

Abstract
Cannabis and analogs of Δ9-tetrahydrocannabinol have been used for therapeutic purposes, but their therapeutic use remains limited because of various adverse effects. Endogenous cannabinoids have been discovered, and dysregulation of endocannabinoid signaling is implicated in the pathophysiology of major depressive disorder (MDD). Recently, endocannabinoid hydrolytic enzymes such as fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) have become new therapeutic targets in the treatment of MDD. Several FAAH or MAGL inhibitors are reported to have no cannabimimetic side effects and, therefore, are new potential therapeutic options for patients with MDD who are resistant to first-line antidepressants (selective serotonin and serotonin-norepinephrine reuptake inhibitors). In this review, we focus on the possible relationships between MDD and the endocannabinoid system as well as the inhibitors’ therapeutic potential. MAGL inhibitors may reduce inflammatory responses through activation of cannabinoid receptor type 2. In the hypothalamic–pituitary–adrenal axis, repeated FAAH inhibitor administration may be beneficial for reducing circulating glucocorticoid levels. Both FAAH and MAGL inhibitors may contribute to dopaminergic system regulation. Recently, several new inhibitors have been developed with strong potency and selectivity. FAAH inhibitor, MAGL inhibitor, or dual blocker use would be promising new treatments for MDD. Further pre-clinical studies and clinical trials using these inhibitors are warranted.
Keywords: Antidepressants, Chronic inflammation, Dopamine, Endocannabinoids, Fatty acid amide hydrolase, Hypothalamic–pituitary–adrenal axis, Major depressive disorder, Monoacylglycerol lipase
1. INTRODUCTION
In the Global Burden of Disease Study 2010, major depressive disorder (MDD) was estimated as the second largest cause of years lived with disability [1]. MDD is a common disease with an estimated global point prevalence of 4.4% in 2010 (95% confidence interval: 4.1–4.4), 1990 (4.2–4.7), and 2005 (4.1–4.7) [2]. Unfortunately, however, a substantial proportion of patients with MDD failed to respond to first-line therapeutic drugs such as selective serotonin reuptake inhibitors (SSRI) and serotonin-norepinephrine reuptake inhibitors [3]. Therefore, different classes of antidepressants are much needed.
“Cannabinoids” is a generic term for chemical constituents contained in Cannabis (C. sativa subsp. sativa, C. sativa subsp. indica, C. ruderalis). Cannabinoids consist of over 60 chemicals, which include Δ9-tetrahydrocannabinol (THC), cannabinol, and cannabidiol. Mechoulam et al. isolated THC from cannabis in 1964 [4], and it was later found to be the major psychoactive constituent among the 60 cannabinoids. Cannabis has been used for medical purposes in the treatment of pain, nausea, or seizure. It also promotes sociability, rewarding feelings, and relaxation [5]. However, therapeutic use of cannabis and analogs of THC remain limited because of their various side effects such as disturbing sensorimotor and/or cognitive ability or influencing affection [6]. When used as psychomimetics, direct agonists of cannabinoid receptors can cause panic or psychosis, while their antagonists can lead to depression or anxiety symptoms [5].
Endogenous cannabinoids have been discovered, and endocannabinoid signaling dysregulation has been implicated in the pathophysiology of MDD. Therefore, the endocannabinoid hydrolytic enzymes have been regarded as new therapeutic targets for the treatment of MDD. Several inhibitors of these enzymes are reported to have no cannabimimetic side effects [7-10]. Here, we review the literature on the possible relationships between the endocannabinoid system and MDD pathophysiology as well as the implication of the inhibitors of endocannabinoid-degrading enzymes for the amelioration of MDD symptoms.
1.1. Discovery of the Endocannabinoid System
THC acts through G protein-coupled cannabinoid receptor type 1 (CB1), which was discovered in 1988 [11] and cloned in a rat cDNA library in 1990 [12]. Subsequently, G protein-coupled cannabinoid receptor type 2 (CB2) was also identified in a human promyelocytic leukemia cell cDNA library [13]. These findings promoted investigations to seek endogenous ligands for these receptors.
In 1992, N-arachidonoylethanolamine (AEA, also called “anandamide”) was found to be an endogenous ligand for cannabinoid receptors from the porcine brain [14]. Subsequently, another endocannabinoid, 2-arachidonoylglycerol (2-AG), was identified by two groups, one from a rat brain [15] and another from a canine intestine [16]. According to Sugiura et al., 2-AG’s binding affinity to the cannabinoid receptor was 24 times lower than that of AEA (Ki = 99 nM and 2.4 μM, respectively). However, concentrations of 2-AG were observed to be about 800 times higher than those of AEA in the rat brain. AEA was reported to account for 0.7% of the total N-acylethanolamines (NAEs) (597.3 pmol/g [rat brain tissue]) [17], while 2-AG accounted for 47.4% of the total monoacylglycerols (6.86 nmol/g [rat brain tissue]) [15]. Therefore, 2-AG may play more essential physiological roles than AEA in the brain endocannabinoid system [18].
However, the function of endocannabinoids in synaptic transmission remains unclear. In the 1990s, it was discovered that depolarization of the hippocampal CA1 pyramidal neurons or cerebellum Purkinje cells inhibits presynaptic γ-amino butyric acid (GABA) release onto these cells [19], which was termed as “depolarization-induced suppression of inhibition” [20]. In 2001, three groups discovered that postsynaptic release of endocannabinoids plays a role in mediating retrograde signaling through the presynaptic CB1, which inhibits presynaptic neurotransmitter release [21-23]. When the retrograde signaling occurs at the excitatory (glutamatergic) synapses, it is called “depolarization-induced suppression of excitation” [21]. These findings revealed that endocannabinoids behave as retrograde messengers in modulating transmitter release at synaptic sites and are key regulators in the central nervous system (CNS). Several reviews have described endocannabinoid systems in detail [6, 18, 24].
1.2. Characteristics of Endocannabinoid Receptors and Ligands
CB1 receptors are distributed abundantly in the CNS (cerebral cortex, hippocampus, amygdala, striatum, substantia nigra) and peripheral tissues (lung, small intestine, uterus, testis) [25], and the functional roles of CB1 are relevant to motor regulation, control of appetites, and memory processing [26]. CB2 receptors are mainly expressed in the peripheral tissues (spleen, tonsil, lymph nodes) and immune cells (macrophages, monocytes, B lymphocytes, natural killer cells) [25]. A study using postmortem brains reported that these receptors are expressed only in microglial cells in the CNS [27]. Subsequently, CB2 receptors were found to be expressed in neurons in specific brain regions [28]. CB2 receptors are considered to play roles in the regulation of inflammatory and immune responses. Several recent studies have shown that activation of the CB2 receptor causes reversal of experimental memory deficiency in rats [29], promotes neuroprotective effects in brain infarction model mice [30], and ameliorates induced inflammatory responses [31, 32] and experimental autoimmune encephalomyelitis [33]. However, inconsistent results have also been reported [34].
The endocannabinoid 2-AG is a full agonist for CB1 and CB2 receptors, while AEA and THC have less affinity for the CB2 receptor than for the CB1 receptor [35, 36]. Furthermore, AEA is a known ligand for transient receptor potential vanilloid type 1 (TRPV1). TRPV1 co-localizes with the CB1 receptor in neurons in several brain regions [37], and both proteins are upregulated during inflammation [38]. TRPV1 also co-localizes with CB1 and CB2 in the mouse bone marrow-derived dendritic cells and in human skeletal muscle cells, myometrial smooth muscle cells, osteoclasts, proximal tubular cells of the kidney, keratinocytes, melanocytes, and dental pulp cells [37]. Recently, it was discovered that 2-AG acts as a ligand for TRPV1 [39].
1.3. Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase
AEA and 2-AG are released by Ca2+-driven (postsynaptic Ca2+ elevation) or Ca2+ independent receptor-driven mechanisms at the synaptic sites. These endocannabinoids are produced on demand and are degraded by hydrolysis and oxidation. AEA is hydrolyzed by fatty acid amide hydrolase (FAAH, also called FAAH-1), which was identified in 1993 [40], followed by FAAH-2 in 2006 [41]. From the analysis of the Expressed Sequence Tags public database, FAAH-1 gene transcript expressions were observed in humans, rats, mice, cows, and frogs, but FAAH-2 expression was in a limited number of species such as humans and frogs but not mice, rats, or cows [41]. In humans, FAAH-1 is distributed in the brain, kidney, liver, small intestine, lung, prostate, and testis while FAAH-2 is distributed in the heart, kidney, liver, lung, and prostate but not in the brain [41]. It was suggested that 85% of 2-AG is hydrolyzed by monoacylglycerol lipase (MAGL), and the remaining 15% is catalyzed by abhydrolase domain-containing protein 6 (ABHD6) or 12 [42]. MAGL was discovered in 1976. In rats, MAGL mRNA is widely expressed in the adipose tissue, adrenal gland, ovary, heart, lung, liver, skeletal muscle, kidney, testis, and brain [43]. Both AEA and 2-AG are oxidized by cyclooxygenases (COX) or lipoxygenases (LOX). Cyclooxygenase-2 (COX-2) acts on AEA and 2-AG, which produce prostaglandin (PG) ethanolamide and prostaglandin glycerol ester, respectively.
We summarized the pathways with a focus on FAAH and MAGL in Fig. (1). In these pathways, AEA also plays the role of a weak ligand for peroxisome proliferator-activated receptor (PPAR) α [44] and PPAR-γ [45]. The relationships between PPARs and NAEs have been discussed in other reviews [46, 47].
Fig. (1).
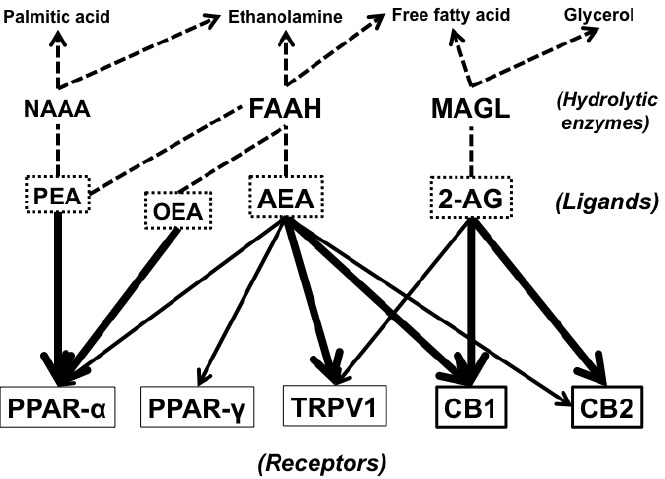
Endocannabinoids, their target receptors, hydrolytic enzymes, and metabolites. Abbreviations: NAAA, N-acylethanolaminehydrolyzing acid amidase; FAAH, fatty acid amide hydrolase; MAGL, monoacylglycerol lipase; PEA, N-palmitoylethanolamine; OEA, N-oleoylethanolamine; AEA, N-arachidonoylethanolamine; 2-AG, 2-arachidonoylglycerol; PPAR-α, peroxisome proliferator-activated receptor α; PPAR-γ, peroxisome proliferator-activated receptor γ; TRPV1, transient receptor potential vanilloid type 1; CB1, cannabinoidreceptor type 1; CB2, cannabinoid receptor type 2.
2. THE ENDOCANNABINOID SYSTEM AND MAJOR DEPRESSIVE DISORDER
2.1. Involvement of Endocannabinoids in MDD
Endocannabinoid signaling modulation is implicated in MDD pathophysiology. The elevation of CB1 receptor expression levels and CB1 receptor-stimulated [35S]GTPγS binding reactivity were observed in the prefrontal cortex of the postmortem brains of MDD patients who had committed suicide [48]. Hill et al. imposed a stress task on 15 women with MDD and 15 healthy women and examined whether they showed endocannabinoid alterations after the task. Compared with the control group at baseline, patients with MDD showed decreased serum levels of AEA and 2-AG but not of N-oleoylethanolamine (OEA) and N-palmitoylethanolamine (PEA). Increase in serum 2-AG levels—but not AEA—was higher in patients during the stress tasks than in controls. After the stress task, serum OEA and PEA levels were decreased compared with controls [49]. According to another study by Hill et al., 16 female patients with MDD showed significantly lower serum 2-AG concentrations compared with 28 healthy female controls at baseline [50]. Serum 2-AG levels in the patients with MDD were negatively correlated with current depressive episode length. Furthermore, patients with MDD showed significantly negative correlations between serum AEA levels and both “Cognitive anxiety” and “Somatic anxiety” subscale scores on the Hamilton Rating Scale for Depression (HRSD) [50]. We reported that the cerebrospinal fluid (CSF) level of ethanolamine (EA), which is a metabolite of AEA, was lower in patients with MDD than in healthy controls [51]. Additionally, in accordance with studies by Hill et al., MDD patients with lower EA concentrations in the CSF scored higher on the “Somatic anxiety” HRSD subscale. Additionally, those with lower CSF EA in our study had a higher total HRSD score than did those with higher CSF EA levels [51].
In an animal study, Hill et al. demonstrated that mice lacking FAAH showed antianxiety-like behavior and increased AEA levels in the amygdala compared with wild-type (WT) mice [52]. In humans [53], the possible association of a missense single nucleotide polymorphism C385A (rs324420) of the FAAH gene, which reduces FAAH activity and expression [54], with stress reactivity was investigated. Individuals with A/A genotype (homozygosity for the low-activity allele) showed significantly lower stress reactivity Z score on the Multidimensional Personality Questionnaire than did those with A/C or C/C genotypes, indicating that genetically determined reduction in FAAH activity is associated with attenuated stress response [53]. Taken together, these results suggest that the AEA pathway is involved in anxiety—in particular, somatic anxiety among MDD symptoms. Ho et al. revealed that diastolic blood pressure is significantly correlated with both serum AEA and 2-AG levels in individuals with depression but not in controls [55]. This finding also suggests that endocannabinoids are involved in autonomic dysregulation in MDD. Since Hill et al. and Ho et al. employed only female participants, further investigations should be conducted in male participants.
However, contradictive negative results have been reported in several studies. Giuffrida et al. found no significant differences in the CSF AEA levels between 12 patients with depression and healthy controls, although the levels were elevated in un-medicated patients with schizophrenia (SZ) [56]. Among these 12 patients with depression, eight had bipolar disorder, and all were medicated with psychotropic drugs. Further exploration of endocannabinoid dynamics in non-medicated patients with MDD will be warranted.
2.2. Neuroinflammation and Endocannabinoids
Inflammatory responses are implicated in MDD pathophysiology. Recently, we reported that MDD and SZ patient groups showed significantly higher CSF Interleukin-6 (IL-6) concentrations compared to a healthy control group [57]. Although patients with MDD and SZ in our study did not show significant differences in IL-6 blood serum levels compared to controls [57], a meta-analysis of 99 studies investigating serum or plasma IL-6 levels clearly showed elevated circulating IL-6 concentrations in patients with MDD [58]. In another meta-analysis, certain pro-inflammatory cytokine and related proteins, such as soluble IL-2 receptor [59] and tumor necrosis factor-α (TNF-α) [59, 60], were elevated in MDD, but other cytokines such as IL-1β [59, 60], IL-10 [58, 60], IL-2 [60], and interferon-γ were not [60]. Still another meta-analysis reported that antidepressant SSRI treatment recovered serum IL-6 levels, and other antidepressant treatments recovered serum IL-1β levels [61]. Taken together, these results seem to indicate that particular cytokines are significantly related to MDD. We recently reported in a meta-analysis that plasma concentrations of L-tryptophan, which is a precursor for serotonin, were decreased in those with MDD [62]. The mechanism is assumed to function as follows: pro-inflammatory cytokines induce indoleamine 2,3-dioxygenase expressed in the epithelial cells, endothelial cells, macrophages, and dendritic cells, converting tryptophan to kynurenine. Pro-inflammatory cytokines may be involved in the decrease of serotonin caused by tryptophan deficiency. In microglial cells, kynurenine is predominantly converted to quinolinic acid or 3-hydroxykynurenine, causing a neurotoxic effect by agonizing the NMDA receptor [63].
Recently, Hodes et al. reported that peripheral IL-6 is highly relevant to stress vulnerability in mice [64]. Even before the stress paradigm, leukocytes collected from stress-susceptible mice produced higher IL-6 than those from resilient mice when the leukocytes were stimulated with lipopolysaccharide (LPS). Mice with transplanted hematopoietic progenitor cells from stress-susceptible mice showed significantly greater depression-like behaviors compared to the those with transplanted cells from control mice [64]. IL-6 is produced by macrophages, endothelial cells, and microglial cells [65], and the CB2 distribution site in the CNS overlaps with IL-6 expression. As we mentioned above, CB2 agonist suppresses the stress-induced inflammatory response in mice [32]. In vitro studies revealed that a CB2 blocker reversed the THC effects on pro-inflammatory cytokines such as IL-6, IL-8, IL-1β, and LPS-induced TNF-α [66], and a selective agonist for CB2, HU-308, reduced cytokine productions such as IL-6, IL-1β, and LPS-induced TNF-α [67]. Collectively, CB2 is involved in the production of pro-inflammatory cytokines and may be a therapeutic target to alter the neuroinflammatory state in MDD. Since 2-AG—and not AEA and THC—is a full agonist for CB2, inhibition of hydrolysis of 2-AG may be beneficial for suppressing CNS inflammation through CB2 signaling. Therefore, MAGL inhibitors may ameliorate symptoms of neuroinflammation-related MDD.
COX-2 is also relevant to inflammation as a key enzyme for producing PGs. In the brain, COX-2 is usually expressed at undetectable levels in the endothelial cells [68]; however, COX-2 is strongly induced in the brain endothelial cells, and the number of COX-2 immunoreactive microglial cells is increased in an inflammatory status [69]. Celecoxib, which is one of the COX-2 specific non-steroidal anti-inflammatory drugs (NSAIDs), showed antidepressive effects when combined with reboxetine (a selective noradrenaline reuptake inhibitor) [70]. Later, celecoxib’s add-on effects were confirmed by a meta-analysis [71]. In animal studies, celecoxib’s monotherapeutic effects on depression-like behavior were examined [72, 73], and celecoxib showed equivalent effects to other antidepressants in the rat forced swim test (FST) [73]. Zoppi et al. reported that mice treated with JWH-133, a selective agonist for CB2, or CB2 over-expressing mice showed no significant changes in COX-2 expression levels but showed significantly low PGE2 levels compared to controls after a four-day stress procedure, whereas CB2 knock-out (KO) mice showed increased COX-2 expressions and higher PGE2 levels in the brain [32]. Taken together, these results indicate that endocannabinoid signaling, especially of CB2, could affect inflammatory responses, and could be a therapeutic target for inflammation-related depression.
2.3. Hypothalamic–Pituitary–adrenal (HPA) Axis and Endocannabinoids
Dysregulation of the HPA axis, which is considered to be related to chronic stress, is implicated in MDD pathophysiology. Both psychological and physiological stressors activate the HPA axis, followed by an increase in corticotropin-releasing hormone (CRH) and arginine-vasopressin production from the paraventricular nuclei (PVN) of the hypothalamus. So far, several studies have demonstrated that patients with MDD show hypercortisolism [74, 75], and HPA axis restoration occurs after MDD remission [74]. Therefore, HPA axis function can be a state-dependent marker of MDD. We also reported that a single nucleotide polymorphism of the IL-1β gene is associated with glucocorticoid levels after dexamethasone (DEX) but not CRH administration [76].
In previous studies in rodents, intracerebroventricular (i.c.v.) or direct injection of URB597, an FAAH inhibitor, to the brain parenchyma showed significant decrease in blood corticosterone in an acute stress paradigm [77, 78]. Although a single intraperitoneal (i.p.) administration (0.1–1.0 mg/kg) of URB597 resulted in no significant decrease in blood corticosterone in an acute stress paradigm [79-81], significantly decreased mRNA expressions of pituitary proopiomelanocortin (POMC), PVN CRH, and c-Fos in the amygdala and PVN were observed after administration of URB597 in an acute stress paradigm [79]. These results suggest that URB597 could alter HPA axis functions. Hill et al. showed that, in nine days of a 30-min repeated stress paradigm, basal levels of AEA in several brain regions (i.e., frontal cortex, hippocampus, hypothalamus, and amygdala) decreased significantly [82]. Repeated administration of URB597 (0.3 mg/kg i.p.) significantly decreased basal levels of corticosterone after a repeated stress paradigm [82].
These nine days of repeated 30-min restraint stress also resulted in increased 2-AG levels, but not in the unstressed or basal state [82]. The corticosterone levels after repeated stress were significantly lower than after single stress. This “adaptive” decrease in corticosterone response was blocked by the CB1 antagonist, AM251, but not by the FAAH inhibitor, URB597. Furthermore, the 2-AG levels in the amygdala after 30 min of repeated restraint stress was significantly and negatively correlated with blood corticosterone levels [82]. In another report, CB1 KO mice or CB1 antagonist-treated mice also showed significantly elevated blood corticosterone levels responding to restraint stress [80]. These results suggest that the adaptation to stress (i.e., decreased response of corticosterone after repeated stress administration) or response of corticosterone levels after repeated stress is related to brain CB1 and 2-AG rather than AEA. However, since AM251 also works as a CB2 antagonist [83], we cannot rule out the possibility that CB2 partly contributes to the adaptation effect of corticosterone levels after a repeated stress paradigm. However, CB1 KO mice or CB1 antagonist-treated mice also showed significantly elevated blood corticosterone levels [80].
Contradictorily, Roberts et al. showed that male mice that received JZL184 (16 mg/kg i.p.) showed significantly enhanced blood corticosterone levels. Similar results were observed by Aliczki et al. in mice treated with JZL184 (16 mg/kg i.p.) [84]. Roberts et al. argued that high-concentration 2-AG causes rapid desensitization of CB1 signaling. In addition, we consider several possibilities for JZL184-induced increase in corticosterone levels. First, as shown in Table 1, JZL184 has a strong sub-ten nanomolar affinity for the carboxylesterase family of enzymes such as esterase-1 or triglyceride hydrolase 2 (both 5 nM) [85]. To rule out this possibility of off-target effects, other selective and potent inhibitors of MAGL, such as MJN110 or KML29, should be used to confirm MAGL inhibition on circulating corticosterone. Second, effects of chronic and acute administration of MAGL inhibitors may have different effects on circulating corticosterone. Therefore, it is also possible that chronic administration of the drug results in different effects on blood corticosterone levels compared to acute administration.
Table 1.
Potencies of leading FAAH and MAGL inhibitors with sub-hundred nanomolar potency.
| Name | FAAH Inhibition (IC50, nM) | Reversibility | MAGL Inhibition (IC50, nM) |
Reversibility | Inhibition of other Enzymes or Receptors (IC50, nM) | In Vivo Efficacy on Brain |
|---|---|---|---|---|---|---|
| AM3506 [123] | 48 (h) [123] 2.8 ± 0.3 (r) [112] 5.1 ± 0.5 (h) [112] |
Irreversible [112] | 11,000 (h) [123] 383.1 ± 42.3 (h) [112] |
N.A. | N.A. | Yes [123] |
| AM374 (hexadecylsulfonyl fluoride) [124] | 13 (r) [124] 10.2 ± 0.1 (r) [125] |
Irreversible [124] | 6,200 ± 100 (r) [126] 241 ± 17 (r) [127] |
N.A. | 520 (462–586) (r CB1 agonist binding) [124] | N.A. |
| AM6701 [118] | 1.2 ± 0.13 (h) [128] | N.A. | 1.7 (r) [118] 0.9 (h) [118] 20 (monkey) [115] 1.2 ± 0.35 (h) [128] |
Slowly reversible [129] | 7.2 ± 2.5 (h NAAA) [130] 7.7 ± 0.2 (h NAAA) [130] |
N.A. |
| CAY10435 [131] | 0.81 (0.56–1.1) (m) [132] 17 (h) [133] |
Reversible [133] | N.A. | N.A. | 50,000 (21,000–120,000) (m KIAA1363) [132] 83 (69–100) (m TGH) [132] |
Yes [134] |
| CAY10499 [135] | 76 (h) [135] 86 (70–110) (r) [136] 1,300 (r) [114] |
Irreversible [135] | 500 ± 30 (h) [135] 92 (82–100) (h) [136] 480 (h) [114] 144 ± 3 (h) [137] |
Irreversible [137, 138] | 90 (h HSL) [136] | N.A. |
| IDFP [139] | 3 ± 2 (m) [139] | N.A. | 0.8 ± 0.2 (m) [139] 0.107 (r) [140] 0.219 (m) [140] 1.5 (h) [140] |
N.A. | 6,300 ± 2,300 (m AChE) [139] 2 ± 1 (m CB1 agonist binding) [141] |
Yes [139, 141] |
| JJKK-048 [140] | 2,400 (r) [140] 4,800 (h) [140] |
N.A. | 0.214 (r) [140] 0.275 (m) [140] 0.363 (h) [140] |
N.A. | 229 (h ABHD6) [140] | N.A. |
| JNJ-1661010 (Takeda-25) [142, 143] | 10 (r FAAH) [142] 12 (h FAAH) [142] 15–388 (h FAAH) [142] 34 ± 6.5 (r) [143] 33 ± 2.1 (h) [143] |
Partially reversible (80% recovery) [142] | No inhibition (r) [142] | N.A. | 5,700 (h FAAH-2) [142] No inhibition (r COX-1) [142] No inhibition (r COX-2) [142] No inhibition (r NAAA) [142] |
Yes [142] |
| JNJ-40355003 (JNJ-5003) [144] | 1.4 ± 0.41 (h) [144] 33 ± 8.7 (r) [144] |
N.A. | N.A. | N.A. | >10,000 (h FAAH-2) [144] | Yes [144] |
| JW618 [145] | >50,000 (m) [145] >50,000 (r) [145] >50,000 (h) [145] |
N.A. | 123 (91–160) (m) [145] 385 (329–451) (r) [145] 6.9 (4.4–11) (h) [145] |
N.A. | 38 (29–50) (m ABHD6) [145] 13 (8.9–18) (r ABHD6) [145] |
N.A. |
| JW651 [146] | >50,000 (m) [146] | N.A. | 4.5 (m) [146] 38 (23–64) (m) [146] |
N.A. | 10,380 (4,612–23,400) (m ABHD6) [146] | Yes [146] |
| JZL184 [147] |
4,000 (m) [147] 4,690 (m) [148] 3,570 (2,540–5,020) (r) [145] >50,000 (h) [145] 36,308 (h) [149] |
N.A. | 8 (m) [147] 6 (m) [147] 2 (m) [85] 25 (r) [85] 2 (h) [85] 10 (m) [148] 262 (188–363) (r) [145] 3.9 (1.8–8.1) (h) [145] 37 (h) [149] 2,700 (r) [140] 95 (m) [140] |
Irreversible [85, 138] | 3,270 (m ABHD6) [148] 2,940 (1,441–6,010) (r ABHD6) [145] 5 (u ES1) [85] 5 (u TGH2) [85] >100,000 (u HSL) [85] |
Yes [145, 147] |
| Name | FAAH Inhibition (IC50, nM) | Reversibility | MAGL Inhibition (IC50, nM) |
Reversibility | Inhibition of other Enzymes or Receptors (IC50, nM) | In Vivo Efficacy on Brain |
| JZL195 [150] | 13 (m) [150] 12 (r) [150] 110 (h) [150] |
N.A. | 19 (m) [150] 100 (r) [150] 5 (h) [150] 3,700 (r) [140] 3,300 (m) [140] |
N.A. | 50 (m ABHD6) [150] >5,000 (m NTE) [150] 50 (r ABHD6) [150] >5,000 (r NTE) [150] >5,000 (h NTE) [150] >20,000 (h CB1 agonist binding) [150] >20,000 (h CB2 agonist binding) [150] |
Yes [150] |
| JZP-327A [133] | 11 (h) [133] | Slowly reversible [133] | 16% inhibition at 10 μM (h) [133] | N.A. | No inhibition at 10 μM (bovine COX-1) [133] 20% inhibition at 10 μM (h COX-2) [133] |
N.A. |
| KML29 [145] | >50,000 (m) [145] >50,000 (r) [145] >50,000 (h) [145] |
N.A. | 2.5 (m) [145] 15 (11–21) (m) [145] 43 (36–52) (r) [145] 5.9 (4.0–9.9) (h) [145] 0.14 (h) [145] 2.5 (r) [140] 0.851 (m) [140] 3.6 (h) [140] |
N.A. | 4,870 (4,120–5,760) (m ABHD6) [145] 1,600 (1,260–2,040) (r ABHD6) [145] |
Yes [145, 151] |
| LY2183240 [152] | 2.1 (r) [115] 12.4 (8.3–18.6) (m) [153] 14.0 ± 0.662 (r) [154] 37.3 ± 5.4 (h) [155] 5 (r) [114] |
Irreversible [153] | 8,100 (monkey) [115] 5.3 (3.9–7.3) (u) [153] 54.4 ± 5.2 (h) [155] 290 (h) [114] |
N.A. | 0.27 ± 0.029 (AEA uptake in RBL-2H3 cells) [152] 0.09 (0.07–0.10) (u ABHD6) [153] 8.2 (6.1–11) (u KIAA1363) [153] |
Yes [152, 153] |
| MAFP [156, 157] | 2.5 (m) [157] 1–3 (m, r) [156] 6 (porcine) [158] 0.10 ± 0.02 (m) [159] 0.33 ± 0.07 (h) [155] |
Irreversible [156, 157] | 2–3 (porcine) [158] 800 ± 50 (r) [126] 2.2 ± 0.3 (r) [127] 76 ±4 (h) [135] 26.3 ± 9.95 (h) [155] 62–160 (h) [160] 1.0 (r) [140] 1.8 (m) [140] |
Irreversible [138, 155] | 20 (r CB1 agonist binding) [157] 0.60 ± 0.10 (m NTE) [113] 124 ± 17 (m AChE) [159] |
Yes [134] |
| MJN110 [161] | >50,000 (m) [161] >50,000 (h) [161] >100,000 (m) [146] |
N.A. | 2.1 (m) [161] 9.1 (h) [161] 0.43 (h) [161] 9.5 (5.7–15.8) (m) [146] |
N.A. | 260 (174–394) (m ABHD6) [146] | Yes [161] |
| ML30 [149] | 562 (h) [149] | N.A. | 0.54 (h) [149] 4.4 (r) [140] 1.9 (m) [140] 1.5 (h) [140] |
Irreversible [149] | N.A. | N.A. |
| N-arachidonoyl maleimide [162] | >10,000 (r) [114] 2,180 ± 620 (h) [155] |
N.A. | 140 (r) [162] 10,500 ± 3,800 (h) [135] 2,800 (h) [118] 1,120 ± 140 (h) [155] 46 ± 7 (r) [163] No inhibition at 10 μM (h) [114] 148 (r) [140] 324 (m) [140] |
Irreversible [138, 164] | N.A. | Yes [10] |
| Name | FAAH Inhibition (IC50, nM) |
Reversibility | MAGL Inhibition (IC50, nM) |
Reversibility | Inhibition of other Enzymes or Receptors (IC50, nM) | In Vivo Efficacy on Brain |
| OL-135 [165] | 206 (126–336) (h FAAH-1) [41] 208–297 (h) [166] 2.1 (1.5–3.0) (m) [120] 1,100 (h FAAH-HEK whole cell assay) [167] |
Reversible [165, 166] | >100,000 (m) [120] | N.A. | 13.4 (11.3–15.9) (h FAAH-2) [41] 4 (h FAAH-2) [142] 620 (490–780) (m TGH) [120] >100,000 (m AAD) [120] >100,000 (m CE-1) [120] >100,000 (m LPL) [120] >100,000 (m KIAA1363) [120] 22% inhibition at 10 μM (r CB1) [120] 30% inhibition at 10 μM (h CB2) [120] |
Yes [120] |
| OL-92 [165] | 0.28 (0.15–0.52) (m) [120] | Reversible [120] | >100,000 (m) [120] | N.A. | >100,000 (m TGH) [120] >100,000 (m AAD) [120] >100,000 (m CE-1) [120] >100,000 (m LPL) [120] 17000 (9500–28000) (m KIAA1363) [120] |
No, not active in vivo [120] |
| PF-04457845 (PF-7845) [168] | 7.2–50.4 (h) [169] 7.4–43.1 (r) [169] 2.5 (m) [170] 1.0 (h) [170] |
Irreversible [168] | No inhibition at 10 μM (m) [170] No inhibition at 10 μM (h) [170] |
N.A. | 30,000 (h CYP1A2) [168] 9,800 (h CYP2C9) [168] 12600 (h CYP2C19) [168] 14,500 (h CYP2D6) [168] 30,000 (h CYP3A4) [168] No inhibition at 10 μM (m ABHD6) [170] |
Yes [169, 170] |
| PF-3845 [171] | 18 (14–23) (h FAAH-1) [171] | Irreversible [171] | N.A. | N.A. | >10,000 (h FAAH-2) [171] | Yes [86, 171] |
| PF-750 [166] | 16.2–595 (h) [166] 320 (h) [133] |
Irreversible [166] | N.A. | N.A. | No inhibition at 50 μM (m liver serine hydrolases) [166] | Yes [166] |
| SA-57 [170] | 3.2 (m) [170] 1.9 (h) [170] |
Irreversible [170]. | 410 (m) [170] 1,400 (h) [170] |
Irreversible [170] | 850 (m ABHD6) [170] | Yes [170] |
| ST4070 [172] | 9 (m) [172] | Reversible [172] | >10,000 (m) [172] | N.A. | >10,000 (m CB1 agonist binding) [172] >10,000 (m CB2 agonist binding) [172] >10,000 (m TRPV1) [172] 100 (h AMT) [172] >10,000 (m NAPE-PLD) [172] >10,000 (m DAGL) [172] |
Yes [173] |
| URB597 (KDS-4103) [7] | 3.8 (2.9–5.0) (r) [127] 45 (31–65) (m) [174] 33.5 (h) [175] 26.3 (h) [175] 109 (h) [176] 119 (r) [176] 4.6 ± 1.6 (r) [7] 0.5 ± 0.05 (r) [7] 113 (79–170) (m) [120] 101–505 (h FAAH-1) [41] 157 ± 5.23 (r) [154] 109–986 (h) [166] 7.7 ± 1.5 (r) [177] 31 (h FAAH-HEK whole cell assay) [167] |
Irreversible [7, 142, 166] | >100,000 (m) [120] >30,000 (r) [7] |
N.A. | 196 (119–322) (m CE-6) [174] >100,000 (m ES31) [174] 5.0–26.3 (h FAAH-2) [41] 7 (h FAAH-2) [142] 192 (96–320) (m TGH) [120] >100,000 (m AAD) [120] >100,000 (m CE-1) [120] >100,000 (m LPL) [120] >100,000 (m KIAA1363) [120] >30,000 (h AMT) [7] >100,000 (r CB1 agonist binding) [7] >100,000 (u CB2 agonist binding) [7] |
Yes [153, 166, 174, 177] |
| URB880 [117] | 0.63 ± 0.04 (r) [117] | N.A. | No inhibition at 100 μM (r) [117] | N.A. | N.A. | N.A. |
| URB937 [178] | 26.8 ± 4.9 (r) [178] | Irreversible [119] | >100,000 (r) [178] | N.A. | N.A. | No, peripherally restricted effects [178] |
Abbreviations: FAAH, fatty acid amide hydrolase; MAGL, monoacylglycerol lipase; IC50, 50% inhibitory concentration; N.A., not available; h, human; r, rat; m, mouse; u, undescribed species; CB, cannabinoid receptor; NAAA, N-acylethanolamine-hydrolyzing acid amidase; HSL, hormone-sensitive lipase; AChE, acetylcholinesterase; ABHD, α/β-hydrolase domain 6; COX, cyclooxygenase; ES, esterase; TGH, triacylglycerol hydrolase; NTE, neuropathy target esterase; KIAA1363, ether-lipid metabolic enzyme; RBL, rat basophilic leukemia; HEK, human embryonic kidney; AAD, arylacetamide deacetylase; CE, carboxylesterase; LPL, lipoprotein lipase; CYP, cytochrome P450; AMT, anandamide membrane transporter The data are presented as mean, mean ± standard deviation, or mean (range).
Wiebelhaus et al. reported that URB597 administration increased other NAEs (e.g., PEA and OEA) as well as AEA in several brain regions [86]. Hence, NAEs might play a role in the regulation of HPA axis functions through their receptors (e.g., PPARs) [87]) rather than endocannabinoid receptors. Moreover, since AEA is a ligand for TRPV1 (Fig. (1)), the observed effect of URB597 on basal corticosterone might be caused by TRPV1 signaling. To clarify the individual roles of these factors in HPA axis regulation, further studies are warranted.
Endocannabinoids may be correlated to stress responses and may alter HPA axis responses to psychological or physiological stressors. Repeated administration of FAAH inhibitors may modulate HPA axis function, but effects of MAGL inhibitors are still elusive. The effects of chronic administration of FAAH/MAGL inhibitors and off-target effects of the inhibitors must also be confirmed extensively. CB1 may profoundly relate with the stress responsive corticosterone release and stress adaptation.
2.4. The Dopaminergic System and Endocannabinoids
Abnormality of the dopaminergic system has been implicated in MDD pathophysiology. Klimek et al. showed reduced dopamine transporter levels and increasing numbers of D2/D3 receptors in the amygdala of postmortem brain samples from MDD patients [88]. Another study investigating CSF monoamine metabolite levels showed a decrease in CSF levels of homovanillic acid (HVA), the primary dopamine metabolite, implying that dopamine turnover is blunted [89]. Sulpiride exerts rapid antidepressive effects at low doses by activating dopamine transmission through dopamine autoreceptor blockades [90]. Other studies exist in which MDD patients received electroconvulsive therapy or vagus nerve stimulation and showed increases in CSF HVA levels [91, 92]. We showed that adjunctive administration of a dopamine agonist pramipexole significantly ameliorated depression severity in MDD patients who had failed to respond to treatment with SSRIs [93]. Different classes of antidepressants are reported to have common effects: inhibition of dopamine reuptake at synaptosome [94] and increase of extracellular dopamine levels [95]. Collectively, these facts support the dopaminergic dysregulation in MDD pathophysiology.
A study using a social isolation paradigm, an animal model of depression [96], in adolescent rats showed decrease in dopamine D2 receptor expressions in the adult rat brain [97]. In our study, the dopamine D2 receptor agonist, cabergoline, exerted antidepressant-like effects upon acute and sub-chronic administration in Wistar rats and Wistar Kyoto rats [98]. Mesocortical and mesolimbic pathways are considered to be related to depression, and these pathways originate in the ventral tegmental area (VTA). Since VTA regulation was related to the projection of brain-derived neurotrophic factor (BDNF) positive glutamatergic neurons [99], BDNF expression levels in the other regions that innervate the VTA (e.g., cortex, hippocampus, and amygdala) are also thought to be important for the dopaminergic system. Our study using cabergoline revealed significant increases in hippocampal BDNF levels after a two-week administration of the drug [98]. Murillo-Rodriguez et al. reported that male Wistar rats infused with URB597, OEA, or PEA by i.c.v injection [100] or microinjection into the lateral hypothalamus or dorsal raphe nuclei [101] showed significantly elevated extracellular dopamine concentrations in the nucleus accumbens (NAc) and wakefulness as well as significantly decreased slow-wave sleep [100, 101], suggesting activated dopaminergic functions. As shown in Fig. (1), both OEA and PEA are catalyzed by FAAH (PEA is also selectively catalyzed by NAE-hydrolyzing acid amidase), suggesting the presence of a mediating mechanism other than cannabinoid receptors, such as PPARs, whose agonists are OEA and PEA (Fig. (1)). In our study, CSF EA levels showed significantly positive correlations with CSF HVA concentrations [51]. Since EA is a component of NAEs, CSF EA levels might reflect central NAEs or dopamine turnover.
Wiebelhaus et al. reported that, even in systemic injection, inhibition of FAAH increased AEA, PEA, and OEA levels in several brain regions. On the other hand, MAGL inhibitors increased 2-AG in the NAc and VTA [86]. Notably, when MAGL inhibitors were administered, AEA concentrations in the NAc decreased significantly.
Nader et al. found that URB597 or JZL184 administration prevented methamphetamine-induced neurotoxicity. CB2 antagonist blocked such neuroprotective effects, but CB1 antagonist in mice did not [102]. Zhang et al. reported that CB2 receptors were expressed in the VTA dopamine neurons, and CB2 agonists inhibited the VTA dopamine neuron firing in WT or CB1-/- mice but not in CB2-/- mice [28]. These results suggest that CB2 signaling may prevent excitotoxicity in the VTA dopaminergic neurons. In the intracranial self-stimulation (ICSS) paradigm using an electrode implanted at the right middle forebrain bundle, the FAAH inhibitor, MAGL inhibitors, and FAAH/MAGL co-inhibitor all caused a dose-dependent reduction in the number of ICSS in mice while cocaine induced a dose-dependent increase [86]. In the food restriction paradigm, extracellular dopamine levels increased drastically (about 400%) [103], and the elevation of dopamine levels was completely blocked by CB1 antagonist SR141716, suggesting that CB1 is essential for dopamine elevation in food restriction.
These indicate that administrations of FAAH inhibitors might elevate dopaminergic transmission in the mesolimbic/mesocortical pathways. CB2 may have protective effects against excitotoxicity in dopamine neurons and addictive behavior. In other words, CB2 may have suppressive effects on the dopaminergic system. However, for “triggered” induction of dopamine, CB1 plays a pivotal role. CB1 and CB2 might work in the opposite direction, in which case the balance of both receptors might be important.
3. PHARMACOLOGICAL APPLICATION
3.1. Future Prospects of FAAH and MAGL Inhibitors
To date, numerous compounds have been synthesized as FAAH/MAGL inhibitors, and several of these have been used for pre-clinical studies. Vinod et al. reported that sub-chronic URB597 administration (0.3 mg/kg i.p. for seven days) exerts antidepressant-like effects on the Wistar Kyoto rat, which is a genetic animal model for MDD [104]. Adamczyk et al. reported that a single administration of URB597 (0.03–0.3 mg/kg i.p.) significantly reduced immobility time in the FST, and this reduction was nullified by the CB1 antagonist rimonabant [105]. Bortolato et al. reported that chronic URB597 (0.3 mg/kg i.p. for five weeks) reversed decrease of sucrose intake induced by chronic unpredictable mild stress in the sucrose preference (or consumption) test [106]. The same research group reported that repeated administration of JZL184 (8 mg/kg i.p. every two days for three or four weeks) rescued the increase in immobility time in the FST induced by chronic unpredictable mild stress [107, 108]. Naidu et al. reported that a single administration of URB597 (0.1 mg/kg i.p.) significantly decreased immobility time in a modified tail suspension test in mice [109]. However, contradictive negative results have been reported. Lomazzo et al. reported that a repeated administration (one week or more) of URB597 (1 mg/kg/day i.p.) and/or JZL184 (8 mg/kg/day i.p.) significantly increased immobility time in the FST in mice [110]. Since chronic, but not acute, administration of URB597 reduced basal corticosterone levels [82], chronic administration of inhibitors seems to be more effective in inducing their antidepressant-like effects. Because Bortolato et al. showed that URB597 required at least five weeks to show its antidepressant-like effect, we expect that the FAAH and/or MAGL inhibitors may require several weeks for clear antidepressant-like effects.
We summarize in Table 1 the inhibitors of FAAH and/or MAGL with the sub-hundred nanomolar potencies that are currently available and have general names. The detailed potencies of first-generation inhibitors were described in Minkkilä’s review [111]. Hundreds of chemicals other than the compounds presented in Table 1 were developed as analogs of previously developed or originally synthesized compounds [111-117]. We also excluded those compounds with no confirmed detailed information from this table such as SSR411298, MM-433593, and JNJ-42165279. Among the compounds listed in Table 1, URB597 and JZL184 are the most frequently used drugs in pre-clinical studies. Recently, inhibitors have been developed with higher potencies and selectivity, and these newly developed drugs can be useful tools for clinical or preclinical studies. Among MAGL inhibitors, for example, KML29, JW651, and MJN110 showed superior potency and selectivity for MAGL over FAAH compared with previously developed compounds (e.g., JZL184). Among FAAH inhibitors, OL-135, JNJ-1661010 (Takeda-25), and ST4070 showed highly selective potentials for FAAH over MAGL. URB597 inhibits FAAH in an irreversible manner, whereas OL-135 and ST4070 inhibition is reversible, and JNJ-1661010 (Takeda-25) inhibition is covalent but partially reversible. Although both URB597 and OL-135 inhibit FAAH-2 as well as FAAH-1, JNJ-1661010 has a hundred times more selectivity for FAAH-1 over FAAH-2. Therefore, JNJ-1661010 (Takeda-25) can be a useful tool for testing functions of FAAH-1 and -2. Selectivity of ST4070 for FAAH-2 should be confirmed. Many compounds inhibit both FAAH and MAGL and are high potency drugs that can be used as “dual blockers” for FAAH and MAGL. Fluorophosphonates such as MAFP and IDFP have strong inhibitory potency for FAAH and MAGL. However, these fluorophosphonates also inhibit CB1 receptor agonist binding and other serine hydrolases, as shown in Table 1. These characteristics could be complicating factors in experiments. AM6701 [118] and N,N-dimethyl-5-(4-phenoxyphenyl)-2H-tetrazole-2-carboxamide [114] were developed as potent dual blockers of FAAH and MAGL. Selectivity of these new inhibitors toward other serine hydrolases needs to be clarified. In addition, for other FAAH/MAGL inhibitors, information regarding potencies toward enzymes other than target enzymes remains insufficient.
Inhibitors belong to two large classes: reversible and irreversible action. Covalent, irreversible inhibitors generally have strong potency but also have a greater risk of off-target effects. Currently, non-covalent and reversible or covalent but partially reversible inhibitors with high potency and selectivity are being developed. These may reduce the risks of off-target effects and side effects due to enzyme inhibition; therefore, reversible drugs have advantages in drug development. In addition, in vivo efficacies of all compounds have not been determined for the brain. For example, URB937 has good potency for FAAH in the periphery but not in the brain because of ATP-binding cassette sub-family G member 2 (ABCG2) transporter functions at the blood–brain barrier [119]. OL-92 also showed good potency for FAAH in vitro; however, there was no evidence for its pharmacological action in the brain [120]. Therefore, confirmation of brain efficacy of compounds in vivo is important.
In humans, a clinical trial using an FAAH inhibitor for MDD was performed from 2008 to 2010 by Sanofi (Sanofi-Aventis) in which older patients with MDD were treated orally with the FAAH inhibitor SSR411298 for eight weeks [121]. Of these patients, 106 were in the placebo arm, 316 the SSR411298 arms (10 mg arm: 105 patients; 50 mg arm: 106; 200 mg arm: 105), and 103 the 10 mg escitalopram arm. After the trial period, the severity of depression in patients with MDD was assessed using the 17-item HRSD. According to the results published on Sanofi’s website [122], no significant differences were observed in the changes of mean HRSD scores between any doses of SSR411298 and placebo from baseline to Day 56. However, we do not know detailed information of SSR411298 and statistical outcomes of the clinical trial. AEA could alter anxiety symptoms, and FAAH inhibitors may have anxiolytic effects in MDD. Moreover, AEA could affect blood glucocorticoid levels in response to psychological or physiological stress, and FAAH inhibitors may be effective in altering stress responses. Since dominant endocannabinoid is 2-AG, MAGL may exert more potent effects on MDD symptoms than FAAH. Clinical trials using MAGL inhibitor for MDD should be performed. In parallel with the pre-clinical studies using FAAH or MAGL inhibitors, further clinical trials are needed.
CONCLUSION
We have reviewed possible relationships between MDD and the endocannabinoid system and potential therapeutic effects of FAAH or MAGL inhibitors on the illness. However, the number of relevant studies is still limited in both humans and animals. Accumulation of further extensive evidence is needed. Since AEA has low affinity for CB2, and the main physiological role for CB2 is through 2-AG, MAGL inhibitor can be considered to play a principal role with CB2. On the other hand, since FAAH inhibitors also suppress degradation of other NAEs, the efficacy of FAAH may be affected by the elevation of NAEs. TRPV1 might also mediate the effects of FAAH/MAGL inhibitors because both AEA and 2-AG are ligands for TRPV1. Further investigation into their detailed roles is needed in the future.
We have discussed several hypotheses for the pathophysiology of MDD. First, since the levels of several pro-inflammatory cytokines such as IL-6 and TNF-α are elevated in patients with MDD, CB2 activation may reduce stress-induced inflammatory responses. Therefore, MAGL inhibitors may alter inflammation-related MDD symptoms. Second, patients with MDD show disturbance in the HPA axis compared to healthy participants. Repeated administration of FAAH inhibitors may be beneficial for alteration of HPA axis functions, but details are elusive. In future research, the effects of drugs (i.e., FAAH and MAGL inhibitors) with high selectivity toward HPA axis functions in repeated and acute administrations should be investigated. Third, the pathophysiology of MDD is known to be related to dopaminergic dysregulation. Several lines of evidence suggest that endocannabinoids regulate the dopaminergic system by suppression and/or activation. CB1 plays a key role in activating the dopaminergic system, while CB2 may prevent excessive activation of dopaminergic neurons. The use of selective inhibitors may be beneficial for regulating the dopaminergic system through activation or suppression.
Although several new inhibitors with strong potency and selectivity have been developed as summarized in Table 1, more non-covalent reversible and potent inhibitors should be developed and tested in pre-clinical and clinical studies. Not only FAAH inhibitors but also MAGL inhibitors or dual blockers should be examined in future clinical trials.
ACKNOWLEDGEMENTS
This study was supported by the Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (24-11), Grant-in-Aid for Scientific Research (A) (25253075) from the Japan Society for the Promotion of Science (JSPS), Health and Labour Sciences Research Grants (KHC1214; H24-seishin-ippan-001), and grants from the SENSHIN Medical Research Foundation.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Ferrari A.J., Charlson F.J., Norman R.E., Patten S.B., Freedman G., Murray C.J., Vos T., Whiteford H.A. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547. doi: 10.1371/journal.pmed.1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrari A.J., Charlson F.J., Norman R.E., Flaxman A.D., Patten S.B., Vos T., Whiteford H.A. The epidemiological modelling of major depressive disorder: application for the Global Burden of Disease Study 2010. PLoS One. 2013;8(7):e69637. doi: 10.1371/journal.pone.0069637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rush A.J., Trivedi M.H., Wisniewski S.R., Stewart J.W., Nierenberg A.A., Thase M.E., Ritz L., Biggs M.M., Warden D., Luther J.F., Shores-Wilson K., Niederehe G., Fava M., STAR*D Study Team Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N. Engl. J. Med. 2006;354(12):1231–1242. doi: 10.1056/NEJMoa052963. [DOI] [PubMed] [Google Scholar]
- 4.Gaoni Y., Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964;86(8):1646–1647. doi: 10.1021/ja01062a046. [DOI] [Google Scholar]
- 5.Moreira F.A., Grieb M., Lutz B. Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: focus on anxiety and depression. 2009. [DOI] [PubMed]
- 6.Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat. Rev. Drug Discov. 2008;7(5):438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 7.Kathuria S., Gaetani S., Fegley D., Valiño F., Duranti A., Tontini A., Mor M., Tarzia G., La Rana G., Calignano A., Giustino A., Tattoli M., Palmery M., Cuomo V., Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9(1):76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 8.Piomelli D., Tarzia G., Duranti A., Tontini A., Mor M., Compton T.R., Dasse O., Monaghan E.P., Parrott J.A., Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006;12(1):21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ignatowska-Jankowska B.M., Ghosh S., Crowe M.S., Kinsey S.G., Niphakis M.J., Abdullah R.A., Tao Q., O’ Neal S.T., Walentiny D.M., Wiley J.L., Cravatt B.F., Lichtman A.H. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 2014;171(6):1392–1407. doi: 10.1111/bph.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burston J.J., Sim-Selley L.J., Harloe J.P., Mahadevan A., Razdan R.K., Selley D.E., Wiley J.L. N-arachidonyl maleimide potentiates the pharmacological and biochemical effects of the endocannabinoid 2-arachidonylglycerol through inhibition of monoacylglycerol lipase. J. Pharmacol. Exp. Ther. 2008;327(2):546–553. doi: 10.1124/jpet.108.141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devane W.A., Dysarz F.A., III, Johnson M.R., Melvin L.S., Howlett A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988;34(5):605–613. [PubMed] [Google Scholar]
- 12.Matsuda L.A., Lolait S.J., Brownstein M.J., Young A.C., Bonner T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 13.Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 14.Devane W.A., Hanus L., Breuer A., Pertwee R.G., Stevenson L.A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 15.Sugiura T., Kondo S., Sukagawa A., Nakane S., Shinoda A., Itoh K., Yamashita A., Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215(1):89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 16.Mechoulam R., Ben-Shabat S., Hanus L., Ligumsky M., Kaminski N.E., Schatz A.R., Gopher A., Almog S., Martin B.R., Compton D.R., Pertwee R.G., Griffin G., Bayewitch M., Barg J., Vogel Z. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50(1):83–90. doi: 10.1016/0006-2952(95)00109-D. [DOI] [PubMed] [Google Scholar]
- 17.Sugiura T., Kondo S., Sukagawa A., Tonegawa T., Nakane S., Yamashita A., Ishima Y., Waku K. Transacylase-mediated and phosphodiesterase-mediated synthesis of N-arachidonoylethanolamine, an endogenous cannabinoid-receptor ligand, in rat brain microsomes. Comparison with synthesis from free arachidonic acid and ethanolamine. Eur. J. Biochem. 1996;240(1):53–62. doi: 10.1111/j.1432-1033.1996.0053h.x. [DOI] [PubMed] [Google Scholar]
- 18.Sugiura T., Kobayashi Y., Oka S., Waku K. Biosynthesis and degradation of anandamide and 2-arachidonoylglycerol and their possible physiological significance. Prostaglandins Leukot. Essent. Fatty Acids. 2002;66((2-3)):173–192. doi: 10.1054/plef.2001.0356. [DOI] [PubMed] [Google Scholar]
- 19.Llano I., Leresche N., Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6(4):565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- 20.Pitler T.A., Alger B.E. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13(6):1447. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 21.Kreitzer A.C., Regehr W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29(3):717–727. doi: 10.1016/S0896-6273(01)00246-X. [DOI] [PubMed] [Google Scholar]
- 22.Ohno-Shosaku T., Maejima T., Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29(3):729–738. doi: 10.1016/S0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 23.Wilson R.I., Nicoll R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410(6828):588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 24.Kano M., Ohno-Shosaku T., Hashimotodani Y., Uchigashima M., Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009;89(1):309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 25.Sugiura T., Waku K. Cannabinoid receptors and their endogenous ligands. J. Biochem. 2002;132(1):7–12. doi: 10.1093/oxfordjournals.jbchem.a003200. [DOI] [PubMed] [Google Scholar]
- 26.Grotenhermen F. Pharmacology of cannabinoids. Neuroendocrinol. Lett. 2004;25(1-2):14–23. [PubMed] [Google Scholar]
- 27.Núñez E., Benito C., Pazos M.R., Barbachano A., Fajardo O., González S., Tolón R.M., Romero J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004;53(4):208–213. doi: 10.1002/syn.20050. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H.Y., Gao M., Liu Q.R., Bi G.H., Li X., Yang H.J., Gardner E.L., Wu J., Xi Z.X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA. 2014;111(46):E5007–E5015. doi: 10.1073/pnas.1413210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J., Bie B., Yang H., Xu J.J., Brown D.L., Naguib M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging. 2013;34(3):791–804. doi: 10.1016/j.neurobiolaging.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Zarruk J.G., Fernandez-Lopez D., Garcia-Yebenes I., Garcia-Gutierrez M.S., Vivancos J., Nombela F., Torres M., Burguete M.C., Manzanares J., Lizasoain I., Moro M.A. 2012. [DOI] [PubMed]
- 31.Merighi S., Gessi S., Varani K., Fazzi D., Mirandola P., Borea P.A. Cannabinoid CB(2) receptor attenuates morphine-induced inflammatory responses in activated microglial cells. Br. J. Pharmacol. 2012;166(8):2371–2385. doi: 10.1111/j.1476-5381.2012.01948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zoppi S., Madrigal J.L., Caso J.R., García-Gutiérrez M.S., Manzanares J., Leza J.C., García-Bueno B. Regulatory role of the cannabinoid CB2 receptor in stress-induced neuroinflammation in mice. Br. J. Pharmacol. 2014;171(11):2814–2826. doi: 10.1111/bph.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong W., Li H., Tuma R.F., Ganea D. Selective CB2 receptor activation ameliorates EAE by reducing Th17 differentiation and immune cell accumulation in the CNS. Cell. Immunol. 2014;287(1):1–17. doi: 10.1016/j.cellimm.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmöle A.C., Lundt R., Ternes S., Albayram O., Ulas T., Schultze J.L., Bano D., Nicotera P., Alferink J., Zimmer A. 2014. [DOI] [PubMed]
- 35.Gonsiorek W., Lunn C., Fan X., Narula S., Lundell D., Hipkin R.W. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol. Pharmacol. 2000;57(5):1045–1050. [PubMed] [Google Scholar]
- 36.Sugiura T., Kondo S., Kishimoto S., Miyashita T., Nakane S., Kodaka T., Suhara Y., Takayama H., Waku K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 2000;275(1):605–612. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- 37.Di Marzo V., De Petrocellis L. Why do cannabinoid receptors have more than one endogenous ligand? 2012. [DOI] [PMC free article] [PubMed]
- 38.Amaya F., Shimosato G., Kawasaki Y., Hashimoto S., Tanaka Y., Ji R.R., Tanaka M. Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain. 2006;124(1-2):175–183. doi: 10.1016/j.pain.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Zygmunt P.M., Ermund A., Movahed P., Andersson D.A., Simonsen C., Jönsson B.A., Blomgren A., Birnir B., Bevan S., Eschalier A., Mallet C., Gomis A., Högestätt E.D. Monoacylglycerols activate TRPV1--a link between phospholipase C and TRPV1. PLoS One. 2013;8(12):e81618. doi: 10.1371/journal.pone.0081618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deutsch D.G., Chin S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. 1993. [DOI] [PubMed]
- 41.Wei B.Q., Mikkelsen T.S., McKinney M.K., Lander E.S., Cravatt B.F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006;281(48):36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- 42.Blankman J.L., Simon G.M., Cravatt B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007;14(12):1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karlsson M., Contreras J.A., Hellman U., Tornqvist H., Holm C. 1997. [DOI] [PubMed]
- 44.Sun Y., Alexander S.P., Garle M.J., Gibson C.L., Hewitt K., Murphy S.P., Kendall D.A., Bennett A.J. 2007. [DOI] [PMC free article] [PubMed]
- 45.Bouaboula M., Hilairet S., Marchand J., Fajas L., Le Fur G., Casellas P. Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 2005;517(3):174–181. doi: 10.1016/j.ejphar.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 46.O’Sullivan S.E. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007;152(5):576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Sullivan S.E., Kendall D.A. Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiology. 2010;215(8):611–616. doi: 10.1016/j.imbio.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Hungund B.L., Vinod K.Y., Kassir S.A., Basavarajappa B.S., Yalamanchili R., Cooper T.B., Mann J.J., Arango V. 2004. [DOI] [PubMed]
- 49.Hill M.N., Miller G.E., Carrier E.J., Gorzalka B.B., Hillard C.J. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology. 2009;34(8):1257–1262. doi: 10.1016/j.psyneuen.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill M.N., Miller G.E., Ho W.S., Gorzalka B.B., Hillard C.J. Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry. 2008;41(2):48–53. doi: 10.1055/s-2007-993211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogawa S., Hattori K., Sasayama D., Yokota Y., Matsumura R., Matsuo J., Ota M., Hori H., Teraishi T., Yoshida S., Noda T., Ohashi Y., Sato H., Higuchi T., Motohashi N., Kunugi H. Reduced cerebrospinal fluid ethanolamine concentration in major depressive disorder. 2015. [DOI] [PMC free article] [PubMed]
- 52.Hill M.N., Kumar S.A., Filipski S.B., Iverson M., Stuhr K.L., Keith J.M., Cravatt B.F., Hillard C.J., Chattarji S., McEwen B.S. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. 2013. [DOI] [PMC free article] [PubMed]
- 53.Gunduz-Cinar O., MacPherson K.P., Cinar R., Gamble-George J., Sugden K., Williams B., Godlewski G., Ramikie T.S., Gorka A.X., Alapafuja S.O., Nikas S.P., Makriyannis A., Poulton R., Patel S., Hariri A.R., Caspi A., Moffitt T.E., Kunos G., Holmes A. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol. Psychiatry. 2013;18(7):813–823. doi: 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang K.P., Gerber A.L., Sipe J.C., Cravatt B.F. Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum. Mol. Genet. 2004;13(18):2113–2119. doi: 10.1093/hmg/ddh216. [DOI] [PubMed] [Google Scholar]
- 55.Ho W.S., Hill M.N., Miller G.E., Gorzalka B.B., Hillard C.J. Serum contents of endocannabinoids are correlated with blood pressure in depressed women. Lipids Health Dis. 2012;11:32. doi: 10.1186/1476-511X-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giuffrida A., Leweke F.M., Gerth C.W., Schreiber D., Koethe D., Faulhaber J., Klosterkotter J., Piomelli D. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. 2004. [DOI] [PubMed]
- 57.Sasayama D., Hattori K., Wakabayashi C., Teraishi T., Hori H., Ota M., Yoshida S., Arima K., Higuchi T., Amano N., Kunugi H. Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J. Psychiatr. Res. 2013;47(3):401–406. doi: 10.1016/j.jpsychires.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 58.Hiles S.A., Baker A.L., de Malmanche T., Attia J. A meta-analysis of differences in IL-6 and IL-10 between people with and without depression: exploring the causes of heterogeneity. Brain Behav. Immun. 2012;26(7):1180–1188. doi: 10.1016/j.bbi.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y., Ho R.C., Mak A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J. Affect. Disord. 2012;139(3):230–239. doi: 10.1016/j.jad.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 60.Dowlati Y., Herrmann N., Swardfager W., Liu H., Sham L., Reim E.K., Lanctôt K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry. 2010;67(5):446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 61.Hannestad J., DellaGioia N., Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology. 2011;36(12):2452–2459. doi: 10.1038/npp.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogawa S., Fujii T., Koga N., Hori H., Teraishi T., Hattori K., Noda T., Higuchi T., Motohashi N., Kunugi H. Plasma L-tryptophan concentration in major depressive disorder: new data and meta-analysis. J. Clin. Psychiatry. 2014;75(9):e906–e915. doi: 10.4088/JCP.13r08908. [DOI] [PubMed] [Google Scholar]
- 63.Schwarcz R., Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. 2002. [DOI] [PubMed]
- 64.Hodes G.E., Pfau M.L., Leboeuf M., Golden S.A., Christoffel D.J., Bregman D., Rebusi N., Heshmati M., Aleyasin H., Warren B.L., Lebonté B., Horn S., Lapidus K.A., Stelzhammer V., Wong E.H., Bahn S., Krishnan V., Bolaños-Guzman C.A., Murrough J.W., Merad M., Russo S.J. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl. Acad. Sci. USA. 2014;111(45):16136–16141. doi: 10.1073/pnas.1415191111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Erta M., Quintana A., Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012;8(9):1254–1266. doi: 10.7150/ijbs.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang L., Li F.F., Han Y.C., Jia B., Ding Y. Cannabinoid Receptor CB2 Is Involved in Tetrahydrocannabinol-Induced Anti-Inflammation against Lipopolysaccharide in MG-63 Cells. 2015. [DOI] [PMC free article] [PubMed]
- 67.Qian H., Yi J., Zhou J., Zhao Y., Li Y., Jin Z., Ding Y. Activation of cannabinoid receptor CB2 regulates LPS-induced pro-inflammatory cytokine production and osteoclastogenic gene expression in human periodontal ligament cells. Sci. Res. 2013;3(1):44–51. doi: 10.4236/ojst.2013.31009. [DOI] [Google Scholar]
- 68.Warner T.D., Mitchell J.A. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 2004;18(7):790–804. doi: 10.1096/fj.03-0645rev. [DOI] [PubMed] [Google Scholar]
- 69.Röhrenbeck A.M., Bette M., Hooper D.C., Nyberg F., Eiden L.E., Dietzschold B., Weihe E. Upregulation of COX-2 and CGRP expression in resident cells of the Borna disease virus-infected brain is dependent upon inflammation. Neurobiol. Dis. 1999;6(1):15–34. doi: 10.1006/nbdi.1998.0225. [DOI] [PubMed] [Google Scholar]
- 70.Müller N., Schwarz M.J., Dehning S., Douhe A., Cerovecki A., Goldstein-Müller B., Spellmann I., Hetzel G., Maino K., Kleindienst N., Möller H.J., Arolt V., Riedel M. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry. 2006;11(7):680–684. doi: 10.1038/sj.mp.4001805. [DOI] [PubMed] [Google Scholar]
- 71.Faridhosseini F., Sadeghi R., Farid L., Pourgholami M. Celecoxib: a new augmentation strategy for depressive mood episodes. A systematic review and meta-analysis of randomized placebo-controlled trials. Hum. Psychopharmacol. 2014;29(3):216–223. doi: 10.1002/hup.2401. [DOI] [PubMed] [Google Scholar]
- 72.Kurhe Y., Mahesh R., Gupta D. Effect of a selective cyclooxygenase type 2 inhibitor celecoxib on depression associated with obesity in mice: an approach using behavioral tests. 2014. [DOI] [PubMed]
- 73.Santiago R.M., Barbiero J., Martynhak B.J., Boschen S.L., da Silva L.M., Werner M.F., Da Cunha C., Andreatini R., Lima M.M., Vital M.A. Antidepressant-like effect of celecoxib piroxicam in rat models of depression. J Neural Transm (Vienna) 2014;121(6):671–682. doi: 10.1007/s00702-014-1159-5. [DOI] [PubMed] [Google Scholar]
- 74.Kunugi H., Ida I., Owashi T., Kimura M., Inoue Y., Nakagawa S., Yabana T., Urushibara T., Kanai R., Aihara M., Yuuki N., Otsubo T., Oshima A., Kudo K., Inoue T., Kitaichi Y., Shirakawa O., Isogawa K., Nagayama H., Kamijima K., Nanko S., Kanba S., Higuchi T., Mikuni M. Assessment of the dexamethasone/CRH test as a state-dependent marker for hypothalamic-pituitary-adrenal (HPA) axis abnormalities in major depressive episode: a Multicenter Study. Neuropsychopharmacology. 2006;31(1):212–220. doi: 10.1038/sj.npp.1300868. [DOI] [PubMed] [Google Scholar]
- 75.Parker K.J., Schatzberg A.F., Lyons D.M. Neuroendocrine aspects of hypercortisolism in major depression. 2003. [DOI] [PubMed]
- 76.Sasayama D., Hori H., Iijima Y., Teraishi T., Hattori K., Ota M., Fujii T., Higuchi T., Amano N., Kunugi H. Modulation of cortisol responses to the DEX/CRH test by polymorphisms of the interleukin-1beta gene in healthy adults. Behav. Brain Funct. 2011;7:23. doi: 10.1186/1744-9081-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hill M.N., McLaughlin R.J., Morrish A.C., Viau V., Floresco S.B., Hillard C.J., Gorzalka B.B. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34(13):2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruginsk S.G., Uchoa E.T., Elias L.L., Antunes-Rodrigues J. Anandamide modulates the neuroendocrine responses induced by extracellular volume expansion. Clin. Exp. Pharmacol. Physiol. 2013;40(10):698–705. doi: 10.1111/1440-1681.12155. [DOI] [PubMed] [Google Scholar]
- 79.Bedse G., Colangeli R., Lavecchia A.M., Romano A., Altieri F., Cifani C., Cassano T., Gaetani S. Role of the basolateral amygdala in mediating the effects of the fatty acid amide hydrolase inhibitor URB597 on HPA axis response to stress. 2014. [DOI] [PubMed]
- 80.Roberts C.J., Stuhr K.L., Hutz M.J., Raff H., Hillard C.J. Endocannabinoid signaling in hypothalamic-pituitary-adrenocortical axis recovery following stress: effects of indirect agonists and comparison of male and female mice. Pharmacol. Biochem. Behav. 2014;117:17–24. doi: 10.1016/j.pbb.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kerr D.M., Burke N.N., Ford G.K., Connor T.J., Harhen B., Egan L.J., Finn D.P., Roche M. Pharmacological inhibition of endocannabinoid degradation modulates the expression of inflammatory mediators in the hypothalamus following an immunological stressor. Neuroscience. 2012;204:53–63. doi: 10.1016/j.neuroscience.2011.09.032. [DOI] [PubMed] [Google Scholar]
- 82.Hill M.N., McLaughlin R.J., Bingham B., Shrestha L., Lee T.T., Gray J.M., Hillard C.J., Gorzalka B.B., Viau V. Endogenous cannabinoid signaling is essential for stress adaptation. 2010. [DOI] [PMC free article] [PubMed]
- 83.New D.C., Wong Y.H. BML-190 and AM251 act as inverse agonists at the human cannabinoid CB2 receptor: signalling via cAMP and inositol phosphates. FEBS Lett. 2003;536(1-3):157–160. doi: 10.1016/S0014-5793(03)00048-6. [DOI] [PubMed] [Google Scholar]
- 84.Aliczki M., Zelena D., Mikics E., Varga Z.K., Pinter O., Bakos N.V., Varga J., Haller J. Monoacylglycerol lipase inhibition-induced changes in plasma corticosterone levels, anxiety and locomotor activity in male CD1 mice. Horm. Behav. 2013;63(5):752–758. doi: 10.1016/j.yhbeh.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 85.Long J.Z., Nomura D.K., Cravatt B.F. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009;16(7):744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wiebelhaus J.M., Grim T.W., Owens R.A., Lazenka M.F. ; Sim-Selley L.J., Abdullah R.A., Niphakis M.J., Vann R.E., Cravatt B.F., Wiley J.L., Negus S.S., Lichtman A.H. Delta9-tetrahydrocannabinol and endocannabinoid degradative enzyme inhibitors attenuate intracranial self-stimulation in mice. 2015. [DOI] [PMC free article] [PubMed]
- 87.Fidaleo M., Fanelli F., Ceru M.P., Moreno S. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARα) and its lipid ligands. Curr. Med. Chem. 2014;21(24):2803–2821. doi: 10.2174/0929867321666140303143455. [DOI] [PubMed] [Google Scholar]
- 88.Klimek V., Schenck J.E., Han H., Stockmeier C.A., Ordway G.A. Dopaminergic abnormalities in amygdaloid nuclei in major depression: a postmortem study. Biol. Psychiatry. 2002;52(7):740–748. doi: 10.1016/S0006-3223(02)01383-5. [DOI] [PubMed] [Google Scholar]
- 89.Reddy P.L., Khanna S., Subhash M.N., Channabasavanna S.M., Rao B.S. CSF amine metabolites in depression. 1992. [DOI] [PubMed]
- 90.Serra G., Forgione A., D’Aquila P.S., Collu M., Fratta W., Gessa G.L. Possible mechanism of antidepressant effect of L-sulpiride. Clin. Neuropharmacol. 1990;13(Suppl. 1):S76–S83. doi: 10.1097/00002826-199001001-00009. [DOI] [PubMed] [Google Scholar]
- 91.Carpenter L.L., Moreno F.A., Kling M.A., Anderson G.M., Regenold W.T., Labiner D.M., Price L.H. Effect of vagus nerve stimulation on cerebrospinal fluid monoamine metabolites, norepinephrine, and gamma-aminobutyric acid concentrations in depressed patients. Biol. Psychiatry. 2004;56(6):418–426. doi: 10.1016/j.biopsych.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 92.Nikisch G., Mathé A.A. CSF monoamine metabolites and neuropeptides in depressed patients before and after electroconvulsive therapy. Eur. Psychiatry. 2008;23(5):356–359. doi: 10.1016/j.eurpsy.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 93.Hori H., Kunugi H. 2012.
- 94.Randrup A., Braestrup C. Uptake inhibition of biogenic amines by newer antidepressant drugs: relevance to the dopamine hypothesis of depression. Psychopharmacology (Berl.) 1977;53(3):309–314. doi: 10.1007/BF00492370. [DOI] [PubMed] [Google Scholar]
- 95.Tanda G., Carboni E., Frau R., Di Chiara G. Increase of extracellular dopamine in the prefrontal cortex: a trait of drugs with antidepressant potential? Psychopharmacology (Berl.) 1994;115(1-2):285–288. doi: 10.1007/BF02244785. [DOI] [PubMed] [Google Scholar]
- 96.Djordjevic J., Djordjevic A., Adzic M., Radojcic M.B. Effects of chronic social isolation on Wistar rat behavior and brain plasticity markers. Neuropsychobiology. 2012;66(2):112–119. doi: 10.1159/000338605. [DOI] [PubMed] [Google Scholar]
- 97.Fitzgerald M.L., Mackie K., Pickel V.M. The impact of adolescent social isolation on dopamine D2 and cannabinoid CB1 receptors in the adult rat prefrontal cortex. Neuroscience. 2013;235:40–50. doi: 10.1016/j.neuroscience.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chiba S., Numakawa T., Ninomiya M., Yoon H.S., Kunugi H. Cabergoline, a dopamine receptor agonist, has an antidepressant-like property and enhances brain-derived neurotrophic factor signaling. Psychopharmacology (Berl.) 2010;211(3):291–301. doi: 10.1007/s00213-010-1894-8. [DOI] [PubMed] [Google Scholar]
- 99.Nestler E.J., Carlezon W.A., Jr The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry. 2006;59(12):1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 100.Murillo-Rodriguez E., Vazquez E., Millan-Aldaco D., Palomero-Rivero M., Drucker-Colin R. Effects of the fatty acid amide hydrolase inhibitor URB597 on the sleep-wake cycle, c-Fos expression and dopamine levels of the rat. 2007. [DOI] [PubMed]
- 101.Murillo-Rodríguez E., Palomero-Rivero M., Millán-Aldaco D., Arias-Carrión O., Drucker-Colín R. Administration of URB597, oleoylethanolamide or palmitoylethanolamide increases waking and dopamine in rats. PLoS One. 2011;6(7):e20766. doi: 10.1371/journal.pone.0020766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nader J., Rapino C., Gennequin B., Chavant F., Francheteau M., Makriyannis A., Duranti A., Maccarrone M., Solinas M., Thiriet N. Prior stimulation of the endocannabinoid system prevents methamphetamine-induced dopaminergic neurotoxicity in the striatum through activation of CB2 receptors. Neuropharmacology. 2014;87:214–221. doi: 10.1016/j.neuropharm.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dazzi L., Talani G., Biggio F., Utzeri C., Lallai V., Licheri V., Lutzu S., Mostallino M.C., Secci P.P., Biggio G., Sanna E. Involvement of the cannabinoid CB1 receptor in modulation of dopamine output in the prefrontal cortex associated with food restriction in rats. PLoS One. 2014;9(3):e92224. doi: 10.1371/journal.pone.0092224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vinod K.Y., Xie S., Psychoyos D., Hungund B.L., Cooper T.B., Tejani-Butt S.M. Dysfunction in fatty acid amide hydrolase is associated with depressive-like behavior in Wistar Kyoto rats. 2012. [DOI] [PMC free article] [PubMed]
- 105.Adamczyk P., Gołda A., McCreary A.C., Filip M., Przegaliński E. Activation of endocannabinoid transmission induces antidepressant-like effects in rats. J. Physiol. Pharmacol. 2008;59(2):217–228. [PubMed] [Google Scholar]
- 106.Bortolato M., Mangieri R.A., Fu J., Kim J.H., Arguello O., Duranti A., Tontini A., Mor M., Tarzia G., Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. 2007. [DOI] [PubMed]
- 107.Zhang Z., Wang W., Zhong P., Liu S.J., Long J.Z., Zhao L., Gao H.Q., Cravatt B.F., Liu Q.S. 2014.
- 108.Zhong P., Wang W., Pan B., Liu X., Zhang Z., Long J.Z., Zhang H.T., Cravatt B.F., Liu Q.S. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology. 2014;39(7):1763–1776. doi: 10.1038/npp.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Naidu P.S., Varvel S.A., Ahn K., Cravatt B.F., Martin B.R., Lichtman A.H. Evaluation of fatty acid amide hydrolase inhibition in murine models of emotionality. Psychopharmacology (Berl.) 2007;192(1):61–70. doi: 10.1007/s00213-006-0689-4. [DOI] [PubMed] [Google Scholar]
- 110.Lomazzo E., Bindila L., Remmers F., Lerner R., Schwitter C., Hoheisel U., Lutz B. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology. 2015;40(2):488–501. doi: 10.1038/npp.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Minkkila A., Saario S., Nevalainen T. Discovery and development of endocannabinoid-hydrolyzing enzyme inhibitors. 2010. [DOI] [PubMed]
- 112.Alapafuja S.O., Nikas S.P., Bharathan I.T., Shukla V.G., Nasr M.L., Bowman A.L., Zvonok N., Li J., Shi X., Engen J.R., Makriyannis A. Sulfonyl fluoride inhibitors of fatty acid amide hydrolase. J. Med. Chem. 2012;55(22):10074–10089. doi: 10.1021/jm301205j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Quistad G.B., Sparks S.E., Segall Y., Nomura D.K., Casida J.E. Selective inhibitors of fatty acid amide hydrolase relative to neuropathy target esterase and acetylcholinesterase: toxicological implications. Toxicol. Appl. Pharmacol. 2002;179(1):57–63. doi: 10.1006/taap.2001.9342. [DOI] [PubMed] [Google Scholar]
- 114.Holtfrerich A., Hanekamp W., Lehr M. (4-Phenoxyphenyl)tetrazolecarboxamides and related compounds as dual inhibitors of fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). Eur. J. Med. Chem. 2013;63:64–75. doi: 10.1016/j.ejmech.2013.01.050. [DOI] [PubMed] [Google Scholar]
- 115.Ortar G., Cascio M.G., Moriello A.S., Camalli M., Morera E., Nalli M., Di Marzo V. Carbamoyl tetrazoles as inhibitors of endocannabinoid inactivation: a critical revisitation. 2008. [DOI] [PubMed]
- 116.Ortar G., Morera E., De Petrocellis L., Ligresti A., Schiano Moriello A., Morera L., Nalli M., Ragno R., Pirolli A., Di Marzo V. Biaryl tetrazolyl ureas as inhibitors of endocannabinoid metabolism: modulation at the N-portion and distal phenyl ring. 2013. [DOI] [PubMed]
- 117.Mor M., Lodola A., Rivara S., Vacondio F., Duranti A., Tontini A., Sanchini S., Piersanti G., Clapper J.R., King A.R., Tarzia G., Piomelli D. Synthesis and quantitative structure-activity relationship of fatty acid amide hydrolase inhibitors: modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J. Med. Chem. 2008;51(12):3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zvonok N., Pandarinathan L., Williams J., Johnston M., Karageorgos I., Janero D.R., Krishnan S.C., Makriyannis A. Covalent inhibitors of human monoacylglycerol lipase: ligand-assisted characterization of the catalytic site by mass spectrometry and mutational analysis. Chem. Biol. 2008;15(8):854–862. doi: 10.1016/j.chembiol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moreno-Sanz G., Sasso O., Guijarro A., Oluyemi O., Bertorelli R., Reggiani A., Piomelli D. Pharmacological characterization of the peripheral FAAH inhibitor URB937 in female rodents: interaction with the Abcg2 transporter in the blood-placenta barrier. 2012. [DOI] [PMC free article] [PubMed]
- 120.Lichtman A.H., Leung D., Shelton C.C., Saghatelian A., Hardouin C., Boger D.L., Cravatt B.F. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. 2004. [DOI] [PubMed]
- 121.National Institute of Health
- 122.Sanofi Corporation
- 123.Godlewski G., Alapafuja S.O., Bátkai S., Nikas S.P., Cinar R., Offertáler L., Osei-Hyiaman D., Liu J., Mukhopadhyay B., Harvey-White J., Tam J., Pacak K., Blankman J.L., Cravatt B.F., Makriyannis A., Kunos G. Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem. Biol. 2010;17(11):1256–1266. doi: 10.1016/j.chembiol.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Deutsch D.G., Lin S., Hill W.A., Morse K.L., Salehani D., Arreaza G., Omeir R.L., Makriyannis A. Fatty acid sulfonyl fluorides inhibit anandamide metabolism and bind to the cannabinoid receptor. Biochem. Biophys. Res. Commun. 1997;231(1):217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- 125.Dinh T.P., Freund T.F., Piomelli D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem. Phys. Lipids. 2002;121(1-2):149–158. doi: 10.1016/S0009-3084(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 126.Dinh T.P., Carpenter D., Leslie F.M., Freund T.F., Katona I., Sensi S.L., Kathuria S., Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA. 2002;99(16):10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Saario S.M., Savinainen J.R., Laitinen J.T., Järvinen T., Niemi R. Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem. Pharmacol. 2004;67(7):1381–1387. doi: 10.1016/j.bcp.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 128.Naidoo V., Karanian D.A., Vadivel S.K., Locklear J.R., Wood J.T., Nasr M., Quizon P.M., Graves E.E., Shukla V., Makriyannis A., Bahr B.A. 2012. [DOI] [PMC free article] [PubMed]
- 129.Karageorgos I., Wales T.E., Janero D.R., Zvonok N., Vemuri V.K., Engen J.R., Makriyannis A. Active-site inhibitors modulate the dynamic properties of human monoacylglycerol lipase: a hydrogen exchange mass spectrometry study. Biochemistry. 2013;52(29):5016–5026. doi: 10.1021/bi400430k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.West J.M., Zvonok N., Whitten K.M., Vadivel S.K., Bowman A.L., Makriyannis A. Biochemical and mass spectrometric characterization of human N-acylethanolamine-hydrolyzing acid amidase inhibition. PLoS One. 2012;7(8):e43877. doi: 10.1371/journal.pone.0043877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Boger D.L., Sato H., Lerner A.E., Hedrick M.P., Fecik R.A., Miyauchi H., Wilkie G.D., Austin B.J., Patricelli M.P., Cravatt B.F. Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc. Natl. Acad. Sci. USA. 2000;97(10):5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leung D., Hardouin C., Boger D.L., Cravatt B.F. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat. Biotechnol. 2003;21(6):687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 133.Patel J.Z., Parkkari T., Laitinen T., Kaczor A.A., Saario S.M., Savinainen J.R., Navia-Paldanius D., Cipriano M., Leppänen J., Koshevoy I.O., Poso A., Fowler C.J., Laitinen J.T., Nevalainen T. Chiral 1,3,4-oxadiazol-2-ones as highly selective FAAH inhibitors. J. Med. Chem. 2013;56(21):8484–8496. doi: 10.1021/jm400923s. [DOI] [PubMed] [Google Scholar]
- 134.Glaser S.T., Gatley S.J., Gifford A.N. Ex vivo imaging of fatty acid amide hydrolase activity and its inhibition in the mouse brain. J. Pharmacol. Exp. Ther. 2006;316(3):1088–1097. doi: 10.1124/jpet.105.094748. [DOI] [PubMed] [Google Scholar]
- 135.Muccioli G.G., Labar G., Lambert D.M. CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. ChemBioChem. 2008;9(16):2704–2710. doi: 10.1002/cbic.200800428. [DOI] [PubMed] [Google Scholar]
- 136.Minkkila A., Savinainen J.R., Kasnanen H., Xhaard H., Nevalainen T., Laitinen J.T., Poso A., Leppanen J., Saario S.M. Screening of various hormone-sensitive lipase inhibitors as endocannabinoid-hydrolyzing enzyme inhibitors. 2009. [DOI] [PubMed]
- 137.Tuccinardi T., Granchi C., Rizzolio F., Caligiuri I., Battistello V., Toffoli G., Minutolo F., Macchia M., Martinelli A. Identification and characterization of a new reversible MAGL inhibitor. Bioorg. Med. Chem. 2014;22(13):3285–3291. doi: 10.1016/j.bmc.2014.04.057. [DOI] [PubMed] [Google Scholar]
- 138.Savinainen J.R., Yoshino M., Minkkilä A., Nevalainen T., Laitinen J.T. Characterization of binding properties of monoglyceride lipase inhibitors by a versatile fluorescence-based technique. Anal. Biochem. 2010;399(1):132–134. doi: 10.1016/j.ab.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 139.Nomura D.K., Blankman J.L., Simon G.M., Fujioka K., Issa R.S., Ward A.M., Cravatt B.F., Casida J.E. Activation of the endocannabinoid system by organophosphorus nerve agents. Nat. Chem. Biol. 2008;4(6):373–378. doi: 10.1038/nchembio.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Aaltonen N., Savinainen J.R., Ribas C.R., Rönkkö J., Kuusisto A., Korhonen J., Navia-Paldanius D., Häyrinen J., Takabe P., Käsnänen H., Pantsar T., Laitinen T., Lehtonen M., Pasonen-Seppänen S., Poso A., Nevalainen T., Laitinen J.T. Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem. Biol. 2013;20(3):379–390. doi: 10.1016/j.chembiol.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 141.Nomura D.K., Hudak C.S., Ward A.M., Burston J.J., Issa R.S., Fisher K.J., Abood M.E., Wiley J.L., Lichtman A.H., Casida J.E. Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorg. Med. Chem. Lett. 2008;18(22):5875–5878. doi: 10.1016/j.bmcl.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Karbarz M.J., Luo L., Chang L., Tham C.S., Palmer J.A., Wilson S.J., Wennerholm M.L., Brown S.M., Scott B.P., Apodaca R.L., Keith J.M., Wu J., Breitenbucher J.G., Chaplan S.R., Webb M. Biochemical and biological properties of 4-(3-phenyl-[1,2,4] thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a mechanism-based inhibitor of fatty acid amide hydrolase. Anesth. Analg. 2009;108(1):316–329. doi: 10.1213/ane.0b013e31818c7cbd. [DOI] [PubMed] [Google Scholar]
- 143.Keith J.M., Apodaca R., Xiao W., Seierstad M., Pattabiraman K., Wu J., Webb M., Karbarz M.J., Brown S., Wilson S., Scott B., Tham C.S., Luo L., Palmer J., Wennerholm M., Chaplan S., Breitenbucher J.G. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2008;18(17):4838–4843. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- 144.Keith J.M., Apodaca R., Tichenor M., Xiao W., Jones W., Pierce J., Seierstad M., Palmer J., Webb M., Karbarz M., Scott B., Wilson S., Luo L., Wennerholm M., Chang L., Brown S., Rizzolio M., Rynberg R., Chaplan S., Breitenbucher J.G. Aryl Piperazinyl Ureas as Inhibitors of Fatty Acid Amide Hydrolase (FAAH) in Rat, Dog, and Primate. ACS Med. Chem. Lett. 2012;3(10):823–827. doi: 10.1021/ml300186g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chang J.W., Niphakis M.J., Lum K.M., Cognetta A.B., III, Wang C., Matthews M.L., Niessen S., Buczynski M.W., Parsons L.H., Cravatt B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012;19(5):579–588. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chang J.W., Cognetta A.B., III, Niphakis M.J., Cravatt B.F. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 2013;8(7):1590–1599. doi: 10.1021/cb400261h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Long J.Z., Li W., Booker L., Burston J.J., Kinsey S.G., Schlosburg J.E., Pavon F.J., Serrano A.M., Selley D.E., Parsons L.H., Lichtman A.H., Cravatt B.F. 2009. [DOI] [PMC free article] [PubMed]
- 148.Long J.Z., Jin X., Adibekian A., Li W., Cravatt B.F. Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J. Med. Chem. 2010;53(4):1830–1842. doi: 10.1021/jm9016976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morera L., Labar G., Ortar G., Lambert D.M. Development and characterization of endocannabinoid hydrolases FAAH and MAGL inhibitors bearing a benzotriazol-1-yl carboxamide scaffold. 2012. [DOI] [PubMed]
- 150.Long J.Z., Nomura D.K., Vann R.E., Walentiny D.M., Booker L., Jin X., Burston J.J., Sim-Selley L.J., Lichtman A.H., Wiley J.L., Cravatt B.F. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. USA. 2009;106(48):20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pasquarelli N., Porazik C., Hanselmann J., Weydt P., Ferger B., Witting A. Comparative biochemical characterization of the monoacylglycerol lipase inhibitor KML29 in brain, spinal cord, liver, spleen, fat and muscle tissue. Neuropharmacology. 2015;91:148–156. doi: 10.1016/j.neuropharm.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 152.Moore S.A., Nomikos G.G., Dickason-Chesterfield A.K., Schober D.A., Schaus J.M., Ying B.P., Xu Y.C., Phebus L., Simmons R.M., Li D., Iyengar S., Felder C.C. Identification of a high-affinity binding site involved in the transport of endocannabinoids. Proc. Natl. Acad. Sci. USA. 2005;102(49):17852–17857. doi: 10.1073/pnas.0507470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alexander J.P., Cravatt B.F. The putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J. Am. Chem. Soc. 2006;128(30):9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- 154.Dickason-Chesterfield A.K., Kidd S.R., Moore S.A., Schaus J.M., Liu B., Nomikos G.G., Felder C.C. Pharmacological characterization of endocannabinoid transport and fatty acid amide hydrolase inhibitors. Cell. Mol. Neurobiol. 2006;26(4-6):407–423. doi: 10.1007/s10571-006-9072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Matuszak N., Muccioli G.G., Labar G., Lambert D.M. Synthesis and in vitro evaluation of N-substituted maleimide derivatives as selective monoglyceride lipase inhibitors. J. Med. Chem. 2009;52(23):7410–7420. doi: 10.1021/jm900461w. [DOI] [PubMed] [Google Scholar]
- 156.De Petrocellis L., Melck D., Ueda N., Maurelli S., Kurahashi Y., Yamamoto S., Marino G., Di Marzo V. Novel inhibitors of brain, neuronal, and basophilic anandamide amidohydrolase. Biochem. Biophys. Res. Commun. 1997;231(1):82–88. doi: 10.1006/bbrc.1997.6000. [DOI] [PubMed] [Google Scholar]
- 157.Deutsch D.G., Omeir R., Arreaza G., Salehani D., Prestwich G.D., Huang Z., Howlett A. Methyl arachidonyl fluorophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem. Pharmacol. 1997;53(3):255–260. doi: 10.1016/S0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- 158.Goparaju S.K., Ueda N., Taniguchi K., Yamamoto S. 1999.
- 159.Quistad G.B., Sparks S.E., Casida J.E. Fatty acid amide hydrolase inhibition by neurotoxic organophosphorus pesticides. Toxicol. Appl. Pharmacol. 2001;173(1):48–55. doi: 10.1006/taap.2001.9175. [DOI] [PubMed] [Google Scholar]
- 160.Holtfrerich A., Makharadze T., Lehr M. High-performance liquid chromatography assay with fluorescence detection for the evaluation of inhibitors against human recombinant monoacylglycerol lipase. Anal. Biochem. 2010;399(2):218–224. doi: 10.1016/j.ab.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 161.Niphakis M.J., Cognetta A.B., III, Chang J.W., Buczynski M.W., Parsons L.H., Byrne F., Burston J.J., Chapman V., Cravatt B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013;4(9):1322–1332. doi: 10.1021/cn400116z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Saario S.M., Salo O.M., Nevalainen T., Poso A., Laitinen J.T., Järvinen T., Niemi R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem. Biol. 2005;12(6):649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 163.King A.R., Lodola A., Carmi C., Fu J., Mor M., Piomelli D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. 2009. [DOI] [PMC free article] [PubMed]
- 164.Blankman J.L., Cravatt B.F. Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 2013;65(2):849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boger D.L., Miyauchi H., Du W., Hardouin C., Fecik R.A., Cheng H., Hwang I., Hedrick M.P., Leung D., Acevedo O., Guimarães C.R., Jorgensen W.L., Cravatt B.F. Discovery of a potent, selective, and efficacious class of reversible alpha-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J. Med. Chem. 2005;48(6):1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ahn K., Johnson D.S., Fitzgerald L.R., Liimatta M., Arendse A., Stevenson T., Lund E.T., Nugent R.A., Nomanbhoy T.K., Alexander J.P., Cravatt B.F. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. 2007. [DOI] [PubMed]
- 167.Scott C.W., Tian G., Yu X.H., Paschetto K.A., Wilkins D.E., Meury L., Cao C.Q., Varnes J., Edwards P.D. Biochemical characterization and in vitro activity of AZ513, a noncovalent, reversible, and noncompetitive inhibitor of fatty acid amide hydrolase. Eur. J. Pharmacol. 2011;667(1-3):74–79. doi: 10.1016/j.ejphar.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 168.Johnson D.S., Stiff C., Lazerwith S.E., Kesten S.R., Fay L.K., Morris M., Beidler D., Liimatta M.B., Smith S.E., Dudley D.T., Sadagopan N., Bhattachar S.N., Kesten S.J., Nomanbhoy T.K., Cravatt B.F., Ahn K. 2011.
- 169.Ahn K., Smith S.E., Liimatta M.B., Beidler D., Sadagopan N., Dudley D.T., Young T., Wren P., Zhang Y., Swaney S., Van Becelaere K., Blankman J.L., Nomura D.K., Bhattachar S.N., Stiff C., Nomanbhoy T.K., Weerapana E., Johnson D.S., Cravatt B.F. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. 2011. [DOI] [PMC free article] [PubMed]
- 170.Niphakis M.J., Johnson D.S., Ballard T.E., Stiff C., Cravatt B.F. O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2012;3(5):418–426. doi: 10.1021/cn200089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ahn K., Johnson D.S., Mileni M., Beidler D., Long J.Z., McKinney M.K., Weerapana E., Sadagopan N., Liimatta M., Smith S.E., Lazerwith S., Stiff C., Kamtekar S., Bhattacharya K., Zhang Y., Swaney S., Van Becelaere K., Stevens R.C., Cravatt B.F. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009;16(4):411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gattinoni S., De Simone C., Dallavalle S., Fezza F., Nannei R., Amadio D., Minetti P., Quattrociocchi G., Caprioli A., Borsini F., Cabri W., Penco S., Merlini L., Maccarrone M. Enol carbamates as inhibitors of fatty acid amide hydrolase (FAAH) endowed with high selectivity for FAAH over the other targets of the endocannabinoid system. ChemMedChem. 2010;5(3):357–360. doi: 10.1002/cmdc.200900472. [DOI] [PubMed] [Google Scholar]
- 173.Caprioli A., Coccurello R., Rapino C., Di Serio S., Di Tommaso M., Vertechy M., Vacca V., Battista N., Pavone F., Maccarrone M., Borsini F. The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. 2012. [DOI] [PubMed]
- 174.Alexander J.P., Cravatt B.F. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 2005;12(11):1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ramarao M.K., Murphy E.A., Shen M.W., Wang Y., Bushell K.N., Huang N., Pan N., Williams C., Clark J.D. A fluorescence-based assay for fatty acid amide hydrolase compatible with high-throughput screening. Anal. Biochem. 2005;343(1):143–151. doi: 10.1016/j.ab.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 176.Zhang D., Saraf A., Kolasa T., Bhatia P., Zheng G.Z., Patel M., Lannoye G.S., Richardson P., Stewart A., Rogers J.C., Brioni J.D., Surowy C.S. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52(4):1095–1105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 177.Clapper J.R., Vacondio F., King A.R., Duranti A., Tontini A., Silva C., Sanchini S., Tarzia G., Mor M., Piomelli D. A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem. 2009;4(9):1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Clapper J.R., Moreno-Sanz G., Russo R., Guijarro A., Vacondio F., Duranti A., Tontini A., Sanchini S., Sciolino N.R., Spradley J.M., Hohmann A.G., Calignano A., Mor M., Tarzia G., Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 2010;13(10):1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]