

Abstract
Phospholipase C-related catalytically inactive protein (PRIP) was first identified as an inositol 1,4,5-trisphosphate-binding protein, and was later found to be involved in a variety of cellular events, particularly those related to protein phosphatases. We previously reported that Prip knock-out (KO) mice exhibit a lean phenotype with a small amount of white adipose tissue. In the present study, we examined whether PRIP is involved in energy metabolism, which could explain the lean phenotype, using high-fat diet (HFD)-fed mice. Prip-KO mice showed resistance to HFD-induced obesity, resulting in protection from glucose metabolism dysfunction and insulin resistance. Energy expenditure and body temperature at night were significantly higher in Prip-KO mice than in wild-type mice. Gene and protein expression of uncoupling protein 1 (UCP1), a thermogenic protein, was up-regulated in Prip-KO brown adipocytes in thermoneutral or cold environments. These phenotypes were caused by the promotion of lipolysis in Prip-KO brown adipocytes, which is triggered by up-regulation of phosphorylation of the lipolysis-related proteins hormone-sensitive lipase and perilipin, followed by activation of UCP1 and/or up-regulation of thermogenesis-related genes (e.g. peroxisome proliferator-activated receptor-γ coactivator-1α). The results indicate that PRIP negatively regulates UCP1-mediated thermogenesis in brown adipocytes.
Keywords: adipocyte, adipose tissue metabolism, lipolysis, obesity, uncoupling protein, thermogenesis
Introduction
Obesity, which develops due to chronic excess food intake that exceeds total energy expenditure, is becoming an epidemic worldwide. Obesity is a risk factor for many chronic diseases, such as type 2 diabetes mellitus, cardiovascular disease, dyslipidemia, obstructive sleep apnea, and certain forms of cancer, which is decreasing both the quality and length of life, and increasing individual and national healthcare costs (1). To maintain energy homeostasis at the appropriate level for a given environmental condition, a complex interplay exists between the central nervous system and the peripheral organs. In the periphery, nutrient levels are regulated in key storage organs (e.g. fat in adipose tissue and glycogen in the liver and elsewhere) as well as in the blood (e.g. blood glucose) (2).
In mammals, adipose tissue exists as mainly as two different types: white adipose tissue (WAT)3 and brown adipose tissue (BAT). As the major form of energy storage, fat in white adipocytes provides a buffer for energy imbalances when energy intake is not equal to energy output; i.e. excessive energy is stored as triglyceride (TAG) and is supplied to the body by lipolysis in a nutrient-starved state. Brown adipocytes directly dissipate the chemical energy in fatty acids through uncoupling protein 1 (UCP1); i.e. the enzyme uncouples respiration from ATP synthesis and dissipates the energy as heat (3).
Recently, many studies have shown that BAT participates in adult human obesity, and activation of UCP1-mediated thermogenesis in BAT prevents obesity and diabetes (3–5). Therefore, BAT has attracted much attention as a target for the treatment of obesity, diabetes mellitus, and other metabolic diseases. In mammals, sympathetic hyperactivity causes β-adrenergic receptor (β-AR)-induced cAMP-dependent protein kinase (PKA) activation, followed by activation of lipolysis in brown adipocytes. Consequently, elevated intracellular fatty acids activate UCP1-mediated heat generation (6, 7). PKA activation also regulates thermogenesis through transcriptional control in BAT (8).
PKA triggers lipolysis in white and brown adipocytes by indirectly activating adipose triglyceride lipase (ATGL) through phosphorylation of perilipin (9) and direct phosphorylation and activation of hormone-sensitive lipase (HSL) (10). Stepwise breakdown of TAG is catalyzed by a series of enzymes: ATGL, HSL, and monoacylglycerol lipase, and produces free fatty acids (FFAs) and glycerol. In the regulation of thermogenic gene expression, PKA mediates the phosphorylation of cAMP response element-binding protein (CREB) and p38 mitogen-activated protein kinase (p38 MAPK), followed by activation of the gene expression of peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC1α) (3, 11, 12). Mottillo et al. (8) recently reported that β-AR-induced lipolysis promotes PPARα and PPARδ activation followed by Ucp1 gene expression in brown adipocytes.
The dephosphorylation of perilipin and HSL, catalyzed by protein phosphatase 1 (PP1) or protein phosphatase 2A (PP2A), inhibits lipolysis (13–15). We first identified phospholipase C-related catalytically inactive protein (PRIP) as a signaling protein (16–23); it was later found to be involved in intracellular trafficking of the type-A γ-aminobutyric acid receptor by regulating protein phosphatases (24–30). Furthermore, by analyzing WAT from Prip knock-out (Prip-KO) mice, we recently demonstrated that PRIP enhances PP1 and PP2A translocation to lipid droplets and negatively regulates lipolysis through dephosphorylation of HSL and perilipin (31). Prip-KO mice showed a lean phenotype, even though their food intake was similar to or even greater than that of wild-type mice. Hence, in the present study, we investigated how PRIP regulates whole body energy metabolism, with special attention to UCP1-mediated thermogenesis in BAT.
Experimental Procedures
Animals
There are two homologs of PRIP in mammals, PRIP1 and PRIP2. Prip1-KO and Prip2-KO mouse strains (25, 32) were mated to produce Prip1 and Prip2 double knock-out (Prip-KO) mice, and corresponding wild-type mice as described previously (27, 28). Briefly, heterozygous (Prip1+/−, Prip2+/−) mice were mated to generate a Prip-KO strain and corresponding wild-type strain. Each strain of littermates was mated inter se, and F1 generations of Prip-KO or wild-type homozygotes were obtained. To obtain the required number of experimental mice, each strain of mice were mated inter se, and F2 to F5 generations were used for the experiments. Mice were housed under specific pathogen-free conditions at the Kasumi Laboratory Animal Center of Hiroshima University under a 12-h light/dark cycle at 23 ± 2 °C. Mice had ad libitum access to water and either a regular diet (RD: MF, 3.59 kcal/g, 12.5% of calories from fat) or a high-fat diet (HFD: HFD-60, 5.06 kcal/g, 60% of calories from fat; both from Oriental Yeast Co. Ltd., Tokyo, Japan). Mice were fed either RD or HFD from the age of 5 to 20 weeks, and male mice were used in all experiments. This study was approved by the Animal Care and Use Committee of Hiroshima University (permission numbers: A12-98, A14-70) and was performed in accordance with the Guide for Hiroshima University Animal Experimentation Regulation.
Measurement of Blood Glucose and Plasma Insulin
Blood glucose concentrations were measured in tail vein blood with a blood glucose meter (Glutest-Neo; Sanwa Kagaku, Nagoya, Japan). Plasma insulin concentration was measured using an insulin ELISA kit (Mercodia, Uppsala, Sweden). For the glucose tolerance test, mice were injected intraperitoneally with glucose (2 g/kg of body weight) after fasting for 12 h. For the insulin tolerance test, mice were injected intraperitoneally with human regular insulin (0.5 unit/kg of body weight; Eli Lilly, Kobe, Japan) after fasting for 4 h. Blood glucose levels were measured at 0, 30, 60, 90, and 120 min after injection.
Analysis of Energy Metabolism
Twenty-week-old wild-type and Prip-KO mice fed either RD or HFD were subjected to metabolic analyses. Oxygen consumption (VO2) and carbon dioxide production (VCO2) were measured using a computer-controlled open-circuit calorimetry system (Oxymax System, Columbus Instruments, Columbus, OH). All mice were acclimatized for 24 h before measurements, and VO2 and VCO2 data were recorded for 2 days. The respiratory exchange ratio was calculated as the ratio of VCO2 to VO2. Energy expenditure was also calculated using the equation heat = CV × VO2, where CV = 3.815 + 1.232 × respiratory exchange ratio (CV, calorific value based on the observed respiratory exchange ratio) (33).
Locomotor Activity Analysis and Body Temperature Measurement
Locomotor activity was recorded using a video camera (HDR-XR550V; Sony, Tokyo, Japan) and analyzed with an ANY-maze video tracking system (Stoelting Co., Wood Dale, IL). Mice fed either RD or HFD were maintained in a cage (a square arena, 30 cm × 30 cm, with 40-cm high opaque walls) for 1 week before 20 weeks of age, and then the total distance traveled was measured for 24 h. The rectal temperature of wild-type and Prip-KO mice was measured at 18 weeks of age using a digital thermometer (BDT-100; Bioresearch Center Co., Ltd., Nagoya, Japan), at 2 p.m. (day) and 2 a.m. (night).
Primary Culture of Mouse Brown Adipocytes and Oil Red O Staining
Brown adipocyte precursors were isolated using a previously reported method with some modifications (34). Briefly, interscapular BAT from 4-week-old wild-type and Prip-KO mice was minced and digested with collagenase for 30–40 min in an isolation buffer (123 mm NaCl, 5 mm KCl, 1.3 mm CaCl2, 5 mm glucose, 100 mm HEPES, and 4% BSA) with 0.2% collagenase type I (Worthington Biochemical Co., Freehold, NJ). The digested tissues were filtered through 500- and 100-μm nylon mesh filters. The cells were centrifuged for 7 min at 500 × g and washed once with the isolation buffer. The pellets were resuspended in culture medium (Dulbecco's modified Eagle's medium, containing 10% newborn calf serum, 4.5 g/liter of glucose, 10 mm HEPES, 100 units/ml of penicillin, 0.5 mg/ml ofstreptomycin, 3 nm insulin, and 15 μm ascorbic acid), seeded on 6- or 12-well plates, and grown in a humidified atmosphere of 5% CO2 and 95% air. The culture medium was changed on day 1 and every second day thereafter. For differentiation, after reaching confluence, the cells were incubated for 2 days in a differentiation medium (the culture medium supplemented with 30 nm insulin, 0.5 mm 1-methyl-3-isobutyl-1-methylxanthine, and 0.5 μm dexamethasone). Two days later, the differentiation medium was replaced with fresh culture medium. The culture medium was changed every 2 days until the differentiated cells resembled brown adipocytes. The primary brown adipocytes were stimulated with 10 μm isoproterenol, a β-adrenergic agonist, and intracellular lipid droplets were visualized by Oil Red O staining and imaged by light microscopy.
Histochemistry
Epididymal and inguinal white adipose tissues (eWAT and iWAT, respectively) and the liver were fixed with 10% formalin in phosphate-buffered saline (PBS) and then embedded in paraffin. Sections were subjected to standard hematoxylin-eosin staining. Cell area was measured using ImageJ software, a freeware image analysis program developed at the National Institutes of Health.
Immunoblot Analysis
Interscapular BAT or cultured primary brown adipocytes were homogenized in a lysis buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm NaF, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 500 μm Na3VO4, and protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 100 μm (p-amidinophenyl)methanesulfonyl fluoride hydrochloride, 10 μg/ml of leupeptin, 10 μg/ml of pepstatin A, and 3.4 μg/ml of aprotinin). After 30 min of incubation on ice, cell lysates were centrifuged at 20,000 × g for 30 min, and supernatants were collected. Protein concentration was measured using the Protein Assay Rapid Kit (Wako, Osaka, Japan), and the lysate samples were used for immunoblotting. For phosphoprotein detection, blots were blocked with Blocking One-P solution (Nacalai Tesque Inc., Kyoto, Japan). The primary antibodies used were as follows: anti-β-actin antibody (IMG-5142A; Imgenex, San Diego, CA); anti-ATGL (number 2138), anti-HSL (number 4107), anti-p-HSL Ser-660 (number 4126), anti-p-HSL Ser-563 (number 4139), anti-perilipin (number 9349), anti-p-CREB Ser-133 (number 9198), and anti-p38 MAPK (number 8690) antibodies (all from Cell Signaling Technology, Inc., Danvers, MA); anti-p-perilipin Ser-497 antibody (number 4855; Ser-497 in human perilipin 1A is equivalent to Ser-492 in murine perilipin 1A; Vala Sciences, San Diego, CA); anti-CGI-58 (also known as Abhd5, ab58283) and anti-β3AR (ab59685) antibodies (both from Abcam, Cambridge, UK); anti-monoacylglycerol lipase antibody (LS-C138957; Lifespan Biosciences, Seattle, WA); anti-p-p38 MAPK antibody (TB262; Promega, Madison, WI); and anti-CREB (number 06-863) and anti-UCP1 (AB1426) antibodies (both from Merck Millipore, Darmstadt, Germany). Production of the anti-PRIP1 and anti-PRIP2 antibodies was described previously (25, 32). For the secondary antibodies, horseradish peroxidase-conjugated antibodies against rabbit (Dako, Glostrup, Denmark) or mouse (GE Healthcare Life Sciences, Uppsala, Sweden) IgG was used. An enhanced chemiluminescence Western detection reagent (Nacalai Tesque) was used for signal development. Resultant signals were captured using an ImageQuant LAS 4000 Mini Detection System (GE Healthcare Life Sciences). The density of each band was analyzed using ImageJ software.
RNA Preparation and Quantitative Real-time PCR
Total RNA was extracted from interscapular BAT or cultured primary brown adipocytes using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was generated by reverse transcription using 0.1 μg of isolated RNA with the Verso cDNA Synthesis Kit (Thermo Scientific Inc., Waltham, MA). Quantitative real-time PCR (qRT-PCR) was performed using Fast SYBR Green Master Mix in the StepOnePlus Real Time PCR system (Applied Biosystems, Carlsbad, CA). Ct values were normalized to 36B4, a reference gene whose expression is unaffected by adipogenesis, and relative gene expression was calculated by the ΔΔCT method. The sequences of the primers used in qRT-PCR are listed in supplemental Table S1.
Statistical Analysis
Data are expressed as the mean ± S.E. Statistical significance was assessed using Student's t test or analysis of variance, followed by a contrast test with Tukey's error protection. A p value less than 0.05 was considered statistically significant.
Results
Protection against HFD-induced Obesity in Prip-KO Mice
To observe the anti-obesity effects in Prip-KO mice, we fed a HFD from the 5 to 20 weeks of age to induce obesity. During weeks 9 to 20, Prip-KO mice showed less of a body weight increase, and the body weight at 20 weeks of age was about 20% lower than that of wild-type mice, whereas the anal-nasal body length of the Prip-KO and wild-type mice was similar (Fig. 1, A and B). Food intake in the Prip-KO mice was significantly increased (Fig. 1C). However, the Prip-KO mice were obviously leaner than the wild-type mice, due to smaller-sized WAT (Fig. 1, D and E). The eWAT and iWAT masses in the Prip-KO mice were about half of those in the wild-type mice, and the BAT weight was also lower in the Prip-KO mice. Other examined tissue weights, except the kidney, were similar in mice with the two genotypes (Fig. 1F). Histological analyses showed that the size of Prip-KO adipocytes in eWAT and iWAT were significantly lower (∼40%) than that of the control (Fig. 1, G and H). Importantly, ectopic lipid accumulation in the liver was strongly decreased in HFD-fed Prip-KO mice (Fig. 1G). These results suggest protection against HFD-induced obesity in Prip-KO mice.
FIGURE 1.

Altered fat metabolism in HFD-fed Prip-KO mice. Wild-type (WT) and Prip-KO (KO) mice were fed a HFD (A–H) or a RD (I–K) ad libitum for 16 weeks starting at 5 weeks of age. A, change in body weight of WT (n = 14) and KO (n = 14) mice. B, body length of 20-week-old WT (n = 9) and KO (n = 9) mice. C, daily food intake of 18-week-old WT (n = 7) and KO (n = 7) mice. D–H, 20-week-old WT and KO mice were used for the experiments. Representative images of overall appearance (D), epididymal WAT (E), and hematoxylin and eosin-stained sections of epididymal WAT (eWAT), inguinal WAT (iWAT), and the liver (G) are shown. Similar data were obtained from the 8 mice of each genotype (D and E). Mean tissue weights for WT (n = 8) and KO (n = 8) mice were measured (F). Mean cell sizes in eWAT (WT and KO, n = 3 sections) and iWAT (WT and KO, n = 3 sections) were calculated. Sections from 3 different mice comprised 30 cells (H). I–K, representative images of overall appearance and epididymal WAT (I), and mean epididymal WAT weights (J; WT and KO, n = 6) of RD-fed mice. Similar data were obtained from the 12 mice of each genotype (I). Representative images of hematoxylin and eosin-stained sections of the liver are shown (K). Similar images were obtained from 3 independent experiments. Scale bar, 50 μm (G and K). The data represent the mean ± S.E. **, p < 0.01; ***, p < 0.001 versus the corresponding WT value; n.s., not significant.
Consistent with our previous report (31), RD-fed Prip-KO mice, a control group for the HFD feeding experiment, exhibited a lean phenotype with smaller WAT (size and weight) than RD-fed wild-type mice (Fig. 1, I and J). In addition, although Prip-KO mice showed slightly more food intake than wild-type mice (data not shown), ectopic lipid accumulation was not observed in the liver of Prip-KO mice (Fig. 1K).
Glucose Metabolic Analyses in HFD-fed Prip-KO Mice
The observation that Prip-KO mice were resistant to HFD-induced obesity suggests that PRIP deficiency may affect glucose metabolism. We performed glucose tolerance and insulin tolerance tests using HFD-fed mice at 16 weeks of age. Blood glucose and plasma insulin levels were increased by 1.4- and 2.5-fold, respectively, in HFD-fed wild-type mice compared with the levels in RD-fed groups after 12 h of starvation; however, these increases were not observed in Prip-KO mice (Fig. 2, A and B). The glucose tolerance test showed that the increase in blood glucose levels of HFD-fed Prip-KO mice was lower than that in wild-type mice (Fig. 2C). Similarly, the insulin tolerance test showed that Prip-KO mice were more sensitive to insulin than wild-type mice (Fig. 2D). These results indicate that HFD-fed Prip-KO mice have better glucose tolerance and higher insulin sensitivity.
FIGURE 2.

Resistance against glucose metabolism dysfunction and maintenance of insulin sensitivity in HFD-fed Prip-KO mice. Twenty-week-old wild-type (WT) and Prip-KO (KO) mice, which were fed a RD or a HFD from 5 weeks of age, were fasted for 12 (A–C) or 4 h (D) before blood sample collection. A, blood glucose concentration in WT (RD, n = 9; HFD, n = 12) and KO (RD, n = 9; HFD, n = 12) mice. B, plasma insulin concentration in WT (RD and HFD, n = 4) and KO (RD and HFD, n = 4) mice. C and D, glucose tolerance test and insulin tolerance test in HFD-fed mice (WT and KO, n = 8). Blood glucose levels were measured after intraperitoneal injection of glucose (C) or insulin (D). Graphs in D show the percentages of initial blood glucose levels. The data represent the mean ± S.E. *, p < 0.05; **, p < 0.01 versus RD (A and B) or WT (C and D); n.s., not significant.
Increased Energy Expenditure in Prip-KO Mice
We next measured oxygen consumption and carbon dioxide production to estimate energy expenditure of mice with both genotypes using an indirect calorimetric system. The equation-estimated respiratory exchange ratio and energy expenditure from VO2 and VCO2 are shown in Fig. 3 A and B. The cumulative energy expenditure in the nighttime was significantly higher in Prip-KO mice than wild-type mice in both the RD- and HFD-feeding experiments (Fig. 3C). The rectal temperature of Prip-KO mice at night showed a significantly higher core body temperature than wild-type mice in both the RD- and HFD-feeding conditions (Fig. 3D). However, in the daytime, the rest period for mice, Prip-KO mice did not display high energy expenditure and thermogenesis (Fig. 3, C and D).
FIGURE 3.

Increased energy expenditure in Prip-KO mice. In 20-week-old wild-type (WT; RD and HFD, n = 7) and Prip-KO (KO; RD and HFD, n = 7) mice, oxygen consumption (VO2) and carbon dioxide production (VCO2) were assessed by an indirect calorimetric system over a 24-h period with a 12-h light/dark cycle (day from 8 a.m. to 8 p.m.). A–C, calculated respiratory exchange ratio (RER) (A), calculated energy expenditure (B), and cumulative energy expenditure during daytime and nighttime (C) are shown. D, rectal temperature of 18-week-old WT (RD and HFD, n = 19) and KO (RD and HFD, n = 19) mice. The body temperature was measured at 2 p.m. (for day) and 2 a.m. (for night). E, locomotor activity of 20-week-old WT (RD and HFD, n = 6) and KO (RD and HFD, n = 6) mice. Total traveled distance (day from 8 a.m. to 8 p.m.; night from 8 p.m. to 8 a.m.) was analyzed with an ANY-maze video tracking system. The data represent the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the corresponding WT value.
We then measured locomotor activity of wild-type and Prip-KO mice fed a RD or HFD for 16 weeks from 5 weeks of age. Although the total distances traveled by the mice over 24 h were much less in the HFD-fed groups than in the RD-fed groups, there were no significant differences between the genotypes (Fig. 3E). These results suggest that, despite the similar energy consumption levels due to physical activity of both phenotypes, Prip-KO mice release energy through heat generation at nighttime; this is likely due to thermogenesis stimulated by activation of the sympathetic nervous system.
Enhanced HSL Phosphorylation and Ucp1 Gene Expression in Prip-KO BAT at 30 °C, a Thermoneutral Temperature
BAT is a heat-generating apparatus expressing UCP1, an essential protein for adaptive adrenergic non-shivering thermogenesis. Because wild-type BAT expresses PRIP1 and PRIP2 (Fig. 4A), PRIP deficiency may affect UCP1-mediated thermogenesis. Therefore, we analyzed the expression of thermogenesis-related genes in BAT from RD-fed mice kept at 30 °C, a thermoneutral temperature that does not induce thermogenesis in BAT, for 24 h (35, 36). Prip-KO mice showed higher Ucp1 mRNA expression and slightly higher protein expression than wild-type mice (Fig. 4, B and C). HSL phosphorylation (Ser-660) was significantly higher in Prip-KO BAT (Fig. 4D), suggesting that up-regulation of HSL phosphorylation in Prip-KO BAT may be involved in UCP1-mediated thermogenesis in a thermoneutral environment.
FIGURE 4.

Enhanced expression of UCP1 mRNA and protein and elevated lipolysis in Prip-KO mouse BAT. RD-fed 10-week-old wild-type (WT) and Prip-KO (KO) mice kept at 30 °C for 24 h were used for these experiments. A, expression of PRIP1 and PRIP2 in BAT. Arrowhead shows the specific PRIP1 band. B, Ucp1 mRNA expression in BAT was analyzed by qRT-PCR (WT and KO, n = 6). C and D, UCP1 protein expression (C) and HSL phosphorylation (p-HSL S660) (D) in BAT were analyzed by immunoblotting (WT and KO, n = 6). β-Actin (C) and HSL (D) were used as the loading controls. Bands were quantified using ImageJ software. Mean values normalized to the loading control are shown. Representative images from 6 independent experiments are shown. E and F, Pgc1α, Dio2, Pparα, Pparδ, and Pparγ mRNA expression in BAT was analyzed by qRT-PCR (30 °C, WT and KO, n = 6). The WT value is set to 1. The data represent the mean ± S.E. *, p < 0.05; ***, p < 0.001 versus the corresponding WT value.
PGC1α, DIO2, PPARα, PPARδ, and PPARγ are known to be involved in the regulation of Ucp1 gene expression by activating the sympathetic nervous system (3, 8). The mRNAs of the thermogenic genes, Pgc1α and Dio2, were higher in Prip-KO mice than in wild-type mice, but not the mRNAs of Pparα, Pparδ, and Pparγ (Fig. 4, E and F).
Cold Stimulation-induced UCP1 Activation in Prip-KO BAT
To investigate cold exposure-induced UCP1 expression, mice were exposed to a cold environment (4 °C) for 6 h. RD-fed Prip-KO mice showed significantly higher mRNA expression of Ucp1 and the thermogenic genes, Pgc1α, Dio2, Pparα, Pparδ, and Pparγ, in BAT than wild-type mice (Fig. 5A). UCP1 protein expression was also significantly higher in Prip-KO BAT (Fig. 5B), suggesting that Prip deficiency induces UCP1 expression, resulting in thermogenesis via sympathetic nerve activation. HSL phosphorylation (Ser-660) was slightly higher in Prip-KO BAT than in wild-type BAT (Fig. 5C). However, the levels were apparently lower than those at 30 °C, suggesting that the effect of lipolysis in BAT after cold (4 °C) exposure for 6 h faded.
FIGURE 5.

Cold stimulation enhanced the expression of UCP1 mRNA and protein in Prip-KO BAT. RD-fed 10-week-old wild-type (WT) and Prip-KO (KO) mice kept at 4 °C for 6 h (A–D) or 30 °C for 24 h (C and D) were used for the experiments. A, Ucp1, Pgc1α, Dio2, Pparα, Pparδ, and Pparγ mRNA expression in BAT was analyzed by qRT-PCR (WT and KO, n = 3). The WT value is set to 1. B, UCP1 protein expression in BAT was analyzed by immunoblotting (WT and KO, n = 3). β-Actin was used as the loading control. Bands were quantified using ImageJ software. Mean values normalized to β-actin are shown. The data represent the mean ± S.E. *, p < 0.05; **, p < 0.01 versus the corresponding WT value. C, phosphorylation of HSL (p-HSL S660) in BAT at 30 and 4 °C. A set of representative images from 3 independent experiments is shown. HSL was used as the loading control. D, Ucp3 mRNA expression in the skeletal muscle at 30 and 4 °C was analyzed by qRT-PCR (WT and KO, n = 3). Data are expressed as the mean ± S.E. of relative gene expression.
Skeletal muscle is also a major energy expenditure organ, and UCP3 is believed to be involved in energy metabolism in skeletal muscle and participate in glucose and lipid metabolism (37). The mRNA expression of Prip1 and Prip2 was detected in skeletal muscle by PCR. Ucp3 mRNA expression in the skeletal muscle of wild-type and Prip-KO at 30 (24 h) or 4 °C (6 h) was similar (Fig. 5D), suggesting that energy expenditure in skeletal muscle is similar between the two genotypes.
Increased UCP1 Expression and Lipolysis following β-AR Stimulation in Cultured Primary Brown Adipocytes from Prip-KO Mice
Finally, we analyzed cultured primary brown adipocytes to exclude the systemic effect of PRIP deficiency in the regulation of thermogenic gene expression and lipid metabolism. The protein expression levels of several lipid metabolism-related proteins, including HSL, perilipin, monoacylglycerol lipase, ATGL, comparative gene identification-58 (CGI-58), and β3-AR, in wild-type and Prip-KO brown adipocytes were similar (Fig. 6A).
FIGURE 6.

Enhanced expression of thermogenesis-related genes and lipolysis in the cultured primary brown adipocytes from Prip-KO mice. Cultured primary brown adipocytes from wild-type (WT) and Prip-KO (KO) mice were stimulated with 10 μm isoproterenol (Iso, +) or PBS (Vehicle, −) for 4 (B, F, and G) or 6 h (C–E and H). A, comparison of lipid metabolism-related proteins. Whole lysates from the cultured cells were analyzed by immunoblotting. Similar results were obtained from 3 independent experiments, and representative images are shown. B, expression of Ucp1 mRNA in the isoproterenol-stimulated cells was analyzed by qRT-PCR. The WT value in vehicle is set to 1. C and D, the expression of UCP1, PRIP1, and PRIP2, and the phosphorylation of HSL (p-HSL S660, p-HSL S563) and perilipin (p-perilipin S497) in cell lysates were analyzed by immunoblotting. Assessment of UCP1 protein expression is shown in C using β-actin as the loading control. Arrowheads in D indicate the specific PRIP1 bands. HSL and perilipin were used as the loading controls (D). Similar data were obtained from 3 independent experiments, and representative images are shown (C and D). Bands were quantified using ImageJ software. The mean value in WT is set to 1. E, brown adipocytes stimulated with or without isoproterenol were stained with Oil Red O. A set of typical images from 3 independent experiments is shown. Scale bar, 50 μm. Each lower panel shows a magnified view of the framed area in the upper panel. F–H, expression of Pgc1α, Pparα, Pparδ, Dio2, and Pparγ mRNA in the isoproterenol-stimulated cells was analyzed by qRT-PCR (F and H). The quantity in WT cells treated with vehicle is set to 1. Phosphorylation of CREB (p-CREB S133) and p38 MAPK (p-p38 MAPK T198) in cell lysates were analyzed by immunoblotting (G). Total amounts of CREB and p38 MAPK were used as the controls. Similar data were obtained from 3 independent experiments, and representative images are shown. The data represent the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the corresponding WT value.
In response to β-AR stimulation, UCP1 mRNA and protein expression was increased by 2.7- and 2.0-fold, respectively, in Prip-KO brown adipocytes compared with the levels in wild-type cells (Fig. 6, B and C). HSL phosphorylation at Ser-660 and Ser-563 and perilipin phosphorylation at Ser-497 were significantly higher in Prip-KO brown adipocytes than in wild-type adipocytes (Fig. 6D). Consistently, the lipid droplets in β-AR-stimulated Prip-KO brown adipocytes appeared to be smaller than those in wild-type cells (Fig. 6E), suggesting high lipolytic activity in Prip-KO brown adipocytes. In β-AR-stimulated cultured brown adipocytes from Prip-KO mice, the mRNA expression of Pgc1α, Pparα, and Pparδ was significantly higher (1.2-, 1.7-, and 1.3-fold, respectively) than that in cells from wild-type mice (Fig. 6F), suggesting that lipolytic products regulate the expression of these mRNAs (see “Discussion”).
The stimulation of brown adipocytes with isoproterenol induced the phosphorylation of CREB and p38 MAPK in mice of the two genotypes. However, these phosphorylation increases were less dramatic in Prip-KO mice than in wild-type mice (Fig. 6G). Consistently, the mRNA expression of the downstream signaling molecules of CREB and p38 MAPK, Dio2, and Pparγ, was similar in mice of both genotypes (Fig. 6H). These results show that PRIP deficiency increases UCP1 expression and enhances lipolysis in brown adipocytes; therefore, activated UCP1 promotes thermogenesis, resulting in the promotion of systemic energy expenditure.
Discussion
BAT acts as a crucial regulator of energy metabolism through β-AR-induced liberation of FFAs from TAG by lipolysis. As shown in Fig. 7, FFAs are used not only as substrates for oxidative respiration but also as allosteric activators of UCP1. Adrenergic signaling also stimulates Ucp1 transcription through thermogenesis-related molecules, including CREB, p38 MAPK, PPARs, and PGC-1α. Thus, elucidation of the molecular regulatory mechanism underlying thermogenic programs in BAT is important for developing new prophylaxes and therapeutic treatments for obesity. The present study demonstrates that PRIP regulates β-AR signaling-induced UCP1-dependent thermogenesis in BAT through phosphoregulation of HSL and perilipin.
FIGURE 7.
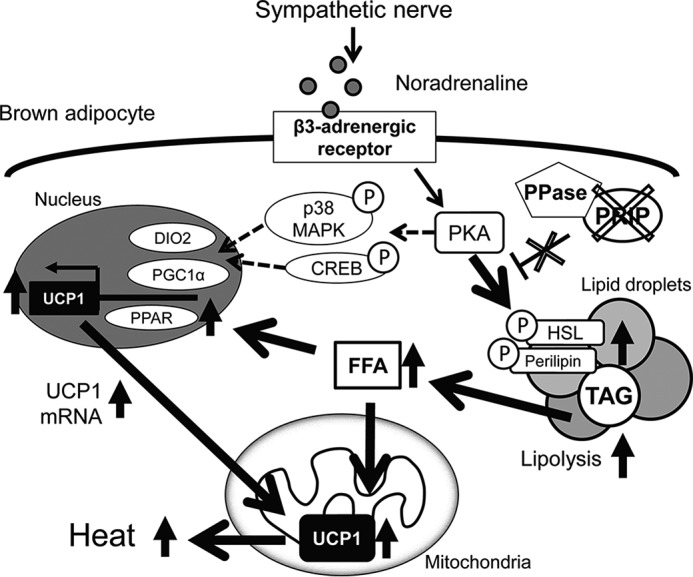
Schematic representation of the regulation of brown fat thermogenesis by PRIP. Sympathetic nerve stimulation enhances receptor-mediated PKA activation in brown adipocytes. PKA induces UCP1 activation by at least two pathways. (i) PKA facilitates the phosphorylation of HSL and perilipin, which promotes lipolysis. The resulting FFAs enhance UCP1 enzymatic activation and Ucp1 gene expression through PPAR-mediated PGC1α activation (the pathway represented by the solid arrows). (ii) PKA also induces phosphorylation of CREB and p38 MAPK, which activate PGC1α and DIO2 (represented by dashed-line arrows). Consequently, Ucp1 gene expression and activity are elevated. Our data show that Prip gene ablation (represented by the X-marks) leads to elevation of HSL and perilipin phosphorylation-mediated lipolytic cascades followed by FFA-mediated UCP1-induced heat production. Upward arrows denote up-regulation of expression or activity. Encircled Ps indicate phosphoproteins. DIO2, type II iodothyronine 5′ deiodinase; FFA, free fatty acid; PKA, cAMP-dependent protein kinase; p38 MAPK, p38 mitogen-activated protein kinase; PGC1α, peroxisome proliferator-activated receptor γ co-activator 1α; PPase, protein phosphatase.
Energy balance is a complex process involving caloric intake, storage, and expenditure, which is essentially the sum of the internal heat produced and external work. Prip-KO mice fed a HFD exhibited little fat accumulation in WAT, albeit with greater caloric intake and similar physical activity. Therefore, these data indicate that internal heat production is up-regulated in Prip-KO mice. BAT is found in small animals, including rodents, and burns calories to produce heat (3). An increase in the metabolic rate allows the burning of ingested calories and promotes the removal of body fat.
Thermal stress can be eliminated under thermoneutral conditions, which in mice is 30 °C (35, 36); therefore, at this temperature, the consequences of lipolysis signaling in Prip-KO BAT can be observed. The levels of HSL phosphorylation were significantly higher in Prip-KO BAT than in wild-type BAT (Fig. 4D). Because elevated intracellular fatty acids activate UCP1-mediated heat generation (6, 7), lipolysis-mediated non-shivering thermogenesis in BAT may be enhanced in Prip-KO mice. Therefore, we investigated the regulatory mechanism of β-AR signaling-induced UCP1-dependent thermogenesis in BAT via PRIP using cultured primary brown adipocytes to exclude a systemic effect. PRIP promotes the translocation of PP1 and PP2A to lipid droplets to trigger the dephosphorylation of HSL and perilipin, thus reducing PKA-mediated fatty acid production in WAT (31) and probably in BAT. Therefore, PRIP deficiency led to the reduced dephosphorylation of HSL and perilipin (Fig. 6D). Phosphorylated perilipin releases CGI-58, and CGI-58 activates ATGL, which drives TAG hydrolysis. Phosphorylated perilipin also retains phosphorylated (activated) HSL on lipid droplets (for a review, see Ref. 38). Consequently, lipolysis is up-regulated in Prip-KO BAT (Fig. 6E), leading to increased thermogenesis.
In brown adipocytes, non-shivering thermogenesis occurs via UCP1 activation, and it dissipates energy as heat. The activity of UCP1 is regulated by FFAs; therefore, UCP1 activity in BAT is regulated by lipases, including HSL (7). Similar to WAT, non-shivering thermogenesis in BAT is activated by the cAMP/PKA pathway through β-ARs (39). PKA activation leads to phosphorylation of HSL and perilipin, which promotes lipolysis and results in the release of FFAs. In the resting state, UCP1 is inhibited by purine nucleotides, such as ATP (40). However, when brown adipocytes are activated by the sympathetic nervous system, β-AR-induced FFAs directly activate UCP1 with long-chain fatty acids, the most efficient activators of UCP1 in vitro (6, 7). Thus, Prip-KO mice display a high body temperature and expend energy in the nighttime (Fig. 3, C and D).
The Ucp1 and Pgc1α genes contain binding sites for PPARs (41, 42), and β-AR agonism can increase PPARα and PPARδ activation in brown adipocytes (8). Thus, lipolytic products can activate Ucp1 gene expression in brown adipocytes (43). In Prip-KO cultured primary brown adipocytes, high amounts of lipolytic products induced Pparα and Pparδ gene expression. Thus, one aspect of Ucp1 gene expression is dependent on the regulation of these genes (Fig. 6F). Multiple transcriptional regulatory cascades have been reported to be involved in Ucp1 gene transcription. PPARγ is a master transcriptional factor required for both white and brown adipocyte differentiation (44). Several transcriptional factors and cofactors affect brown adipogenic and thermogenic gene expression. However, PPARγ antagonists do not affect the induction of thermogenic genes, including Ucp1, which is induced by β-AR agonism (8). This supports our results showing that β-AR-mediated PPARγ expression in both wild-type and Prip-KO brown adipocytes is similar (Fig. 6H).
Ucp1 gene expression is also controlled by transcriptional regulators such as PGC1α and DIO2, which are in turn modulated by phosphorylation of cellular transcription factors, including CREB (45) and p38 MAPK (46). In the cultured primary brown adipocytes, the β-AR agonist, isoproterenol, stimulated PKA-induced phosphorylation of CREB and p38 MAPK in the two genotypes. However, the phosphorylation increases in Prip-KO mice were not as prominent as in wild-type mice (Fig. 6G). These results suggest that PRIP has little effect on CREB and p38 MAPK phosphorylation-mediated Ucp1 gene expression. In contrast, Pgc1α gene expression was increased in Prip-KO brown adipocytes. Because PPARα affects PGC1α expression (42), up-regulation of PPARα in Prip-KO brown adipocytes may induce Pgc1α gene expression, and subsequent Ucp1 gene up-regulation. Therefore, UCP1 protein expression was up-regulated in Prip-KO BAT. However, in vivo experiments under thermoneutral conditions showed up-regulation of Pgc1α and Dio2 gene expression without enhancement of PPARs (Fig. 4, E and F), suggesting that an unknown mechanism may be still involved.
Non-shivering thermogenesis is a defense mechanism against environmental cold stress in mammals, including humans. Acute non-shivering thermogenesis and chronic cold exposure induce UCP1 up-regulation, which maintains body temperature via activation of the sympathetic nerve system (6, 11). The principal energy source for this process is fatty acids that are either synthesized de novo in BAT or imported from circulation through fatty acid transport protein 1 (47). Cold exposure accelerates plasma clearance of triglycerides as a result of increased uptake into BAT (48). In wild-type mice, the phosphorylation levels of HSL were greatly decreased during a 4-h exposure to 4 °C compared with exposure to 30 °C (Fig. 5C), which suggests that the severe cold exposure necessitates importation of fatty acids probably by depletion of de novo synthesized fatty acids in BAT. Although HSL activity is already low in Prip-KO BAT as well as in wild-type mice in the 4 °C environment, imported fatty acid facilitates UCP1 expression through a genetic regulatory pathway in Prip-KO BAT (Fig. 5, A and B); i.e. PRIP is involved in the pathway of UCP1 gene expression.
PRIP is similar to phospholipase C-δ1 (PLC-δ1); however, PRIP has no phospholipase C activity (16–18, 20). PLC-δ1−/− mice also showed an anti-obesity phenotype in HFD feeding experiments (49). However, PLC-δ1 expression levels in Prip-KO BAT and brown adipocytes were similar or higher to those in wild-type tissues and cells (supplemental Fig. S1). Therefore, the enhanced thermogenesis observed in Prip-KO mice occurs independent of the PLC-δ1-mediated signaling pathway in BAT.
In conclusion, PRIP is a modulator of fat lipolysis and thermogenesis in adipocytes, suggesting the utility of PRIP silencing as a potential anti-obesity therapy. Further comprehensive studies of PRIP signaling will improve our understanding of the etiopathology of obesity and contribute to the development of new therapeutic targets aimed at tackling excess body fat accumulation.
Author Contributions
K. O. performed the experiments and wrote a draft of the manuscript. J. Z. performed and analyzed the experiments together with K. O. K. H., S. A., Y. Y., and M. Ha. each contributed to data collection and discussion. H. F. and T. T. supported the histopathological analyses. M. I. was involved in the study design. M. Hi. reviewed and edited the manuscript. T. K. conceived and coordinated the study and wrote the paper. All authors commented on the manuscript.
Acknowledgments
We thank the staff of the Natural Science Center for Basic Research and Development (NBARD) at Hiroshima University for assistance with mouse breeding. We thank Editage, Cactus Communications Ltd., for help with the final editing of the manuscript.
This work was supported in part by Funding Program for Next Generation World-Leading Researchers Grant LS087 (to T. K.) and JSPS KAKENHI Grants 266708090 (to T. K.), 15H06433 (to O. K.), and 24229009 (to M. H.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. S1 and Table S1.
- WAT
- white adipose tissue
- ATGL
- adipocyte triglyceride lipase
- β-AR
- β-adrenergic receptor
- BAT
- brown adipose tissue
- CGI-58
- comparative gene identification-58
- CREB
- cAMP response element-binding protein
- HFD
- high-fat diet
- HSL
- hormone-sensitive lipase
- iWAT
- inguinal white adipose tissue
- eWAT
- epididymal WAT
- PGC1α
- PPAR-γ coactivator-1α
- PP1
- protein phosphatase 1
- PLC-δ1
- phospholipase C-δ1
- PP2A
- protein phosphatase 2A
- PPAR
- peroxisome proliferator-activated receptor
- PRIP
- phospholipase C-related catalytically inactive protein
- Prip-KO
- PRIP1 and PRIP2 double-knockout
- RD
- regular diet
- TAG
- triacylglycerol
- UCP1
- uncoupling protein 1
- VCO2
- carbon dioxide production
- VO2
- oxygen consumption
- qRT
- quantitative RT.
References
- 1. Spiegelman B. M., and Flier J. S. (2001) Obesity and the regulation of energy balance. Cell 104, 531–543 [DOI] [PubMed] [Google Scholar]
- 2. Woods S. C., and D'Alessio D. A. (2008) Central control of body weight and appetite. J. Clin. Endocrinol. Metab. 93, S37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harms M., and Seale P. (2013) Brown and beige fat: development, function and therapeutic potential. Nat. Med. 19, 1252–1263 [DOI] [PubMed] [Google Scholar]
- 4. van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., and Teule G. J. (2009) Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508 [DOI] [PubMed] [Google Scholar]
- 5. Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T., Taittonen M., Laine J., Savisto N. J., Enerbäck S., and Nuutila P. (2009) Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 6. Cannon B., and Nedergaard J. (2004) Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 [DOI] [PubMed] [Google Scholar]
- 7. Fedorenko A., Lishko P. V., and Kirichok Y. (2012) Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mottillo E. P., Bloch A. E., Leff T., and Granneman J. G. (2012) Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 287, 25038–25048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Granneman J. G., Moore H. P., Krishnamoorthy R., and Rathod M. (2009) Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 284, 34538–34544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anthonsen M. W., Rönnstrand L., Wernstedt C., Degerman E., and Holm C. (1998) Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 273, 215–221 [DOI] [PubMed] [Google Scholar]
- 11. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., and Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 12. Cao W., Daniel K. W., Robidoux J., Puigserver P., Medvedev A. V., Bai X., Floering L. M., Spiegelman B. M., and Collins S. (2004) p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 24, 3057–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wood S. L., Emmison N., Borthwick A. C., and Yeaman S. J. (1993) The protein phosphatases responsible for dephosphorylation of hormone-sensitive lipase in isolated rat adipocytes. Biochem. J. 295, 531–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clifford G. M., McCormick D. K., Londos C., Vernon R. G., and Yeaman S. J. (1998) Dephosphorylation of perilipin by protein phosphatases present in rat adipocytes. FEBS Lett. 435, 125–129 [DOI] [PubMed] [Google Scholar]
- 15. Kinney B. P., Qiao L., Levaugh J. M., and Shao J. (2010) B56α/protein phosphatase 2A inhibits adipose lipolysis in high-fat diet-induced obese mice. Endocrinology 151, 3624–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanematsu T., Takeya H., Watanabe Y., Ozaki S., Yoshida M., Koga T., Iwanaga S., and Hirata M. (1992) Putative inositol 1,4,5-trisphosphate binding proteins in rat brain cytosol. J. Biol. Chem. 267, 6518–6525 [PubMed] [Google Scholar]
- 17. Yoshida M., Kanematsu T., Watanabe Y., Koga T., Ozaki S., Iwanaga S., and Hirata M. (1994) d-myo-Inositol 1,4,5-trisphosphate-binding proteins in rat brain membranes. J. Biochem. 115, 973–980 [DOI] [PubMed] [Google Scholar]
- 18. Kanematsu T., Misumi Y., Watanabe Y., Ozaki S., Koga T., Iwanaga S., Ikehara Y., and Hirata M. (1996) A new inositol 1,4,5-trisphosphate binding protein similar to phospholipase C-δ 1. Biochem. J. 313, 319–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeuchi H., Kanematsu T., Misumi Y., Yaakob H. B., Yagisawa H., Ikehara Y., Watanabe Y., Tan Z., Shears S. B., and Hirata M. (1996) Localization of a high-affinity inositol 1,4,5-trisphosphate/inositol 1,4,5,6-tetrakisphosphate binding domain to the pleckstrin homology module of a new 130 kDa protein: characterization of the determinants of structural specificity. Biochem. J. 318, 561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kanematsu T., Yoshimura K., Hidaka K., Takeuchi H., Katan M., and Hirata M. (2000) Domain organization of p130, PLC-related catalytically inactive protein, and structural basis for the lack of enzyme activity. Eur. J. Biochem. 267, 2731–2737 [DOI] [PubMed] [Google Scholar]
- 21. Kitayama T., Morita K., Sultana R., Kikushige N., Mgita K., Ueno S., Hirata M., and Kanematsu T. (2013) Phospholipase C-related but catalytically inactive protein modulates pain behavior in a neuropathic pain model in mice. Mol. Pain. 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Umebayashi H., Mizokami A., Matsuda M., Harada K., Takeuchi H., Tanida I., Hirata M., and Kanematsu T. (2013) Phospholipase C-related catalytically inactive protein, a novel microtubule-associated protein 1 light chain 3-binding protein, negatively regulates autophagosome formation. Biochem. Biophys. Res. Commun. 432, 268–274 [DOI] [PubMed] [Google Scholar]
- 23. Harada-Hada K., Harada K., Kato F., Hisatsune J., Tanida I., Ogawa M., Asano S., Sugai M., Hirata M., and Kanematsu T. (2014) Phospholipase C-related catalytically inactive protein participates in the autophagic elimination of Staphylococcus aureus infecting mouse embryonic fibroblasts. PLoS ONE 9, e98285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshimura K., Takeuchi H., Sato O., Hidaka K., Doira N., Terunuma M., Harada K., Ogawa Y., Ito Y., Kanematsu T., and Hirata M. (2001) Interaction of p130 with, and consequent inhibition of, the catalytic subunit of protein phosphatase 1α. J. Biol. Chem. 276, 17908–17913 [DOI] [PubMed] [Google Scholar]
- 25. Kanematsu T., Jang I. S., Yamaguchi T., Nagahama H., Yoshimura K., Hidaka K., Matsuda M., Takeuchi H., Misumi Y., Nakayama K., Yamamoto T., Akaike N., Hirata M., and Nakayama K. (2002) Role of the PLC-related, catalytically inactive protein p130 in GABAA receptor function. EMBO J. 21, 1004–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Terunuma M., Jang I. S., Ha S. H., Kittler J. T., Kanematsu T., Jovanovic J. N., Nakayama K. I., Akaike N., Ryu S. H., Moss S. J., and Hirata M. (2004) GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J. Neurosci. 24, 7074–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanematsu T., Yasunaga A., Mizoguchi Y., Kuratani A., Kittler J. T., Jovanovic J. N., Takenaka K., Nakayama K. I., Fukami K., Takenawa T., Moss S. J., Nabekura J., and Hirata M. (2006) Modulation of GABAA receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. J. Biol. Chem. 281, 22180–22189 [DOI] [PubMed] [Google Scholar]
- 28. Mizokami A., Kanematsu T., Ishibashi H., Yamaguchi T., Tanida I., Takenaka K., Nakayama K. I., Fukami K., Takenawa T., Kominami E., Moss S. J., Yamamoto T., Nabekura J., and Hirata M. (2007) Phospholipase C-related inactive protein is involved in trafficking of γ2 subunit-containing GABAA receptors to the cell surface. J. Neurosci. 27, 1692–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujii M., Kanematsu T., Ishibashi H., Fukami K., Takenawa T., Nakayama K. I., Moss S. J., Nabekura J., and Hirata M. (2010) Phospholipase C-related but catalytically inactive protein is required for insulin-induced cell surface expression of γ-aminobutyric acid type A receptors. J. Biol. Chem. 285, 4837–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sugiyama G., Takeuchi H., Nagano K., Gao J., Ohyama Y., Mori Y., and Hirata M. (2012) Regulated interaction of protein phosphatase 1 and protein phosphatase 2A with phospholipase C-related, but catalytically inactive protein. Biochemistry 51, 3394–3403 [DOI] [PubMed] [Google Scholar]
- 31. Okumura T., Harada K., Oue K., Zhang J., Asano S., Hayashiuchi M., Mizokami A., Tanaka H., Irifune M., Kamata N., Hirata M., and Kanematsu T. (2014) Phospholipase C-related catalytically inactive protein (PRIP) regulates lipolysis in adipose tissue by modulating the phosphorylation of hormone-sensitive lipase. PLoS ONE 9, e100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takenaka K., Fukami K., Otsuki M., Nakamura Y., Kataoka Y., Wada M., Tsuji K., Nishikawa S., Yoshida N., and Takenawa T. (2003) Role of phospholipase C-L2, a novel phospholipase C-like protein that lacks lipase activity, in B-cell receptor signaling. Mol. Cell. Biol. 23, 7329–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McLean J. A. (1987) Analysis of gaseous exchange in open-circuit indirect calorimetry. Med. Biol. Eng. Comput. 25, 239–240 [DOI] [PubMed] [Google Scholar]
- 34. Hernandez A., de Mena R. M., Martin E., and Obregon M. J. (2011) Differences in the response of UCP1 mRNA to hormonal stimulation between rat and mouse primary cultures of brown adipocytes. Cell. Physiol. Biochem. 28, 969–980 [DOI] [PubMed] [Google Scholar]
- 35. Feldmann H. M., Golozoubova V., Cannon B., and Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 36. Galmozzi A., Sonne S. B., Altshuler-Keylin S., Hasegawa Y., Shinoda K., Luijten I. H., Chang J. W., Sharp L. Z., Cravatt B. F., Saez E., and Kajimura S. (2014) ThermoMouse: an in vivo model to identify modulators of UCP1 expression in brown adipose tissue. Cell Rep. 9, 1584–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zurlo F., Larson K., Bogardus C., and Ravussin E. (1990) Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Invest. 86, 1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lampidonis A. D., Rogdakis E., Voutsinas G. E., and Stravopodis D. J. (2011) The resurgence of hormone-sensitive lipase (HSL) in mammalian lipolysis. Gene 477, 1–11 [DOI] [PubMed] [Google Scholar]
- 39. Collins S., and Surwit R. S. (2001) The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog. Horm. Res. 56, 309–328 [DOI] [PubMed] [Google Scholar]
- 40. Nicholls D. G. (1974) Hamster brown-adipose-tissue mitochondria. The chloride permeability of the inner membrane under respiring conditions, the influence of purine nucleotides. Eur. J. Biochem. 49, 585–593 [DOI] [PubMed] [Google Scholar]
- 41. Kozak U. C., Kopecky J., Teisinger J., Enerbäck S., Boyer B., and Kozak L. P. (1994) An upstream enhancer regulating brown-fat-specific expression of the mitochondrial uncoupling protein gene. Mol. Cell. Biol. 14, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hondares E., Rosell M., Díaz-Delfin J., Olmos Y., Monsalve M., Iglesias R., Villarroya F., and Giralt M. (2011) Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1alpha (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J. Biol. Chem. 286, 43112–43122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barbera M. J., Schluter A., Pedraza N., Iglesias R., Villarroya F., and Giralt M. (2001) Peroxisome proliferator-activated receptor α activates transcription of the brown fat uncoupling protein-1 gene: a link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 276, 1486–1493 [DOI] [PubMed] [Google Scholar]
- 44. Kajimura S., and Saito M. (2014) A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu. Rev. Physiol. 76, 225–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rim J. S., and Kozak L. P. (2002) Regulatory motifs for CREB-binding protein and Nfe2l2 transcription factors in the upstream enhancer of the mitochondrial uncoupling protein 1 gene. J. Biol. Chem. 277, 34589–34600 [DOI] [PubMed] [Google Scholar]
- 46. Robidoux J., Cao W., Quan H., Daniel K. W., Moukdar F., Bai X., Floering L. M., and Collins S. (2005) Selective activation of mitogen-activated protein (MAP) kinase kinase 3 and p38α MAP kinase is essential for cyclic AMP-dependent UCP1 expression in adipocytes. Mol. Cell. Biol. 25, 5466–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu Q., Kazantzis M., Doege H., Ortegon A. M., Tsang B., Falcon A., and Stahl A. (2006) Fatty acid transport protein 1 is required for nonshivering thermogenesis in brown adipose tissue. Diabetes 55, 3229–3237 [DOI] [PubMed] [Google Scholar]
- 48. Bartelt A., Bruns O. T., Reimer R., Hohenberg H., Ittrich H., Peldschus K., Kaul M. G., Tromsdorf U. I., Weller H., Waurisch C., Eychmüller A., Gordts P. L., Rinninger F., Bruegelmann K., Freund B., Nielsen P., Merkel M., and Heeren J. (2011) Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17, 200–205 [DOI] [PubMed] [Google Scholar]
- 49. Hirata M., Suzuki M., Ishii R., Satow R., Uchida T., Kitazumi T., Sasaki T., Kitamura T., Yamaguchi H., Nakamura Y., and Fukami K. (2011) Genetic defect in phospholipase Cδ1 protects mice from obesity by regulating thermogenesis and adipogenesis. Diabetes 60, 1926–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]