

Abstract
Transient receptor potential vanilloid 1 (TRPV1) channel is a tetrameric protein that acts as a sensor for noxious stimuli such as heat and for diverse inflammatory mediators such as oxidative stress to mediate nociception in a subset of sensory neurons. In TRPV1 oxidation sensing, cysteine (Cys) oxidation has been considered as the principle mechanism; however, its biochemical basis remains elusive. Here, we characterize the oxidative status of Cys residues in differential redox environments and propose a model of TRPV1 activation by oxidation. Through employing a combination of non-reducing SDS-PAGE, electrophysiology, and mass spectrometry we have identified the formation of subunit dimers carrying a stable intersubunit disulfide bond between Cys-258 and Cys-742 of human TRPV1 (hTRPV1). C258S and C742S hTRPV1 mutants have a decreased protein half-life, reflecting the role of the intersubunit disulfide bond in supporting channel stability. Interestingly, the C258S hTRPV1 mutant shows an abolished response to oxidants. Mass spectrometric analysis of Cys residues of hTRPV1 treated with hydrogen peroxide shows that Cys-258 is highly sensitive to oxidation. Our results suggest that Cys-258 residues are heterogeneously modified in the hTRPV1 tetrameric complex and comprise Cys-258 with free thiol for oxidation sensing and Cys-258, which is involved in the disulfide bond for assisting subunit dimerization. Thus, the hTRPV1 channel has a heterogeneous subunit composition in terms of both redox status and function.
Keywords: dimerization, pain, post-translational modification (PTM), reactive oxygen species (ROS), redox regulation, transient receptor potential channels (TRP channels), disulfide bond
Introduction
Ion channels play important roles in sensory systems by detecting diverse changes in the environment and inside the body. Drosophila melanogaster transient receptor potential (TRP)2 channel and its homologs are putative six-transmembrane proteins that assemble into tetramers to form non-selective cation channel biosensors (1–3). One of the intriguing features of TRP channels is their ability to detect changes in the redox environment (4). Indeed, numerous TRP channels including TRPV1, TRPV3, TRPV4, TRPC1, TRPC4, TRPC5, and TRPA1 sense alterations in the redox environment (5–7). The oxidative modification of the cysteine (Cys) residues in these channels mediates their activation and plays an essential role in various physiological and pathological conditions (5, 7–12).
TRPV1 is localized in a subset of sensory neurons such as C and Aδ fibers, which are associated with acute and chronic inflammatory pain. TRPV1 is activated by diverse inflammatory mediators, such as heat, acidity, pungent compounds, endogenous lipid signaling molecules, and oxidative stress, to mediate nociception (5, 7, 13–20). During inflammation, the redox environment surrounding the cells changes due to rapid production of reactive oxygen species. Increased levels of reactive oxygen species are implicated in the etiology of inflammatory hyperalgesia (14, 21–24). Importantly, TRPV1 was found to be responsible for long sustained thermal hypersensitivity in inflammatory hyperalgesia induced by increased levels of hydrogen peroxide (H2O2) (25). It is, therefore, possible that oxidative stress triggers hyperalgesia, at least in part, through activation of TRPV1.
There have been multiple hypotheses proposed for the regulation of TRPV1 by oxidation. We previously reported that oxidizing agents activate rat TRPV1 (rTRPV1) by modifying the extracellular Cys residues, where Cys modification acts as a “switch” for channel activation (7). Another group alternatively hypothesized that H2O2 activates chicken TRPV1 (cTRPV1) via oxidizing multiple Cys residues located on the cytoplasmic side; each Cys residue contributing to the activation in a “graded” fashion (26). The same group also claimed that C-terminal dimerization through disulfide bond formation is essential for oxidation-induced cTRPV1 activation (27). However, a unified view of how oxidizing agents activate the channel has not yet been achieved, although there is a consensus that oxidative modifications of multiple Cys residues are involved in this mode of TRPV1 activation.
There are two crucial caveats in the above hypotheses that limit the elucidation of the mechanism underlying TRPV1 activation by oxidation. First, all of the studies concluding the involvement of oxidation of Cys residues in the activation mechanism are heavily based on site-directed Cys mutagenesis experiments, which are often regarded as an indirect approach to assess functional importance of specific residues. Second, most of these mechanistic models are built based on the simple assumption that Cys residues in TRPV1 have predominantly free thiols, both readily accessible and modifiable, at cellular conditions. Although this is a fair assumption based on studies implicating that the intracellular milieu is primarily reduced (28), previous studies have implicated otherwise. For instance, TRPA1 was demonstrated to possess a disulfide bond between Cys residues facing the intracellular side (29). Moreover, TRPV1 has been hypothesized to possess stable structural disulfide bonds because a reducing agent dithiothreitol (DTT) alters the cooperative binding of TRPV1-specific agonist resiniferatoxin to rat dorsal root ganglion (DRG) and regulates rTRPV1 activity when expressed in HEK293 cells (5, 12, 30, 31).
Here we address these two caveats for the previous approaches to understand the role of Cys residues by characterizing the oxidative status of Cys residues in differential redox environments and propose a refined mechanism of TRPV1 activation by oxidation. In particular, we focus on the activation mechanism of human TRPV1 (hTRPV1), which has not been characterized previously.
Experimental Procedures
Reagents
DTT was purchased from Nacalai Tesque (Kyoto, Japan). Capsaicin, cycloheximide, N-ethylmaleimide (NEM), and iodoacetamide were purchased from Sigma. Fura-2-AM ester, 5-nitro-2-pyridyl disulfide (5-nitro-2-PDS), and 2-pyridyl disulfide were purchased from Dojindo (Kumamoto, Japan). 3-Nitrophenyl disulfide and dipropyl disulfide, dimethyl sulfoxide (DMSO), H2O2, and reduced glutathione were purchased from Wako (Osaka, Japan). 4-Chlorophenyl disulfide and 4-aminophenyl disulfide were purchased from Tokyo Chemical (Tokyo, Japan). NEM, iodoacetamide, cycloheximide, and reactive disulfides were prepared as stock solutions in DMSO and were diluted at working concentrations in aqueous solutions containing 0.01% or 0.1% DMSO. DTT and H2O2 were directly dissolved in aqueous solutions at working concentrations.
Cell Culture
Human embryonic kidney (HEK) 293 and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 30 units/ml penicillin, and 30 μg/ml streptomycin at 37 °C under 5% CO2. HaCaT cells were culture in DMEM containing 10% fetal bovine serum, 30 units/ml penicillin, 30 μg/ml streptomycin, and 2 mm l-glutamate at 37 °C under 5% CO2.
cDNA Cloning and Recombinant Plasmid Construction
Plasmids carrying hTRPV1 and rTRPV1 cDNA were prepared as previously described (7). TRPV1 and hTRPV1 Cys mutants were subcloned into pCI-neo vector (Promega Corp., Madison, WI), pEGFP-N (Clontech Laboratories, Mountain View, CA), or pCMV-Tag2 (Stratagene, Palo Alto, CA).
cDNA Expression in Cells
HEK293 cells and HEK293T cells were transfected with recombinant plasmids using SuperFect Transfection Reagent (Qiagen, Valencia, CA) and Lipofectamine 2000 transfection reagent (Invitrogen), respectively, according to the manufacturer's instructions. For intracellular Ca2+ concentration ([Ca2+]i) measurement and electrophysiological measurements, recombinant plasmids were co-transfected with pEGFP-F and pEGFP-N1, respectively, and HEK293 cells with green fluorescence were analyzed. Transfected cells were grown for 30–40 h before [Ca2+]i measurement and electrophysiological measurements and were grown for 48 h before immunoprecipitation and Western blotting (WB).
Western Blots
Cells were lysed in RIPA buffer containing the 50 mm Tris (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 20 mm NEM. Protein samples were fractioned by electrophoresis through 7.5% SDS-polyacrylamide gels (SDS-PAGE) and analyzed by WB with anti-TRPV1 (Santa Cruz, SC-12498), anti-β-actin (Sigma, A2228), and anti-α-tubulin (Sigma, T6074) antibodies. The chemiluminescence intensities of the bands were measured by Multigauge version 3.0 (Fuji film, Tokyo, Japan).
Blue Native Polyacrylamide Gel Electrophoresis
Blue native polyacrylamide gel electrophoresis was performed according to the method of Schägger and co-workers with some modification (32). HEK293 cells transfected with rTRPV1 or hTRPV1 were suspended in sucrose buffer (83 mm sucrose, 6.6 mm imidazole/HCl, 20 mm NEM, pH 7.0) and homogenized with 20–40 strokes using a Dounce homogenizer. The lysates were centrifuge at 15,000 × g, and the pellet was solubilized in 35 μl of solubilization buffer (50 mm NaCl, 50 mm imidazole/HCl, 6 aminohexanoic acid, 1 mm EDTA, 20 mm NEM, pH 7.0). Subsequently, 5 μl of 20% n-dodecyl β-d-maltoside was added and incubated for 10 min at 4 °C. The sample was then centrifuged at 15,000 × g, and supernatant was collected, mixed with 5 μl of 50% glycerol, and 2.5 μl of 5% Coomassie Brilliant Blue G-250. The supernatant was aliquoted into four samples with increasing concentrations of SDS (0, 0.5, 1.0, and 1.5%) and incubated for 20 min at room temperature. The samples were then subjected to non-denaturing electrophoresis initially at 100 V until the samples were within the stacking gel and were continued with a voltage and current limited to 500 V and 15 mA at 4 °C. After separation, the gel was de-stained for 3 min with 100% methanol and electro-transferred to PVDF membrane (Millipore, Bedford, MA). After the transfer, the PVDF membrane was incubated in denaturing solution containing 50 mm Tris-HCl (pH 7.4) and 2% SDS at 50 °C for 30 min. The blots were then analyzed by anti-TRPV1.
Chemical Cross-linking
HEK293 cells transfected with hTRPV1 were detached from the 6-mm dish using trypsin and washed 3 times with phosphate-buffered saline (PBS). The sample was aliquoted into three samples with the concentrations of disuccinimidyl suberate (Sigma) at final concentration of 0, 25, and 75 μm for 20 min at room temperature. The cross-linking reaction was quenched with 1 m Tris-HCl (pH 8.0), and samples were mixed with SDS sample buffer and then analyzed by 3–18% polyacrylamide SDS-PAGE and WB with an antibody to TRPV1.
Cell Surface Labeling Experiments
The cell surface hTRPV1 was measured by biotinylation as previously described with some modifications (7). Cells grown in 60-mm dishes were washed with HEPES-buffered saline (HBS) containing 107 mm NaCl, 6 mm KCl, 1.2 mm MgSO4, 2 mm CaCl2, 11.5 mm glucose, and 20 mm HEPES (pH adjusted to 7.4 with NaOH). After incubation of the cells with 10 mm DTT, dishes were washed with HBS twice and then incubated with sulfo-NHS-SS-biotin (Thermo Fisher Scientific, San Jose, CA) for 30 min at 4 °C. The cells were washed with PBS containing 10 mm glycine 3 times to stop the biotinylation reaction. The cells were then lysed with 300 μl of RIPA buffer with 20 mm NEM and rotated for 2 h at 4 °C. After centrifugation, the supernatant was collected and incubated with NeutrAvidin-Plus beads (Thermo Fisher Scientific) for 2 h at 4 °C. The proteins were washed with RIPA buffer containing 50 mm NEM 3 times and resuspended in SDS sample buffer and subjected to SDS-PAGE and WB.
Protein Stability Assay
HEK293 cells were transfected with hTRPV1 or hTRPV1 Cys mutants and cultured for 24 h. The cycloheximide was added to a final concentration of 50 μg/ml to culture media, and cells were washed twice with PBS and recovered with RIPA buffer after the indicated times. The samples were mixed with reducing sample buffer and analyzed by SDS-PAGE and WB using antibody to TRPV1 and β-actin. Quantification of hTRPV1 expression was achieved by normalization with β-actin.
Isolation of Rat DRG Neurons
DRG neurons were prepared from 4-week-old male Sprague-Dawley rats as described previously (6). The DRG neurons were subjected to WB. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committees of the Graduate School of Engineering, Kyoto University.
[Ca2+]i Measurement
Transfected HEK293 cells were subjected to [Ca2+]i measurement 3–16 h after plating onto poly-l-lysine-coated glass coverslips. The fura-2 fluorescence was measured in HBS. Fluorescence images of the cells were recorded and analyzed with the video image analysis system AQUACOSMOS (Hamamatsu Photonics, Shizuoka, Japan) according to the manufacturer's instructions. Fura-2 measurements were carried out at room temperature in HBS. The 340:380-nm ratio images were obtained on a pixel-by-pixel basis and converted to Ca2+ concentrations by in vivo calibrations using 40 μm ionomycin (33).
Electrophysiology
For electrophysiological measurements, coverslips with cells were placed in dishes containing bath solutions. Currents from cells were recorded at room temperature using patch-clamp techniques of whole-cell mode with EPC-10 (Heka Elektronik, Lambrecht/Pfalz, Germany) patch clamp amplifier as previously described (34). The patch electrode prepared from borosilicate glass capillaries had a resistance of 2–4 megaohms. Current signals were filtered at 2.9 kHz with a 4-pole Bessel filter and digitized at 10 kHz. Patchmaster (Heka Elektronik) software was used for command pulse control, data acquisition, and data analysis. The series resistance was compensated (50–70%) to minimize voltage errors. A holding potential of −80 mV was used for all experiments. The external solution contained 100 mm NaCl, 5 mm KCl, 2 mm BaCl2, 5 mm MgCl2, 25 mm HEPES, and 30 mm glucose (pH 7.3 adjusted with NaOH, and osmolarity adjusted to 320 mosm with d-mannitol). The pipette solution contained 140 mm CsCl, 4 mm MgCl2, 10 mm EGTA, 10 mm HEPES (pH 7.3 adjusted with CsOH and osmolarity adjusted to 300 mosm with d-mannitol). For the DTT treatment, liquid junction potential was adjusted before each experiment.
Liquid Chromatography-Mass Spectrometry
FLAG-hTRPV1or FLAG-hTRPV1 Cys mutants were transfected into HEK293T cells. After 48 h the cells were lysed with buffer containing 50 mm Tris (pH 7.4), 2% SDS, 50 mm NEM, 150 mm NaCl, 1% Nonidet P-40, and 0.5% sodium deoxycholate. The cell lysates were sonicated, centrifuged at 15,000 × g, and 10× diluted in RIPA buffer containing 50 mm NEM. FLAG-hTRPV1 was immunoprecipitated with M2 monoclonal antibody conjugated to protein A-agarose breads (Sigma) and rocked overnight at 4 °C. The complex was washed 8 times with RIPA buffer containing 50 mm NEM for 5 min at room temperature and resuspended into SDS sample buffer and rocked for 30 min. The protein samples were fractionated by 7.5% SDS-PAGE. The bands corresponding to the molecular weight of dimeric or monomeric FLAG-hTRPV1 were excised and reduced with 10 mm DTT, alkylated with 55 mm iodoacetamide, and digested in-gel by treatment with trypsin (12.5 ng/μl) or chymotrypsin (10 ng/μl) or glu-C (50 ng/μl) in 50 mm ammonium bicarbonate for overnight at 37 °C as previously described (35). The samples were analyzed by reverse phase liquid chromatography tandem mass spectrometry using a LTQ-orbitrap-XL hybrid mass spectrometer (Thermo Fisher Scientific) as follows.
A capillary reverse phase HPLC-MS/MS system composed of an Agilent 1100 series gradient pump equipped with Valco C2 valves with 150-μm pots, and LTQ-orbitrap XL hybrid mass spectrometer equipped with an XYZ nano-electrospray ionization source (AMR, Tokyo, Japan) were employed. Samples were automatically injected using PAL system (CTC analytics, Zwingen, Switzerland) into a peptide L-trap column (Chemical Evaluation Research Institute, Tokyo, Japan) attached to an injector valve for desalinating and concentrating peptides. After washing the trap with water containing 0.1% trifluoroacetic acid and 2% acetonitrile, the peptides were separated on a NTCC-360/100-3-123 column (100 μm diameter, 3 μm C18 particles, Nikkyo Technos Co. Ltd., Tokyo, Japan) using a gradient of A (0.1% formic acid) and B (80% acetonitrile, 0.1% formic acid). The elution was carried out at 500 nl/min by running from 5% to 45% over 45 min (as shown in Figs. 5 and 6) or 25 min (as shown in Figs. 8 and 9), 95% for 6 min, 5% for 9 min for selected reaction monitoring (SRM) transitions for the peptide containing Cys-158, -258, -363, and -742. The elution was carried out from 25% to 80% over 20 min, 95% for 6 min, and 5% for 9 min for SRM transition for the peptides containing Cys-127 (as shown in Fig. 8). SRM was carried out with collision-induced dissociation (35% normal collision energy), 1 microscan, 100-ms ion time on LTQ linear ion trap. Peak area was calculated using Xcaliber 3.0 software version 2.2.0.48 (Thermo Fisher Scientific). For the shotgun proteomics experiment, the LTQ orbitrap XL was operated in a data-dependent mode, selecting three precursors for fragmentation by collision-induced dissociation. The survey scan was performed in the orbitrap at 30,000 resolutions from 300 to 2000 m/z. Precursors were isolated with a width of 2 m/z. Single and assigned charge states were ignored for precursor selection. Proteome discoverer 1.3 with Sequest (36) was used for protein identification against protein database containing the hTRPV1 sequence (accession number Q8NER1). Precursor mass tolerance was set to 10 ppm and 0.8 Da for orbitrap and linear ion trap, respectively. Oxidized methionine (M), carbamidomethylation (C), and NEM (C) were set as dynamic modification. Trypsin, chymotrypsin, or trypsin/glu-C were chosen as the enzymes for digestion.
FIGURE 5.

Quantitative mass spectrometric analyses of disulfide bond pairing. A, disulfide bond pairings that were tested. B, schematic description of the protocol employed for elucidation of disulfide pairing. C, non-reducing SDS-PAGE and WB analysis of WT and K156Q hTRPV1-expressing HEK293 cells. D, maximal Ca2+ responses (Δ[Ca2+]i) to 10 μm 3-nitrophenyl disulfide stimulation (n = 19–33) were normalized to responses to 10 μm capsaicin (n = 28–42) for K156Q hTRPV1 mutant expressed in HEK293 cells. Differences not statistically significant are labeled as ns. E and F, MS/MS spectra of CAM (E)- and NEM (F)-labeled GRPGFYFGELPLSLAACTNQLGIVK (C is Cys-258) peptide, which were generated from tryptic digestion of FLAG-hTRPV1 purified from HEK293T cells. The asterisks (*) denote peaks used in multiple reaction monitoring transitions. G and H, representative multiple reaction monitoring analysis of CAM (black)- and NEM (gray)-labeled Cys-258-containing peptides.
FIGURE 6.

An intersubunit disulfide bond between Cys-258 and Cys-742 in hTRPV1 channel. A and B, representative SRM experiments of peptides containing Cys-258 and Cys-363, which were generated from tryptic digestion of purified WT and C742S FLAG-hTRPV1. C, SRM experiments for peptides containing Cys-158, which were generated from tryptic digestion of purified FLAG-hTRPV1 mutants, K156Q and K156Q/C742S FLAG-hTRPV1. The transitions used to quantify CAM (black)- and NEM (gray)-labeled peptides containing Cys-258, Cys-363, and Cys-158 are shown below. The mode of modification is followed by transitions for the peptides containing the respective Cys residues: CAM (1354.72(z = +2) > 1124.55(b10+)) and NEM (1388.73(z = +2) > 1124.55(b10+)) for Cys-258, CAM (530.74(z = +2) > 561.25(y4+)) and NEM (564.75(z = +2) > 629.27(y4+)) for Cys-363, and CAM (749.36(z = +3) > 1146.58(y10+)) and NEM (772.04(z = +3) > 1214.61(y10+)) for Cys-158. The K156Q mutation is labeled as lowercase q. The extent of Cys oxidation is represented by %CAM labeled peptide, which is the peak area of CAM-labeled peptide/(peak area of CAM-labeled peptide + peak area of NEM-labeled peptide) (left). D, SRM experiments shown in A–C are summarized as % change in %CAM labeled peptide between WT and C742S for Cys-258 and Cys-363 (between K156Q and K156Q/C742S for Cys-158). Data points are the mean ± S.D. of three analytical replicates.
FIGURE 8.

Cys residues at the interface are accessible by oxidants. A–E, Cys oxidation of WT or K156Q FLAG-hTRPV1 induced by 500 μm 5-nitro-2-PDS treatment for 30 min at 37 °C. Oxidized and reduced Cys of WT or K156Q FLAG-hTRPV1 (for Cys-158 only) treated with or without 500 μm 5-nitro-2-PDS were masked with CAM (black) and NEM (gray), respectively. The samples were subjected to SRM analysis after purification from HEK293T cells and trypsinization. %CAM-labeled peptides were determined for Cys-127, Cys-158, Cys-258, Cys-363, and Cys-742 using the following SRM transitions: Cys-127 CAM (894.13(z = +3) > 1017.52(y172+)) and NEM (916.80(z = +3) > 1051.53(y172+)) for A; Cys-158 CAM (749.36(z = +3) > 1146.58(y10+)) and NEM (772.04(z = +3) > 1214.61(y10+)) for B; Cys-258 CAM (1354.72(z = +2) > 1124.55(b10+)) and NEM (1388.73(z = +2) > 1124.55(b10+)) for C; Cys-363 CAM (530.74(z = +2) > 561.25(y4+)) and NEM (564.75(z = +2) > 629.27(y4+)) for D; Cys-742 CAM (797.05(z = +3) > 968.43(y152+)) and NEM (819.73(z = +3) > 1002.44(y152+)) for E. F, summary of %CAM-labeled peptides for the respective Cys residues. Data points are the mean ± S.D. of three analytical replicates.
FIGURE 9.

Quantification of hTRPV1 Cys oxidation induced by H2O2. A, maximal Ca2+ responses (Δ[Ca2+]i) to 1 mm H2O2 stimulation at 37 °C (n = 22–100) were normalized to responses to 100 nm capsaicin (n = 12–46) for disulfide-less hTRPV1 mutants expressed in HEK293 cells. *, p < 0.01 compared with WT. B, oxidation sensitivity of disulfide-less hTRPV1 mutants. Plots of Δ[Ca2+]i evoked by 10 μm concentrations of the congeneric series of reactive disulfides (see Fig. 1A) in HEK293 cells expressing hTRPV1 or hTRPV1 Cys mutants (n = 5–37). p < 0.05 compared with vector for hTRPV1 WT (*) and C363S mutant (#). Data points are the means ± S.E. C–E, oxidized and reduced Cys of FLAG-hTRPV1 (for Cys-258 and Cys-363) or FLAG-hTRPV1 K156Q (for Cys-158) treated with various doses of H2O2 for 30 min at 37 °C were masked with CAM and NEM, respectively. The samples were subjected to SRM analysis after purification from HEK293T cells and trypsinization. The half-max was derived by fitting the plots to the Hill equation (Cys-258half-max = 0.7 mm, Cys-363half-max = 2.2 mm): Hill equation, ƒ(x) = A0 + (Amax − A0)/(1 + (EC50/x)n, where A0 is then basal %CAM-labeled peptide, Amax is maximum %CAM-labeled peptide, x is the H2O2 concentration, and n is the Cyshalf-max. Data points are the means ± S.D. of three analytical replicates. F, the half-maximal saturation of H2O2 for Cys-258 and Cys-363 is indicated as dotted lines on a dose-response curve of H2O2 analyzed at 37 °C stimulated for 8 min (n = 41–115). Data points are the mean ± S.E. *, p < 0.01 compared with vector.
Structural Modeling
Three-dimensional structure of rTRPV1(PDB code 3J5P) was used to estimate the distance between the Cys residues. Structure was visualized using CCP4 mg software version 2.9.0 (37).
Statistical Analysis
All data are expressed as the means ± S.E. or ± S.D. We accumulated the data for each condition from at least three independent experiments. The statistical analyses were performed using Student's t test. A value of p < 0.05 was considered significant.
Results
Characterization of Redox Modification in TRPV1
Activation of cTRPV1 under oxidative stress via dimerization has been described previously (27). To test whether oxidizing agents activate mammalian TRPV1 via dimerization similar to cTRPV1, the protein multimerization of hTRPV1 and rTRPV1 expressed in HEK293 cells treated with a congeneric series of reactive disulfides with differential redox potentials, was assessed by non-reducing SDS-PAGE (Fig. 1A) (38). Once the cells were lysed, sample preparation was carried out in the presence of NEM, an irreversible Cys alkylating agent that prevents post-lysis oxidation and thiol-disulfide shuffling (39). hTRPV1 and rTRPV1 showed significant differences in their mobility even in the absence of exogenous oxidizing agents. Although the rTRPV1 band was observed at ∼100 kDa as predicted from its molecular mass (95 kDa), hTRPV1 was predominantly observed at ∼245 kDa (Fig. 1B). Recombinant expression of hTRPV1 and rTRPV1 elicited robust [Ca2+]i increases in response to the congeneric series of reactive disulfides in HEK293 cells, which were not observed in the vector-transected cells (Fig. 1C). Notably, the reactive disulfides that activated the channels did not alter the band patterns of human or rat TRPV1 on the non-reducing SDS-PAGE (Fig. 1, B and C). These data suggest that oxidative modification elicited by reactive disulfides do not affect the multimeric patterns of mammalian TRPV1 channels.
FIGURE 1.

Oxidative modification and its impacts on human and rat TRPV1. A, congeneric series of reactive disulfides with their redox potentials (mV). Compounds with less negative redox potentials have stronger electrophilicity. B, non-reducing SDS-PAGE and WB analysis of hTRPV1- or rTRPV1-transfected HEK293 cells treated with congeneric series of reactive disulfides (10 μm) for 10 min at room temperature. WB for β-actin are also shown (bottom). C, averaged time courses of [Ca2+]i rises evoked by reactive disulfides with indicated redox potentials for 8 min in HEK293 cells expressing hTRPV1, rTRPV1, or vector (left). Plots of maximal rise of Ca2+ (Δ[Ca2+]i) induced by 10 μm concentrations of reactive disulfides for 8 min in HEK293 cells expressing hTRPV1 or rTRPV1 (n = 10–52) (right). Data points are the mean ± S.E. *, p < 0.01 compared with rTRPV1. D, Non-reducing SDS-PAGE and WB analysis of hTRPV1-transfected HEK293 cells treated with various concentrations of DTT for 10 min at 37 °C. E, hTRPV1-transfected HEK293 cells were treated with 10 mm DTT for 10 min, washed with HBS twice, and recovered as lysates after the indicated time points to be analyzed with non-reducing SDS-PAGE and WB. F, identification of the FLAG-hTRPV1 245-kDa band isolated from HEK293T cells by mass spectrometric analysis. The excised 245-kDa band was subjected to enzymatic digestion (combination of trypsin or chymotrypsin or trypsin/glu-C) and analyzed with liquid chromatography coupled with tandem mass spectrometry. The amino acids in black were identified.
Incubation of hTRPV1-expressing HEK293 cells with DTT shifted the 245-kDa band to a monomeric position in a dose-dependent manner (Fig. 1D), suggesting that the formation of disulfide bonds may be responsible for the band shift observed under the non-reducing conditions. To examine the reversibility of the 245-kDa band in a cellular environment, we incubated the hTRPV1-expressing HEK293 cells with DTT for 10 min followed by washes, and re-formation of the band was monitored. The 245-kDa band reappeared after 5 min in a time-dependent manner, demonstrating that the monomeric hTRPV1 has high propensity to form a 245-kDa complex when expressed in cells (Fig. 1E). To confirm the identity of the 245-kDa band, a purified FLAG-hTRPV1 245-kDa band was analyzed by mass spectrometry after SDS-PAGE and enzymatic digestions. We obtained an 81% amino acid sequence coverage of hTRPV1, including the C and N terminus, revealing that the dimeric band indeed consists of full-length hTRPV1 (Fig. 1F). Thus, hTRPV1 appears to possess a stable intersubunit disulfide bond between two subunit proteins that is unaffected by oxidative conditions.
To further test whether hTRPV1 forms a complex with other proteins or hTRPV1 multimer, we co-expressed hTRPV1-GFP and hTRPV1 and assessed whether an intermediate band that consists of hTRPV1-GFP+hTRPV1 could be observed under non-reducing conditions. We observed three distinct bands that corresponded to the molecular weights of hTRPV1+hTRPV1, hTRPV1-GFP+hTRPV1-GFP, and hTRPV1+hTRPV1-GFP complexes, suggesting that hTRPV1 forms an intersubunit disulfide bond (Fig. 2A). To confirm the stoichiometry of disulfide-linked hTRPV1, the band position of the disulfide-linked hTRPV1 complex was compared with disuccinimidyl suberate cross-linked hTRPV1 bands. The position of the disulfide-linked hTRPV1 corresponded to two disuccinimidyl suberate cross-linked hTRPV1, revealing the disulfide linkage between two hTRPV1 subunits (Fig. 2B). To test whether disulfide-linked hTRPV1 subunits are incorporated into the tetramer, hTRPV1 was subjected to non-reducing blue native PAGE. rTRPV1 was also analyzed in parallel as a control for disulfide-less TRPV1. hTRPV1 and rTRPV1 showed a marked difference in the mobility; rTRPV1 showed a tetramer to monomer transition as a function of increasing concentrations of SDS, whereas hTRPV1 showed an absence of monomeric band but increased intensity in the dimeric band (Fig. 2C). These results suggest that hTRPV1 has a unique intersubunit disulfide bond.
FIGURE 2.

The hTRPV1 channel has an intersubunit disulfide bond between two subunits. A, hTRPV1 and hTRPV1-GFP were transfected individually and co-transfected to HEK293 cells. The lysates were analyzed with reducing and non-reducing SDS-PAGE and WB. B, cell lysates from hTRPV1-transfected HEK293 cells with and without treatment of indicated concentrations of disuccinimidyl suberate (DSS) were subjected to reducing SDS-PAGE and WB. C, cell lysates of hTRPV1- or rTRPV1-expressing HEK293 cells were treated with or without various concentration of SDS and subjected to non-reducing blue native PAGE. D and E, reducing and non-reducing SDS-PAGE and WB analysis of native hTRPV1 and rTRPV1 in HaCaT cells (D) and rat DRG neurons (E), respectively.
To examine whether the intersubunit disulfide bond of hTRPV1 could be observed in an endogenous system, HaCaT cells, human keratinocytes known to express TRPV1 (40–42) were subjected to non-reducing SDS-PAGE. TRPV1 expressed in rat DRG was also examined as a negative control. Although hTRPV1 intersubunit formation was less pronounced than in the recombinant system, a DTT-sensitive hTRPV1 dimeric band was clearly observed in HaCaT cells (Fig. 2D), whereas DTT-sensitive bands were not observed with rTRPV1 expressed in rat DRG (Fig. 2E).
hTRPV1 Has an Intersubunit Disulfide Bond between N- and C-terminal Cys Residues at the Subunit Interface
An alignment of rat and human TRPV1 sequences revealed two differences in Cys residues (Fig. 3A). rTRPV1 has two extra Cys residues compared with hTRPV1, whereas the remaining Cys residues are conserved among the two groups. To test whether substitution of these rTRPV1 Cys residues into hTRPV1 alters the disulfide bond status of the hTRPV1, S31C and W616C hTRPV1 mutants were subjected to non-reducing SDS-PAGE. Neither S31C nor W616C substitution disrupted the intersubunit disulfide bond formation of hTRPV1 (Fig. 3A). However, to our surprise we found that rTRPV1-mimicking double amino acid substitutions, S31C and W616C, disrupted the intersubunit disulfide bond of hTRPV1 (Fig. 3B), suggesting that the difference in the disulfide bond pattern between rat and human species is affected by the difference in Cys homology. This is an intriguing case where presence, not absence, of additional Cys residues disrupts the formation of the disulfide bond.
FIGURE 3.

Formation of the intersubunit disulfide bond between C and N termini in the hTRPV1 channel. A, alignments of partial amino acid sequences of rat with human TRPV1 where Cys residues are not conserved (GenBankTM accession number NM_018727 and NM_031982, respectively). Non-reducing SDS-PAGE and WB analysis of HEK293 cells expressing WT, S31C, W616C, or S31C + W616C hTRPV1 is shown. B, non-reducing SDS-PAGE and WB analysis of HEK293 cells expressing WT or double S31C + W616C hTRPV1 mutant. C, non-reducing SDS-PAGE and WB analysis of hTRPV1 mutants with single Cys substitution transiently expressed in HEK293T cells. The amounts of transfected vectors were adjusted to elicit comparative expression level. The extent of intersubunit disulfide bond formation was quantified by the ratio between the dimer and monomer band (top) (n = 5 independent experiments). Quantitative densitometric analysis of bands shown in the top panel (bottom). Data points are the mean ± S.E. *, p < 0.05 and **, p < 0.01, compared with WT. D, hTRPV1 Cys mutants with significant reductions in dimer/monomer ratio are indicated as black circles on a schematic topology of hTRPV1 (left). Shown is a ribbon structure model of rTRPV1 adapted from Liao et al. (3) showing views from side (right). Cys residues are marked with green, and the residues implicated in intersubunit disulfide bond are marked black and indicated with arrows. Unresolved C-terminal electron density that is predicted to contain Cys-742 is also highlighted in black.
To identify Cys residues that form intersubunit disulfide bonds, 16 individual Cys residues in hTRPV1 were mutated to serines, and the dimer/monomer ratios were tested using non-reducing SDS-PAGE. Mutation of Cys-158, -258, -363, and -742 significantly reduced the dimer/monomer ratios (Fig. 3C). The former three Cys residues are within or vicinal to an ankyrin repeat domain (ARD), and Cys-742 was previously predicted to interface the neighboring subunit (3, 43). ARD and the C terminus were hypothesized to mediate subunit interactions based on the high resolution structure (3), where ARD interfaces the ARD-S1 (S1: the first transmembrane region) linker region and the C-terminal β-strands of the neighboring subunit (Fig. 3D).
To delineate the disulfide pairing of the hTRPV1 intersubunit disulfide bond, electrophysiological analysis was conducted to categorize the functional phenotypes of hTRPV1 mutants with disrupted intersubunit disulfide bonds. First, a surface-labeling assay confirmed the intersubunit disulfide bond formation of channel protein complexes at the plasma membrane (Fig. 4A). The presence of DTT, which efficiently disrupts dimerization of hTRPV1 (Fig. 1D), in the external solution caused an increase in the capsaicin EC50 of hTRPV1, suggesting that the disruption of disulfide bond formation reduced the sensitivity to capsaicin (Fig. 4, B and C). The C258S and C742S mutations of hTRPV1 caused an increase in the EC50 to capsaicin, whereas C158S and C363S mutations caused no such change (Fig. 4D). Importantly, the C258S and C742S mutations caused a similar change in the EC50 as the DTT treatment, suggesting a Cys-258–Cys-742 disulfide bond pairing between hTRPV1 proteins.
FIGURE 4.
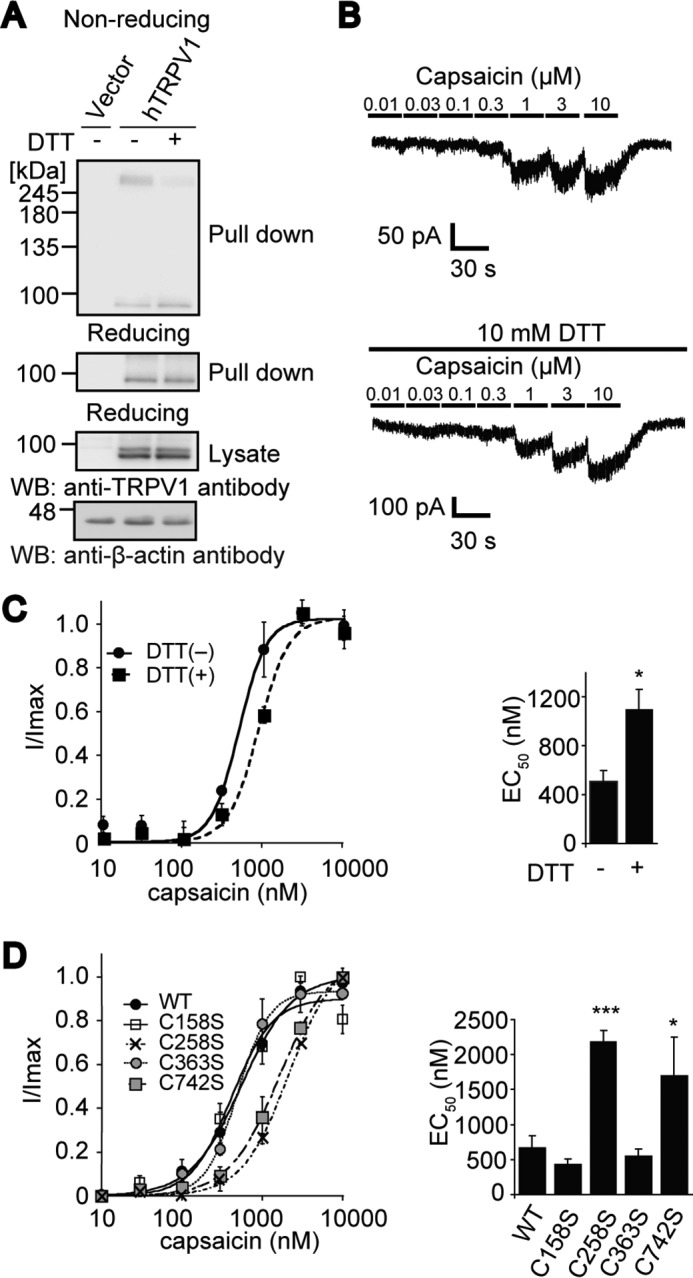
Electrophysiology analysis of hTRPV1 with the disrupted intersubunit disulfide bond. A, cell surface expression of hTRPV1-expressing HEK293 cells treated with or without 10 mm DTT for 10 min. Cell lysates prepared after exposure to sulfo-NHS-SS-biotin were incubated with NeutrAvidin beads and subjected to non-reducing SDS-PAGE and WB. B, whole-cell currents from hTRPV1-expressing HEK293 cells in response to various concentration of capsaicin in the presence or absence of 10 mm DTT. C, relative amplitudes of whole-cell currents of experiments shown in B (n = 3–4). EC50 are summarized on the right. *, p < 0.05 compared with DTT (−). D, relative amplitudes of whole-cell currents in hTRPV1 mutants-transfected HEK293 cells in response to various doses of capsaicin (n = 3–7). EC50 are summarized on the right. *, p < 0.05 and ***, p < 0.001, compared with WT. The plots were fitted to the Hill equation ƒ(x) = A0 + (Amax − A0)/(1 + (EC50/x)n, where A0 is the basal response, Amax is the maximum response, x is the capsaicin concentration, and n is the Hill coefficient. A holding potential of −80 mV was used, and the current amplitudes were normalized with the maximum current amplitudes obtained by capsaicin concentration that elicited the largest response (I/I max). Data points are the mean ± S.E.
Mass Spectrometric Analysis of the hTRPV1 Intersubunit Disulfide Bond between Cys-258 and Cys-742
To further delineate the disulfide bond pairing at the N and C termini interface, quantitative mass spectrometric analysis was carried out. We reasoned that if a disulfide bond is formed between Cys-258 and Cys-742, mutation of C742S should reduce the oxidative status of Cys-258 because the oxidizing partner Cys-742 is not present in proximity. In contrast, we supposed that the oxidative status of the other Cys residues that do not form a disulfide pair should remain relatively unaffected (Fig. 5A). The quantitation of Cys oxidation for Cys-158, -258, and -363 were carried out by masking the free (reduced) thiol with NEM and oxidized thiol with carbamidomethyl group (CAM) and determining the peak area for the peptides with the respective chemical modifications using SRM (Fig. 5B). Cys-158 is spanned by proximal lysine (Lys) residues, which results in short peptides upon tryptic cleavage, hindering identification and quantitation by SRM. The Lys residue at position 156 was, therefore, mutated to glutamine (K156Q) to optimize the protein sequence for quantitation by SRM. The intersubunit disulfide bond of hTRPV1 and the response against reactive disulfide were largely unchanged by the K156Q mutation (Fig. 5, C and D).
After non-reducing SDS-PAGE, the dimeric hTRPV1 band and monomeric hTRPV1 C742S band (or the respective K156Q mutants) were excised and subjected to trypsin digestion and quantification of the CAM- and NEM-labeled peptides with SRM. The transitions for SRM were selected based on MS/MS spectra for both CAM- and NEM-labeled peptides (Fig. 5, E and F), and the largest peak area was quantified by multiple reaction monitoring (Fig. 5, G and H). The mutation of C742S substantially reduced the oxidation status of Cys-258, as demonstrated by a decreased percentage of CAM-labeled Cys-258-containing peptides (Fig. 6A), whereas % CAM-labeled peptide for the Cys residues Cys-363 and Cys-158 (Fig. 6, B and C) were relatively unaffected (Fig. 6D). We selected to test the effect of C742S mutation on the oxidative status of Cys-258 instead of the effect of C258S mutation on the oxidative status of Cys-742 because the short tryptic peptide containing Cys-742 was not competent for optimization for detection by mass spectrometry due to the disruption of hTRPV1 dimer after mutation of proximal arginine residue. These results strongly suggest the intersubunit disulfide bond formation occurs between Cys-258 and Cys-742 of hTRPV1.
Disruption of the Intersubunit Disulfide Bond Reduces Channel Stability
The primary function of a disulfide bond is to provide structural support, increasing the stability of a protein. The stability of the disulfide-less C258S and C742S hTRPV1 mutants were examined by culturing the transfected HEK293T cells for various times in the presence of cycloheximide, an inhibitor of protein synthesis (44, 45), and monitoring the expression level of the proteins. We found that the disulfide-less C258S and C742S hTRPV1 had shorter protein lifespans than WT hTRPV1; protein half-life (t½) values for C258S, C742S, and WT were 67, 19, and 122 min, respectively (Fig. 7, A and B). This result suggests that the intersubunit disulfide bond of hTRPV1 enhance protein stability.
FIGURE 7.
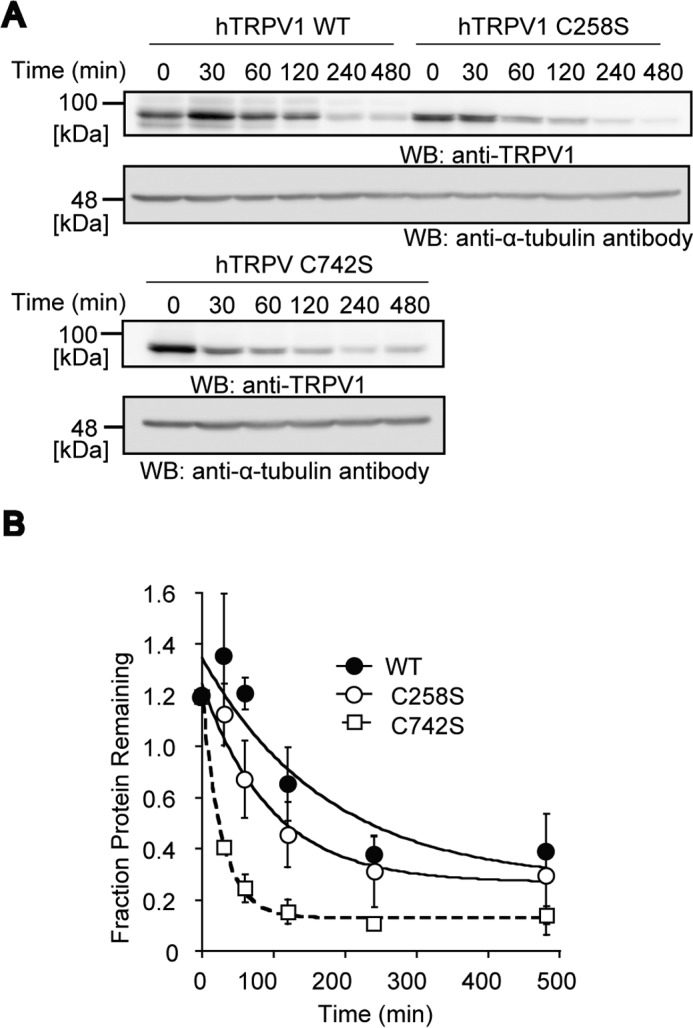
Decreased protein stability of C258S and C742S hTRPV1 mutants. A, HEK293T cells expressing WT, C258S, and C742S hTRPV1 were cultured in the presence of cycloheximide for the indicated time periods. The cell lysates were subjected to reducing SDS-PAGE and WB. The levels of hTRPV1 were quantified through normalization by those of α-tubulin. B, quantitative densitometric analysis of the protein levels of WT, C258S, and C742S normalized to those of α-tubulin (n = 3). Data points are the mean ± S.E. The plots were fitted to the exponential decay equation ƒ(t) = C + A0e−λt, where C is the constant, A0 is the initial amount, x is the time after cycloheximide administration, and λ is the decay constant. The half-life (t½) was calculated by ln(2)/λ.
Cys Residues Involved in Intersubunit Disulfide Bond Formation Are Also Reactive to Oxidation
Having elucidated the presence of the structural disulfide bond at the subunit interface, we next asked whether this same region plays a role in sensing oxidation as the ARD and C terminus of TRPV1 is known to be a target of diverse modulators such as ATP and calmodulin (46). First, the accessibility of Cys residues localized at the subunit interface was assessed. FLAG-hTRPV1-expressing HEK293 cells were treated with 500 μm 5-nitro-2-PDS and reduced, and modified Cys residues were masked with NEM and CAM, respectively, followed by quantification by SRM. We found that Cys-127, -158, -258, -363, and -742 are oxidized upon treatment with 5-nitro-2-PDS, suggesting that Cys residues localized at the cytoplasmic N- and C-terminal interface are at least in part accessible by reactive disulfide oxidizing agents (Fig. 8, A–F). Considering that oxidizing agents do not affect the intersubunit disulfide bond formation of hTRPV1 (Fig. 1B), these data also suggest that Cys-258 and Cys-742 of the hTRPV1 dimer possess free thiols on one subunit and intersubunit disulfide bond on the adjacent subunit.
To examine whether Cys residues involved in intersubunit disulfide bond formation are implicated in sensing oxidation, the activity of C158S, C258S, C363S, and C742S hTRPV1 against H2O2 was tested. C258S and C363S hTRPV1 mutants showed a significant reduction in Ca2+ rises (Δ[Ca2+]i) evoked by H2O2 when normalized to capsaicin responses, suggesting that Cys-258 and Cys-363 play a role in sensing oxidation (Fig. 9A). We next quantified the oxidation sensitivity of these mutants by systematically comparing the responses of WT, C258S, and C363S hTRPV1 to a congeneric series of reactive disulfides with differential redox potentials (Fig. 1A). Consistent with the response to H2O2, C258S and C363S mutants showed significant impairment in oxidation sensitivity, reflected by their activating redox potentials (redox potentials where the channel begins to respond); ∼−2000 mV for WT, ∼−1300 mV for C363S, and no response for C258S (Fig. 9B).
To explore the hTRPV1 structure-function relationship, the oxidation sensitivity of Cys-258 and its neighboring Cys residues (Cys-158 and Cys-363) were assessed by determining the half-maximum saturation (half-max) of H2O2 for each Cys residues by SRM analysis. The Cys-258 and Cys-363 showed a dose-dependent relationship between H2O2 concentrations and %CAM-labeled peptide, revealing they are indeed oxidized upon treatment with H2O2 (Fig. 9, C and D). In contrast, Cys-158 was not susceptible to oxidation at the same dosage of H2O2 as Cys-258 or Cys-363 (Fig. 9E). Notably, Cys-258 showed the highest sensitivity to oxidation among the neighboring Cys residues, suggesting Cys-258 is the prime target of oxidation. These data are consistent with functional analysis where C258S and C363S hTRPV1 showed impaired responses against oxidation, whereas C158S showed intact responses (Fig. 9, A and B). Moreover, these data may implicate one of the Cys-258 in the hTRPV1 disulfide-linked dimer is involved in the disulfide bond formation, whereas the other Cys-258 retains a free thiol that is modified by oxidizing agents. Furthermore, Cys-258half-max and Cys-363half-max lie in the range of hTRPV1 activation by H2O2 and are oxidized differentially at varying H2O2 concentrations, confirming the hypothesis that the activation of hTRPV1 by oxidants is “graded,” as previously proposed (Fig. 9F) (26). These results suggest that Cys-258 may not only contribute to the stability of the ion channel by forming a structural disulfide bond but also acts as an allosteric switch for channel activation (Fig. 10).
FIGURE 10.
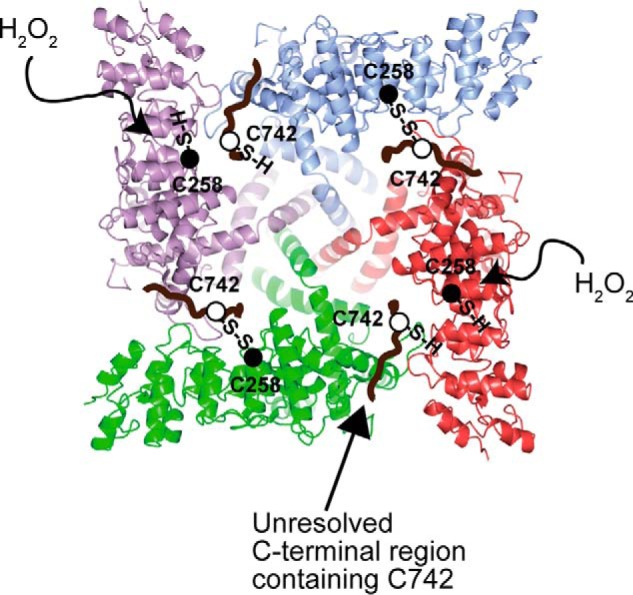
Schematic representation of hTRPV1 activation by oxidants. Schematic of hTRPV1 tetrameric complex viewed from the cytoplasmic side (assuming identical structure with the rTRPV1 structure reported by Liao et al. (3)). Four subunits of hTRPV1 are shown in different colors (green, red, purple, and blue) and the respective Cys-258 (black circle) and the unresolved C-terminal electron density (brown) predicted to contain Cys-742 (white circle) are indicated.
Discussion
Our combined approach of using non-reducing SDS-PAGE, electrophysiology, and mass spectrometry consistently suggest disulfide bond formation between Cys-258 and Cys-742 from two adjacent subunits. Non-reducing SDS-PAGE demonstrated that C158S, C258S, C363S, and C742S hTRPV1 mutants have significantly impaired intersubunit disulfide bond formation (Fig. 3C). We aimed to determine the details of Cys-based mechanisms of hTRPV1 subunits dimerization and oxidation sensing through Cys-258 and Cys-742. C258S and C742S mutations in hTRPV1 caused a similar increase in the EC50 of capsaicin as DTT treatment, which efficiently disrupts disulfide bonds (Fig. 4, C and D). Quantitative mass spectrometric analysis revealed that insertion of the C742S mutation into hTRPV1 substantially reduced the oxidative status of Cys-258, corroborating that idea that Cys-258–Cys-742 are the disulfide bond-forming pair (Fig. 6D). In addition, a high resolution EM structure of rTRPV1 also supports disulfide bond formation between Cys-258 and Cys-742. Cys-258 and Cys-742 in hTRPV1 subunits are likely to be proximal to each other, considering that C-terminal β-strands (predicted to be residues within 719–764) are interfacing finger three and four of the ARD where Cys-257 (rTRPV1 counterpart of hTRPV1 Cys-258) is positioned in rTRPV1 (3). It is highly unlikely that a disulfide bond could form between any combinations of Cys-157 (rTRPV1 counterpart of hTRPV1 Cys-158), Cys-257, and Cys-362 (rTRPV1 counterpart of hTRPV1 Cys-363) from two adjacent subunits, where the proximity of 2.05 Å is required (Fig. 3D) and the shortest distance, estimated from the near atomic structure of rTRPV1, among them is ∼31.3 Å (Cys-157–Cys-362). However, we do not rule out the possibility of other intersubunit disulfide bonds formed by either Cys-158 or Cys-363. In fact, the mutation of any one of Cys-158, -258, -363, and -742 do not eliminate the intersubunit disulfide bond of hTRPV1, although a significant reduction in the dimer/monomer ratios was observed (Fig. 3C). Moreover, our stability data (Fig. 7B) indicates that C258S hTRPV1 mutant display reduced stability than WT hTRPV1 but considerably higher stability than C742S hTRPV1 mutant, suggesting that an absence of Cys-258 could be compensated through Cys-742 interacting with other Cys residues, whereas Cys-742 is necessary for stable channel expression. Further work is necessary to fully appreciate the complexity of oxidative modification in hTRPV1 at the subunit interface.
Interestingly, the C258S hTRPV1 mutant showed no response to oxidants (Fig. 9, A and B). Moreover, mass spectrometric analysis of Cys residues of hTRPV1 treated with H2O2 showed that Cys-258 is highly sensitive to oxidation, demonstrating that Cys-258 confers oxidation sensitivity to hTRPV1 (Fig. 9C). These results showed that Cys-258 residues are heterogeneously modified in the hTRPV1 tetrameric complex and comprise Cys-258 with a free thiol for oxidation sensing and Cys-258 involved in the disulfide bond formation for assisting in subunit dimerization. Thus, the homotetrameric hTRPV1 channel is essentially a heterotetrameric channel in terms of both redox status and function (Fig. 10).
The TRPV1 tetrameric complex is thought to have a 4-fold symmetry centering the channel pore under the reducing conditions (3). If this is true, all four Cys-258 on each TRPV1 subunit should have an equal chance of being oxidized and thus should result in disulfide-linked tetramer. However, our data show that TRPV1 forms disulfide-linked dimer. To obtain a tetrameric complex consisting of two disulfide-linked TRPV1 dimers, the intersubunit disulfide bonds must be arranged in the opposing position (Fig. 10). Considering that TRPV1 do not form disulfide-linked tetramer even under the extreme oxidizing environment (such as treatment with oxidizing agent (Fig. 1B)), it is possible that 4-fold symmetry of TRPV1 is not present in human species and that TRPV1 possesses heterogeneity in subunit orientation. Although our current data are limited in drawing a definitive conclusion concerning the three-dimensional structural orientation underlying heterogeneous oxidation, we speculate that hTRPV1 is susceptible to heterogeneous oxidation due to heterogeneity in orientation of Cys-258 on each subunit. Ion channels are known to possess disulfide bonds to provide a structural scaffold or redox sensitivity. These include ion channels such as NMDA receptor, CLIC1, Kir2.2, cardiac ryanodine receptor, TWIK-1 channel, and TRPC5 (47–53). However, to our knowledge this is the first observation in which an ion channel uses the redox heterogeneity of a single specific Cys residue (Cys-258) to provide both oxidation sensitivity and protein stability. Because of the unexplored field of Cys biochemistry in membrane proteins, we believe that this type of functional bifurcation is likely to be found in other channels in the near future.
The intersubunit disulfide bond of hTRPV1 was highly stable, demonstrated by rapid reformation after reduction by DTT (Fig. 1E). This may explain how the cytoplasmic intersubunit disulfide bond may have survived the reductive intracellular milieu. Consistent with our observation, Tao et al. (54) report that the preparation of Kir2.2 crystal structure in the presence of 20 mm DTT and 3 mm tris-(2-carboxyethyl) phosphine (TCEP) still yielded Kir2.2 with a disulfide bond, suggesting that disulfide bonds can be a stable structural feature of ion channels.
In our study it is also important to note that the oxidative status of Cys residues was characterized based on the reactivity of free Cys residues with NEM and the reversibility of oxidized Cys residues with DTT. As a consequence, oxidative Cys modifications including sulfenic acid, disulfides, glutathionylation, and nitrosylation were not distinguished (55). In addition, because the irreversible oxidative modification such as sulfonic or sulfinic acid cannot be detected with the methodology employed, these modifications were also not accounted for in our analysis.
In the current study we focused primarily on the elucidation of an oxidant-induced activation mechanism at the N- and C-terminal interface of hTRPV1. However, as described previously by others for cTRPV1, other domains in the TPRV1 channels are likely to be involved in sensing oxidation, and the complete picture of oxidation sensing requires characterization of Cys residues in these domains (26). Although this study describes Cys modification as the primary mechanism of activation by oxidants, it is highly likely that other residues, such as the Lys residue and methionine residue, are also targeted by oxidants. In fact, a Lys residues in TRPA1 is a known target of electrophiles, and methionine residues in TRPM2 is implicated in sensing H2O2 (56, 57).
We found that oxidative post-translational modifications between hTRPV1 and rTRPV1 are significantly different, as rTRPV1 lacks the propensity to form an intersubunit disulfide bond (Fig. 1B). This suggests that the structure and function between hTRPV1 and rTRPV1 is not completely conserved for the oxidizing stimuli. Thus, one may need to be cautious when applying a conclusion to both rTRPV1 and hTRPV1. This is also true for cTRPV1, where oxidation-induced activation was correlated with dimerization (27). Interestingly, hTRPV1 endogenously expressed in HaCaT cells is subjected to multiple redox modifications (as implicated by multiple DTT-sensitive bands), in stark contrast with rTRPV1 expressed in DRG neurons where only the monomeric protein was observed under the non-reducing SDS-PAGE (Fig. 2, D and E). Functional characterization and clarification of the significance of unique redox modifications in hTRPV1 are largely unexplored and appear to be a fertile field for further investigation.
To our surprise we found that rTRPV1-mimicking double amino acid substitutions, S31C and W616C, disrupted the intersubunit disulfide bond of hTRPV1 (Fig. 3B), suggesting that the difference in the disulfide bond pattern between rat and human species is affected by the difference in Cys homology. This is an intriguing case where presence, not absence, of additional Cys residues disrupts the formation of disulfide bond. There could be two possibilities underlying this phenomenon; 1) the presence of Cys-31 and Cys-616 alters the oxidative folding at the endoplasmic reticulum and consequent disulfide bond patterns, or 2) Cys-31 and Cys-616 react with Cys residues that otherwise form disulfide bond and prevent the formation of intersubunit disulfide bond. In the near-atomic EM structure of TRPV1, both Cys-31 and Cys-616 are not resolved, preventing us from drawing inferences from the current available structural information (3). Further investigation of the mechanism underlying the disruption of disulfide bond by insertion of Cys-31 and Cys-616 will enhance our understanding of biochemistry of TRPV1. TRPV1 is a polymodal sensor that has diverse physiological functions in addition to nociception, and general blockage of TRPV1 function for treatment of pain has caused serious side effects in vivo (58). Detailed understanding of the activation mechanisms is necessary to achieve selective regulation of TRPV1 in a modality-specific manner. Our findings improve the understanding of activation of hTRPV1 by oxidation and provide new insight to enable the design of modality-specific drugs for treating pain.
Author Contributions
N. O. designed the study, conducted most of the experiments, analyzed the results, and wrote most of the paper. K. F. conducted and analyzed the experiments on electrophysiology. O. K. P. conducted the Ca2+ imaging experiment shown in Fig. 9B. H. B. conducted a portion of the Ca2+ imaging experiment shown in Fig. 9A. N. T. collected preliminary data and constructed vectors for the expression of mutant proteins. T. K. and Y. M. directed the research, wrote the manuscript, analyzed, interpreted the data, and critically reviewed the manuscript.
Acknowledgments
We thank H. Atomi, M. Hirano, M.X. Mori, and R. Sakaguchi for experimental advice and helpful discussions.
This work was supported by Grant-in-aid for Scientific Research on Innovative Areas “Oxygen biology: a new criterion for integrated understanding of life” 26111004 of the Ministry of Education, Culture, Sports, Science, and Technology, Japan and Grant-in-aid for Scientific Research (A) 24249017 of the Japan Society for the Promotion of Science. The authors declare that they have no conflicts of interest with the contents of this article.
- TRP
- transient receptor potential
- SRM
- selected reaction monitoring
- TRPV1
- transient receptor potential vanilloid 1
- WB
- Western Blotting
- rTRPV1
- rat TRPV1
- cTRPV1
- chicken TRPV1
- hTRPV1
- human TRPV1
- DRG
- dorsal root ganglion
- NEM
- N-ethylmaleimide
- 5-nitro-2-PDS
- 5-nitro-2-pyridyl disulfide
- RIPA
- radioimmune precipitation assay buffer
- HBS
- HEPES-buffered saline
- ARD
- ankyrin repeat domain
- CAM
- carbamidomethyl group
- half-max
- half-maximal.
References
- 1. Clapham D. E. (2003) TRP channels as cellular sensors. Nature 426, 517–524 [DOI] [PubMed] [Google Scholar]
- 2. Clapham D. E., Julius D., Montell C., and Schultz G. (2005) International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol. Rev. 57, 427–450 [DOI] [PubMed] [Google Scholar]
- 3. Liao M., Cao E., Julius D., and Cheng Y. (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kozai D., Ogawa N., and Mori Y. (2014) Redox regulation of transient receptor potential channels. Antioxid. Redox. Signal. 21, 971–986 [DOI] [PubMed] [Google Scholar]
- 5. Susankova K., Tousova K., Vyklicky L., Teisinger J., and Vlachova V. (2006) Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol. Pharmacol. 70, 383–394 [DOI] [PubMed] [Google Scholar]
- 6. Takahashi N., Mizuno Y., Kozai D., Yamamoto S., Kiyonaka S., Shibata T., Uchida K., and Mori Y. (2008) Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels 2, 287–298 [DOI] [PubMed] [Google Scholar]
- 7. Yoshida T., Inoue R., Morii T., Takahashi N., Yamamoto S., Hara Y., Tominaga M., Shimizu S., Sato Y., and Mori Y. (2006) Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2, 596–607 [DOI] [PubMed] [Google Scholar]
- 8. Xu S. Z., Sukumar P., Zeng F., Li J., Jairaman A., English A., Naylor J., Ciurtin C., Majeed Y., Milligan C. J., Bahnasi Y. M., Al-Shawaf E., Porter K. E., Jiang L. H., Emery P., Sivaprasadarao A., and Beech D. J. (2008) TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersson D. A., Gentry C., Moss S., and Bevan S. (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J. Neurosci. 28, 2485–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bessac B. F., Sivula M., von Hehn C. A., Escalera J., Cohn L., and Jordt S. E. (2008) TRPA1 is a major oxidant sensor in murine airway sensory neurons. J. Clin. Invest. 118, 1899–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu B. Y., Tsai T. L., Ho C. Y., Lu S. H., Lai C. J., and Kou Y. R. (2013) Role of TRPA1 and TRPV1 in the reactive oxygen species-dependent sensory irritation of superior laryngeal capsaicin-sensitive afferents by cigarette smoke in anesthetized rats. Pulm. Pharmacol. Ther. 26, 364–372 [DOI] [PubMed] [Google Scholar]
- 12. Vyklický L., Lyfenko A., Susánková K., Teisinger J., and Vlachová V. (2002) Reducing agent dithiothreitol facilitates activity of the capsaicin receptor VR-1. Neuroscience 111, 435–441 [DOI] [PubMed] [Google Scholar]
- 13. Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., and Julius D. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 [DOI] [PubMed] [Google Scholar]
- 14. Tominaga M., Caterina M. J., Malmberg A. B., Rosen T. A., Gilbert H., Skinner K., Raumann B. E., Basbaum A. I., and Julius D. (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 [DOI] [PubMed] [Google Scholar]
- 15. Xu H., Blair N. T., and Clapham D. E. (2005) Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J. Neurosci. 25, 8924–8937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang S. W., Cho H., Kwak J., Lee S. Y., Kang C. J., Jung J., Cho S., Min K. H., Suh Y. G., Kim D., and Oh U. (2000) Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. U.S.A. 97, 6155–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Premkumar L. S., and Ahern G. P. (2000) Induction of vanilloid receptor channel activity by protein kinase C. Nature 408, 985–990 [DOI] [PubMed] [Google Scholar]
- 18. Zygmunt P. M., Petersson J., Andersson D. A., Chuang H., Sørgård M., Di Marzo V., Julius D., and Högestätt E. D. (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452–457 [DOI] [PubMed] [Google Scholar]
- 19. Caterina M. J., Leffler A., Malmberg A. B., Martin W. J., Trafton J., Petersen-Zeitz K. R., Koltzenburg M., Basbaum A. I., and Julius D. (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 [DOI] [PubMed] [Google Scholar]
- 20. Davis J. B., Gray J., Gunthorpe M. J., Hatcher J. P., Davey P. T., Overend P., Harries M. H., Latcham J., Clapham C., Atkinson K., Hughes S. A., Rance K., Grau E., Harper A. J., Pugh P. L., Rogers D. C., Bingham S., Randall A., and Sheardown S. A. (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187 [DOI] [PubMed] [Google Scholar]
- 21. Wang Z. Q., Porreca F., Cuzzocrea S., Galen K., Lightfoot R., Masini E., Muscoli C., Mollace V., Ndengele M., Ischiropoulos H., and Salvemini D. (2004) A newly identified role for superoxide in inflammatory pain. J. Pharmacol. Exp. Ther. 309, 869–878 [DOI] [PubMed] [Google Scholar]
- 22. Ndengele M. M., Cuzzocrea S., Esposito E., Mazzon E., Di Paola R., Matuschak G. M., and Salvemini D. (2008) Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J. 22, 3154–3164 [DOI] [PubMed] [Google Scholar]
- 23. Khattab M. M. (2006) TEMPOL, a membrane-permeable radical scavenger, attenuates peroxynitrite- and superoxide anion-enhanced carrageenan-induced paw edema and hyperalgesia: a key role for superoxide anion. Eur. J. Pharmacol. 548, 167–173 [DOI] [PubMed] [Google Scholar]
- 24. Bezerra M. M., Brain S. D., Girão V. C., Greenacre S., Keeble J., and Rocha F. A. (2007) Neutrophils-derived peroxynitrite contributes to acute hyperalgesia and cell influx in zymosan arthritis. Naunyn. Schmiedebergs Arch. Pharmacol. 374, 265–273 [DOI] [PubMed] [Google Scholar]
- 25. Keeble J. E., Bodkin J. V., Liang L., Wodarski R., Davies M., Fernandes E. S., Coelho C. de F., Russell F., Graepel R., Muscara M. N., Malcangio M., and Brain S. D. (2009) Hydrogen peroxide is a novel mediator of inflammatory hyperalgesia, acting via transient receptor potential vanilloid 1-dependent and independent mechanisms. Pain 141, 135–142 [DOI] [PubMed] [Google Scholar]
- 26. Chuang H. H., and Lin S. (2009) Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc. Natl. Acad. Sci. U.S.A. 106, 20097–20102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang S., and Chuang H. H. (2011) C-terminal dimerization activates the nociceptive transduction channel transient receptor potential vanilloid 1. J. Biol. Chem. 286, 40601–40607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hansen R. E., Roth D., and Winther J. R. (2009) Quantifying the global cellular thiol-disulfide status. Proc. Natl. Acad. Sci. U.S.A. 106, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang L., Cvetkov T. L., Chance M. R., and Moiseenkova-Bell V. Y. (2012) Identification of in vivo disulfide conformation of TRPA1 ion channel. J. Biol. Chem. 287, 6169–6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szallasi A., and Blumberg P. M. (1993) [3H]resiniferatoxin binding by the vanilloid receptor: species-related differences, effects of temperature and sulfhydryl reagents. Naunyn. Schmiedebergs Arch. Pharmacol. 347, 84–91 [DOI] [PubMed] [Google Scholar]
- 31. Szallasi A., Lewin N. A., and Blumberg P. M. (1993) Vanilloid (capsaicin) receptor in the rat: positive cooperativity of resiniferatoxin binding and its modulation by reduction and oxidation. J. Pharmacol. Exp. Ther. 266, 678–683 [PubMed] [Google Scholar]
- 32. Wittig I., Braun H. P., and Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 33. Nishida M., Nagao T., and Kurose H. (1999) Activation of Rac1 increases c-Jun NH2-terminal kinase activity and DNA fragmentation in a calcium-dependent manner in rat myoblast cell line H9c2. Biochem. Biophys. Res. Commun. 262, 350–354 [DOI] [PubMed] [Google Scholar]
- 34. Okada T., Inoue R., Yamazaki K., Maeda A., Kurosaki T., Yamakuni T., Tanaka I., Shimizu S., Ikenaka K., Imoto K., and Mori Y. (1999) Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 274, 27359–27370 [DOI] [PubMed] [Google Scholar]
- 35. Grønborg M., Bunkenborg J., Kristiansen T. Z., Jensen O. N., Yeo C. J., Hruban R. H., Maitra A., Goggins M. G., and Pandey A. (2004) Comprehensive proteomic analysis of human pancreatic juice. J. Proteome Res. 3, 1042–1055 [DOI] [PubMed] [Google Scholar]
- 36. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 37. McNicholas S., Potterton E., Wilson K. S., and Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takahashi N., Kuwaki T., Kiyonaka S., Numata T., Kozai D., Mizuno Y., Yamamoto S., Naito S., Knevels E., Carmeliet P., Oga T., Kaneko S., Suga S., Nokami T., Yoshida J., and Mori Y. (2011) TRPA1 underlies a sensing mechanism for O-2. Nat. Chem. Biol. 7, 701–711 [DOI] [PubMed] [Google Scholar]
- 39. Ryle A. P., and Sanger F. (1954) Disulphide interchange reactions. Biochem. J. 58, v–vi [PubMed] [Google Scholar]
- 40. Lee Y. M., Li W. H., Kim Y. K., Kim K. H., and Chung J. H. (2008) Heat-induced MMP-1 expression is mediated by TRPV1 through PKCα signaling in HaCaT cells. Exp. Dermatol. 17, 864–870 [DOI] [PubMed] [Google Scholar]
- 41. Bodó E., Bíró T., Telek A., Czifra G., Griger Z., Tóth B. I., Mescalchin A., Ito T., Bettermann A., Kovács L., and Paus R. (2005) A hot new twist to hair biology: involvement of vanilloid receptor-1 (VR1/TRPV1) signaling in human hair growth control. Am. J. Pathol. 166, 985–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tóth B. I., Dobrosi N., Dajnoki A., Czifra G., Oláh A., Szöllosi A. G., Juhász I., Sugawara K., Paus R., and Bíró T. (2011) Endocannabinoids modulate human epidermal keratinocyte proliferation and survival via the sequential engagement of cannabinoid receptor-1 and transient receptor potential vanilloid-1. J. Invest. Dermatol. 131, 1095–1104 [DOI] [PubMed] [Google Scholar]
- 43. Flynn R., Chapman K., Iftinca M., Aboushousha R., Varela D., and Altier C. (2014) Targeting the transient receptor potential vanilloid type 1 (TRPV1) assembly domain attenuates inflammation-induced hypersensitivity. J. Biol. Chem. 289, 16675–16687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whiffen A. J. (1948) The production, assay, and antibiotic activity of actidione, an antibiotic from Streptomyces griseus. J. Bacteriol. 56, 283–291 [DOI] [PubMed] [Google Scholar]
- 45. Kerridge D. (1958) The effect of actidione and other antifungal agents on nucleic acid and protein synthesis in Saccharomyces carlsbergensis. J. Gen. Microbiol. 19, 497–506 [DOI] [PubMed] [Google Scholar]
- 46. Lishko P. V., Procko E., Jin X., Phelps C. B., and Gaudet R. (2007) The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54, 905–918 [DOI] [PubMed] [Google Scholar]
- 47. Lesage F., Reyes R., Fink M., Duprat F., Guillemare E., and Lazdunski M. (1996) Dimerization of TWIK-1 K+ channel subunits via a disulfide bridge. EMBO J. 15, 6400–6407 [PMC free article] [PubMed] [Google Scholar]
- 48. Hong C., Kwak M., Myeong J., Ha K., Wie J., Jeon J. H., and So I. (2015) Extracellular disulfide bridges stabilize TRPC5 dimerization, trafficking, and activity. Pflugers Arch. 467, 703–712 [DOI] [PubMed] [Google Scholar]
- 49. Cho H. C., Tsushima R. G., Nguyen T. T., Guy H. R., and Backx P. H. (2000) Two critical cysteine residues implicated in disulfide bond formation and proper folding of Kir2.1. Biochemistry 39, 4649–4657 [DOI] [PubMed] [Google Scholar]
- 50. Littler D. R., Harrop S. J., Fairlie W. D., Brown L. J., Pankhurst G. J., Pankhurst S., DeMaere M. Z., Campbell T. J., Bauskin A. R., Tonini R., Mazzanti M., Breit S. N., and Curmi P. M. (2004) The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J. Biol. Chem. 279, 9298–9305 [DOI] [PubMed] [Google Scholar]
- 51. Zissimopoulos S., Viero C., Seidel M., Cumbes B., White J., Cheung I., Stewart R., Jeyakumar L. H., Fleischer S., Mukherjee S., Thomas N. L., Williams A. J., and Lai F. A. (2013) N-terminal oligomerization regulates the function of cardiac ryanodine receptors. J. Cell Sci. 126, 5042–5051 [DOI] [PubMed] [Google Scholar]
- 52. Wo Z. G., and Oswald R. E. (1996) Ligand-binding characteristics and related structural features of the expressed goldfish kainate receptors: identification of a conserved disulfide bond and three residues important for ligand binding. Mol. Pharmacol. 50, 770–780 [PubMed] [Google Scholar]
- 53. Sullivan J. M., Traynelis S. F., Chen H. S., Escobar W., Heinemann S. F., and Lipton S. A. (1994) Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron 13, 929–936 [DOI] [PubMed] [Google Scholar]
- 54. Tao X., Avalos J. L., Chen J., and MacKinnon R. (2009) Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reisz J. A., Bechtold E., King S. B., Poole L. B., and Furdui C. M. (2013) Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 280, 6150–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hinman A., Chuang H. H., Bautista D. M., and Julius D. (2006) TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. U.S.A. 103, 19564–19568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kashio M., Sokabe T., Shintaku K., Uematsu T., Fukuta N., Kobayashi N., Mori Y., and Tominaga M. (2012) Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. U.S.A. 109, 6745–6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Patapoutian A., Tate S., and Woolf C. J. (2009) Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug Discov. 8, 55–68 [DOI] [PMC free article] [PubMed] [Google Scholar]