriorating heart. In line with the observed severe contractile dysfunction, and cardiac hypertrophy, we also observed excessive fibrosis in Mzp−/− compared with the control mice group in response to TAC (Fig. 4, M and N). Consistently, we observed an increased expression of prototypic fibrotic markers like collagen III and PAI-I (Fig. 4O).
FIGURE 4.

Biomechanical stress accelerated cellular hypertrophy in myozap-deficient mice. Heart slices obtained from SHAM or TAC-operated wild-type (WT) or myozap-KO (Mzp−/−) mice were stained with FITC-conjugated lectin (A), and cell surface area of the cardiomyocytes was measured (B) showing accelerated hypertrophy in Mzp−/− mice compared with the WT littermates. This was further confirmed by super induction of fetal genes Nppa/Nppb determined by quantitative real time PCR (C), marked up-regulation of β-myosin heavy chain (Myh7, D–F), and reduced expression of alpha-myosin heavy chain (Myh6, G–I), or Serca2a (J–L), both at transcript and protein levels. M, heart sections obtained from SHAM or TAC operated wild-type (WT) or Mzp−/− mice were stained with Sirius-red/Fast-green (upper panel), and the representative fibrotic area is highlighted in the lower panel. N, fibrotic lesions stained in red were measured using Keyence's HybridCellCount software module in bright field single-extraction mode, which confirms the increased fibrosis in Mzp−/− mice in comparison with WT littermates. O, increased expression of fibrotic markers collagen III (Col-III) and plasminogen activator inhibitor I (PAI-I) further supports the increased fibrosis in Mzp−/− mice. n = 6. Statistical significance was calculated by two-way ANOVA. Error bars show mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Biomechanical Stress Causes Junctional Remodeling in Mice Lacking Myozap
Myozap is an ID protein that interacts with other ID proteins, including ZO-1 and desmoplakin. ID proteins are known to (partially) compensate for the gain or loss of other ID molecules by “junctional” remodeling (39, 40). Given the dramatic response to biomechanical stress, we were interested in testing whether these findings are also associated with any structural and/or junctional effects. We thus studied the ultrastructure of myozap-deficient hearts in comparison with wild-type littermates after TAC. Biomechanical stress caused structural alterations, particularly in myozap-KO mice. The prominent difference observed between genotypes was disarrangement of the intercellular junctions in Mzp−/− mice (Fig. 5, A and B, panels I and II). Very occasionally, focal widening of the intercellular space within junctions was also observed in the Mzp−/− aortic banded mice, which was absent in wild-type counterparts (data not shown). In the myozap knock-out mouse, which showed the most severe functional defects after TAC, some myocytes displayed large cytoplasmic fields without myofilaments but filled with autophagic vacuoles, polyribosomes, and other cell organelles. Such fields were detectable even by light microscopy (Fig. 5B, panel III) and suggested the beginning of myocyte degeneration (indicated with an arrow). In contrast, wild-type TAC animals showed only small cytoplasmic fields with slightly increased numbers of cell organelles near the nuclear pole, as usual. Additionally, we examined the expression of other ID proteins that are direct or indirect interaction partners of myozap to assess whether these structural defects also correspond to altered levels of junctional proteins. In fact, we found a striking up-regulation (∼10–15-fold, p < 0.001) of desmoplakin in Mzp−/− mice both at baseline and in response to the TAC operation (Fig. 5, C and D). Likewise, expression of desmin was significantly increased after TAC in myozap-null compared with wild-type control mice (Fig. 5, C and E). In contrast, expression of N-cadherin and connexin-43 was drastically reduced after the induction of biomechanical stress in Mzp−/− mice (Fig. 5, F–H). Finally, ZO-1 was up-regulated after TAC in wild-type mice but significantly reduced in myozap-deficient mice (Fig. 5, I and J). Delmar and co-workers (41) recently demonstrated direct structural and functional interaction between connexin-43 and the voltage-gated sodium channel (Nav1.5) highlighting the cross-talk between gap junction and the sodium channel complex with respect to cardiac arrhythmias. Nevertheless, expression of Nav1.5 was unchanged in either of the conditions, although there was a similar trend of its up-regulation after TAC in both genotypes (Fig. 5, K and L). Furthermore, a similar effect, i.e. drastic reduction of connexin-43 and unchanged Nav1.5, was observed in isolated NRVCMs upon myozap knockdown under the influence of biomechanical stress (Fig. 5, M–O). Taken together, these data support the concept of a requirement for the ID protein myozap to prevent adverse junctional remodeling in response to biomechanical stress.
FIGURE 5.

Biomechanical stress causes junctional remodeling in mice lacking myozap. Electron micrographs showing the ID structure of wild-type (A, panels I and II) and myozap-KO (B, panels I and II) mice after TAC indicating disorganized IDs. Light microscopy images of wild-type (A, panel III) and myozap-KO (B, panel III) mice after TAC exhibiting cardiomyocyte degeneration in myozap-KO mice. Expression of intercalated disc proteins that directly or indirectly interact with myozap was determined by Western blotting and showed strong up-regulation of desmoplakin and desmin (C–E), although N-cadherin, connexin-43 (F–H), and ZO-1 (I and J) were strongly reduced after the induction of biomechanical stress due to TAC in Mzp−/− mice. Expression of Nav1.5 was unaltered in mice or rat cardiomyocytes (K–N), whereas connexin-43 was again down-regulated after myozap knockdown in stressed or unstressed cardiomyocytes (M and O). n = 6. Statistical significance was calculated by two-way ANOVA. Error bars show mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Actin Cytoskeleton and Connexin-43 Localization in Vitro and in Vivo
To gain additional insight into the behaviors of connexin-43 and the actin cytoskeleton in the absence of myozap with or without extrinsic stress both in vivo and in vitro, we performed immunofluorescence imaging. Our data here confirmed the marked reduction in connexin-43 expression, and its altered localization in the absence of myozap (Fig. 6, A, C, and D). Connexin-43 was ubiquitously distributed throughout the cell in wild-type sham or TAC mice, although it was present at quite low levels and, most importantly, in an ID pattern in Mzp−/− mice (Fig. 6A, panels III and IV). Similarly, connexin-43 is widely expressed both in control NRVCMs and neonatal mouse ventricular cardiomyocytes (NMVCM) isolated from wild-type mice (Fig. 6, C and D). In interconnected cardiomyocytes or after PE treatment, it is redistributed strongly to cell-cell contact junctions and the perinuclear region (Fig. 6, C and D, panel I). However, in NRVCMs with adenovirus-mediated knockdown of myozap, or in NMVCMs isolated from Mzp−/− mice, it was only modestly expressed and was mainly visible at cell edges and cell-cell junctions. When observed in singly isolated mouse cardiomyocytes, a similar pattern was observed (Fig. 6D, panel II). We additionally stained heart sections and NRVCMs/NMVCMs with α-actinin for the sarcomere and phalloidin for filamentous actin. In line with the EM data after TAC, Mzp−/− mice show a disorganized sarcomeric pattern (Fig. 6A). A similar disorganization was also observed in myozap-deprived NRVCMs or NMVCMs after PE treatment (data not shown). Furthermore, an interesting finding with actin staining indicated its distribution to the ID region in sham Mzp−/− but not in sham wild-type mice (Fig. 6B, see arrows in panel II), which otherwise was observed in both wild-type and Mzp−/− mice after TAC (Fig. 6B).
FIGURE 6.

Overview of connexin-43 and actin cytoskeleton in myozap-deficient mice or rat cardiomyocytes. Expression and localization of connexin-43 and filamentous actin and sarcomere were studied by immunoimaging with respective antibodies and phalloidin (for filamentous actin) in mouse heart cryosections (n = 5, A and B), neonatal rat cardiomyocytes (C), or neonatal mouse cardiomyocytes isolated from wild-type or Mzp−/− mice (D). Images clearly indicate visible differences between both genotypes after TAC with respect to the localization of connexin-43, altered sarcomere, and actin cytoskeleton. Experiments for cultured NRVCMs or NMVCMs were repeated three times in triplicate.
Myozap Binds to Actin and Is Required for Cardiac SRF and MAPK Signaling in Response to Pressure Overload
Actin dynamics and RhoA-dependent SRF activation are functionally linked where actin polymerization in response to RhoA signaling stimulates SRF activity (42, 43). In this context, we previously reported that myozap co-localizes with actin and activates Rho-dependent SRF signaling (35). Moreover, in this study, we observed actin cytoskeleton defects in myozap-deficient mice. Therefore, to gain further insight into this mechanism, we performed actin co-sedimentation assays with purified myozap. Myozap, which was exclusively detected in the supernatant in the absence of F-actin, partially co-sedimented with actin in the pellet fraction upon addition of F-actin (data not shown), confirming the direct interaction of myozap and F-actin. Along these lines, we have earlier shown that overexpression of myozap in vitro activates Rho-dependent SRF signaling (35). Conversely, we now found that the knockdown of myozap in cardiomyocytes resulted in reduced activation of SRF-dependent gene expression as shown by reduced luciferase activity of constructs containing its respective enhancer elements (Fig. 7A). Consistent with the inhibition of SRF signaling by myozap knockdown, we detected a reduction in the expression of several SRF target genes, including c-fos, Myl2, and Serca2a in Mzp−/− mice after TAC (Fig. 7B, Fig. 4, J–L) and in aged mice (Fig. 7C). In line with reduced SRF signaling, we also observed a marked inhibition of MAPK signaling (Fig. 7, D–G). Specifically and counter-intuitively, ERK1/2 (Fig. 7, D and E) and p38 (Fig. 7, F and G) were found to be less phosphorylated in myozap-deficient mice despite pressure overload. Moreover, expression of MKL1, which is an upstream activator of SRF, was found highly reduced in these mice after TAC (Fig. 7, H and I).
FIGURE 7.

Myozap deficiency inhibits cardiac SRF and MAPK signaling due to pressure overload. A, bar graph showing reduced SRF-RE-driven luciferase activity upon myozap knockdown in cardiomyocytes. Expression levels of selected SRF target gene skeletal muscle α-actin (Acta1), cardiac muscle α-actin (Actc1), c-Fos, and myosin light chain 2 (Myl2) were detected in SHAM/TAC-operated (B), or 1-year-old mice (C). Proteins extracted from post-operative hearts were immunoblotted for ERK1/2 and phospho-ERK1/2 (p-ERK1/2; D), MAPK p38, and phospho-p38 (p-p38; F), and MKL1 (H). Respective densitometric analysis indicates the inhibition of ERK1/2 (E), p38 (G), as well as down-regulation of MKL1 (I) after TAC operations exclusively in Mzp−/− mice. n = 6. Statistical significance was calculated by two-way ANOVA. Error bars show mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cardiac Hypertrophy in Mzp−/− Mice Because of Biomechanical Stress Is Attributed to Calcineurin-NFAT and β-Catenin/GSK-3β Signaling
Our current data demonstrate accelerated hypertrophy in Mzp−/− mice upon pressure overload, yet SRF and MAPK signaling was significantly repressed. Therefore, to address this seeming disconnect, and to find the underlying mechanistic cause, we next evaluated several alternative signaling pathways known to mediate cardiac hypertrophy and cardiomyopathy, such as GSK-3β, PI3K/AKT, and calcineurin/NFAT. There was no significant difference between wild-type and Mzp−/− mice in terms of the activation of AKT signaling (Fig. 8, A and B). Yet, we observed a significant increase in the phosphorylation of GSK-3β in Mzp−/− mice after TAC compared with SHAM-operated Mzp−/− animals or wild-type TAC-operated mice (Fig. 8, C and D). In addition to these findings, we also noticed increased activation of calcineurin signaling as shown by increased levels of RCAN1–4 (Fig. 8, E–G). Moreover, likely as a result of increased phosphorylation of GSK-3β, the active form of β-catenin was found significantly increased after TAC in Mzp−/− mice (Fig. 8, H and I), providing a potential explanation for the observed pathological hypertrophy even in the context of inhibited MAPK signaling. These findings, i.e. inhibition of MAPKs and hypertrophic activation of GSK-3β, were reciprocated in vitro in NRVCMs under the influence of biomechanical stress (Fig. 8, J–N). Cultured cardiomyocytes also allowed us to study calcineurin signaling using luciferase reporter assays. First, we confirmed the up-regulation of Rcan1–4 after stretch in myozap knocked down NRVCMs (Fig. 8, O and P). Second, we studied the effect of increased or reduced expression of myozap on NFAT-RE-driven firefly luciferase, which is an indirect measure of calcineurin activation. Overexpression of myozap did not show any significant impact, neither at baseline nor in the presence of constitutively active calcineurin, although there was a trend toward reduced activation (Fig. 8Q). Nevertheless, knockdown or absence of myozap significantly activated the luciferase activity at baseline in NRVCMs (Fig. 8R) and NMVCMs (Fig. 8S), respectively. The presence of constitutively active calcineurin A further exacerbated this effect, strongly indicating that the knockdown of myozap is sufficient to activate calcineurin signaling.
FIGURE 8.

Cardiac hypertrophy in myozap-deletion mutant mice due to biomechanical stress is attributed to β-catenin/GSK-3β signaling. Proteins extracted from post-operative hearts were immunoblotted for Akt and phospho-Akt (p-Akt; A and B), GSK3α/3β and phospho-GSK3β (p-GSK3β; C and D), Rcan1–4 at protein (E and F) and transcript level (G), and β-catenin and phospho-β-catenin (p-β-catenin) (H and I). Respective densitometric analysis revealed no significant difference in Akt activation (B); nevertheless, expression of p-GSK3β (D), Rcan1–4 (F), and β-catenin (I) was significantly increased in Mzp−/− mice after the induction of biomechanical stress (n = 6). These findings were recapitulated in rat cardiomyocytes after myozap knockdown under mechanical stress with inhibition or MAPKs (J–L), increased phosphorylation of GSK-3β (M and N), and up-regulation of Rcan1–4 (O and P). Effect of myozap expression was also studied on NFAT reporter-mediated firefly luciferase activity by either its overexpression or knockdown in NRVCM or in NMVCM isolated from wild-type or myozap-null mice. Overexpression of myozap had no effect (Q), whereas reduction or the absence of myozap significantly accelerated the luciferase activity in NRVCMs (R), or NMVCMs (S), respectively. Experiments for cultured NRVCMs or NMVCMs were repeated three times in triplicate (hexaplicate for luciferase assay). Statistical significance was calculated by two-way ANOVA. Error bars show mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Myozap is a cardiac-enriched protein that localizes to the ID (35). Our earlier gain-of-function studies in vitro and in vivo indicated that myozap activates Rho-dependent SRF signaling (35, 36). Knockdown of the myozap ortholog in zebrafish led to severe contractile dysfunction and cardiomyopathy (35). In this study we now show that myozap is dispensable for proper vertebrate development. However, its deficiency in vivo leads to a maladaptive response to increased biomechanical stress associated with adverse cardiac remodeling, contractile dysfunction, ultrastructural defects, heart failure, and premature death. Mechanistically, activation of SRF and MAPK signaling is severely impaired, whereas maladaptive, pro-hypertrophic calcineurin/NFAT and GSK-3β/β-catenin signaling pathways are enhanced in myozap-deficient mice after pressure overload.
The ID is a unique morphological feature of cardiac muscle and a highly specialized cell-cell contact structure that both mechanically connects individual cardiomyocytes but also is essential for intercellular signal transduction and electromechanical coupling (44). ID components not only provide structural and functional integrity to the myocardium but are also essential for proper development of the heart during embryogenesis. Loss of several ID proteins causes lethal cardiac abnormalities in mice, whereas genetic mutations may lead to cardiomyopathy in humans. For example, systemic deletion of N-cadherin resulted in embryonic lethality due to improper development of the heart tube, accompanied by multiple embryonic abnormalities, including severe cardiovascular defects (8). Similarly, loss of plakoglobin resulted in embryonic lethality as a result of severe cardiac dysfunction, reduced desmosomes, and deformed ID structure (6, 45). We showed earlier that knockdown of the myozap ortholog in zebrafish also led to cardiomyopathy and contractile dysfunction associated with pericardial edema and blood pooling at the venous sinus as signs of heart failure (35). Despite the functional impairment, myozap morphant hearts displayed normal morphology in zebrafish (35). In agreement with those findings, our present data suggest no developmental role for myozap, as its complete loss did not affect cardiac development nor did it alter baseline ultrastructural ID architecture in mice. However, we observed signs of mild hypertrophy in myozap-deficient mice upon aging. Similarly, it has been shown that ablation of mXinα, a Xin repeat-containing ID protein, does not affect heart development (46). However, loss of mXinα resulted in ID disruption and myofilament disarray followed by late-onset cardiac hypertrophy and cardiomyopathy along with conduction defects (46). Also, deletion of lysosomal integral membrane protein 2 (LIMP-2), another ID component, did not affect cardiac development (47). ID components like these, demarcate the developmental constituents from those involved in other functional aspects of the heart independent of cardiac development, which altogether further emphasize the functional complexity and multiple roles of the intercalated disc.
ID proteins quickly respond to perturbed expression of the companion proteins with their own up- or down-regulation as an effect or as a compensatory mechanism (48). For example, a significant decrease in the gap junction proteins connexin-43 and connexin-40 was observed in N-cadherin-depleted myocytes, which was accompanied by the development of dilated cardiomyopathy and impaired left ventricular function (40). Similarly, deletion of α-T-catenin resulted in the reduced expression of connexin-43 and plakophilin-2 (39). In contrast, cardiac-specific deletion of β-catenin in adult mice caused up-regulation of γ-catenin, suggesting that γ-catenin can compensate for the loss of β-catenin in cardiomyocytes to maintain normal cardiac structure and function (49). In this study, we observed significant alterations in the expression of some of the important ID proteins like β-actin, connexin-43, desmin, desmoplakin, N-cadherin, and ZO-1, which are either direct or indirect binding partners of myozap. Altered expression of N-cadherin and β-catenin might also be involved in the cytoskeletal defects that we observed in TAC-operated Mzp−/− mice, because N-cadherin attachment to cytoskeletal F-actin through β-catenin is necessary for cell adhesion and mechanosensory function (50, 51). The observed direct interaction between myozap and actin also supports this notion. Earlier reports also suggest a connection between reduced expression of connexin-43, which is a hallmark of failing hearts, with that of lowered levels of N-cadherin and ZO-1, which is also seen in myozap-KO mice after TAC. Of note, molecular remodeling at the ID is involved in many human cardiac diseases and the pathogenesis of heart failure, especially ARVC. Therefore, a similar finding in yet another ID protein highlights the importance of understanding the cross-talk between the junctional proteins of the ID and its implications for cardiomyocyte signaling and cardiomyopathy.
The lack of myozap in unstressed mouse hearts did not alter ultrastructural architecture of the myocardium as a whole; nor did it affect the ID structure, as IDs were normal and intact. In contrast, pressure overload greatly altered ultrastructural architecture of myozap-deficient mouse hearts with disorganized and disoriented IDs, elongation of ID convolutions, and an occasional widening of interjunctional gap, similar to the phenotype of ARVC. A conditional knock-out of N-cadherin has been reported previously to cause dissolution of the ID structure and sarcomeric defects like decreased sarcomeric length and altered Z-line thickness (52). Another interesting study by Rizzo et al. (53) using a desmoglein-2 mutation, which has been linked to ARVC, also exhibited widening of the intercellular space at gap junction and desmosomes along with lowered Na+ current density and reduced action potential upstroke velocity. An important study by transmission electron microscopy of endomyocardial biopsies from ARVC patients revealed disorganization of IDs and increased widening of the interjunctional gap (54). They also found a decrease in the number but an increase in the length of desmosomes. Our data along with these studies point toward the importance of the ID structural integrity in maintaining normal cardiac function. Our data suggest that the presence of myozap is also necessary to cope with biomechanical stress due to pressure overload. Although myozap deficiency did not cause arrhythmias in 1-year-old unstressed experimental animals, it will be interesting to see if, how, and when these mice develop arrhythmias after TAC.
At the molecular and phenotypic level, myozap deficiency did not affect the baseline cardiac phenotype, yet we observed excessive cardiac remodeling with increased cellular and cardiac hypertrophy in response to pressure overload. Specifically, Mzp−/− mouse hearts were more susceptible to pressure overload and reacted by robust induction of cardiac hypertrophy both at the morphological and cellular level with further up-regulation of Nppa and Nppb. This in turn caused severe contractile dysfunction associated with signs of heart failure such as increased lung weights and enhanced cardiac fibrosis. Ultimately, maladaptive remodeling in myozap-deficient mice also led to an increase in mortality after the induction of biomechanical stress. These data imply that Myozap is one of the few ID proteins that are important for proper adaptation to biomechanical stress.
In pursuit of underlying signaling pathway(s) responsible for the cardiac hypertrophy detected in myozap-deficient mice, we first focused on the SRF signaling as we earlier reported the activation of this signaling pathway via the RhoA axis (36). As expected, we found reduced SRF signaling in terms of reduced SRF-RE-driven luciferase activity, reduced expression of SRF targets like Serca2a, Myh6, c-Fos, etc., and a striking reduction in MKL1 expression in myozap-deficient mice after TAC. Moreover, there was an apparent inhibition of ERK1/2 and p38 MAPKs that are known to be linked with SRF signaling. Kalita et al. (57) proposed that MKL1 is a mediator of the signaling between ERK1/2 and SRF, which was further supported by Muehlich et al. (58) when they showed that MKL1 is phosphorylated and regulated by ERK1/2. Likewise, p38 also phosphorylates and activates MAPKAPK2, which is another upstream modulator of SRF (59). Despite significant inhibition of p38 in Mzp−/− mice, we did not observe any changes in MAPKAPK2 activation (data not shown), suggesting that this branch of SRF signaling is not relevant for myozap-mediated SRF activation. As SRF signaling, typically a rather pro-hypertrophic pathway (60), was inhibited, we next aimed to identify the molecular basis of excessive hypertrophy in Mzp−/− mice upon pressure overload. One important clue was the inhibition of p38 MAPK, as inhibition of p38 is known to promote hypertrophic cardiomyopathy through activation of calcineurin-NFAT signaling (61). We thus studied the calcineurin-NFAT signaling axis and found a marked up-regulation of Rcan1–4 expression, a sign of increased activation of calcineurin-NFAT signaling (62). Along these lines, we also observed a strong increase in the phosphorylation of GSK-3β, which is downstream of calcineurin in the calcineurin-NFAT signaling cascade promoting cardiac hypertrophy. Up-regulation of phosphorylated GSK-3β is also involved in increasing cytosolic β-catenin levels as a consequence of decreased GSK-3β-mediated degradation (34, 55). Of note, β-catenin is essentially an ID protein necessary for cardiac development that also serves as an activator of transcription factors from the T-cell factor/lymphocyte enhancer factor (Tcf/Lef) family, eventually enhancing cardiac hypertrophy in stress conditions (30, 56). Activation of calcineurin and GSK-3β-prohypertrophic signaling was validated in isolated cardiomyocytes with reporter assays and immunoblotting further strengthening the proposed mechanism. These results imply that the activation of GSK-3β/β-catenin together with activated calcineurin-NFAT signaling translates into the observed cardiac hypertrophy phenotype in myozap-deficient mice due to pressure overload.
To summarize, this study is the first in vivo characterization of the ID protein myozap using loss-of-function approach. In contrast to several other major ID proteins, like cadherins, plakoglobin, etc., myozap is not essential for ID morphology or cardiac development during embryogenesis. Instead, myozap appears to play an important role in cardiac stress-signaling, as myozap deficiency in vivo led to a maladaptive response to pressure overload associated with cardiac remodeling, as well as cardiomyopathy, heart failure. and premature death. Mechanistically, the lack of myozap elicited junctional remodeling followed by discordant regulation of ERK/SRF, calcineurin-NFAT, and GSK-3β/β-catenin signaling pathways. This study also highlights the complex role of the ID protein myozap, which differentially affects two major cardiac prohypertrophic pathways, e.g. activation of SRF signaling upon up-regulation via the RhoA axis, although its deficiency leads to the activation of calcineurin-NFAT/GSK-3β/β-catenin-driven prohypertrophic signaling (Fig. 9). Viewed in a wider perspective, our data expand the notion of an essential role of the ID not only in structural integrity of the heart but also in hypertrophic signaling. Further understanding of the cross-talk between its individual constituents in cardiac adaptation and remodeling is likely to yield information that will be useful in understanding the pathogenesis of cardiomyopathy and heart failure.
FIGURE 9.
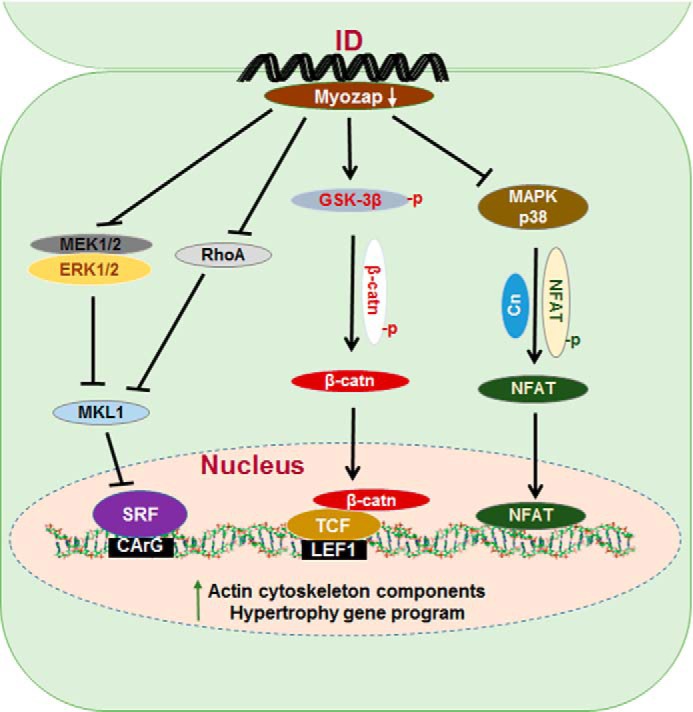
Simplified pictorial representation of effects of myozap deficiency on cardiac signaling. Myozap deficiency on the one hand inhibits SRF signaling via inhibition of MAPK ERK1/2, RhoA, and MKL1. On the other hand, it activates prohypertrophic calcineurin-NFAT via inhibition of MAPK p38 and interconnected GSK-3β/β-catenin-driven signaling cascade in response to biomechanical stress. Cn, calcineurin; β-catn, β-catenin.
Author Contributions
A. Y. R. co-designed the experimental strategy with D. F. and N. F., conducted most of the experiments, analyzed and interpreted the data, and wrote the first draft of the manuscript. M. E. conducted echocardiography and associated data analysis of TAC/isoproterenol-treated mice. R. P. performed TAC operations. C. K. performed some TAC and implanted isoproterenol mini-pumps. K. S. performed some of the immunoblotting and data analysis experiments from Figs. 7 and 8. F. D. prepared samples for electron microscopy. A. B. standardized and performed immunofluorescence experiments for cell size measurements. R. L. conducted transmission electron microscopy and ultrastructural analysis. H. W. helped in generating myozap-KO mouse line. P. K. performed and analyzed ECG and electrophysiology data. D. F. designed the study, interpreted some of the data, and critically revised manuscript. N. F. devised the strategy and generated myozap-KO mice with the help of H. W., designed the study, wrote some parts of the manuscript, and critically revised the manuscript. All authors approved final version of the manuscript.
Acknowledgments
We thank G. Brunke, V. Mangels, and C. Tannert for excellent technical assistance in NRVCM preparation and maintenance of cell lines used in this study. We also thank D. Niemeir for technical assistance in EM studies.
This work was supported in part by Bundesministerium für Bildung and Forschung (BMBF/NGFNplus) Grant BMBF/01GS0902. The authors declare that they have no conflicts of interest with the contents of this article.
- ID
- intercalated disc
- ARVC
- arrhythmogenic right ventricular cardiomyopathy
- NFAT
- nuclear factor of activated T-cell
- TRITC
- tetramethylrhodamine isothiocyanate
- ANOVA
- analysis of variance
- m.o.i.
- multiplicity of infection
- NMVCM
- neonatal mouse ventricular cardiomyocyte
- TAC
- transverse aortic constriction
- SRF
- serum-response factor
- NRVCM
- neonatal rat cardiomyocyte
- PE
- phenylephrine
- qRT-PCR
- quantitative real time PCR.
References
- 1. Forbes M. S., and Sperelakis N. (1985) Intercalated discs of mammalian heart: a review of structure and function. Tissue Cell 17, 605–648 [DOI] [PubMed] [Google Scholar]
- 2. Joshi-Mukherjee R., Coombs W., Musa H., Oxford E., Taffet S., and Delmar M. (2008) Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)-related plakophilin-2 (PKP2) mutations. Heart Rhythm 5, 1715–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McKoy G., Protonotarios N., Crosby A., Tsatsopoulou A., Anastasakis A., Coonar A., Norman M., Baboonian C., Jeffery S., and McKenna W. J. (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355, 2119–2124 [DOI] [PubMed] [Google Scholar]
- 4. Norgett E. E., Hatsell S. J., Carvajal-Huerta L., Cabezas J. C., Common J., Purkis P. E., Whittock N., Leigh I. M., Stevens H. P., and Kelsell D. P. (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 9, 2761–2766 [DOI] [PubMed] [Google Scholar]
- 5. Saffitz J. E. (2011) The pathobiology of arrhythmogenic cardiomyopathy. Annu. Rev. Pathol. 6, 299–321 [DOI] [PubMed] [Google Scholar]
- 6. Ruiz P., Brinkmann V., Ledermann B., Behrend M., Grund C., Thalhammer C., Vogel F., Birchmeier C., Günthert U., Franke W. W., and Birchmeier W. (1996) Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol. 135, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haegel H., Larue L., Ohsugi M., Fedorov L., Herrenknecht K., and Kemler R. (1995) Lack of β-catenin affects mouse development at gastrulation. Development 121, 3529–3537 [DOI] [PubMed] [Google Scholar]
- 8. Radice G. L., Rayburn H., Matsunami H., Knudsen K. A., Takeichi M., and Hynes R. O. (1997) Developmental defects in mouse embryos lacking N-cadherin. Dev. Biol. 181, 64–78 [DOI] [PubMed] [Google Scholar]
- 9. Bisping E., Wakula P., Poteser M., and Heinzel F. R. (2014) Targeting cardiac hypertrophy: toward a causal heart failure therapy. J. Cardiovasc. Pharmacol. 64, 293–305 [DOI] [PubMed] [Google Scholar]
- 10. Cohn J. N., Ferrari R., and Sharpe N. (2000) Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 35, 569–582 [DOI] [PubMed] [Google Scholar]
- 11. Kehat I., and Molkentin J. D. (2010) Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122, 2727–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burchfield J. S., Xie M., and Hill J. A. (2013) Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation 128, 388–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Opie L. H., Commerford P. J., Gersh B. J., and Pfeffer M. A. (2006) Controversies in ventricular remodelling. Lancet 367, 356–367 [DOI] [PubMed] [Google Scholar]
- 14. Pfeffer J. M., Pfeffer M. A., and Braunwald E. (1985) Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ. Res. 57, 84–95 [DOI] [PubMed] [Google Scholar]
- 15. Pfeffer M. A., and Braunwald E. (1990) Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81, 1161–1172 [DOI] [PubMed] [Google Scholar]
- 16. Frey N., and Olson E. N. (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79 [DOI] [PubMed] [Google Scholar]
- 17. Gerdes A. M., Kellerman S. E., Moore J. A., Muffly K. E., Clark L. C., Reaves P. Y., Malec K. B., McKeown P. P., and Schocken D. D. (1992) Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 86, 426–430 [DOI] [PubMed] [Google Scholar]
- 18. Selvetella G., Hirsch E., Notte A., Tarone G., and Lembo G. (2004) Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence. Cardiovasc. Res. 63, 373–380 [DOI] [PubMed] [Google Scholar]
- 19. Bueno O. F., De Windt L. J., Tymitz K. M., Witt S. A., Kimball T. R., Klevitsky R., Hewett T. E., Jones S. P., Lefer D. J., Peng C. F., Kitsis R. N., and Molkentin J. D. (2000) The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cantley L. C. (2002) The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 [DOI] [PubMed] [Google Scholar]
- 21. Garrington T. P., and Johnson G. L. (1999) Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 11, 211–218 [DOI] [PubMed] [Google Scholar]
- 22. Molkentin J. D., Lu J. R., Antos C. L., Markham B., Richardson J., Robbins J., Grant S. R., and Olson E. N. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oudit G. Y., Sun H., Kerfant B. G., Crackower M. A., Penninger J. M., and Backx P. H. (2004) The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 37, 449–471 [DOI] [PubMed] [Google Scholar]
- 24. Shioi T., Kang P. M., Douglas P. S., Hampe J., Yballe C. M., Lawitts J., Cantley L. C., and Izumo S. (2000) The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugden P. H., and Clerk A. (1998) Cellular mechanisms of cardiac hypertrophy. J. Mol. Med. 76, 725–746 [DOI] [PubMed] [Google Scholar]
- 26. Sugden P. H., and Clerk A. (1998) Regulation of mitogen-activated protein kinase cascades in the heart. Adv. Enzyme Regul. 38, 87–98 [DOI] [PubMed] [Google Scholar]
- 27. Sugden P. H., and Clerk A. (1998) “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ. Res. 83, 345–352 [DOI] [PubMed] [Google Scholar]
- 28. Wilkins B. J., and Molkentin J. D. (2004) Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 322, 1178–1191 [DOI] [PubMed] [Google Scholar]
- 29. Bergmann M. W. (2010) WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ. Res. 107, 1198–1208 [DOI] [PubMed] [Google Scholar]
- 30. Chen X., Shevtsov S. P., Hsich E., Cui L., Haq S., Aronovitz M., Kerkelä R., Molkentin J. D., Liao R., Salomon R. N., Patten R., and Force T. (2006) The β-catenin/T-cell factor/lymphocyte enhancer factor signaling pathway is required for normal and stress-induced cardiac hypertrophy. Mol. Cell. Biol. 26, 4462–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deb A. (2014) Cell-cell interaction in the heart via Wnt/β-catenin pathway after cardiac injury. Cardiovasc. Res. 102, 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J., Swope D., Raess N., Cheng L., Muller E. J., and Radice G. L. (2011) Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of β-catenin signaling. Mol. Cell. Biol. 31, 1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zelarayan L., Gehrke C., and Bergmann M. W. (2007) Role of β-catenin in adult cardiac remodeling. Cell Cycle 6, 2120–2126 [DOI] [PubMed] [Google Scholar]
- 34. Haq S., Michael A., Andreucci M., Bhattacharya K., Dotto P., Walters B., Woodgett J., Kilter H., and Force T. (2003) Stabilization of β-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc. Natl. Acad. Sci. U.S.A. 100, 4610–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seeger T. S., Frank D., Rohr C., Will R., Just S., Grund C., Lyon R., Luedde M., Koegl M., Sheikh F., Rottbauer W., Franke W. W., Katus H. A., Olson E. N., and Frey N. (2010) Myozap, a novel intercalated disc protein, activates serum response factor-dependent signaling and is required to maintain cardiac function in vivo. Circ. Res. 106, 880–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frank D., Rangrez A. Y., Poyanmehr R., Seeger T. S., Kuhn C., Eden M., Stiebeling K., Bernt A., Grund C., Franke W. W., and Frey N. (2014) Mice with cardiac-restricted overexpression of Myozap are sensitized to biomechanical stress and develop a protein-aggregate-associated cardiomyopathy. J. Mol. Cell. Cardiol. 72, 196–207 [DOI] [PubMed] [Google Scholar]
- 37. Rangrez A. Y., Bernt A., Poyanmehr R., Harazin V., Boomgaarden I., Kuhn C., Rohrbeck A., Frank D., and Frey N. (2013) Dysbindin is a potent inducer of RhoA-SRF-mediated cardiomyocyte hypertrophy. J. Cell Biol. 203, 643–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuhn C., Frank D., Dierck F., Oehl U., Krebs J., Will R., Lehmann L. H., Backs J., Katus H. A., and Frey N. (2012) Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res. Cardiol. 107, 262. [DOI] [PubMed] [Google Scholar]
- 39. Li J., Goossens S., van Hengel J., Gao E., Cheng L., Tyberghein K., Shang X., De Rycke R., van Roy F., and Radice G. L. (2012) Loss of αT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J. Cell Sci. 125, 1058–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J., Patel V. V., Kostetskii I., Xiong Y., Chu A. F., Jacobson J. T., Yu C., Morley G. E., Molkentin J. D., and Radice G. L. (2005) Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ. Res. 97, 474–481 [DOI] [PubMed] [Google Scholar]
- 41. Sato P. Y., Coombs W., Lin X., Nekrasova O., Green K. J., Isom L. L., Taffet S. M., and Delmar M. (2011) Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Posern G., Sotiropoulos A., and Treisman R. (2002) Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol. Biol. Cell 13, 4167–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hill C. S., Wynne J., and Treisman R. (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 44. Noorman M., van der Heyden M. A., van Veen T. A., Cox M. G., Hauer R. N., de Bakker J. M., and van Rijen H. V. (2009) Cardiac cell-cell junctions in health and disease: electrical versus mechanical coupling. J. Mol. Cell. Cardiol. 47, 23–31 [DOI] [PubMed] [Google Scholar]
- 45. Bierkamp C., Mclaughlin K. J., Schwarz H., Huber O., and Kemler R. (1996) Embryonic heart and skin defects in mice lacking plakoglobin. Dev. Biol. 180, 780–785 [DOI] [PubMed] [Google Scholar]
- 46. Gustafson-Wagner E. A., Sinn H. W., Chen Y. L., Wang D. Z., Reiter R. S., Lin J. L., Yang B., Williamson R. A., Chen J., Lin C. I., and Lin J. J. (2007) Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am. J. Physiol. Heart Circ. Physiol. 293, H2680–H2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schroen B., Leenders J. J., van Erk A., Bertrand A. T., van Loon M., van Leeuwen R. E., Kubben N., Duisters R. F., Schellings M. W., Janssen B. J., Debets J. J., Schwake M., Høydal M. A., Heymans S., Saftig P., and Pinto Y. M. (2007) Lysosomal integral membrane protein 2 is a novel component of the cardiac intercalated disc and vital for load-induced cardiac myocyte hypertrophy. J. Exp. Med. 204, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perriard J. C., Hirschy A., and Ehler E. (2003) Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc. Med. 13, 30–38 [DOI] [PubMed] [Google Scholar]
- 49. Zhou J., Qu J., Yi X. P., Graber K., Huber L., Wang X., Gerdes A. M., and Li F. (2007) Upregulation of γ-catenin compensates for the loss of β-catenin in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 292, H270–H276 [DOI] [PubMed] [Google Scholar]
- 50. Linask K. K., Ludwig C., Han M. D., Liu X., Radice G. L., and Knudsen K. A. (1998) N-cadherin/catenin-mediated morphoregulation of somite formation. Dev. Biol. 202, 85–102 [DOI] [PubMed] [Google Scholar]
- 51. Imanaka-Yoshida K., Knudsen K. A., and Linask K. K. (1998) N-cadherin is required for the differentiation and initial myofibrillogenesis of chick cardiomyocytes. Cell Motil. Cytoskeleton 39, 52–62 [DOI] [PubMed] [Google Scholar]
- 52. Kostetskii I., Li J., Xiong Y., Zhou R., Ferrari V. A., Patel V. V., Molkentin J. D., and Radice G. L. (2005) Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 96, 346–354 [DOI] [PubMed] [Google Scholar]
- 53. Rizzo S., Lodder E. M., Verkerk A. O., Wolswinkel R., Beekman L., Pilichou K., Basso C., Remme C. A., Thiene G., and Bezzina C. R. (2012) Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 95, 409–418 [DOI] [PubMed] [Google Scholar]
- 54. Basso C., and Thiene G. (2006) The pathophysiology of myocardial reperfusion: a pathologist's perspective. Heart 92, 1559–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Naito A. T., Akazawa H., Takano H., Minamino T., Nagai T., Aburatani H., and Komuro I. (2005) Phosphatidylinositol 3-kinase-Akt pathway plays a critical role in early cardiomyogenesis by regulating canonical Wnt signaling. Circ. Res. 97, 144–151 [DOI] [PubMed] [Google Scholar]
- 56. Brembeck F. H., Rosário M., and Birchmeier W. (2006) Balancing cell adhesion and Wnt signaling, the key role of β-catenin. Curr. Opin. Genet. Dev. 16, 51–59 [DOI] [PubMed] [Google Scholar]
- 57. Kalita K., Kharebava G., Zheng J. J., and Hetman M. (2006) Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J. Neurosci. 26, 10020–10032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Muehlich S., Wang R., Lee S. M., Lewis T. C., Dai C., and Prywes R. (2008) Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol. Cell. Biol. 28, 6302–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heidenreich O., Neininger A., Schratt G., Zinck R., Cahill M. A., Engel K., Kotlyarov A., Kraft R., Kostka S., Gaestel M., and Nordheim A. (1999) MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J. Biol. Chem. 274, 14434–14443 [DOI] [PubMed] [Google Scholar]
- 60. Zhang X., Azhar G., Chai J., Sheridan P., Nagano K., Brown T., Yang J., Khrapko K., Borras A. M., Lawitts J., Misra R. P., and Wei J. Y. (2001) Cardiomyopathy in transgenic mice with cardiac-specific overexpression of serum response factor. Am. J. Physiol. Heart Circ. Physiol. 280, H1782–H1792 [DOI] [PubMed] [Google Scholar]
- 61. Braz J. C., Bueno O. F., Liang Q., Wilkins B. J., Dai Y. S., Parsons S., Braunwart J., Glascock B. J., Klevitsky R., Kimball T. F., Hewett T. E., and Molkentin J. D. (2003) Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Invest. 111, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Molkentin J. D. (2004) Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 63, 467–475 [DOI] [PubMed] [Google Scholar]