

Abstract
Arachidonic acid (AA)-derived lipid mediators are called eicosanoids. Eicosanoids have emerged as key regulators of a wide variety of physiological responses and pathological processes, and control important cellular processes. AA can be converted into biologically active compounds by metabolism by cyclooxygenases (COX). Beneficial effect of COX-2 inhibitor celecoxib add-on therapy has been reported in early stage of schizophrenia. Moreover, add-on treatment of celecoxib attenuated refractory depression and bipolar depression. Further, the COX/prostaglandin E pathway play an important role in synaptic plasticity and may be included in pathophysiology in autism spectrum disorders (ASD). In this regard, plasma transferrin, which is an iron mediator related to eicosanoid signaling, may be related to social impairment of ASD. COX-2 is typically induced by inflammatory stimuli in the majority of tissues, and the only isoform responsible for propagating the inflammatory response. Thus, COX-2 inhibitors considered as the best target for Alzheimer’s disease.
Keywords: Alzheimer’s disease, arachidonic acid, autism spectrum disorder, cyclooxygenases-1 inhibitors, cyclooxygenases-2 inhibitors, depression, eicosanoids, schizophrenia.
1. INTRODUCTION
Arachidonic acid (AA) is an abundant polyunsaturated fatty acid of the membrane phospholipids, where it is stored in the sn-2 position of phosphatidylinositol and/or phosphatidylcholine [1]. AA-derived eicosanoids belong to a complex family of lipid signaling mediators that regulate a wide variety of physiological responses and pathological processes [2], and control important cellular processes, including cell proliferation, apoptosis, metabolism and migration [3]. Eicosanoids perform numerous regulatory functions in the brain and throughout the rest of the body, in particular for the regulation of immune and inflammatory responses [4]. In addition, eicosanoids play a role in regulating mainly immunopathological processes of inflammatory responses and chronic tissue remodeling [5]. Eicosanoid biosynthesis is usually initiated by the activation of phospholipase A2 (PLA2) from membrane phospholipids in mammalian cells [5] (Fig. 1). There are four families of eicosanoids—the prostaglandins (PG), prostacyclins (PGI), the thromboxanes (TX) and the leukotrienes (LT) [3]. The AA is transformed by cyclooxygenase (COX) and lipoxygenase (LO) pathways to prostaglandins, thromboxane and leukotriene [6]. AA-derived eicosanoids from COX and lipoxygenases are important lipid mediators involved in numerous homeostatic and patho-physiological processes [7], including neurodegenerative and neuropsychiatric conditions [8-10] such as schizophrenia [11], autism spectrum disorders (ASD) [12], refractory major depression [13] and Alzheimer’s disease (AD) [14, 15]. COX-2 induction and resultant prostaglandin synthesis are often implicated in neurodegeneration [16]. The brain is known to be sensitive to oxidative stress and lipid peroxidation. Lipid peroxidation has been shown to contribute to pathophysiology of many psychiatric disease including neurodegenerative disorders [17] and autism spectrum disorder (ASD) [18, 19]. In this regard, PLA2 activation induced by oxidative stress contributes to cognitive impairment, neuronal dysfunction and disease [20] and neurodegenerative diseases [21].
Fig. (1).
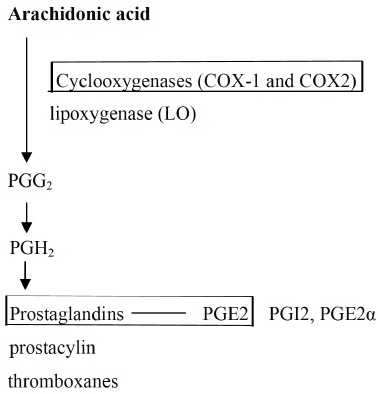
Synthesis pathways for eicosanoids from arachidonic acid (Harizi et al., 2008. [5] ). Arachidonic acid (AA) is released from membrane phopholipids by phospholipases, especially cytosolic phospholipase A2 (cPLA2). The AA is transformed by cyclooxygenase (COX) and lipoxygenase (LO) pathways to prostaglandins, thromboxane and leukotriene. This review addressed the role of COX, and prostaglandins and PGE2.
Among prostaglandins, prostaglandin E (PGE) has closely related to Wnt signaling, which regulates crucial aspects of cell fate determination, cell migration, cell polarity, neural patterning and organogenesis during embryonic development [22] in prenatal development of the nervous systems [23], and a predictive biomarker in ASD [24]. Moreover, PLA2 activity may play a crucial role in neurodegeneration and also be essential in prevention of neuropsychiatric diseases [25]. Additionally, animal studies reported that PLA2-related signaling pathway mediated long-term potentiation induction [26].
Eicosanoids families are modulated by neuroglia such as microglia and astrocyte. Eicosanoids are produced by microglia [27]. Microglia are a type of glial cell that are the resident macrophages of the brain and spinal cord, and thus act as the first and main form of have active immune defense in the central nervous system. PGE synthesis may be related to activation of microglia [28]. Microglia are a major source of PGE2 production through the COX-2 pathway [29]. PLA2 enhances PG synthesis via increased availability of AA, the through the COX pathway in astrocyte [30]. Astrocytes in the hypothalamus release PGE2 in response to cell–cell signaling initiated by neurons and glial cells in human brain [31]. PGD2 mediates microglia/astrocyte interaction [32].
The perspective provided in this review addressed the role of COX in neuropsychiatric disorders such as schizophrenia, major depression, ASD and Alzheimer disease.
2. THE ROLE OF AA-DERIVED EICOSANOIDS IN NRURONAL FUNCTION AND DISEASE
2.1. The Role of Eicosanoids in Neuronal Systems
Downstream targets of the activity of a key set of lipid-modifying enzymes, such as prostaglandin synthase, constitute a complex lipid signaling network with multiple nodes of interaction and cross-regulation. Imbalances in this network contribute to the pathogenesis of human diseases including psychiatric disorders [3].
2.2. The Relationship between Phospholipase A2 and Schizophrenia
Neurotransmitter systems of schizophrenia were modulated by the activation of PLA2 [33]. PLA2 enzyme activities and in vivo brain membrane breakdown specific to schizophrenia are biological processes mediated by three PLA2 isomers such as PLA2G6A, PLA2G4A and PLA2G2A [33]. These three PLA2 enzymes lie downstream of activation of neurotransmitter pathways implicated in schizophrenia, such as dopaminergic, serotoninergic or glutamatergic systems [33].
A previous study on the efficacy of PLA2 in schizophrenia revealed that baseline intracellular PLA2 activity was significantly increased in 35 young drug-naïve and treated first episode patients as compared to 22 healthy controls. Baseline intracellular PLA2 activity was also associated with severity of negative symptoms and lower functioning at baseline [34]. Furthermore, baseline intracellular PLA2 activity was associated with improvement in negative symptoms and functioning within the first 12 weeks of treatment with second-generation antipsychotics [34]. Thus, intracellular PLA2 activity has been considered as a potential predictor of treatment response for different antipsychotic agents [34].
2.3. Efficacy of COX Inhibitors in Psychiatric Disorders
COX is the key enzyme that converts AA to PG play an important role in the nervous system via production of downstream signalling molecules such PGE [12]. COX-2/ PG2 pathway play an important function in synaptic plasticity and refining of mature neuronal connections [12]. Altered COX-2 levels have been found in psychiatric disorders [12]. COX inhibitors are known to influence the immune system in a way that may redirect imbalance of this system [35] and might have additive or even synergistic neuroprotectant effect [28]. Thus, COX inhibitors have therapeutic potential for schizophrenia [36, 37].
2.3.1. Efficacy of COX-inhibitors in Schizophrenia
Increasing evidence from clinical research studies on COX-2 inhibitors points to an advantageous effect of add-on therapy to atypical antipsychotic neuroleptics in schizophrenia, especially in the early stages of the disease [38] through their effects in reducing PGE2, type-2 cytokine and kynurenic acid production and strengthening glutamatergic neuro-transmission [39]. In previous clinical study, COX-2 inhibitor such as celecoxib in addition to amisulpiride, which is atypical antipsychotic, induced a significantly better outcome in the 25 patients with schizophrenia (mean age ± SD: 26.2 ± 7.7 years) compared to 25 patients (mean age ± SD: 30.9 ± 8.1 years) treated with amisulpride plus placebo [36]. Particularly, a significantly superior therapeutic effect was observed in negative symptoms in the celecoxib plus amisulpride group [36]. Update, five clinical studies reported efficacy of COX-2 inhibitor such as celecoxib in schizophrenia. Table 1 summarized these double-blind randomized-placebo-controlled trials. Taking a view of these clinical studies, celecoxib has been reported to be effective add-on treatment to atypical neuroleptics such as risperidone and amisluiride for 5-8 weeks in patients with acute exacerbation [40, 41] or chronic schizophrenia [36, 42] (Table 1). Whereas, another double-blind placebo-controlled trial did not find improvement of celecoxib (400 mg/day for 8 weeks) add-on atypical neuroleptics such as risperidone (3.0 mg/day) or olanzapine (12.6 mg/day) in 18 outpatients with schizophrenia [43]. In this regard, a recent review article commented that results on efficacy of colecoxib in schizophrenia were very heterogeneous, ranging from negative to strong positive effects [44]. The treatment cohorts did not differ on any clinical outcome measures. Review of the data of the previous add-on therapy [36, 40-42] suggests that the psychopathology ratings for the celecoxib augmentation group and placebo group were converging with time. Celecoxib augmentation might potentiate the speed of response but not the overall magnitude of response [43]. Future studies are necessary to elucidate the relationship between doses and treatment periods of atypical neuroleptics and the augmentation effects using a large sample size.
Table 1.
Summaries of findings of clinical trial of celecoxib.
| Design | Treatment, Subject Characteristics | Duration | Efficacy | Interlukin | References |
|---|---|---|---|---|---|
| RCT | celecoxib + risperidone (2-6 mg/day) or placebo (n=25) Acute exacerbation |
5 weeks | PANSS, significant group effect | Soluble IL-2 no significant different | Muller (n=25) et al., 2004 |
| RCT | celecoxib + risperidone (3.0 mg/day) (n=25) or placebo (n=25) out patients with chizophrenia | 8 weeks | PANSS, no significant group effects | NA | Rapaport et al., 2004 |
| RCT | celecoxib (400 mg/day+risperidone (2-6 mg/day) (n=25) or placebo (n=25) Acute exacerbation |
5 weeks | PANSS, significant effect on cognition | NA | Muller et al., 2005 |
| RCT | celecoxib (400mg) + risperidone (6 mg) (n=30) or placebo (n=30) Chronic | 8 weeks | PANSS-positive symptoms and general psycho-psychopathology, significant effect. | NA | Akhondzadeh et al., 2007 |
| RCT | celecoxib (400 mg) + amisulporide amisulporide (200-1000mg or placebo (n=30) Chronic |
8 weeks | PANSS-negative symptoms and general psycho-psychopathology, significant effect | NA | Muller et al., 2010 |
RCT: Randomized Controlled Trial; PANSS: Positive and Negative Syndrome Scale; NA: no available.
2.3.2. Mechanisms of Action of COX-2 Inhibitors in Schizophrenia
The main mechanism of action underlying the efficacy of COX-2 inhibitors in schizophrenia is inhibiting conversion of AA into active prostanoids such as PGE2, prostatanoids D2, prostatanoids 12 and others [45]. Increases or imbalances in cytokines may be related to produce vulnerability to schizophrenia [46]. Thus, cytokine modulation may offer the promise of tailored psychopharmacologic intervention based on individual peripheral and central neuroimmune biomarker profiles [47] (Table 2). Therapeutic effects of COX-2 inhibitors may depend on reducing PGE2, type-2 cytokine and kynurenic acid production and strengthening glutamatergic neurotransmission [39]. Moreovrer, the immunological imbalance in an inflammatory state combined with increased PLA2 production and increased COX-2 expression may be involved in the mechanisms of action of COX-2 inhibitors [48]. The immune response in schizophrenia is confounded by factors partly disease-inherent such as disease duration, chronicity, prevailing symptoms and response to medical treatment [36]. Whereas, another study reported that celecoxib (400 mg/day) add-on treatment to risperidone (mean dose, 3.6 mg/day) or olanzapine (mean dose, 15.0 mg/day) for 8 weeks did not alter any of the cytokine parameters such as IL-6 and TNF-a in 14 patients with schizophrenia compared to 14 normal healthy controls [49]. The relationship between the long duration of disease, treatment resistance and higher IL-6 levels may be related to different immune response possibly linking place in different stages of diseases [36]. In this regard, the blunted type-1 response, which promotes the cell-mediated immune directly against intracellular pathogens, is found primarily in early stages of schizophrenia. While, in later stages of disease, an autoimmune process might additionally play a role in immune function [36]. The findings of the previous many studies shown in Table 2 suggest that schizophrenia is associated with a systemic imbalance in the plasma or serum levels of pro-inflammatory/anti-inflammatory prostaglandins in favor of former [50].
Table 2.
Biomarkers of treatment with COX-2 inhibitors in schizophrenia.
| Subjects Characteristics | Results of Biomarkers | |
|---|---|---|
| Chronic schizophrenia (n = 25) Healthy normal controls (n = 25) |
Plasma levels of IL-2: significantly increased Plasma levels of IL-6: no significantly Correlated to SANS and duration of illness |
Kim et al. 2000 |
| Paranoid schizophrenia (n = 24) Healthy normal controls (n = 24) |
Serum levels of cytokines (IL-6, IL-8 and IFN-γ): significantly increased Serum levels of IL-2, IL-4 and IFN-α: no significant changes |
Kamińska et al., 2001 |
| Schizophrenia (n = 2298) Healthy controls (n = 1858) A research of computerized Literature databases PubMed and EMBASE |
Periopheral levels of IL-IRA, sIL-2R and IL-6: significantly increased Periopheral levels of IL-2: significantly deceased Periopheral levels of IFN-γ, IL-4, IL-1RA, IL-2, soluble IL-2receptor and IL-β: no significant changes |
Potvin et al., 2008 |
| Schizophrenia (n = 28) Colecoxi 400 mg/day vs. olanzapine |
No alterations of blood TNF-alpha and IL-2 levels | Bresee et al., 2006 |
SANS: The Scale for the Assessment of Negative Symptoms. IL-2, IL-6 and IFN-γ are known as cytokines.
2.4. The Efficacy of COX-2 Inhibitors in Major Depression, Bipolar Depression and Refractory Major Depression
2.4.1. Summaries of the Efficacy of COX-2 Inhibitors in Major Depression
There are two review articles on the efficacy of COX-2 inhibitors or non-selective COX inhibitors. Five randomized controlled trials (four unipolar depression studies and one bipolar depression study) has reported that the add-on celecoxib treatment had a statistically significant decrease in means of the Hamilton Depression Rating Scale score at week 4 and week 6. The add-on celecoxib treatment also showed higher remission rates compared with the placebo group [51]. Thus, celecoxib can be considered as an effective add-on treatment for unipolar depressive patients [51]. While, a current review article has reported that the efficacy of COX-2 and non-selective COX inhibitors on depressive symptoms appears to be inconsistent [52]. According to this review article, 4 of a total of 6 randomized controlled trials showed a significant effect of COX-2 inhibitors [52]. In addition, in a total of 5 studies exploring the efficacy of non-selective COX inhibitor, the randomized controlled trials failed to show a significant result [52]. Among the 3 cohort studies, one showed a positive result and the other 2 showed no effect [52]. With regard to these inconsistent findings, significant methodological heterogeneity (i.e. age range, sex, presence of antidepressant use, method of depression measure, severity of depressive symptoms, duration and study design (randomized controlled trial vs. cohort) has been suggested. Further high quality research is needed to explore the effects of COX-2 inhibitors and non-selective COX inhibitors as monotherapy or add-on treatment to various antidepressants [52].
2.4.2. The Efficacy of COX-2 Inhibitors in Refractory Major Depression
It is well known that number of patients with major depression do not respond adequately to current antidepressant pharmacological treatments [53]. Recent studies suggested that inflammatory processes may contribute to patho-physiological processes, since increased levels of pro-inflammatory cytokines and PGE2 have been found in a subset of patients with major depression [53]. In this regard, add-on treatment with COX-2 inhibitors enhance the efficacy of both reboxetine and fluoxetine in treatment-resistant depression. A previous study examined the acute effects of a combined treatment with celecoxib and reboxetine and fluoxetine on noradrenaline, dopamine and 5-HT output in the medial prefrontal cortex in rats [53]. Acute effects of a combined treatment with celecoxib significantly potentiated the efficacy of reboxetine and fluoxetine on increases in cortical noradrenaline and 5-HT output [53]. Thus, the clinical utility of combined treatment with antidepressants in refractory depression may be evident [53]. A previous study reported the case of an elderly depressed woman with acute cognitive deficit who was refractory to multiple antidepressants but only responsive to celecoxib in acute treatment and sustaining remission for a 5-year treatment course [54]. Depressed patients showed lower production of cytokine responses, indicating that their immune cells are in a refractory phase, induced by a pre-existing pro-inflammatory state [55]. Thus, a drug effect could only be shown for imipramine and celecoxib, which were beneficial in terms of re-balancing the immune function [55].
While, in another previous clinical study in 1,245 individuals (mean ± SD age, 53 ± 15.3 years old) who remained depressed despite two or more antidepressant treatments, treatment outcomes with anti-inflammatory drugs (e.g., cytokines) was associated with a greater likelihood of depression classified as treatment resistant depression [56]. This association was apparent in the anti-inflammatory drug group but not in those using other agents with NSAID-like mechanisms such as COX-2 inhibitors [56]. With regard to no effects of COX-2 inhibitors, the authors suggested that general medical comorbidity and painful symptoms seems to be associated with poorer outcomes [56].
2.4.3. The Efficacy of COX -2 Inhibitors in Bipolar Depression
There are few studies on clinical effects of COX-2 inhibitors in bipolar depression. A previous double-blind placebo-controlled trial examined efficacy of celecoxib (400 mg/day) for 6 weeks in 28 patient with DSM-IV bipolar depression 1 and II [57]. In this trial, 28 patients were included if they were experiencing a major depression or mixed episode, with a Hamilton score > 18 and if they had previously been on therapeutic and regular doses of a mood stabilizer or atypical antipsychotic for at least 1 month [57]. The 14 celecoxib treated patients (mean ± SD age, 42.3 ± 10.4 years old) showed significant decreased score in the Hamilton Depression Rating Scale in the first week of treatment as compared to 14 placebo-controlled patients (mean ± SD age, 41.1 ± 9.5 years old). The two groups did not differ significantly on depressive or manic symptoms at any later week [57]. These findings suggest that adjunctive treatment with celecoxib may produce a rapid-onset antidepressant effect in patients with bipolar depression [57].
A previous systemic review of the literature summarized the effects of COX-2 inhibitors on bipolar depression based on articles published from 1950 to April 2008 [58] reported that celecoxib may offer antidepressant effects because inflammation is closely linked with behavioral parameters such as exercise, sleep, alcohol abuse, and smoking, as well as with medical comorbidities including coronary artery disease, obesity and insulin resistance, osteoporosis, and pain [58]. However, methodological limitations precluding definitive conclusions are heterogeneity in sample composition, cytokine assessment procedures, and treatment regimens [58].
2.5. Eicosanoids in Alzheimer’s Disease
2.5.1. The Role of COX in Alzheimer’s Disease
AD is a neurodegenerative disorder characterized by a progressive decline of memory and cognition. The subtlety and variability of the earliest amnestic symptoms, occurring in the absence of any other clinical signs of brain injury, suggest that something is discretely, perhaps intermittently, interrupts the function of synapses that help encode new declarative memories [59]. Although the most common form of dementia, characterized by the excessive accumulation of amyloid-β peptide leads to neurofibrillary tangles composed of aggregated hyperphosphorylated tau, several epidemiological and preclinical studies indicated the pathological role of COX based on the neuroinflammation theory. In this regard, elevated levels of several proinflammatory factors including cytokines and peroxidants in the central nervous system have been detected in patients with AD [60]. These proinflammatory factors act as potent stimuli in brain inflammation through upregulation of cytosolic PLA2 and COX-2 [60]. Moreover, the mechanisms underlying the intracellular signaling pathways may be involved in the expression of several inflammatory proteins induced by proinflammatory factors in AD [60].
In an early stage of AD pathology, when low-fibrillar Abeta deposits are present and only very few neurofibrillary tangles are observed in the cortical areas, COX-2 expression is increased in neurons. COX-1 is primarily expressed in microglia, and associated with fibrillar Abeta deposits. Thus, COX-1 and COX-2 are involved in inflammatory and regenerating pathways respectively in AD pathology [61]
2.5.2. Efficacy of COX-2 and COX-1 Inhibitors in Alzheimer’s Disease
Because COX-2 is typically induced by inflammatory stimuli in the majority of tissues, it was thought to be the only isoform responsible for propagating the inflammatory response and thus, considered as the best target for anti-inflammatory drugs [62]. In this regard, deficits in spatial working memory in female but not male mice were abolished by COX-2 inhibitor celecoxib with alteration of Aβpeptides [63]. Another animal study found that COX-2 inhibitor NS-398 protected memory deficit in mice [64]. According to another animal study, COX-2 inhibitors such as rofecoxib (5 and 10 mg/kg, ip) or valdecoxib (5 and 10 mg/kg, ip) improved memory deficit [65]. With regard to clinical effects of COX-2 inhibitor, a 52-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study in patients aged 50 years or older reported that at the final 52 weeks, change in Assesment Scale-Cognitive Behavior scores from baseline was not significantly different between placebo and celecoxib 200 mg treated groups (5.00 and 4.39, respectively) [66]. According to a randomized, double-blind, multicenter clinical trial of naproxen or celecoxib vs placebo (1:1:1.5 assignment ratio), which conducted between 2001 and 2004 at six U.S.-based clinics in 2528 people with age of 70 years or older, there was no significant effects of celecoxib treatment in these elder patients with AD [67]. In similar line of evidence, according to a double-blinded, multicenter trial in 692 patients with mild or moderate AD aged 50 years or older treated with 25 mg/day COX-2 inhibitor rofecoxib or placebo for 12 months, there is no significant improvement in the rofecoxib treatment group compared with placebo control group [68]. In another double-blind study to investigate whether rofecoxib could delay a diagnosis of AD in patients with mild cognitive impairment aged 65 years or older, rofecoxib 25 mg/day (n = 725) or placebo (n = 732) were treated for up to 4 years [69]. This study did not demonstrate differences between treatment groups. This study reported no consistent evidence that rofecoxib differed from placebo in post hoc analyses comparing the AD assessment scales [69]. Thus, this study did not support the hypothesis that rofecoxib would delay a diagnosis of AD [69]. In conjunction with the lack of effects observed in previous AD studies, the findings suggest that inhibition of COX-2 is not a useful therapeutic approach in AD [69]. Drawing these strands together, COX-2 inhibitors improved memory deficits in animal model of AD, however, several clinical studies did not support the hypothesis that celecoxib prevent progress of adult AD patients. Further precise clinical trial will be needed to study the clinical effects of COX-2 inhibitors.
In regard to efficacy of COX-1 inhibitors, animal studies reported preferential effects of COX-1inhibitors, rather than COX-2 inhibitors [70, 71]. Mice with the lateral ventricle of COX-1-deficient (COX-1(-/-)) induced by injection with beta-amyloid (Abeta(1-42)) exhibited neurodegeneration compared to their wild-type (WT) mice, indicating that inhibition of COX-1 activity may be valid therapeutic strategy to reduce brain inflammatory response and neurodegeneration [70]. Similarly, triple transgenic AD (3 × Tg-AD) mice treated with the COX-1 selective inhibitor SC-560 improved spatial learning and memory, and reduced amyloid deposits and tau hyperphosphorylation [71]. These findings may demonstrate that selective COX-1 inhibition reduces neuroinflammation and neuropathology, and improves cognitive function in 3 × Tg-AD mice [71]. Thus, selective COX-1 inhibition might be a potential therapeutic approach for AD. However, there is few clinical trial with COX-1 inhibitors. Further clinical trials will be needed to demonstrate the efficacy of COX-1 inhibitors in AD.
2.5.3. Differences in Action between COX-1 and COX-2 Inhibitors in Alzheimer’s Disease
The expression levels of COX-1 and COX-2 change in the different stages of AD pathology. In an early stage, COX-2 is increased in neurons with increased neuronal COX-2 expression and with the expression of cell cycle proteins [61]. COX-2 is found in post synaptic dendrites and excitatory terminals, particularly in the cortex, hippocampus and amygdala, with both neuronal and vascular associations [62]. Moreover, COX-2 has a compartmental distribution in somatosensory cortical neurons consistent with the local neuronal synthesis of prostanoids that are involved in neurovascular coupling [72]. While, COX-1 plays anti-inflammatory role [73] and primarily expressed in microglia [61]. Thus, COX-1 and COX-2 play each role in the different stages of AD pathology [61]. Because COX-2 is typically induced by inflammatory stimuli in the majority of tissues, it was thought to be the only isoform responsible for propagating the inflammatory response and thus, considered as the best target for anti-inflammatory drugs [62].
2.6. Eicosanoids in Autism Spectrum Disorders
2.6.1. The Role of Eicosanoids in Autism Spectrum Disorders
The COX/PEG2 pathway play an important role in synaptic plasticity and also may be included in pathophysiology of ASD [12]. Accumulating evidence indicated that PGE2 signalling interacts with another crucial developmental pathway such as the Wnt (ewingless-related MMTV integration site) signalling pathway [74]. The Wnt signalling also regulates neuronal connectivity by controlling axon pathfinding, axon remodelling, dendrite morphogenesis and synapse formation [75]. The canonical Wnt signaling pathway is composed of a network of proteins that modify cell communication and interaction with other cells [76]. The Wnt signaling pathway therefore may be implicated for development of ASD [76]. In most previous studies, changes in families of eicosanoids have reported alterations in antioxidant status in ASD patients [77, 78]. According to a previous study in 44 ASD children aged 3.5-12 years and 44 age-matched healthy matched-children, elevated plasma F2-isoprostane, which is significant risk for antineuronal positivity, was found in 54.5% of ASD children [77]. Families of eicosanoids such as PGE2, leukotrienes and isoprostanes have been reported to be significantly elevated levels in 20 male ASD compared to 19 age and gender-matched controls [78]. Thus, pathophysiology of ASD may be related to eicosanoid regulation [78].
2.6.2. Eicosanoids and AA-Derived Signaling Mediators in Autism Spectrum disorders: Ceruloplasmin
In most previous ASD studies, changes in blood eicosanoid families such as superoxide dismutase (SOD), transferrin (Tf) and ceruloplasmin (Cp) levels indicated alterations in antioxidant status [79-81], vulnerability to oxidative stress [82, 83], and copper dyshomeostasis [84, 85] in ASD patients. It was known that iron and copper can modulates the synthesizing and catabolism of prostaglandins [86]. Furthermore, copper [87] and iron [88] mediators are closely related to AA-derived eicosanoid signaling mediators [89]. In this regard, ceruloplasmin (Cp) is a major copper transport protein in plasma in relation to neurodegenerative conditions, including Alzheimer's disease [90]. Copper supplementation (gycinat, 2 mg/day for 8 weeks) produced a 39 % mean decrease in plasma for eicosanoid family F2α-isoprostanes in young adult women [91]. Superoxide dismutase (SOD), which is a biomarker of copper status that plays a role in signaling [92], play a role in controlling prostaglandin F2 alpha [93]. Further, transferrin (Tf), which carries iron throughout the systemic circulation of tissues [94], may relate to alteration of eicosanoid metabolism [95]. Tf has been reported to have association with COX-2 in liver cancer [96], and COX2 invasion via NF-KB signaling [97]. It is therefore suggested that plasma Tf may be related to eicosanoid signaling. In this regard, a previous 16-week double-blind, randomized placebo-controlled trial examined the role of eicosanoid families estimated the efficacy of supplementation with larger doses of ARA (240 mg/day) added to DHA (240 mg/day) in individuals with ASD (n = 7) (mean age = 14.6 ± 5.9 years) [98]. In this study, plasma Tf levels in the 7 individuals with ASD treated with this supplementation were significantly increased comparted to 6 placebo treated controls [98]. There was also a trend towards significant difference (P = 0.08) in the change in plasma SOD levels [98]. The supplementation significantly improved the Aberrant Behavior Checklist-measured social withdrawal and the Social Responsiveness Scale-measured communication. Considering that mutual relationship between SOD and Tf acts as a mediator in signaling pathways [99], these findings regarding the plasma levels of TF, as well as SOD, suggest that signal transduction was upregulated in the ARA plus DHA supplementation group. Collectively, improvement of social impairment induced by supplementation with larger doses of ARA added to DHA may be related to upregulation of signal transduction [98].
SUMMARY
AA-derived eicosanoids belong to a complex family of lipid signaling mediators that regulate a wide variety of physiological responses and pathological processes, and control important cellular processes, including cell proliferation, apoptosis, metabolism and migration. Moreover, eicosanoids perform numerous regulatory functions in the brain and throughout the rest of the body. AA can be converted into biologically active compounds by COX. The COX pathway are important lipid mediators involved in numerous homeostatic and pathophysiological processes, including neuropsychiatric conditions such as schizophrenia. Celecoxib, a selective COX-2 inhibitor, has been known to have antipsychotic effects in patients with schizophrenia. The main mechanism of action the efficacy of COX-2 inhibitors in early stage of schizophrenia is inhibiting conversion of AA into active prostatanoids such as PLE2, prostatanoids D2, prostatanoids 12 and others. This review summarized beneficial antipsychotic effects of celecoxib add-on therapy compared to atypical antipsychotics such as risperidone or amisulporide. Celecoxib can be considered as an effective add-on treatment for refractory major depression and bipolar depression. The COX/PEG2 pathway an important role in synaptic plasticity and may be included in pathophysiology of ain ASD. In this regard, plasma Tf, which is an iron mediator related to eicosanoid signaling, may be related to social impairment of ASD.
COX-1 and COX-2 are involved in inflammatory and regenerating pathways respectively in AD pathology. COX-1 is typically induced by inflammatory stimuli in the majority of tissues, and the only isoform responsible for propagating the inflammatory response and thus, considered as the best target for AD. However, animal model studies and a few clinical trials demonstrated efficacy of COX-2 inhibitors, further precise clinical studies on efficacy of inhibitors of COX-2 as well as COX-1 are needed to show definitive efficacy of these inhibitors
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (Grant No 21200017) (2009-2012) and a Grant-in-Aid For Scientific Research C (2014-2017) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
CONFICT OF INTEREST
The authors confirm that this article had no conflict of interest.
REFERENCES
- 1.Capra V., Rovati G.E., Mangano P., Buccellati C., Murphy R.C., Sala A. Transcellular biosynthesis of eicosanoid lipid mediators. 2014. [DOI] [PubMed]
- 2.Harizi H., Corcuff J.B., Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. 2008. [DOI] [PubMed]
- 3.Wymann M.P., Roger Schneiter R. Lipid signalling in disease. 2008. [DOI] [PubMed]
- 4.Hallahan B., Garland M.R. Essential fatty acids and mental health. 2005. [DOI] [PubMed]
- 5.Harizi H., Corcuff J.B., Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 2008;14(10):461–469. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Khanapure S.P., Garvey D.S., Janero D.R., Letts L.G. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr. Top. Med. Chem. 2007;7(3):311–340. doi: 10.2174/156802607779941314. [DOI] [PubMed] [Google Scholar]
- 7.Yin H., Zhou Y., Zhu M., Hou S., Li Z., Zhong H., Lu J., Meng T., Wang J., Xia L., Xu Y., Wu Y. Role of mitochondria in programmed cell death mediated by arachidonic acid-derived eicosanoids. 2013. [DOI] [PubMed]
- 8.Ong W.Y., Farooqui T., Farooqui A.A. Involvement of cytosolic phospholipase A(2), calcium independent phospholipase A(2) and plasmalogen selective phospholipase A(2) in neurodegenerative and neuropsychiatric conditions. Curr. Med. Chem. 2010;17(25):2746–2763. doi: 10.2174/092986710791859289. [DOI] [PubMed] [Google Scholar]
- 9.Huber C., Marschallinger J., Tempfer H., Furtner T., Couillard-Despres S., Bauer H.C., Rivera F.J., Aigner L. Inhibition of leukotriene receptors boosts neural progenitor proliferation. Cell. Physiol. Biochem. 2011;28(5):793–804. doi: 10.1159/000335793. [DOI] [PubMed] [Google Scholar]
- 10.Puppolo M., Varma D., Jansen S.A. 2014. [DOI] [PubMed]
- 11.Rao J.S., Kim H.W., Harry G.J., Rapoport S.I., Reese E.A. Increased neuroinflammatory and arachidonic acid cascade markers, and reduced synaptic proteins, in the postmortem frontal cortex from schizophrenia patients. 2013. [DOI] [PMC free article] [PubMed]
- 12.Tamiji J., Crawford D.A. The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals. 2010;18(2):98–112. doi: 10.1159/000323189. [DOI] [PubMed] [Google Scholar]
- 13.Chen C.Y., Tzeng N.S., Chen Y.C. Maintenance therapy of celecoxib for major depression with mimicking neuropsychological dysfunction. Gen. Hosp. Psychiatry. 2010;32(6):647.e7–647.e9. doi: 10.1016/j.genhosppsych.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Wei L.L., Shen Y.D., Zhang Y.C., Hu X.Y., Lu P.L., Wang L., Chen W. Roles of the prostaglandin E2 receptors EP subtypes in Alzheimer’s disease. Neurosci. Bull. 2010;26(1):77–84. doi: 10.1007/s12264-010-0703-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piro J.R., Benjamin D.I., Duerr J.M., Pi Y., Gonzales C., Wood K.M., Schwartz J.W., Nomura D.K., Samad T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Reports. 2012;1(6):617–623. doi: 10.1016/j.celrep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizuno M., Sotoyama H., Narita E., Kawamura H., Namba H., Zheng Y., Eda T., Nawa H. A cyclooxygenase-2 inhibitor ameliorates behavioral impairments induced by striatal administration of epidermal growth factor. J. Neurosci. 2007;27(38):10116–10127. doi: 10.1523/JNEUROSCI.2368-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cahill-Smith S., Li J.M. Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: a role of NADPH oxidase 2. Br. J. Clin. Pharmacol. 2014;78(3):441–453. doi: 10.1111/bcp.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James S.J., Cutler P., Melnyk S., Jernigan S., Janak L., Gaylor D.W., Neubrander J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004;80(6):1611–1617. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- 19.Rose S., Frye R.E., Slattery J., Wynne R., Tippett M., Pavliv O., Melnyk S., James S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. 2014. [DOI] [PMC free article] [PubMed]
- 20.Beaulieu E., Ioffe J., Watson S.N., Hermann P.M., Wildering W.C. Oxidative-stress induced increase in circulating fatty acids does not contribute to phospholipase A2-dependent appetitive long-term memory failure in the pond snail Lymnaea stagnalis. BMC Neurosci. 2014;15:56. doi: 10.1186/1471-2202-15-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagami T., Yamamoto Y., Koma H. The role of secretory phospholipase A₂ in the central nervous system and neurological diseases. Mol. Neurobiol. 2014;49(2):863–876. doi: 10.1007/s12035-013-8565-9. [DOI] [PubMed] [Google Scholar]
- 22.Komiya Y., Habas R. Wnt signal transduction pathways. 2008. [DOI] [PMC free article] [PubMed]
- 23.Wong C.T., Ahmad E., Li H., Crawford D.A. Prostaglandin E2 alters Wnt-dependent migration and proliferation in neuroectodermal stem cells: implications for autism spectrum disorders. Cell Commun. Signal. 2014;12:19. doi: 10.1186/1478-811X-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Ansary A. 2012.
- 25.Ong W.Y., Farooqui T., Farooqui A.A. Involvement of cytosolic phospholipase A(2), calcium independent phospholipase A(2) and plasmalogen selective phospholipase A(2) in neurodegenerative and neuropsychiatric conditions. Curr. Med. Chem. 2010;17(25):2746–2763. doi: 10.2174/092986710791859289. [DOI] [PubMed] [Google Scholar]
- 26.Su L.D., Wang D.J., Yang D., Shen Y., Hu Y.H. Retrograde cPLA2α/arachidonic acid/2-AG signaling is essential for cerebellar depolarization-induced suppression of excitation and long-term potentiation. Cerebellum. 2013;12(3):297–299. doi: 10.1007/s12311-012-0444-9. [DOI] [PubMed] [Google Scholar]
- 27.Matsuo M., Hamasaki Y., Fujiyama F., Miyazaki S. Eicosanoids are produced by microglia, not by astrocytes, in rat glial cell cultures. Brain Res. 1995;685(1-2):201–204. doi: 10.1016/0006-8993(95)00490-H. [DOI] [PubMed] [Google Scholar]
- 28.Singh D.P., Chopra K. Flavocoxid, dual inhibitor of cyclooxygenase-2 and 5-lipoxygenase, exhibits neuroprotection in rat model of ischaemic stroke. Pharmacol. Biochem. Behav. 2014;120:33–42. doi: 10.1016/j.pbb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Badie B., Schartner J.M., Hagar A.R., Prabakaran S., Peebles T.R., Bartley B., Lapsiwala S., Resnick D.K., Vorpahl J. Microglia cyclooxygenase-2 activity in experimental gliomas: possible role in cerebral edema formation. Clin. Cancer Res. 2003;9(2):872–877. [PubMed] [Google Scholar]
- 30.Pistritto G., Ciabattoni G., Mancuso C., Tringali G., Preziosi P., Navarra P. Signaling pathways involved in lipopolysaccharide stimulation of prostaglandin production by rat hypothalamic astroglial cells. J. Endotoxin Res. 2000;6(4):307–311. doi: 10.1177/09680519000060040601. [DOI] [PubMed] [Google Scholar]
- 31.Clasadonte J., Poulain P., Hanchate N.K., Corfas G., Ojeda S.R., Prevot V. Prostaglandin E2 release from astrocytes triggers gonadotropin-releasing hormone (GnRH) neuron firing via EP2 receptor activation. Proc. Natl. Acad. Sci. USA. 2011;108(38):16104–16109. doi: 10.1073/pnas.1107533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohri I., Taniike M., Taniguchi H., Kanekiyo T., Aritake K., Inui T., Fukumoto N., Eguchi N., Kushi A., Sasai H., Kanaoka Y., Ozono K., Narumiya S., Suzuki K., Urade Y. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J. Neurosci. 2006;26(16):4383–4393. doi: 10.1523/JNEUROSCI.4531-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Law M.H., Cotton R.G., Berger G.E. The role of phospholipases A2 in schizophrenia. Mol. Psychiatry. 2006;11(6):547–556. doi: 10.1038/sj.mp.4001819. [DOI] [PubMed] [Google Scholar]
- 34.Smesny S., Kunstmann C., Kunstmann S., Willhardt I., Lasch J., Yotter R.A., Proffitt T.M., Kerr M., Marculev C., Milleit B., Milleit C., Nenadic I., Amminger P., McGorry P.D., Sauer H., Berger G.E. 2011. [DOI] [PubMed]
- 35.Riedel M., Strassnig M., Schwarz M.J., Müller N. COX-2 inhibitors as adjunctive therapy in schizophrenia: rationale for use and evidence to date. CNS Drugs. 2005;19(10):805–819. doi: 10.2165/00023210-200519100-00001. [DOI] [PubMed] [Google Scholar]
- 36.Müller N., Schwarz M.J. Immune System and Schizophrenia. 2010. [DOI] [PMC free article] [PubMed]
- 37.Gałecki P., Talarowska M., Bobińska K., Szemraj J. 2014.
- 38.Müller N., Krause D., Weidinger E., Schwarz M. [Immunological treatment options for schizophrenia]. Fortschr. Neurol. Psychiatr. 2014;82(4):210–219. doi: 10.1055/s-0033-1355776. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt L., Ceglarek U., Kortz L., Hoop M., Kirkby K.C., Thiery J., Himmerich H. Mechanisms of involvement of eicosanoids and their Precursors in the pathophysiology and treatment of schizophrenia. Med. Chem. 2013;9(6):763–773. doi: 10.2174/1573406411309060002. [DOI] [PubMed] [Google Scholar]
- 40.Müller N., Ulmschneider M., Scheppach C., Schwarz M.J., Ackenheil M., Möller H.J., Gruber R., Riedel M. COX-2 inhibition as a treatment approach in schizophrenia: immunological considerations and clinical effects of celecoxib add-on therapy. Eur. Arch. Psychiatry Clin. Neurosci. 2004;254(1):14–22. doi: 10.1007/s00406-004-0478-1. [DOI] [PubMed] [Google Scholar]
- 41.Müller N., Riedel M., Schwarz M.J., Engel R.R. Clinical effects of COX-2 inhibitors on cognition in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2005;255(2):149–151. doi: 10.1007/s00406-004-0548-4. [DOI] [PubMed] [Google Scholar]
- 42.Akhondzadeh S., Tabatabaee M., Amini H., Ahmadi Abhari S.A., Abbasi S.H., Behnam B. Celecoxib as adjunctive therapy in schizophrenia: a double-blind, randomized and placebo-controlled trial. 2007. [DOI] [PubMed]
- 43.Rapaport M.H., Delrahim K.K., Bresee C.J., Maddux R.E., Ahmadpour O., Dolnak D. Celecoxib augmentation of continuously ill patients with schizophrenia. Biol. Psychiatry. 2005;57(12):1594–1596. doi: 10.1016/j.biopsych.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 44.Sommer I.E., van Westrhenen R., Begemann M.J., de Witte L.D., Leucht S., Kahn R.S. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophr. Bull. 2014;40(1):181–191. doi: 10.1093/schbul/sbt139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keller W.R., Kum L.M., Wehring H.J., Koola M.M., Buchanan R.W., Kelly D.L. A review of anti-inflammatory agents for symptoms of schizophrenia. J. Psychopharmacol. (Oxford) 2013;27(4):337–342. doi: 10.1177/0269881112467089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansur R.B., Zugman A., Asevedo E.M., da Cunha G.R., Bressan R.A., Brietzke E. Cytokines in schizophrenia: possible role of anti-inflammatory medications in clinical and preclinical stages. Psychiatry Clin. Neurosci. 2012;66(4):247–260. doi: 10.1111/j.1440-1819.2012.02354.x. [DOI] [PubMed] [Google Scholar]
- 47.Griffin É.W., Skelly D.T., Murray C.L., Cunningham C. Cyclooxygenase-1-dependent prostaglandins mediate susceptibility to systemic inflammation-induced acute cognitive dysfunction. 2013. [DOI] [PMC free article] [PubMed]
- 48.Müller N., Myint A.M., Krause D., Weidinger E., Schwarz M.J. Anti-inflammatory treatment in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;42:146–153. doi: 10.1016/j.pnpbp.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 49.Bresee C.J., Delrahim K., Maddux R.E., Dolnak D., Ahmadpour O., Rapaport M.H. The effects of celecoxib augmentation on cytokine levels in schizophrenia. Int. J. Neuropsychopharmacol. 2006;9(3):343–348. doi: 10.1017/S1461145705005808. [DOI] [PubMed] [Google Scholar]
- 50.Martínez-Gras I., Pérez-Nievas B.G., García-Bueno B., Madrigal J.L., Andrés-Esteban E., Rodríguez-Jiménez R., Hoenicka J., Palomo T., Rubio G., Leza J.C. The anti-inflammatory prostaglandin 15d-PGJ2 and its nuclear receptor PPARgamma are decreased in schizophrenia. Schizophr. Res. 2011;128(1-3):15–22. doi: 10.1016/j.schres.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 51.Faridhosseini F., Sadeghi R., Farid L., Pourgholami M. Celecoxib: a new augmentation strategy for depressive mood episodes. A systematic review and meta-analysis of randomized placebo-controlled trials. Hum. Psychopharmacol. 2014;29(3):216–223. doi: 10.1002/hup.2401. [DOI] [PubMed] [Google Scholar]
- 52.Eyre H.A., Air T., Proctor S., Rositano S., Baune B.T. A critical review of the efficacy of non-steroidal anti-inflammatory drugs in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2015;57:11–16. doi: 10.1016/j.pnpbp.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 53.Johansson D., Falk A., Marcus M.M., Svensson T.H. Celecoxib enhances the effect of reboxetine and fluoxetine on cortical noradrenaline and serotonin output in the rat. 2012. [DOI] [PubMed]
- 54.Chen C.Y., Tzeng N.S., Chen Y.C. Maintenance therapy of celecoxib for major depression with mimicking neuropsychological dysfunction. 2010. [DOI] [PubMed]
- 55.Krause D.L., Riedel M., Müller N., Weidinger E., Schwarz M.J., Myint A.M. Effects of antidepressants and cyclooxygenase-2 inhibitor on cytokines and kynurenines in stimulated in vitro blood culture from depressed patients. Inflammopharmacology. 2012;20(3):169–176. doi: 10.1007/s10787-011-0112-6. [DOI] [PubMed] [Google Scholar]
- 56.Gallagher P.J., Castro V., Fava M., Weilburg J.B., Murphy S.N., Gainer V.S., Churchill S.E., Kohane I.S., Iosifescu D.V., Smoller J.W., Perlis R.H. Antidepressant response in patients with major depression exposed to NSAIDs: a pharmacovigilance study. 2012. [DOI] [PMC free article] [PubMed]
- 57.Nery F.G., Monkul E.S., Hatch J.P., Fonseca M., Zunta-Soares G.B., Frey B.N., Bowden C.L., Soares J.C. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum. Psychopharmacol. 2008;23(2):87–94. doi: 10.1002/hup.912. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein B.I., Kemp D.E., Soczynska J.K., McIntyre R.S. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. 2009. [DOI] [PubMed]
- 59.Selkoe D.J. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 60.Hsieh H.L., Yang C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Res. Int. 2013;2013:484613. doi: 10.1155/2013/484613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoozemans J.J., Rozemuller J.M., van Haastert E.S., Veerhuis R., Eikelenboom P. Cyclooxygenase-1 and -2 in the different stages of Alzheimer’s disease pathology. Curr. Pharm. Des. 2008;14(14):1419–1427. doi: 10.2174/138161208784480171. [DOI] [PubMed] [Google Scholar]
- 62.Aïd S., Bosetti F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie. 2011;93(1):46–51. doi: 10.1016/j.biochi.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Melnikova T., Savonenko A., Wang Q., Liang X., Hand T. ; Melnikova T., Savonenko A., Wang Q., Liang X., Hand T., Wu L., Kaufmann W.E., Vehmas A., Andreasson K.I. Cycloxygenase-2 activity promotes cognitive deficits but not increased amyloid burden in a model of Alzheimer’s disease in a sex-dimorphic pattern. Neuroscience. 2006;141(3):1149–1162. doi: 10.1016/j.neuroscience.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Cakała M., Malik A.R., Strosznajder J.B. Inhibitor of cyclooxygenase-2 protects against amyloid beta peptide-evoked memory impairment in mice. Pharmacol. Rep. 2007;59(2):164–172. [PubMed] [Google Scholar]
- 65.Kumari B., Kumar A., Dhir A. Protective effect of non-selective and selective COX-2-inhibitors in acute immobilization stress-induced behavioral and biochemical alterations. Pharmacol. Rep. 2007;59(6):699–707. [PubMed] [Google Scholar]
- 66.Soininen H., West C., Robbins J., Niculescu L. Long-term efficacy and safety of celecoxib in Alzheimer's disease. 2007. [DOI] [PubMed]
- 67.Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group Results of a follow-up study to the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement. 2013;9(6):714–723. doi: 10.1016/j.jalz.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reines S.A., Block G.A., Morris J.C., Liu G., Nessly M.L., Lines C.R., Norman B.A., Baranak C.C., Rofecoxib Protocol 091 Study Group Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62(1):66–71. doi: 10.1212/WNL.62.1.66. [DOI] [PubMed] [Google Scholar]
- 69.Thal L.J., Ferris S.H., Kirby L., Block G.A., Lines C.R., Yuen E., Assaid C., Nessly M.L., Norman B.A., Baranak C.C., Reines S.A., Rofecoxib Protocol 078 study group A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30(6):1204–1215. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 70.Choi S.H., Aid S., Caracciolo L., Minami S.S., Niikura T., Matsuoka Y., Turner R.S., Mattson M.P., Bosetti F. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J. Neurochem. 2013;124(1):59–68. doi: 10.1111/jnc.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi S.H., Aid S., Caracciolo L., Minami S.S., Niikura T., Matsuoka Y., Turner R.S., Mattson M.P., Bosetti F. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J. Neurochem. 2013;124(1):59–68. doi: 10.1111/jnc.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H., Hitron I.M., Iadecola C., Pickel V.M. Synaptic and vascular associations of neurons containing cyclooxygenase-2 and nitric oxide synthase in rat somatosensory cortex. Cereb. Cortex. 2005;15(8):1250–1260. doi: 10.1093/cercor/bhi008. [DOI] [PubMed] [Google Scholar]
- 73.García-Bueno B., Serrats J., Sawchenko P.E. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. 2009. [DOI] [PMC free article] [PubMed]
- 74.Liu X.H., Kirschenbaum A., Weinstein B.M., Zaidi M., Yao S., Levine A.C. Prostaglandin E2 modulates components of the Wnt signaling system in bone and prostate cancer cells. 2010. [DOI] [PubMed]
- 75.Ciani L., Salinas P.C. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci. 2005;6(5):351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 76.Wong C.T., Ahmad E., Li H., Crawford D.A. Prostaglandin E2 alters Wnt-dependent migration and proliferation in neuroectodermal stem cells: implications for autism spectrum disorders. Cell Commun. Signal. 2014;12:19. doi: 10.1186/1478-811X-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mostafa G.A., El-Hadidi E.S., Hewedi D.H., Abdou M.M. Oxidative stress in Egyptian children with autism: relation to autoimmunity. 2010. [DOI] [PubMed]
- 78.El-Ansary A., Al-Ayadhi L. Lipid mediators in plasma of autism spectrum disorders. Lipids Health Dis. 2012;11:160. doi: 10.1186/1476-511X-11-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yorbik O., Sayal A., Akay C., Akbiyik D.I., Sohmen T. Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot. Essent. Fatty Acids. 2002;67(5):341–343. doi: 10.1054/plef.2002.0439. [DOI] [PubMed] [Google Scholar]
- 80.Söğüt S., Zoroğlu S.S., Ozyurt H., Yilmaz H.R., Ozuğurlu F., Sivasli E., Yetkin O., Yanik M., Tutkun H., Savaş H.A., Tarakçioğlu M., Akyol O. Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin. Chim. Acta. 2003;331(1-2):111–117. doi: 10.1016/S0009-8981(03)00119-0. [DOI] [PubMed] [Google Scholar]
- 81.Zoroglu S.S., Armutcu F., Ozen S., Gurel A., Sivasli E., Yetkin O., Meram I. Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur. Arch. Psychiatry Clin. Neurosci. 2004;254(3):143–147. doi: 10.1007/s00406-004-0456-7. [DOI] [PubMed] [Google Scholar]
- 82.Meguid N.A., Dardir A.A., Abdel-Raouf E.R., Hashish A. Evaluation of oxidative stress in autism: defective antioxidant enzymes and increased lipid peroxidation. Biol. Trace Elem. Res. 2011;143(1):58–65. doi: 10.1007/s12011-010-8840-9. [DOI] [PubMed] [Google Scholar]
- 83.Parellada M., Moreno C., Mac-Dowell K., Leza J.C., Giraldez M., Bailón C., Castro C., Miranda-Azpiazu P., Fraguas D., Arango C. Plasma antioxidant capacity is reduced in Asperger syndrome. J. Psychiatr. Res. 2012;46(3):394–401. doi: 10.1016/j.jpsychires.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 84.Chauhan A., Chauhan V., Brown W.T., Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin-the antioxidant proteins. 2004. [DOI] [PubMed]
- 85.Tórsdóttir G., Hreidarsson S., Kristinsson J., Snaedal J., Jóhannesson T. Ceruloplasmin, superoxide dismutase and copper in autistic patients. Basic Clin. Pharmacol. Toxicol. 2005;96(2):146–148. doi: 10.1111/j.1742-7843.2005.pto960210.x. [DOI] [PubMed] [Google Scholar]
- 86.Sakuma S., Fujimoto Y., Miyata Y., Ohno M., Nishida H., Fujita T. Effects of Fe(2+), Zn(2+), Cu(2+) and Se(4+) on the synthesis and catabolism of prostaglandins in rabbit gastric antral mucosa. Prostaglandins Leukot. Essent. Fatty Acids. 1996;54(3):193–197. doi: 10.1016/S0952-3278(96)90016-2. [DOI] [PubMed] [Google Scholar]
- 87.Ritter A., Goulitquer S., Salaün J.P., Tonon T., Correa J.A., Potin P. Copper stress induces biosynthesis of octadecanoid and eicosanoid oxygenated derivatives in the brown algal kelp Laminaria digitata. New Phytol. 2008;180(4):809–821. doi: 10.1111/j.1469-8137.2008.02626.x. [DOI] [PubMed] [Google Scholar]
- 88.Leblanc C.P., Surette M.E., Fiset S., Turgeon O’Brien H., Rioux F.M. Maternal iron deficiency and its effect on essential fatty acid and eicosanoid metabolism and spatial memory in the guinea pig offspring. Prostaglandins Leukot. Essent. Fatty Acids. 2009;81(1):1–8. doi: 10.1016/j.plefa.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 89.Farooqui A.A. Lipid mediators in the neural cell nucleus: their metabolism, signaling, and association with neurological disorders. Neuroscientist. 2009;15(4):392–407. doi: 10.1177/1073858409337035. [DOI] [PubMed] [Google Scholar]
- 90.Lee K.H., Yun S.J., Nam K.N., Gho Y.S., Lee E.H. Activation of microglial cells by ceruloplasmin. Brain Res. 2007;1171:1–8. doi: 10.1016/j.brainres.2007.07.053. [DOI] [PubMed] [Google Scholar]
- 91.DiSilvestro R.A., Selsby J., Siefker K. A pilot study of copper supplementation effects on plasma F2alpha isoprostanes and urinary collagen crosslinks in young adult women. J. Trace Elem. Med. Biol. 2010;24(3):165–168. doi: 10.1016/j.jtemb.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 92.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011;15(6):1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vu H.V., Lee S., Acosta T.J., Yoshioka S., Abe H., Okuda K. Roles of prostaglandin F2alpha and hydrogen peroxide in the regulation of Copper/Zinc superoxide dismutase in bovine corpus luteum and luteal endothelial cells. Reprod. Biol. Endocrinol. 2012;10:87. doi: 10.1186/1477-7827-10-87. [Erratum in: Reprod. Biol. Endocrinol., 2013, 11, 82. doi: 10.1186/1477-7827-10-87]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kasibhatla S., Jessen K.A., Maliartchouk S., Wang J.Y., English N.M., Drewe J., Qiu L., Archer S.P., Ponce A.E., Sirisoma N., Jiang S., Zhang H.Z., Gehlsen K.R., Cai S.X., Green D.R., Tseng B. A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. 2005. [DOI] [PMC free article] [PubMed]
- 95.Erikson K.M., Pinero D.J., Connor J.R., Beard J.L. Regional brain iron, ferritin and transferrin concentrations during iron deficiency and iron repletion in developing rats. J. Nutr. 1997;127(10):2030–2038. doi: 10.1093/jn/127.10.2030. [DOI] [PubMed] [Google Scholar]
- 96.Hong Y., Yang J., Shen X., Zhu H., Sun X., Wen X., Bian J., Hu H., Yuan L., Tao J., Lei P., Shen G. Sinomenine hydrochloride enhancement of the inhibitory effects of anti-transferrin receptor antibody-dependent on the COX-2 pathway in human hepatoma cells. Cancer Immunol. Immunother. 2013;62(3):447–454. doi: 10.1007/s00262-012-1337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pandey M.K., Sung B., Ahn K.S., Kunnumakkara A.B., Chaturvedi M.M., Aggarwal B.B. Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood. 2007;110(10):3517–3525. doi: 10.1182/blood-2007-03-079616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yui K., Koshiba M., Nakamura S., Kobayashi Y. Effects of large doses of arachidonic acid added to docosahexaenoic acid on social impairment in individuals with autism spectrum disorders: a double-blind, placebo-controlled, randomized trial. 2012. [DOI] [PubMed]
- 99.Ahmed M., Neville M.J., Edelmann M.J., Kessler B.M., Karpe F. Proteomic analysis of human adipose tissue after rosiglitazone treatment shows coordinated changes to promote glucose uptake. Obesity (Silver Spring) 2010;18(1):27–34. doi: 10.1038/oby.2009.208. [DOI] [PubMed] [Google Scholar]