

Abstract
The fast transient outward potassium current (Ito,f) plays a critical role in the electrical and contractile properties of the myocardium. Ito,f channels are formed by the co-assembly of the pore-forming α-subunits, Kv4.2 and Kv4.3, together with the accessory β-subunit KChIP2. Reductions of Ito,f are common in the diseased heart, which is also associated with enhanced stimulation of β-adrenergic receptors (β-ARs). We used cultured neonatal rat ventricular myocytes to examine how chronic β-AR stimulation decreases Ito,f. To determine which downstream pathways mediate these Ito,f changes, adenoviral infections were used to inhibit CaMKIIδc, CaMKIIδb, calcineurin, or nuclear factor κB (NF-κB). We observed that chronic β-AR stimulation with isoproterenol (ISO) for 48 h reduced Ito,f along with mRNA expression of all three of its subunits (Kv4.2, Kv4.3, and KChIP2). Inhibiting either CaMKIIδc nor CaMKIIδb did not prevent the ISO-mediated Ito,f reductions, even though CaMKIIδc and CaMKIIδb clearly regulated Ito,f and the mRNA expression of its subunits. Likewise, calcineurin inhibition did not prevent the Ito,f reductions induced by β-AR stimulation despite strongly modulating Ito,f and subunit mRNA expression. In contrast, NF-κB inhibition partly rescued the ISO-mediated Ito,f reductions in association with restoration of KChIP2 mRNA expression. Consistent with these observations, KChIP2 promoter activity was reduced by p65 as well as β-AR stimulation. In conclusion, NF-κB, and not CaMKIIδ or calcineurin, partly mediates the Ito,f reductions induced by chronic β-AR stimulation. Both mRNA and KChIP2 promoter data suggest that the ISO-induced Ito,f reductions are, in part, mediated through reduced KChIP2 transcription caused by NF-κB activation.
Keywords: adrenergic receptor, Ca2+/calmodulin-dependent protein kinase (CaMK), cardiomyocyte, potassium channel, transcription
Introduction
Fast cardiac transient outward potassium currents are generated by channels comprised of voltage-gated α-pore-forming subunits (which in humans/canines is predominantly Kv4.3, and in rodents is Kv4.2 and Kv4.3) and the accessory β-subunit KChIP2 (1). These currents play a critical role in early cardiac repolarization (1), excitation contraction-coupling (2, 3), and arrhythmias (4). Ito,f and its molecular constituents are invariably reduced in cardiac hypertrophy and disease (1), and heart disease is also characterized by elevations in catecholamines and consequently enhanced activation of β-adrenergic receptors (β-AR)6 (5). Although chronic β-AR stimulation has been shown to decrease Ito,f (6), the molecular mechanisms underlying regulation of Ito,f by β-ARs remain unclear.
Ito,f is regulated by several pathways activated in the diseased myocardium, such as calcineurin, nuclear factor-activated T-cells (NFAT) (6, 7) and mitogen activated protein kinases (8). Although calcineurin/NFAT signaling regulates Ito,f in a manner that is model dependent (6, 9, 10), this pathway does not mediate α-AR-induced changes in Ito,f or its molecular subunits (9, 10). We previously showed that α-AR stimulation activates the transcription factor nuclear factor κB (NF-κB), which mediates reductions in Ito,f via strong repression of KChIP2 expression (9, 10). The results of several studies also suggest that Ca2+/calmodulin-dependent protein kinase (CaMKII) may also regulate Ito,f. For example, mice with transgenic overexpression of CaMKIIδc have reduced levels of Ito,f as well as Kv4.2 and KChIP2 subunits (11), although these changes may be related to concurrent heart disease. In addition, CaMKIIδc activity is increased in human (12) and animal models (13) of heart disease and CaMKII has been shown to mediate NF-κB activation (14). Another cardiac isoform of CaMKII, CaMKIIδb, has also been implicated in disease signaling pathways (15, 16), although its potential regulation of Ito,f has not been investigated.
Although the regulation of Ito,f has been studied in a number of experimental and disease models, the direct link between β-AR stimulation and Ito,f has not been investigated. Therefore, we used cultured neonatal rat ventricular myocytes (NRVM) to examine the effects of the β-AR agonist, isoproterenol (ISO), on Ito,f. Our data show that chronic β-AR stimulation reduces Ito,f as well as mRNA levels for all three of the molecular subunits underlying Ito,f channels. The reductions in Ito,f and KChIP2 mRNA expression were rescued by NF-κB inhibition.
Experimental Procedures
Solutions and Reagents
CBHFF (mmol/liter): 137 NaCl, 5.36 KCl, 0.81 MgSO4·7H2O, 5.55 d-glucose, 0.44 KH2PO4·7H2O, 0.34 Na2HPO2·7H2O, 20 HEPES, pH 7.4 with NaOH; modified Tyrode (mmol/liter): 140 NaCl, 4 KCl, 2 CaCl2·2H2O, 1 MgCl2·6H2O, 10 HEPES, 10 d-glucose, 0.5 CdCl2, pH 7.4 with NaOH; KINT (mmol/liter): 140 KCl, 1 MgCl2, 10 EGTA, 10 HEPES, 5 Mg2ATP, pH 7.25 with KOH.
Stock concentrations of isoproterenol (ISO) (50 μmol/liter) (Sigma) were dissolved in Milli-Q water and stored at −20 °C. Multiple freeze/thawing was avoided by using small aliquots, and new stocks were prepared regularly.
Isolation, Culture, and Treatment of Neonatal Rat Ventricular Myocytes (NRVM)
All studies were in accordance with the guidelines set by the Canadian Council on Animal Care and the University of Toronto. Hearts from 1–2-day-old Sprague-Dawley rats were rapidly excised after cervical dislocation and immediately placed in chilled CBHFF buffer. The ventricles were isolated by removing the atria, and were subsequently washed five times in CBHFF. The ventricles were gradually minced and placed in CBHFF buffer containing 1 mg/ml of trypsin (Life Technologies) at 4 °C for 3 h with gentle rocking. To dissociate myocytes, minced ventricles were washed and placed in 5 ml of pre-warmed buffer containing 1 mg/ml of trypsin plus 0.15 mg/ml collagenase type II (Worthington) and stirred for 10 min 3–5 times. NRVM were pelleted at 1000 rpm for 5 min, resuspended in Dulbecco's Modified Eagle Medium (DMEM) and Ham F12 (1:1 v/v) containing 5% fetal bovine serum (FBS) (Invitrogen), and pre-plated for 1 h at 37 °C, and 4 × 105 myocytes were plated on 35-mm dishes containing glass coverslips coated with 0.6% gelatin for electrophysiology studies. For mRNA experiments, 7.0 × 105 myocytes were plated on 60-mm dishes. NRVMs were cultured in 5% FBS/DMEM/F12 containing 0.1 mmol/liter bromodeoxyuridine (BrdU) (Sigma) and penicillin-streptomycin. The medium was changed to DMEM/F12 (without serum or BrdU), supplemented with 1× insulin-transferrin-selenium-X (Life Technologies), 25 μg/ml ascorbic acid and 1 nmol/liter LiCl after ∼21 h and then treated (or not) with 50 nmol/liter ISO. The medium and treatments were replaced daily, and after 40 h in serum free supplemented media and treatment NRVMs were harvested for quantitative real-time PCR (qRT-PCR) or used for electrophysiology.
CaMKII Activity Assay
CaMKII activity was measured in NRVM homogenate using syntide-2, a synthetic CaMKII-specific substrate peptide. Following homogenization in a lysis buffer (50 mmol/liter HEPES, 10% ethylene glycol, 2 mg/ml BSA, 5mmol/liter EDTA, pH 7.5), ventricular lysates were incubated at 37 °C for 10 min in activity buffer (50 mmol/liter HEPES, 10 mmol/liter magnesium acetate, 1mmol/liter EGTA, 1 mg/ml BSA, 20 μmol/liter Syntide-2, 1 mmol/liter DTT, 400 nmol/liter [γ-32P]ATP, pH 7.5), or buffer to assess maximal activity (50 mmol/liter HEPES, 10 mmol/liter magnesium acetate, 500 nmol/liter calcium chloride, 1μmol/liter calmodulin, 1 mg/ml BSA, 20 μmol/liter syntide-2, 1 mmol/liter DTT, 400 nmol/liter [γ-32P]ATP, pH 7.5). Labeled peptides were blotted onto Whatman P81 filter paper and 32P incorporation was quantified in a scintillation counter. The data were expressed as a percentage of maximal CaMKII activity to normalize differences in total CaMKII protein present in each sample.
Adenoviral Infection
NRVM were infected or 5–6 h with adenoviruses (50 multiplicity of infection) containing green fluorescent protein (GFP) (Adeno-X Expression System 2, Clontech), calcineurin inhibitory peptide (CAIN), the dominant negative kinase dead Ca2+/calmodulin-dependent protein kinases IIδc and δb (CAMKIIδb/c) isoforms, or the IκBα phosphorylation-deficient mutant (IκBαSA) (10) in DMEM/F12 containing 5% FBS, after ∼18 h of plating. NRVM were washed once with DMEM after infection and were cultured in DMEM/F12 serum-free supplemented medium. Measurements were made 48 h after adenoviral transfection.
Electrophysiology
The whole-cell configuration (17) was used to record ionic currents from NRVM and all experiments were performed at room temperature. The electrophysiological setup was as described previously (9, 18). Glass obtained from World Precision Instruments (TW150F-4) was used for patch pipettes to generate a resistance ranging from 2–5 MΩ. Patch pipettes were filled with KINT solution. Modified Tyrode solution (see Solutions) continuously superfused NRVMs and only beating cells were used for recordings. Whole-cell currents were digitized at 10 kHz and filtered at 2 kHz (Molecular Devices, 200B amplifier). Applied voltages were not corrected for a liquid junction potential of −5 mV. The following protocol was designed to exclusively evoke the fast transient outward potassium currents (Ito,f) since Ito,f recovers more quickly from inactivation compared with Ito,s (19). The protocol is as follows: a 150-ms step to −80 mV, from a holding potential of −20 mV, followed by a 50-ms pre-pulse to −40 mV (to discharge the Na+ current) and then a series of 500-ms depolarizing steps from +60 mV to −40 mV. Ito,s was not examined in this study. Subtracting the peak outward current from the current at the end of the depolarizing step was used to determine Ito,f.
Quantitative RT-PCR
MIQE guidelines (20) were followed for sample preparation and qRT-PCR. Each sample group contained 3 replicates and the control samples were infected with GFP only without any drug treatment. Following the manufacturer's recommendations, TRIzol Reagent® (Life Technologies) was used to isolate RNA from cultured NRVM. To assess the quality of the mRNA samples we used a NanoDrop® spectrophotometer (Thermo Scientific) to measure absorption ratios of 260 nm and 280 nm (A260/A280>1.8) and we also examined the 28S/18S rRNA ratio on a 1.2% formaldehyde gel. The cDNA was synthesized from 1–2 μg of RNA(SuperscriptTM III Reverse Transcriptase, Life Technologies) and stored at −80 °C. SYBR green® (Life Technologies) was used to quantify the quantities of Kv4.3 and Kv4.2 as well as GAPDH (reference gene), while a TaqMan master mix (Life Technologies) was used for KChIP2.
The Viia 7 Real-Time thermal cycler PCR System (Life Technologies) was used to amplify the cDNA. The protocol consisted of the following four stages: 50 °C for 2 min, 95 °C for 10 min, 95 °C for 15 s followed by 60 °C for 1 min, repeated 40 times, and a final dissociation step at 95 °C for 15 s followed by 60 °C for 15 s. All “no template control” wells were negative in all PCR runs.
The following primer/probes were used: GAPDH: 5′-AGACAGCCGCATCTTCTTGT-3′ (forward), 5′-CTTGCCGTGGGTAGAGTCAT-3′ (reverse); KChIP2: TaqMan® (Rn01411445_g1) (Life Technologies); Kv4.2: 5′-GTGTCAGGAAGTCATAGA-3′ (forward), 5′-TTACAAAGCAGACACCCT-3′ (reverse); Kv4.3: 5′-CACCACCTGCTACACTGCTTAGAA-3′ (forward), 5′-TCTGCTCATCAATAAACTCGTGGTT-3′ (reverse).
The Viia 7 Software version 1.2 (Life Technologies) was used to analyze qRT-PCR results. The ΔΔCt method with GAPDH as the reference gene was used to quantify relative mRNA levels of the triplicate samples (21). Dissociation curves and agarose electrophoresis were used to confirm specificity. We also used 18S rRNA (Life Technologies) levels to normalize the amplicon levels. The results were identical when normalization was performed using GAPDH compared with 18S rRNA.
Western Blot Analysis
Dishes containing NRVM were washed with PBS and cells were scraped and suspended in the same buffer. The cells were then centrifuged at 1000 × g for 5 min, and the supernatant was discarded. Cells were lysed using buffer consisting of 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10% protease inhibitor mixture (P8340, Sigma) at 4 °C for 1 h. After centrifugation for 15 min at 12,000 × g, supernatants were extracted and protein concentration was determined using DC Protein Assay kit (#500-0112, Bio-Rad). Exactly 20 μg of each sample was resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. Blots were incubated with primary antibodies: Kv4.2 (1:1000, ab123543, abcam), KChIP2 (1:1000, ab99041, abcam), GAPDH (1:2000, abs16, EMD Millipore) for 2 h at room temperature. For Kv4.2 blots, anti-rabbit secondary (1:2000, 7074S, Cell Signaling) was used, and blots were incubated at room temperature for 1 h. For KChIP2 detection, anti-mouse secondary antibody (1:2000, 7076S, Cell Signaling) was used. For GAPDH detection, blots for Kv4.2 and KChIP2 were first stripped using commercial stripping buffer (ST010, GeBa) according to manufacturer's instructions. After incubation with primary antibody for GAPDH, anti-rabbit secondary antibody was used and blots were incubated at room temperature for 1 h. Amersham Biosciences ECL Prime Western blotting Detection Reagent (RPN2232, GE Healthcare) was used for detection and analysis of bands was performed using densitometry plugin for ImageJ software. For statistical analysis, the ISO treated samples were compared with the control (normalized to 1.0) for each experiment. For data presentation, errors are shown for controls to relative inter-experimental variation.
For analysis of p65, cell lysates (30 μg) from NRVMs were resolved on a 4–20% SDS-PAGE gel. Phosphorylated NF-κB p65 protein was detected using abcam anti-NF-κB p65 phospho-S276 antibody (ab106129). Total NF-κB p65 protein was detected by rabbit mAb p65 antibody (Cell Signaling #4764) as we reported (22). Murine antibodies directed toward β-actin (1 μg/ml, Sigma) were used as loading control.
Luciferase Assay
NRVMs were transfected with a KChIP2 promoter luciferase reporter plasmid containing binding elements for KChIP2 (KChIP2 luc) (9) with or without eukaryotic expression plasmids encoding wild type p65 NF-κB or infected with adenovirus encoding a phosphorylation defective mutant of IκBα designated (IκBαSA) as we reported (23). After 24 h transfection or infection, myocytes were treated with isoproterenol (ISO, 10 μmol/liter) for 24 h. Cardiac cell lysates were harvested for luciferase assay. Luciferase activity was normalized to β-galactosidase activity to control for any differences in transfection efficiency among the groups.
Data Analysis and Statistics
Excel 2011 (Microsoft and Clampfit 10.2 (Molecular Devices) were used to analyze data, while Prism 5 (GraphPad) was used for statistical analysis. Statistical tests were considered significant at the p < 0.05 levels, and data were presented as means ± S.E. of the mean. To test for differences between two sets of data, an unpaired Student's t test was used. For multiple comparisons, a one-way analysis of variance (ANOVA) test was performed and, if significant differences were observed, a Bonferroni post-test was used to determine which means differed.
A two-way ANOVA analysis was performed to test for an interaction between the drug (ISO) and a particular inhibitor (e.g. IκBαSA, CAIN, CAMKIIδb, or CAMKIIδc). The two-way ANOVA was set up as follows: GFP (control), GFP+ISO, ISO+inhibitor and inhibitor alone, and a particular parameter (e.g. current or mRNA) was measured for each condition. A significant interaction (p < 0.05) suggested that the effects of ISO in combination with an inhibitor (i.e. ISO+inhibitor) on a particular parameter were different than the effects GFP+ISO and the inhibitor alone on the same parameter.
Results
To understand how β-AR stimulation regulates Ito,f, we treated cultured rat neonatal ventricular myocytes (NRVMs) with 50 nmol/liter isoproterenol (ISO) for 48 h. These NRVMs were infected with adenoviruses expressing either GFP or various constructs to interfere with selected cell signaling pathways (see below). To quantify Ito,f, cells were held at a holding potential of −20 mV and then repolarized to −80 mV for 150 ms prior to the application of depolarizing voltage steps to activate Ito,f. As explained further in the “Discussion,” this protocol successfully permits nearly full inactivation recovery of Ito,f (i.e. >99%) while minimizing the appearance of the overlapping currents generated by Ito,s in these cells, thereby allowing accurate quantification of Ito,f (19). As shown in Fig. 1, ISO decreased (p < 0.001) Ito,f current densities by 62% in association with marked reductions in the mRNA levels of all three of its molecular constituents KChIP2 (p < 0.001), Kv4.2 (p < 0.01), and Kv4.3 (p < 0.001). These changes in Ito,f and mRNA were associated with reductions of Kv4.2 (p < 0.05) and KChIP2 (p = 0.062) protein (Fig. 2). Unfortunately, we could not reliably quantify Kv4.3 protein in NRVMs due to non-specificity of commercially available antibodies.
FIGURE 1.
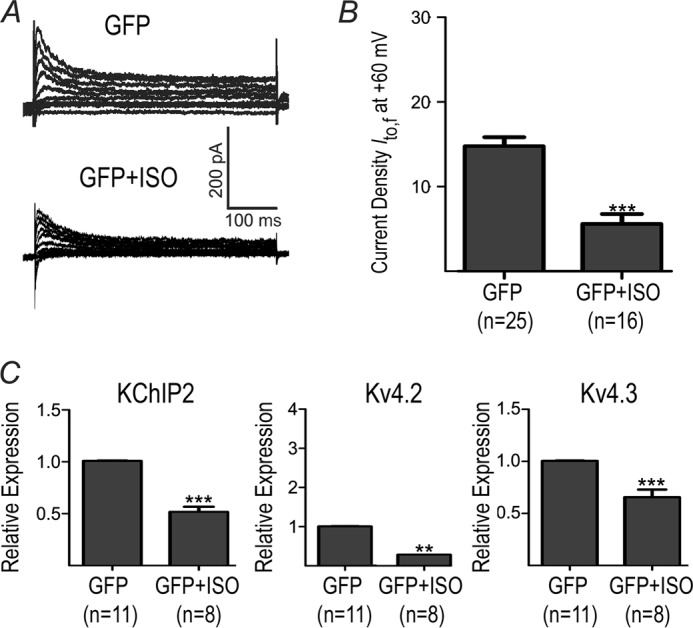
Effects of chronic β-AR stimulation on Ito,f and mRNA. NRVMs were adenovirally infected with GFP and treated (or not) with ISO (50 nm for 48 h). A, representative current Ito,f traces from NRVM. B, averaged Ito,f densities (pA/pF) at +60 mV for GFP and GFP+ISO. C, relative mRNA expression for KChIP2, Kv4.3 and Kv4.2. **, p < 0.01 versus GFP. ***, p < 0.001 versus GFP.
FIGURE 2.
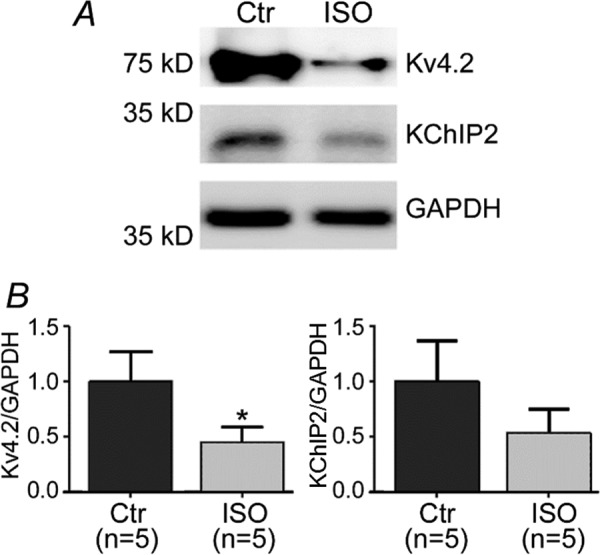
Protein expression quantified by Western blot for Kv4.2 and KChIP2 subunits. A, a representative blot showing Kv4.2, KChIP2 and GAPDH in the presence and absence of chronic ISO (48 h). B, quantifications of the Kv4.2 and KChIP2 show decreases in the presence of ISO compared with control (untreated). *, p < 0.05 versus Control (Ctr).
Previous studies have established that β-AR stimulation leads to CaMKII activation (16, 24, 25) which may underlie the changes in Ito,f seen with CaMKII activation. We found that β-AR stimulation, over a time course of 30 and 60 min (standard for this assay), increases the CaMKII activity in NRVMs (Fig. 3), consistent with previous studies (25). Next, we determined whether CaMKII activation mediates the changes in Ito,f induced by β-AR stimulation. This was initially examined by overexpressing the dominant-negative CaMKIIδc construct (dn-CaMKIIδc) to inhibit CaMKIIδc (26), a major cardiac isozyme of CaMKII (27). Overexpressing dn-CaMKIIδc caused increases in Ito,f (p < 0.01) in the absence of ISO (Fig. 4), establishing that Ito,f is regulated by CaMKIIδc under basal conditions. Interestingly, in the presence of ISO dn-CaMKIIδc caused even further reductions (p < 0.05) in Ito,f (Fig. 4). These findings support the conclusion that CaMKIIδc does not mediate the ISO-induced Ito,f changes, although CaMKIIδc does regulate Ito,f. Indeed, the pattern of changes in the mRNA expression of the molecular constituents of Ito,f, induced by CaMKIIδc inhibition, was similar in the presence and absence of β-AR stimulation (Fig. 4C). Although complex, the link between the changes in Ito,f and its molecular constituents seen in these studies can be readily understood by considering the molecular requirements for channel production, as suggested previously (see “Discussion”) (9, 10).
FIGURE 3.
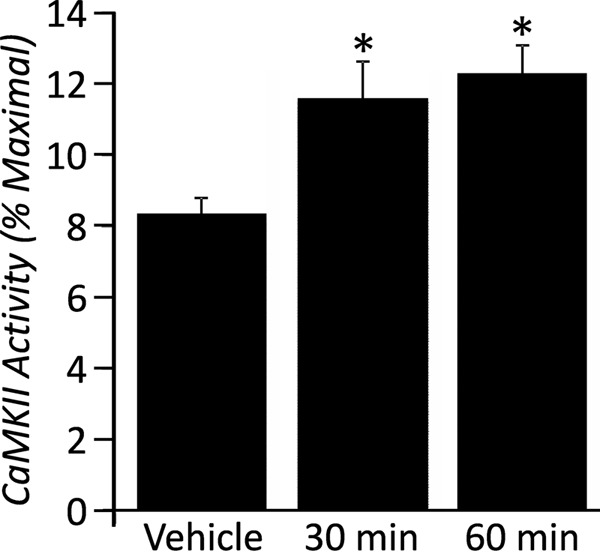
Enhancement of CaMKII activity in NRVM with ISO. Data were expressed as a percentage of the maximal CaMKII activity in order to normalize for differences in total CaMKII protein present in each NRVM sample. Activity of CaMKII is increased in response to ISO at two time-points, 30 and 60 min.*, p < 0.05 versus vehicle.
FIGURE 4.

Effects of chronic β-AR stimulation and CaMKIIδc inhibition. CaMKIIδc was inhibited by adenoviral infection of dn-CaMKIIδc. A, representative current Ito,f traces from NRVM in the presence and absence of ISO (50 nm, 48 h) and with and without dn-CaMKIIδc. B, averaged Ito,f densities (pA/pF) at +60 mV for GFP, GFP+ISO, dn-CAMKIIδc and dn-CAMKIIδc+ISO. C, relative mRNA expression for KChIP2, Kv4.3 and Kv4.2. *, p < 0.05 versus GFP. **, p < 0.01 versus GFP. *** p < 0.001 versus GFP. #, p < 0.05 versus GFP+ISO.
To examine the role of the other major cardiac isoform of CaMKII, CaMKIIδb, we also performed studies using dn-CaMKIIδb to inhibit CaMKIIδb. In contrast to CaMKIIδc, CaMKIIδb inhibition decreased Ito,f (Fig. 5) in the absence of ISO. Moreover, dn-CaMKIIδb also failed to reverse the effects of ISO on Ito,f (p = 0.82) (Fig. 5), suggesting that CaMKIIδb does not mediate the effects of ISO on Ito,f. Similar to CaMKIIδc inhibition, the pattern of mRNA changes of either KChIP2 (which was elevated) or Kv4.2 (which was reduced), induced by dn-CaMKIIδb overexpression, did not depend on whether β-ARs were stimulated. Interestingly, CaMKIIδb did, however, prevent the Kv4.3 and KChIP2 reductions induced by β-AR stimulation when ISO was present, suggesting that CaMKIIδb participates in the ISO-mediated reductions of Kv4.3 and KChIP2. Consistent with this suggestion, a two-way ANOVA analysis revealed positive interactions (p < 0.05) between the actions of ISO and dn-CaMKIIδb for Kv4.3 and KChIP2 subunit expression. Taken together, these findings support the conclusion that the ISO-mediated reductions of Kv4.3 and KChIP2 mRNA involve CaMKIIδb, whose effects appear to impact minimally on the Ito,f reductions induced by β-AR stimulation. Again, the complex dependence of Ito,f channel production on its molecular constituents is discussed more fully in “Discussion” (9, 10).
FIGURE 5.

Chronic β-AR stimulation and CaMKIIδb inhibition. Adenoviral infection of dn-CaMKIIδb was used to inhibit CaMKIIδb. A, representative current Ito,f traces from NRVM in the presence and absence of chronic ISO and with and without dn-CaMKIIδb. B, averaged Ito,f densities (pA/pF) at +60 mV for GFP, GFP+ISO, dn-CAMKIIδb and dn-CAMKIIδb+ISO. For Kv4.3 and KChIP2, there is a positive interaction (p < 0.05) between ISO and dn-CAMKIIδb (two-way ANOVA) C, relative mRNA expression for KChIP2, Kv4.3 and Kv4.2. *, p < 0.05 versus GFP. ***, p < 0.001 versus GFP. #, p < 0.05 versus GFP+ISO.
Although our results with dn-CaMKIIδb and dn-CaMKIIδc support the conclusion that the reductions in Ito,f induced by ISO are not mediated primarily by CaMKII, it is conceivable that these two CaMKII isozymes act together to regulate Ito,f following β-AR stimulation. Therefore, we also co-transfected NRVMs with dn-CaMKIIδc and dn-CaMKIIδb. Unfortunately, dual-infections caused extensive cell death, thus preventing reliable investigations into the changes in Ito,f and its molecular components induced by ISO. Thus, we cannot draw any conclusions regarding the interactions between these two CaMKII isozymes.
To understand the molecular basis for the decreases in Ito,f induced by ISO, we next considered calcineurin, a Ca2+-dependent phosphatase which is activated in response to β-AR stimulation. Consistent with previous studies (9, 10), we found that calcineurin inhibition with CAIN (an endogenous inhibitor), under baseline conditions (i.e. without ISO), caused Ito,f reductions (p < 0.01) in association with reductions in both KChIP2 (p < 0.05) and Kv4.2 (p < 0.01) mRNA expression and slight elevations (p < 0.05) in Kv4.3 expression (Fig. 6). More importantly, when ISO was present, CAIN caused further reductions in Ito,f (p < 0.05), as well as KChIP2 (p < 0.05) with no effect (p = 0.53) on Kv4.3 mRNA levels, and Kv4.2 tended to decrease (p = 0.07). These results demonstrate that the Ito,f reductions seen with β-AR activation are not mediated by calcineurin, even though calcineurin, like CaMKIIδb and CaMKIIδc, can regulate the levels of Ito,f and its molecular constituents in both the presence and absence of β-AR stimulation.
FIGURE 6.

Chronic β-AR stimulation and calcineurin inhibition with CAIN. Adenoviral infection with CAIN was used to inhibit calcineurin. A, representative current Ito,f current traces. B, averaged Ito,f densities (pA/pF) at +60 mV for GFP, GFP+ISO, CAIN and CAIN+ISO. C, relative mRNA expression for KChIP2, Kv4.3 and Kv4.2. **, p < 0.01 versus GFP. *** p < 0.001 versus GFP. #, p < 0.05 versus GFP+ISO.
We previously demonstrated that NF-κB mediates α-AR-induced reductions in Ito,f via down-regulation KChIP2 (10). To explore the potential involvement of NF-κB in β-AR regulation of Ito,f, NF-κB signaling was inhibited using the phosphorylation-deficient mutant of IκBα (IκBαSA) (23). As shown previously (10), NF-κB inhibition with IκBαSA had no effect on baseline Ito,f but caused elevations (p < 0.001) in KChIP2 expression along with reductions (p < 0.001) in Kv4.2 expression and moderate increases (p < 0.01) in Kv4.3 (Fig. 7). In addition, IκBαSA diminished (p < 0.05) the extent of Ito,f reductions induced by ISO in correlation with restoration of KChIP2 mRNA to basal levels (p < 0.05), without affecting either Kv4.2 or Kv4.3 expression (Fig. 7). These results support the hypothesis that NF-κB partly mediates reductions in Ito,f induced by β-AR stimulation. Interestingly, consistent with this conclusion, two-way ANOVA analysis revealed an interaction (p < 0.05) between ISO and IκBαSA for the regulation of KChIP2 mRNA expression, with this trend (p = 0.063) towards an interaction for Ito,f. Furthermore, the KChIP2 promoter activity in NRVMs, assessed via luciferase reporter measurements, was reduced by forced over-expression of the NF-κB subunit p65, (p < 0.001) as well as by ISO treatment (p < 0.05). Conversely, inhibition of p65 NF-κB with IκBαSA increased luciferase activity and abrogated the inhibitory effects of ISO (p < 0.05) (Fig. 8A). ISO treatment also increased the phosphorylation status of p65 subunit (Fig. 8B), which has been shown to increase the transcriptional activity of NF-κB (28, 29).
FIGURE 7.

NF-κB inhibition partly reverses decreases in Ito,f caused by chronic β-AR stimulation. IkBαSA (adenovirally infected) was used to inhibit NF-κB signaling. A, representative current Ito,f current traces. B, averaged Ito,f current densities at +60 mV for GFP, GFP+ISO, IkBαSA and IkBαSA+ISO. C, relative mRNA expression for KChIP2, Kv4.3 and Kv4.2. For KChIP2, there is a positive interaction (p < 0.05) between ISO and IkBαSA (two-way ANOVA). **, p < 0.01 versus GFP. *** p < 0.001 versus GFP. #, p < 0.05 versus GFP+ISO. #, p < 0.05 versus GFP+ISO.
FIGURE 8.
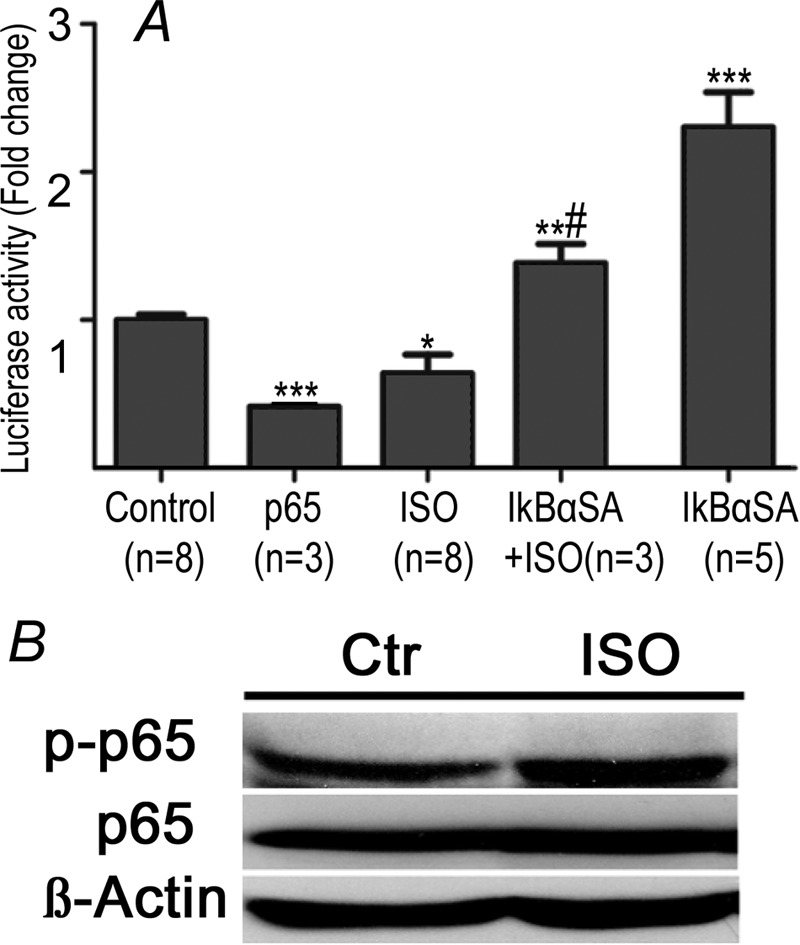
KChIP2 promoter activity and p65 phosphorylation. A, bar graph shows relative changes in luciferase activity of the KChIP2 promoter from baseline in response chronic ISO treatment in the presence and absence of NF-κB inhibitor IkBαSA as well as with IkBαSA alone. Values are relative to control. B, representative Western blot for detection of p65, phosphorylated p65 (p-p65) and β-actin. Incubation with ISO increases phosphorylation of p65. *, p < 0.05 versus Control (Ctr). **, p < 0.01 versus Control (Ctr). *** p < 0.001 versus Control (Ctr). #, p < 0.05 versus ISO. #, p < 0.05 versus ISO.
Discussion
To study the effects of β-AR stimulation on Ito,f, it was essential to quantify and separate Ito,f from other potentially overlapping currents in our patch-clamp studies. To accomplish this goal, we quantified Ito,f using voltage protocols that exploited the vast differences in recovery from inactivation that exist between Ito,f and Ito,s, which have τ-values for recovery from inactivation at ∼45 ms and ∼3800 ms, respectively (19). Specifically, cardiomyocytes were held at −20 mV in order to ensure complete inactivation of both Ito,f and Ito,s after which a brief 150-ms pulse was applied to −80 mV, thereby assuring that >99% of Ito,f has recovered from inactivation with minimal recovery (∼3.8%) of Ito,s (19). Consequently, subsequent depolarization steps (i.e. −40 to +60 mV) to activate Ito,f ensured that the peak currents gave reliable estimations of Ito,f. We contend that these Ito,f measurements, along with Kv4.2/4.3 and KChIP2 mRNA levels, in our cultured NRVMs provide trustworthy data on the regulation of Ito,f by β-AR stimulation.
Previous studies suggest that the Ca2+/calmodulin-dependent protein kinase (CaMKII) may regulate Ito,f (16, 24, 25). In addition, in heart disease that is characterized by enhanced activation of β-adrenergic receptors (β-AR) (5) is associated with both CaMKII activation (12) and reduced Ito,f (1). Consistent with these previous observations, we found that β-AR activation for 48 h in NRVMs caused profound Ito,f reductions, along with decreased mRNA levels of its three molecular subunits (i.e. KChIP2, Kv4.2, and Kv4.3). We also observed ∼50% reductions in both Kv4.2 and KChIP2 protein levels with ISO, although the changes in KChIP2 did not quite reach significance.
Although inhibition of either CaMKII or calcineurin could not rescue the ISO-dependent reductions in Ito,f, despite a clear ability of these pathways to regulate the mRNA expression of the molecular components of Ito,f, NF-κB inhibition did partly reverse the Ito,f reductions induced by ISO, in association with restoration of the KChIP2 subunit mRNA, but not the Kv4.2 or Kv4.3 subunits (summarized in Fig. 9). These results support the conclusion that NF-κB activation contributes to the ISO-mediated Ito,f reductions by suppressing KChIP2 mRNA expression. In support of this, a two-way ANOVA analyses established an interaction between NF-κB inhibition and ISO application in the regulation of KChIP2 expression, while a strong trend toward an interaction between NF-κB inhibition and ISO was also observed for Ito,f currents. Further evidence for our conclusion is provided by the observation that overexpression of the p65 NF-κB subunit reduced KChIP2 promoter activity while β-AR stimulation increased phosphorylation of p65 NF-κB and depressed KChIP2 promoter activity. Notably, the inhibitory effects of NF-κB on KChIP2 transcription were abrogated by inhibition of NF-κB with IκBSA.
FIGURE 9.
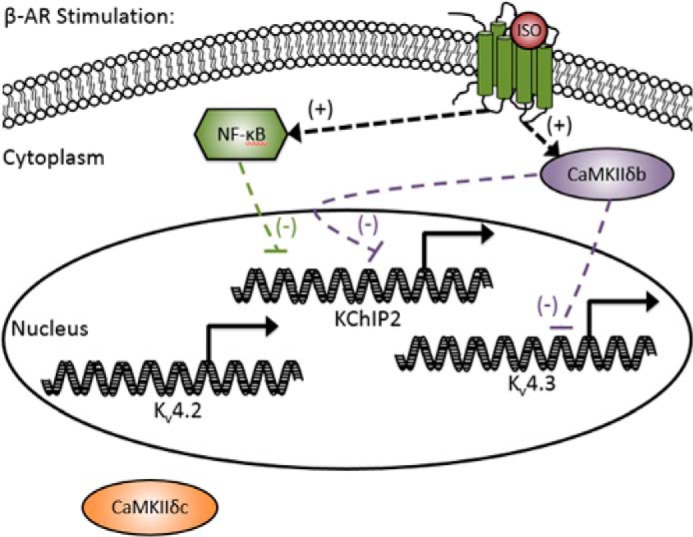
Schematic summary of the principal findings. Regulation of Ito,f subunits by the CaMKIIδ isoforms and NF-κB isoforms in the presence of β-AR stimulation (ISO). For simplicity, calcineurin regulation was not included in the diagram. (+) symbols denote positive regulation and (−) symbols denote negative regulation.
The involvement of NF-κB in β-AR-mediated effects on Ito,f is consistent with previous studies showing that KChIP2 facilitates insertion of Ito,f channels into cell membranes (30) while also limiting channel degradation (31). Importantly, NF-κB inhibition had no measureable effects on Ito,f densities in the absence of β-AR stimulation, despite causing elevations of KChIP2 mRNA levels under these conditions. These differences in Ito,f responses associated with the NF-κB-mediated alterations in KChIP2 mRNA (observed with and without β-AR stimulation) can be explained by considering the connection between channel production and the absolute levels of the various subunits. Specifically, previous studies (10, 32) have concluded that Kv4.2 is the major limiting factor for channel production under baseline conditions while KChIP2 becomes far more influential as a limiting factor when Kv4.2 expression is low (32), as seen with β-AR stimulation. This interpretation is also consistent with our CaMKIIδb/c and calcineurin inhibition studies, as discussed below.
Although acute stimulation of β-ARs activated CaMKII (Fig. 3), we found that inhibition of neither CaMKIIδb nor CaMKIIδc was able to restore the Ito,f reductions caused by chronic β-AR stimulation. Nevertheless, our results establish that CaMKIIδb and CaMKIIδc oppositely regulate Ito,f as well as Kv4.2 and KChIP2 under baseline conditions (i.e. in the absence of ISO). Specifically, CaMKIIδb inhibition caused profound elevations of KChIP2 and reductions in Kv4.2 while CaMKIIδc elevated Kv4.2 and reduced KChIP2 (as well as Kv4.3). Again, largely the same patterns were seen with inhibition of either CaMKIIδb or CaMKIIδc when β-ARs were stimulated. We acknowledge that it is conceivable that CaMKII is not actually activated with chronic β-AR stimulation (48 h), despite being activated following acute ISO treatment (<1 h) (Fig. 3). However, our two-way ANOVA analysis establishes that interactions occur between dn-CaMKIIδb and ISO treatment in the regulation of both Kv4.3, and KChIP2 mRNA levels. Thus, our studies are consistent with the notion that CaMKIIδb participates in the ISO-induced reduction in both the KChIP2 and Kv4.3 subunits (summarized Fig. 9).
The pattern of changes in Ito,f and its molecular constituents associated with CaMKII inhibition warrant further brief discussion. As already mentioned with respects to our NF-κB results, the precise connection between the changes in mRNA expression and Ito,f densities induced by CaMKIIδb or CaMKIIδc inhibition are complex. Nevertheless, our findings are generally consistent with previous studies and show remarkable internal consistency. For example, under basal conditions, the increase in Ito,f seen with CaMKIIδc inhibition correlated with increases in Kv4.2 and occurred despite reductions in KChIP2, which is expected if Kv4.2 subunits are the limiting factors under baseline conditions, as reported previously (9, 32, 33). On the other hand, the Ito,f levels seen in the presence of ISO are actually reduced further by CaMKIIδc inhibition which can be readily explained if KChIP2 becomes the rate-limiting factor for channel production when KChIP2 levels fall excessively, an assumption consistent with previous studies in mice showing that Ito,f is completely eliminated when KChIP2 is ablated (34). Similar patterns are seen with CaMKIIδb inhibition. Specifically, CaMKIIδb inhibition in the absence of ISO results in reduced Ito,f, which is expected from the profound reduction in the mRNA levels of the Kv4.2 subunits (which are channel limiting), despite elevated KChIP2. Similarly, the elevations in KChIP2 induced by CaMKIIδb inhibition are unable to increase Ito,f in the presence of ISO because of the low levels of Kv4.2 mRNA under these conditions.
We also found that calcineurin did not mediate the Ito,f reductions induced by β-AR stimulation, despite the observation that calcineurin regulates Ito,f and its molecular constituents in the presence and absence of β-AR stimulation. Our results confirm previous studies showing that calcineurin and NFAT transcriptional factors regulate Ito,f and its molecular constituents. In particular, we found that calcineurin inhibition with CAIN reduced Ito,f as well as KChIP2 and Kv4.2, but not Kv4.3, in both the presence and absence of ISO. The precise quantitative concordance between changes in Ito,f and its molecular constituents with calcineurin inhibition was complex and, because they essentially followed the patterns discussed for CaMKIIδb/c and NF-κB, are not discussed further. More importantly, the similar responses in the presence and absence of β-AR stimulation when calcineurin was inhibited supports the conclusion that calcineurin does not mediate the Ito,f changes seen with chronic β-AR stimulation.
Despite our very interesting and compelling findings, we cautiously interpreted our data with the following caveats. First, we used NRVMs as a model instead of adult myocytes. While adult myocytes have advantages in terms of maturity, NRVMs were used in our study because they have been utilized previously in many studies for exploring multiple cardiac signaling pathways (8, 9, 18, 30, 33). In addition, NRVMs are more amenable to viral infection and maintenance over a several days without significant de-differentiation, which is the case with adult myocytes. Second, it is important to note that we did not measure Kv1.4 mRNA in these experiments, and Wagner et al. found that Kv1.4 subunits were up-regulated in the hearts of mice overexpressing CaMKIIδc (11). Indeed, Kv1.4 channels form the basis for Ito,s, which recovers slowly from inactivation, compared with Ito,f (Kv4.2/4.3 channels), and thus the modulation of Ito,s by β-AR simulations and CaMKIIs warrants further investigation. Finally, despite the finding of clear correlations between subunit mRNA and protein levels, our major conclusions are based on mRNA measurements alone. This was our goal since the mediators we tested are transcriptional modulators and thus we expect alterations in mRNA of Ito,f subunits. While it is conceivable that some post-translational changes occur with our molecular interventions, the principal points of the study are supported by both Ito,f and subunit mRNA measurements. Additional studies will be needed to determine post-translation changes in Ito,f channel proteins in the setting of CaMKII and NF-κB inhibitions.
In conclusion, several new findings arise from our studies. NF-κB contributes to the Ito,f changes following β-AR stimulation in correlation with changes in KChIP2 mRNA expression. Additional studies will be needed to determine whether modulation of NF-κB will be an effective treatment for prevention of deleterious reductions in Ito,f channels caused by heart disease. We also found, for the first time, that the CaMKII isoforms oppositely regulate Ito,f and its molecular constituents in the absence of β-AR stimulation, while CaMKIIδb contributes partly to changes in Kv4.3 and KChIP2 mRNA expression induced by with ISO.
Author Contributions
P. H. B. and B. K. P. conceived and coordinated the study. L. A. K., J. H. B., C. B. B. G., P. H. B., A. K., and B. K. P. all contributed to writing the manuscript. B. K. P. and A. K. designed, performed, and analyzed the experiments shown in Figs. 1 and 4–7. P. H. B., B. K. P., and A. K. designed Fig. 9. R. A. S. and Y. O. designed, performed, and analyzed the experiments shown in Fig. 2. C. B. B. G. and J. H. B. designed, performed, and analyzed the experiments shown in Fig. 3, as well as provided the CaMKII viral constructs and analysis of the study. L. A. K. and H. G. provided the IκBαSA viral construct and designed, performed, and analyzed the experiments shown in Fig. 8, and provided analysis of the study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. L. Osborne (University of Toronto) for the use of equipment, and we thank Judy Hefferon for assistance with graphics.
This work was supported by Canadian Institutes of Health Research Grant MOP185773 (to P. H. B.) and Grant MOP74456 (to L. A. K.) and by National Institutes of Health Grant P01-HL080101 (to J. H. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- β-AR
- β-adrenergic receptor
- NRVM
- neonatal rat ventricular myocytes
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- IκBαSA
- IκBα phosphorylation-deficient mutant
- CAIN
- calcineurin inhibitor
- NFAT
- nuclear factor of activated T-cells
- ISO
- isoproterenol
- qRT-PCR
- quantitative real time polymerase chain reaction.
References
- 1. Niwa N., and Nerbonne J. M. (2010) Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J. Mol. Cell Cardiol. 48, 12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sah R., Ramirez R. J., Oudit G. Y., Gidrewicz D., Trivieri M. G., Zobel C., and Backx P. H. (2003) Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (I(to)). J. Physiol. 546, 5–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sah R., Ramirez R. J., and Backx P. H. (2002) Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ. Res. 90, 165–173 [DOI] [PubMed] [Google Scholar]
- 4. Calloe K., Cordeiro J. M., Di Diego J. M., Hansen R. S., Grunnet M., Olesen S. P., and Antzelevitch C. (2009) A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc. Res. 81, 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clark A. L., and Cleland J. G. (2000) The control of adrenergic function in heart failure: therapeutic intervention. Heart Fail. Rev. 5, 101–114 [DOI] [PubMed] [Google Scholar]
- 6. Rossow C. F., Dilly K. W., Yuan C., Nieves-Cintrón M., Cabarrus J. L., and Santana L. F. (2009) NFATc3-dependent loss of I(to) gradient across the left ventricular wall during chronic β adrenergic stimulation. J. Mol. Cell Cardiol. 46, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rossow C. F., Minami E., Chase E. G., Murry C. E., and Santana L. F. (2004) NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ. Res. 94, 1340–1350 [DOI] [PubMed] [Google Scholar]
- 8. Jia Y., and Takimoto K. (2006) Mitogen-activated protein kinases control cardiac KChIP2 gene expression. Circ. Res. 98, 386–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gong N., Bodi I., Zobel C., Schwartz A., Molkentin J. D., and Backx P. H. (2006) Calcineurin increases cardiac transient outward K+ currents via transcriptional up-regulation of Kv4.2 channel subunits. J. Biol. Chem. 281, 38498–38506 [DOI] [PubMed] [Google Scholar]
- 10. Panama B. K., Latour-Villamil D., Farman G. P., Zhao D., Bolz S. S., Kirshenbaum L. A., and Backx P. H. (2011) Nuclear factor {κ}B downregulates the transient outward potassium current Ito,f through control of KChIP2 expression. Circ. Res. 108, 537–543 [DOI] [PubMed] [Google Scholar]
- 11. Wagner S., Hacker E., Grandi E., Weber S. L., Dybkova N., Sossalla S., Sowa T., Fabritz L., Kirchhof P., Bers D. M., and Maier L. S. (2009) Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2, 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoch B., Meyer R., Hetzer R., Krause E. G., and Karczewski P. (1999) Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 84, 713–721 [DOI] [PubMed] [Google Scholar]
- 13. Hagemann D., Bohlender J., Hoch B., Krause E. G., and Karczewski P. (2001) Expression of Ca2+/calmodulin-dependent protein kinase II δ-subunit isoforms in rats with hypertensive cardiac hypertrophy. Mol. Cell Biochem. 220, 69–76 [DOI] [PubMed] [Google Scholar]
- 14. Ling H., Gray C. B., Zambon A. C., Grimm M., Gu Y., Dalton N., Purcell N. H., Peterson K., and Brown J. H. (2013) Ca2+/Calmodulin-dependent protein kinase II δ mediates myocardial ischemia/reperfusion injury through nuclear factor-κB. Circ. Res. 112, 935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang T., Johnson E. N., Gu Y., Morissette M. R., Sah V. P., Gigena M. S., Belke D. D., Dillmann W. H., Rogers T. B., Schulman H., Ross J. Jr., and Brown J. H. (2002) The cardiac-specific nuclear δ(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J. Biol. Chem. 277, 1261–1267 [DOI] [PubMed] [Google Scholar]
- 16. Grimm M., Ling H., Willeford A., Pereira L., Gray C. B., Erickson J. R., Sarma S., Respress J. L., Wehrens X. H., Bers D. M., and Brown J. H. (2015) CaMKIIδ mediates β-adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic β-adrenergic stimulation. J. Mol. Cell Cardiol. 85, 282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamill O. P., Marty A., Neher E., Sakmann B., and Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 18. Wickenden A. D., Kaprielian R., Parker T. G., Jones O. T., and Backx P. H. (1997) Effects of development and thyroid hormone on K+ currents and K+ channel gene expression in rat ventricle. J. Physiol. 504, 271–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kassiri Z., Hajjar R., and Backx P. H. (2002) Molecular components of transient outward potassium current in cultured neonatal rat ventricular myocytes. J. Mol. Med. 80, 351–358 [DOI] [PubMed] [Google Scholar]
- 20. Bustin S. A., Benes V., Garson J. A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M. W., Shipley G. L., Vandesompele J., and Wittwer C. T. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 [DOI] [PubMed] [Google Scholar]
- 21. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 22. Dhingra R., Gang H., Wang Y., Biala A. K., Aviv Y., Margulets V., Tee A., and Kirshenbaum L. A. (2013) Bidirectional regulation of nuclear factor-κB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circ. Heart Fail. 6, 335–343 [DOI] [PubMed] [Google Scholar]
- 23. Regula K. M., Baetz D., and Kirshenbaum L. A. (2004) Nuclear factor-κB represses hypoxia-induced mitochondrial defects and cell death of ventricular myocytes. Circulation 110, 3795–3802 [DOI] [PubMed] [Google Scholar]
- 24. Baltas L. G., Karczewski P., Bartel S., and Krause E. G. (1997) The endogenous cardiac sarcoplasmic reticulum Ca2+/calmodulin-dependent kinase is activated in response to β-adrenergic stimulation and becomes Ca2+-independent in intact beating hearts. FEBS Lett. 409, 131–136 [DOI] [PubMed] [Google Scholar]
- 25. Sucharov C. C., Mariner P. D., Nunley K. R., Long C., Leinwand L., and Bristow M. R. (2006) A β1-adrenergic receptor CaM kinase II-dependent pathway mediates cardiac myocyte fetal gene induction. Am. J. Physiol. Heart Circ. Physiol. 291, H1299–1308 [DOI] [PubMed] [Google Scholar]
- 26. MacDonnell S. M., Weisser-Thomas J., Kubo H., Hanscome M., Liu Q., Jaleel N., Berretta R., Chen X., Brown J. H., Sabri A. K., Molkentin J. D., and Houser S. R. (2009) CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ. Res. 105, 316–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maier L. S., Zhang T., Chen L., DeSantiago J., Brown J. H., and Bers D. M. (2003) Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92, 904–911 [DOI] [PubMed] [Google Scholar]
- 28. Li C. C., Dai R. M., Chen E., and Longo D. L. (1994) Phosphorylation of NF-κB1-p50 is involved in NF-κB activation and stable DNA binding. J. Biol. Chem. 269, 30089–30092 [PubMed] [Google Scholar]
- 29. Sasaki C. Y., Barberi T. J., Ghosh P., and Longo D. L. (2005) Phosphorylation of RelA/p65 on serine 536 defines an I{κ}B{α}-independent NF-{κ}B pathway. J. Biol. Chem. 280, 34538–34547 [DOI] [PubMed] [Google Scholar]
- 30. Jin H., Hadri L., Palomeque J., Morel C., Karakikes I., Kaprielian R., Hajjar R., and Lebeche D. (2010) KChIP2 attenuates cardiac hypertrophy through regulation of Ito and intracellular calcium signaling. J. Mol. Cell Cardiol. 48, 1169–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Foeger N. C., Wang W., Mellor R. L., and Nerbonne J. M. (2013) Stabilization of Kv4 protein by the accessory K+ channel interacting protein 2 (KChIP2) subunit is required for the generation of native myocardial fast transient outward K+ currents. J. Physiol. 591, 4149–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaprielian R., Sah R., Nguyen T., Wickenden A. D., and Backx P. H. (2002) Myocardial infarction in rat eliminates regional heterogeneity of AP profiles, I(to) K(+) currents, and [Ca(2+)](i) transients. Am. J. Physiol. Heart Circ. Physiol. 283, H1157–1168 [DOI] [PubMed] [Google Scholar]
- 33. Zobel C., Kassiri Z., Nguyen T. T., Meng Y., and Backx P. H. (2002) Prevention of hypertrophy by overexpression of Kv4.2 in cultured neonatal cardiomyocytes. Circulation 106, 2385–2391 [DOI] [PubMed] [Google Scholar]
- 34. Kuo H. C., Cheng C. F., Clark R. B., Lin J. J., Lin J. L., Hoshijima M., Nguyêñ-Tran V. T., Gu Y., Ikeda Y., Chu P. H., Ross J., Giles W. R., and Chien K. R. (2001) A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107, 801–813 [DOI] [PubMed] [Google Scholar]