

Abstract
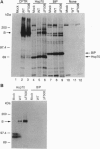
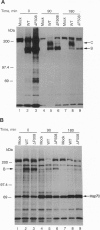
The most common cause of cystic fibrosis is deletion of Phe-508 (delta F508) from the cystic fibrosis transmembrane conductance regulator (CFTR). Previous studies have suggested that delta F508 CFTR is an unstable protein that retains a pattern of glycosylation specific to the endoplasmic reticulum. This report examines the mechanism responsible for the mislocalization of delta F508 CFTR in a human cystic fibrosis epithelial cell line overexpressing recombinant CFTR by virtue of adenovirus-mediated gene transfer. Immunoelectron microscopy confirmed that wild-type CFTR is delivered to the plasma membrane of these cells and that delta F508 CFTR is retained in the endoplasmic reticulum. Pulse-chase studies showed that newly synthesized CFTR complexes with the chaperone hsp70. The wild-type protein dissociates from hsp70 before its transport to the Golgi, and the protein is subsequently degraded in lysosomes. By contrast, the complex formed between delta F508 CFTR and hsp70 is retained in the endoplasmic reticulum and delta F508 CFTR is rapidly degraded in a pre-Golgi nonlysosomal compartment. Thus, hsp70 discriminates between the normal form of CFTR and the form of the protein that most commonly causes cystic fibrosis (delta F508). These findings clarify the mechanism by which mutation causing delta F508 affects the intracellular trafficking of CFTR and suggest another function for hsp70 in ensuring quality control during the biosynthesis of plasma-membrane proteins.
Full text
PDF
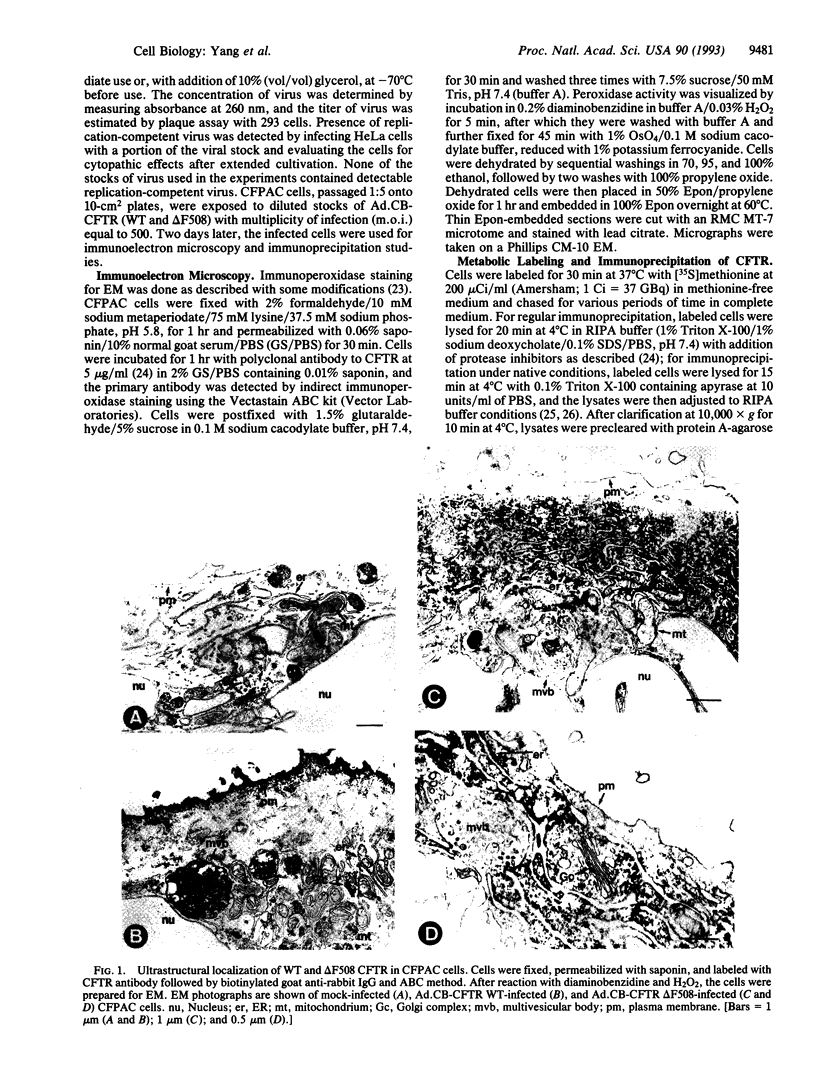


Images in this article
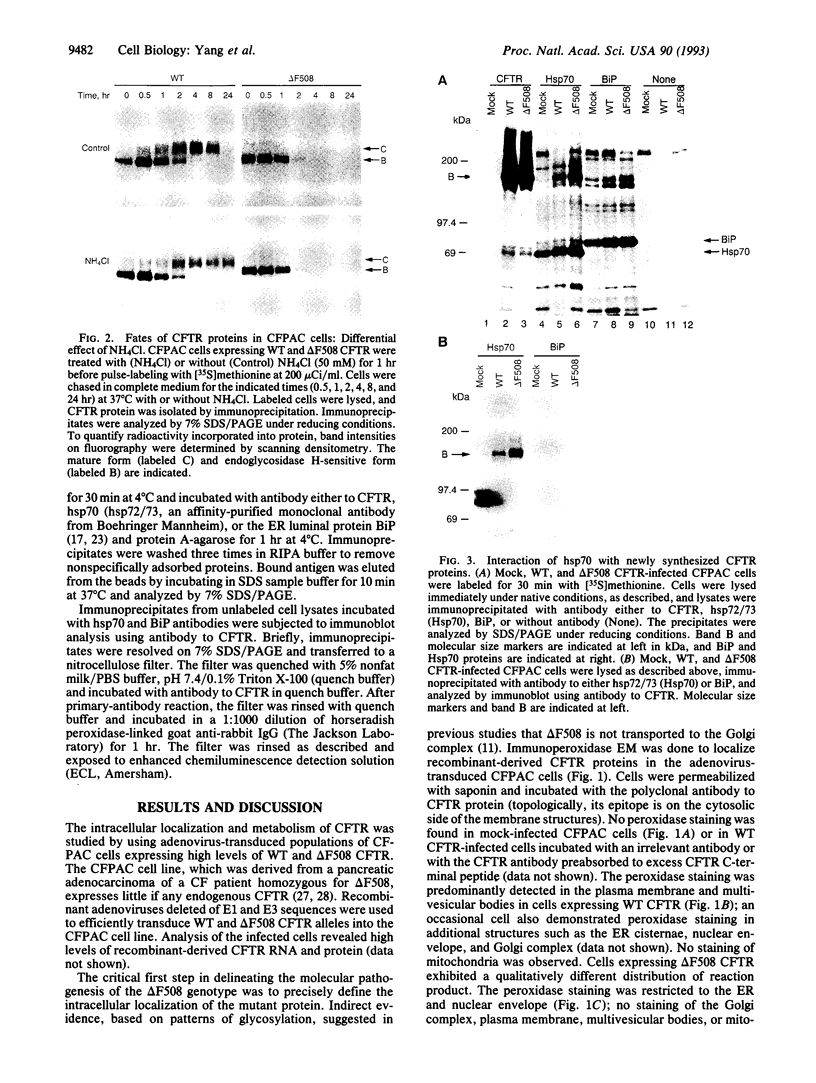
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Accili D., Kadowaki T., Kadowaki H., Mosthaf L., Ullrich A., Taylor S. I. Immunoglobulin heavy chain-binding protein binds to misfolded mutant insulin receptors with mutations in the extracellular domain. J Biol Chem. 1992 Jan 5;267(1):586–590. [PubMed] [Google Scholar]
- Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., Welsh M. J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991 Jul 12;253(5016):202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Barasch J., Kiss B., Prince A., Saiman L., Gruenert D., al-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature. 1991 Jul 4;352(6330):70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- Bear C. E., Li C. H., Kartner N., Bridges R. J., Jensen T. J., Ramjeesingh M., Riordan J. R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell. 1992 Feb 21;68(4):809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Beckmann R. P., Lovett M., Welch W. J. Examining the function and regulation of hsp 70 in cells subjected to metabolic stress. J Cell Biol. 1992 Jun;117(6):1137–1150. doi: 10.1083/jcb.117.6.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann R. P., Mizzen L. E., Welch W. J. Interaction of Hsp 70 with newly synthesized proteins: implications for protein folding and assembly. Science. 1990 May 18;248(4957):850–854. doi: 10.1126/science.2188360. [DOI] [PubMed] [Google Scholar]
- Bole D. G., Dowin R., Doriaux M., Jamieson J. D. Immunocytochemical localization of BiP to the rough endoplasmic reticulum: evidence for protein sorting by selective retention. J Histochem Cytochem. 1989 Dec;37(12):1817–1823. doi: 10.1177/37.12.2685110. [DOI] [PubMed] [Google Scholar]
- Bole D. G., Hendershot L. M., Kearney J. F. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J Cell Biol. 1986 May;102(5):1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshart M., Weber F., Jahn G., Dorsch-Häsler K., Fleckenstein B., Schaffner W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell. 1985 Jun;41(2):521–530. doi: 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- Bradbury N. A., Jilling T., Berta G., Sorscher E. J., Bridges R. J., Kirk K. L. Regulation of plasma membrane recycling by CFTR. Science. 1992 Apr 24;256(5056):530–532. doi: 10.1126/science.1373908. [DOI] [PubMed] [Google Scholar]
- Chappell T. G., Welch W. J., Schlossman D. M., Palter K. B., Schlesinger M. J., Rothman J. E. Uncoating ATPase is a member of the 70 kilodalton family of stress proteins. Cell. 1986 Apr 11;45(1):3–13. doi: 10.1016/0092-8674(86)90532-5. [DOI] [PubMed] [Google Scholar]
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990 Nov 16;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Chirico W. J., Waters M. G., Blobel G. 70K heat shock related proteins stimulate protein translocation into microsomes. Nature. 1988 Apr 28;332(6167):805–810. doi: 10.1038/332805a0. [DOI] [PubMed] [Google Scholar]
- Cohn J. A., Nairn A. C., Marino C. R., Melhus O., Kole J. Characterization of the cystic fibrosis transmembrane conductance regulator in a colonocyte cell line. Proc Natl Acad Sci U S A. 1992 Mar 15;89(6):2340–2344. doi: 10.1073/pnas.89.6.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F. S. Cystic fibrosis: molecular biology and therapeutic implications. Science. 1992 May 8;256(5058):774–779. doi: 10.1126/science.1375392. [DOI] [PubMed] [Google Scholar]
- Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992 Aug 27;358(6389):761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Drumm M. L., Pope H. A., Cliff W. H., Rommens J. M., Marvin S. A., Tsui L. C., Collins F. S., Frizzell R. A., Wilson J. M. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990 Sep 21;62(6):1227–1233. doi: 10.1016/0092-8674(90)90398-x. [DOI] [PubMed] [Google Scholar]
- Engelhardt J. F., Yankaskas J. R., Ernst S. A., Yang Y., Marino C. R., Boucher R. C., Cohn J. A., Wilson J. M. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet. 1992 Nov;2(3):240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- Gething M. J., Sambrook J. Protein folding in the cell. Nature. 1992 Jan 2;355(6355):33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Hainaut P., Milner J. Interaction of heat-shock protein 70 with p53 translated in vitro: evidence for interaction with dimeric p53 and for a role in the regulation of p53 conformation. EMBO J. 1992 Oct;11(10):3513–3520. doi: 10.1002/j.1460-2075.1992.tb05434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P. J., Ostermann J., Shilling J., Neupert W., Craig E. A., Pfanner N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 1990 Nov 8;348(6297):137–143. doi: 10.1038/348137a0. [DOI] [PubMed] [Google Scholar]
- Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat Genet. 1992 Aug;1(5):321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kost T. A., Theodorakis N., Hughes S. H. The nucleotide sequence of the chick cytoplasmic beta-actin gene. Nucleic Acids Res. 1983 Dec 10;11(23):8287–8301. doi: 10.1093/nar/11.23.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman M. A., Schneider W. J., Brown M. S., Davis C. G., Elhammer A., Russell D. W., Goldstein J. L. The Lebanese allele at the low density lipoprotein receptor locus. Nonsense mutation produces truncated receptor that is retained in endoplasmic reticulum. J Biol Chem. 1987 Jan 5;262(1):401–410. [PubMed] [Google Scholar]
- Lomas D. A., Evans D. L., Finch J. T., Carrell R. W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992 Jun 18;357(6379):605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- Machamer C. E., Doms R. W., Bole D. G., Helenius A., Rose J. K. Heavy chain binding protein recognizes incompletely disulfide-bonded forms of vesicular stomatitis virus G protein. J Biol Chem. 1990 Apr 25;265(12):6879–6883. [PubMed] [Google Scholar]
- Poole B., Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981 Sep;90(3):665–669. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 Sep 8;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rommens J. M., Iannuzzi M. C., Kerem B., Drumm M. L., Melmer G., Dean M., Rozmahel R., Cole J. L., Kennedy D., Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 Sep 8;245(4922):1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Rosenfeld M. A., Yoshimura K., Trapnell B. C., Yoneyama K., Rosenthal E. R., Dalemans W., Fukayama M., Bargon J., Stier L. E., Stratford-Perricaudet L. In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium. Cell. 1992 Jan 10;68(1):143–155. doi: 10.1016/0092-8674(92)90213-v. [DOI] [PubMed] [Google Scholar]
- Thomas P. J., Shenbagamurthi P., Sondek J., Hullihen J. M., Pedersen P. L. The cystic fibrosis transmembrane conductance regulator. Effects of the most common cystic fibrosis-causing mutation on the secondary structure and stability of a synthetic peptide. J Biol Chem. 1992 Mar 25;267(9):5727–5730. [PubMed] [Google Scholar]