

Abstract
SHANK3 is a synaptic scaffolding protein and plays an important role in neuronal development. SHANK3 interacts with various synaptic molecules, including post-synaptic density-95 (PSD-95), homer and GluR1 AMPA receptor. SHANK3 gene is a causable gene of the Phelan- McDermid syndrome (also known as the 22q13.3 deletion syndrome), whose manifestation is global developmental delay and autistic behavior, especially shows severe speech and language deficit. Additionally since cumulative gene analysis in autistic subjects identified several mutations in SHANK3 gene, including deletion and duplication in a particular region, abnormality of SHANK3 gene is thought the be related with the neuropathology of autism spectrum disorder (ASD). We here review the recent findings in regard to the roles of SHANK3 in higher brain functions, molecular-biologic studies of the complex expression of Shank3 transcripts and production of SHANK3 isoforms, and behavioral studies of Shank3-mutant mice, including our recent findings, and discuss a novel therapeutic approach for ASD.
Keywords: Autism spectrum disorder, cytokine, microglia, Phelan-McDermid syndrome, SHANK3, synapse.
1. INTRODUCTION
Autism spectrum disorder (ASD) is a neuro-developmental, which shows delays and deficits in the development of multiple brain functions, including social communication and reciprocal interaction, perception, and motor skills, and also shows restricted and stereotyped patterns of interests and behaviors [1, 2]. ASD affects about 1-2 % of the population, with about four times more males diagnosed than females, and are considered to be highly genetic in nature. Although the causative genes of ASD have not been fully understood, structural variations of chromosomes have been identified in some ASD individuals, that is, the most common deletion and duplication have been shown on chromosome 7q, 15q and 22q. Additionally, terminal deletion of the 22q segment causes a syndromic developmental disorder, called the 22q13.3 deletion syndrome that is also called the Phelan-McDermid syndrome. The manifestation of Phelan-McDermid syndrome is severe speech and expressive language deficit, global development delay, and autistic behavior. Furthermore, the patients with Phelan-McDermid syndrome demonstrate hypotonia, normal to advanced growth including dolicocephaly, and minor dysmorphisms [3].
In 2001, Bonaglia et al. performed genetic analysis in the patient with Phelan-McDermid syndrome and found a 46,XY,t(12;22)(q24.1;q13.3) translocation. Since further sequence analysis revealed the breakpoint on chromosome 22 at position 296489/296494, within exon 21 of the SHANK3 gene and identified a recurrent breakpoint within the SHANK3 gene [4, 5], SHANK3 is a strong candidate causative gene of Phelan-McDermid syndrome. In this review, we focus on SHANK3, highlight the defective synapses causing deficits in higher brain functions, and discuss an advanced therapeutic strategy for ASD.
2. ASSOCIATION OF SHANK3 WITH ASD
SHANK3 is one of three members of the SHANK proteins that organizes a cytoskeleton-associated signaling complex at postsynaptic density (PSD) as a scaffolding protein. The SHANK3 gene encodes a multidomain protein containing ankyrin repeats (ANK), a Src homology 3 (SH3) domain, a postsynaptic density 95/discs large/zone occludens-1 (PDZ) domain, a proline-rich region, a homer-binding region, a cortactin-binding region and a sterile alpha motif (SAM) [6]. SHANK3 is mainly localized in the post-synapses, and interacts with various synaptic molecules, for examples GluR1 of a-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPA-R), N-methyl-D-aspartate receptor (NMDA-R) via the postsynaptic density-95 (PSD-95)/guanylate kinase-associated protein (GKAP) complex, and insulin receptor substrate of 53 kDa (IRSp53) [6-11].
Recent human genetic studies carried out in ASD patients strongly support the suspected association between molecular defects of SHANK3 and ASD. Sequence variations of the SHANK3 gene, including missense, frame-shift, insertion and deletion mutations, have been found in ~0.5% of ASD patients [12-17]. Durand et al. showed eight non-synonymous mutations that were observed in ASD individuals, but not in control individuals [12]. Several mutations had effects on the dendritic spine morphology and synaptic transmission [12]. They further found an insertion of a guanidine (G) nucleotide in exon 21 in two brothers with ASD causing a frameshift alteration that resulted in the production of a carboxy (C)-terminus-truncated SHANK3 protein, while Gauthier et al. reported a deletion of a G nucleotide in exon 19, which was also predicted to lead to a frameshift alteration, in a patient with ASD [14]. Gauthier et al. further reported a deletion of a G nucleotide in the highly conserved splicing donor site in intron 19 leading to aberrant splicing that resulted in the production of a truncated SHANK3 protein lacking a large portion of the C-terminal domain [14]. We also performed the genetic analysis of SHANK3 gene in 134 autistic subjects with similar manifestations seen in Phelan-McDermid syndrome patients, especially severe delayed speech development, mental retardation and autistic behavior, and identified 6 patients with Phelan-McDermid syndrome and 7 patients with alterations in the SHANK3 gene, including a novel missense variant in the PDZ domain, an 18-bp nucleotides deletion upstream of the SH3 domain, and intronic insertion and deletion of a 10-bp GC nucleotides repeat located 9-bp downstream from the 3’ end of exon 11 [15]. Thus, alterations in the SHANK3 gene, including 22q13.3 deletion, were detected in about 10% of ASD patients with a particular phenotype (13 patients with SHANK3 gene alterations/134 patients). These findings from genetic studies of ASD patients are thought to suggest a strong association between SHANK3 and ASD [18].
3. SHANK3 TRANSCRIPTS AND SHANK3 ISOFORMS IN THE BRAIN
Recent studies have shown that several Shank3 transcripts are expressed by complicated transcriptional regulation with some intragenic promoters and alternative splicing [19]. Five different intragenic promoters produced various types of Shank3 transcripts and several SHANK3 isoforms (SHANK3a - SHANK3f) (Fig. 1A). We also identified novel transcripts, named Shank3c-3 and Shank3c-4, produced from promoter 3 located in intron 10 (Fig. 1B) [20] (Waga et al., 2014). Although the molecular structures of Shank3 transcripts are becoming increasingly clear, identification of SHANK3 isoforms has remained largely unclear. A western blot analysis, shown in Fig. 2A, indicated the differential expressions of various SHANK3 isoforms in the mouse developing neocortex. Since the anti-Shank3 antibody used was produced against the SHANK3 fragment in exon 21 (amino acids 1016-1357), it was not possible to detect splicing variants lacking exon 21, including SHANK3b, using this anti-Shank3 antibody. Judging from the molecular weight, the largest band of over 200 kDa likely corresponded to SHANK3a, that is, the full-length SHANK3 protein composed of 1730 amino acids. However, it is difficult to determine which band each SHANK3 isoform corresponds to. We recently constructed novel Shank3c-deficient mice by targeting exon 11 and exon 12 (unpublished data). It is expected that experiments using this mouse will help us to identify the several SHANK3 isoforms and their expression profiles in the developing mouse brain. The novel Shank3c-deficient mice express none of SHANK3a, SHANK3b, and SHANK3c, including SHANK3c-3 and SHANK3c-4. In the neocortex, the largest band corresponding to SHANK3a was shown in the wild-type mice, but as expected, it was not observed in the Shank3c-deficient mice (Fig. 2B). Three bands were seen around 140 - 150 kDa in the wild-type mice; since the largest among them could not be observed in the Shank3c-deficient mice, this band is considered to be equivalent to SHANK3c, whose predicted molecular weight is 150 kDa (1322 amino acids). Other bands near 140 kDa that were observed in both wild-type and Shank3c-deficient mice were predicted to be equivalent to SHANK3d and SHANK3f. In the mouse cerebellum, while SHANK3c was clearly expressed, expression of SHANK3a was hardly detected (Fig. 2B). Interestingly, the band near 180 kDa was observed in both the wild-type and Shank3c-deficient mice. While this band was very faint in the neocortex of 8-week-old mice, two bands near 180 kDa were apparently seen in the developing mouse neocortex. Given the notion that the molecular weight of this isoform is greater than that of SHANK3c and that this isoform is expressed in both wild-type and Shank3c-deficient mice constructed by a targeting of exon 11 and exon 12, it is predicted that the SHANK3 isoform corresponding to this band originally lacks exon 11 and exon 12. Judging from the number of nucleotides and codons, the putative SHANK3 isoform will lack three exons, exons 10, 11, and 12, which is 579 bp in length and codes for 193 amino acids. Thus, the predicted molecular weight of the novel SHANK3 isoform is about 180 kDa (1530 amino acids), closely matching the data of the western blot analysis (Fig. 1C and Fig. 2B). We then examined the Shank3 transcripts by RT-PCR method, and confirmed the sequences of the novel Shank3 transcripts lacking exons 10, 11, and 12 in the mouse brain. Recently, Wang et al. also presented the novel Shank3 transcripts produced by alternative splicing in exons 10 to 12 [21]. According to their report, SHANK3a (E10-12S V) lacking exons 10 to 12 is identical to the isoform that we obtained in our study. In addition, they also reported two different Shank3 transcripts and SHANK3 isoforms, SHANK3a/b (E10-12S III/IV) and SHANK3b (E10-12S V).
Fig. (1).
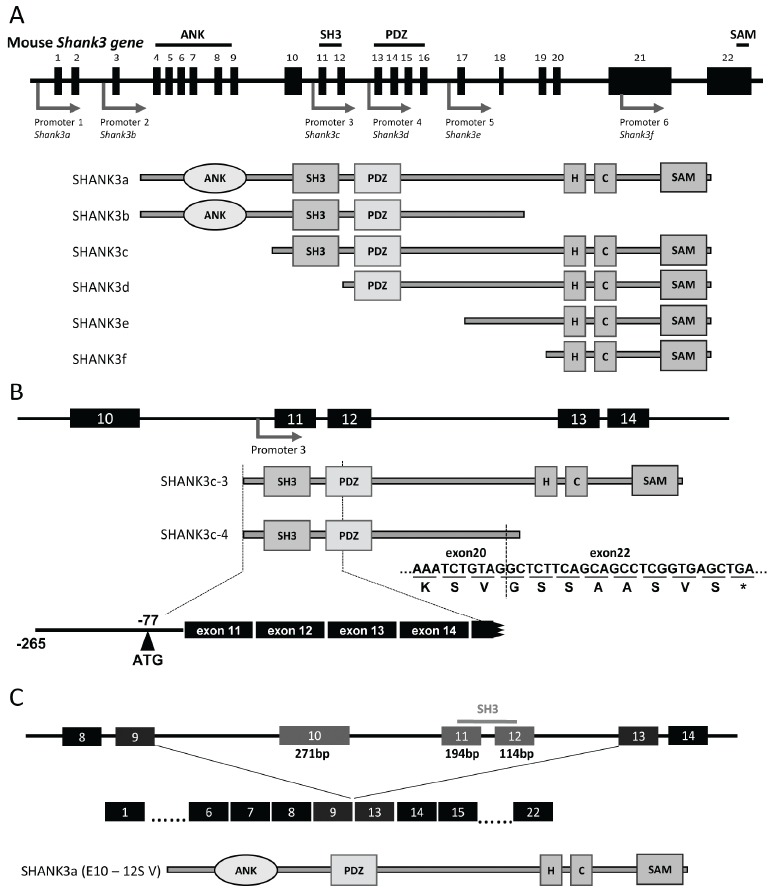
Schematic structures of the mouse Shank3 gene, Shank3 transcripts, and SHANK3 isoforms. (A) Diagram of the Shank3 gene and six SHANK3 isoforms. Top figure shows a schematic diagram of the Shank3 gene. Closed boxes indicate exons (exon 1 - exon 22). Arrows indicate the promoters. Lower figures show schematic structures of six SHANK3 isoforms (SHANK3a - SHANK3f) predicted to be produced from each promoter [19]. (B) Diagram of the Shank3 gene and two SHANK3c isoforms. The SHANK3c-3 consists of a part of intron 10 and all exons from exon 11 to exon 22, and the SHANK3c-4 isoform is an alternatively spliced variant that lacks exon 21. The translational start codon (ATG) is located in intron 10 at position -77 [20]. (C) Diagram of the Shank3 gene and the novel SHANK3 isoform identical to SHANK3a (E10-12S V) [21], which is an alternative splicing variant that lacks exon 10 - exon 12 encoding the SH3 domain. ANK, ankyrin repeats; H, homer-binding region; C, cortactin-binding region.
Fig. (2).
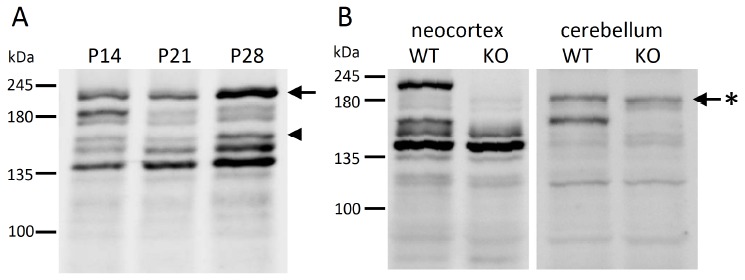
SHANK3 isoforms analyzed by Western blotting. The preparation of the protein samples and the western blotting using anti-Shank3 antibody were in accordance with the methods described by Waga et al. [20]. Molecular weight standards are presented on the left. (A) Expression profiles of the SHANK3 isoforms in the developing neocortex, postnatal day 14 (P14), postnatal day 21 (P21), and postnatal day 28 (P28). Neocortical samples (50 μg) were loaded. The SHANK3a isoform is indicatedd by an arrow and the SHANK3c-3 isoform is indicated by an arrowhead. (B) Expression profiles of the SHANK3 isoforms in the neocortex (left panel) and cerebellum (right panel) of 8-week-old wild-type mice (WT) and Shank3c-deficient mice (KO). Protein samples (50 μg) were loaded. The novel SHANK3 isoform (identical to SHANK3a (E10-12S V) [21]) is indicated by a star mark.
4. SYNAPTIC DEFECTS IN SHANK3-DEFICIENT MICE CAUSED ABNORMAL ASD-LIKE BEHAVIOR
Different Shank3-deficient mice line have been produced and have contributed to investigation of the roles of SHANK3 in synaptic functions and in the neuropathology of ASD. Bozdagi et al. first generated Shank3-deficient mice with targeted disruption from exon 4 to exon 9 [22]. These mice express SHANK3c-SHANK3f produced from downstream of exon 10, but not SHANK3a and SHANK3b, including SHANK3a (E10-12s V). The heterozygous mice (Shank3+/-) showed the low level expression of AMPA-R (GluR1) and impairment in excitatory synaptic transmission and plasticity in hippocampal CA1 pyramidal neurons. In the behavioral analysis, male heterozygotes exhibited less social sniffing and emitted fewer ultrasonic vocalizations interacting with female mice in a state of estrus. In contrast, homozygous mice (Shank3-/-) demonstrated abnormalities in communication patterns and social behaviors, repetitive behaviors and impaired learning and memory, similar to ASD features [23]. Peça et al. generated two different types of Shank3-deficient mice by targeted disruption from exon 4 to exon 7 (Shank3A-/-) and form exon 13 to exon 16 (Shank3B-/-) [24]. The Shank3A-/- mice expressed SHANK3c - SHANK3f, but no SHANK3a and SHANK3b, while the Shank3B-/- mice expressed SHANK3e and SHANK3f, but not SHANK3a - SHANK3d. The Shank3B-/- mice showed the reduction of AMPA-R (GluR2) and NMDA-Rs (GluN2A and GluN2B), PSD-93 and homer, morphological abnormality in the striatal synapses, and reduced cortico-striatal synaptic transmission. Furthermore, the Shank3B-/- mice also showed deficits in social interaction and discrimination of social novelty, while the Shank3A-/- mice displayed normal initiation of social interaction, but impaired recognition of social novelty. Although the Shank3A-/- mice did not show any anxiety-like behavior or lesions, the Shank3B-/- mice also demonstrated anxiety-like behavior, and excessive and self-injurious grooming. These findings suggest that defective SHANK3 expression is probably involved in phenotypic heterogeneity in ASD, and causes synaptic dysfunctions and abnormal neural circuit formation, resulting in various abnormal behaviors resembling the features of ASD. Thus, Shank3-deficient mice are expected as useful mouse models for the study of ASD. To reveal the phenotypic heterogeneity in ASD, however, it is necessary in future studies to clarify the expression profile of each SHANK3 isoform.
5. THERAPEUTIC APPROACHES FOR ASD WITH SHANK3 ABNORMALITY
It is conceivable that restoration of synaptic dysfunction caused by an abnormality of the SHANK3 gene may serve as a useful therapeutic strategy for ASD. According to this concept, insulin-like growth factor-1 (IGF-1) may be an attractive candidate molecule. IGF-1 shows various effects on neuronal development and synaptic functions, and is an especially critical factor for normal dendritic growth [25-27]. The first clinical trial of IGF-1 was carried out for the treatment for Rett syndrome, which is a severe X-linked autistic neurodevelopmental disorder with intellectual disability. Several researchers have reported that IGF-1 is safe and tolerated in individuals with Rett syndrome, and that it ameliorates certain behavioral abnormalities [28, 29]. Recently, Bozdagi et al. reported a successful trial of IGF-1 treatment in Shank3-deficient mice. They reported that daily intraperitoneal injections of IGF-1 for 2 weeks period reversed deficits in hippocampal long-term potentiation, AMPA signaling, and motor performance [30]. Furthermore, Shcheglovitov et al. produced induced pluripotent stem (iPS) cells from Phelan-McDermid syndrome patients and caused them differentiate into neurons showing reduced expressions of SHANK3, GluR1 of AMPA-R, and GluN1 of NMDA-R, which caused major defects in excitatory synaptic transmission. This neuronal abnormality could be corrected by restoring SHANK3 expression, as expected. Interestingly, IGF-1 treatment could also promote the formation of mature excitatory synapses by increasing the number of synaptic AMPA-R and NMDA-R, and correct defects in excitatory synaptic transmission even in the absence of SHANK3 [31]. Since cumulative evidence has shown that the formation and functions of synapses, including transmission and plasticity, are affected by the surrounding glial cells, including astrocytes and microglia, it is conceivable that several intrinsic factors in the brains, especially released factors from glial cells like IGF-1, may restore the defective synapses. Not only neuronal elements between pre- and post-synapses, but also astrocytes, are essential for the proper functioning of synapses, the so-called ‘tripartite synapse’. Gliotransmitters, including glutamate and ATP, released from astrocytes have been shown to control neuronal development and synaptic function, and also to regulate proper formation of neuronal circuits [32-35]. Microglia, which play important roles in active immune defense in the brains, also regulate synapse formation, including synaptic pruning, and neurotransmitter signaling [36-38]. Interestingly, microglial activation and an increased microglial density have been reported to be observed in the brains of patients with ASD [39]. Furthermore, increased expression of several cytokines and their receptors, including chemokine CCL4 and CX3CR1, have been reported in the peripheral blood and cerebral spinal fluid of patients with ASD [40, 41]. Chemokines, which are a family of cytokines released from glial cells, play important roles in neuron-glia interactions [42]. Zhan et al. recently demonstrated that Cx3cr1-deficient mice exhibited a transient reduction of the density of microglia during the early postnatal period and a consequent defect of synaptic pruning. Cx3cr1-deficient mice were reported to show abnormal behavior, that is, impaired social interaction and increased repetitive behavior, resembling ASD features [43]. Thus, glial cell-derived molecules also are expected to pave the way for the development of a novel therapeutic strategy for ASD based on the regulation of synaptic function.
Recent clinical studies on Phelan-McDermid syndrome have shown intriguing aspects. It was reported that several patients of Phelan-McDermid syndrome with SHANK3 deletion also displayed psychiatric features like schizophrenia and bipolar disorder in addition to developmental disabilities [44, 45]. Furthermore, many genetic studies have identified SHANK3 mutations in patients with psychiatric disorders [46-48]. In addition, not only deletion, but also duplication of the terminal 22q segment encompassing the SHANK3 gene has been shown to be associated with various developmental and psychiatric disorders. Durand et al. showed a partial trisomy in terminal 22q in a male subject with Asperger syndrome [12]. We also found two subjects with a microduplication of the 22q13 region; both patients demonstrated moderate delay of developmental and infantile hypotonia but neither had autistic features [49]. A recent comprehensive study on schizophrenia showed an association of the disease with the 22q13 abnormality, including of the SHANK3 gene [50]. As a particular case, Failla et al. found a schizophrenic girl with a 5.4 Mb duplication in the 22q13-qter segment [51]. She developed normally until the age of 13years, but thereafter began to exhibit various psychiatric features, including dysfunction of self-control and self-awareness, auditory hallucinations, psychomotor restlessness and attention deficit. Since duplication of the SHANK3 gene is thought to lead to disruption of the proper SHANK3 dosage and result in higher SHANK3 expression, Shank3-transgenic mice were generated as a model mouse of SHANK3 overexpression. The transgenic mice exhibited manic-like behavior and seizures; interestingly, the manic-like behavior was rescued by treatment with the mood-stabilizing drug valproate, but not with lithium [52].
6. CONCLUSIONS
At present, the major treatments for ASD are behavioral and educational approaches. Recent progression of the understanding of the neuropathology of ASD via molecular-biologic studies have led to improved clarification of the genes associated with ASD. In this review, we focused on the SHANK3 gene that is known to be a causable gene for the Phelan-McDermid syndrome characterized by severe delay of speech and language and autistic features. Cumulative evidence from studies in mice has shown the different Shank3 transcripts and SHANK3 isoforms expressed in the brain, and that deficits in several SHANK3 isoforms can cause synaptic dysfunction and abnormal behaviors similar to ASD features. Based on the results of recent studies, trials of novel therapeutic approaches, such as restoration of deficits in synaptic function, have been started. IGF-1, described in this review, is expected to be one of the potential candidates for the treatment of ASD. In order to develop more effective treatments, the functions and expression profile of each SHANK3 isoform must be clarified and the association between the defective neuronal circuits caused by synaptic dysfunction and the features of ASD need to be elucidated.
ACKNOWLEDGEMENTS
The study carried out our laboratory was supported by grants from the Ministry of Health, Labour and Welfare of Japan, and the Ministry of Education, Culture, Sports, and Science and Technology of Japan. The authors have no conflicts of interest to declare.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Mefford H.C., Batshaw M.L., Hoffman E.P. Genomics, intellectual disability, and autism. N. Engl. J. Med. 2012;366(8):733–743. doi: 10.1056/NEJMra1114194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall C.R., Scherer S.W. Detection and characterization of copy number variation in autism spectrum disorder. Methods Mol. Biol. 2012;838:115–135. doi: 10.1007/978-1-61779-507-7_5. [DOI] [PubMed] [Google Scholar]
- 3.Phelan M.C., Rogers R.C., Saul R.A., Stapleton G.A., Sweet K., McDermid H., Shaw S.R., Claytor J., Willis J., Kelly D.P. 22q13 deletion syndrome. Am. J. Med. Genet. 2001;101(2):91–99. doi: 10.1002/1096-8628(20010615)101:2<91::AID-AJMG1340>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 4.Bonaglia M.C., Giorda R., Borgatti R., Felisari G., Gagliardi C., Selicorni A., Zuffardi O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 2001;69(2):261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonaglia M.C., Giorda R., Mani E., Aceti G., Anderlid B-M., Baroncini A., Pramparo T., Zuffardi O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 2006;43(10):822–828. doi: 10.1136/jmg.2005.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naisbitt S., Kim E., Tu J.C., Xiao B., Sala C., Valtschanoff J., Weinberg R.J., Worley P.F., Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23(3):569–582. doi: 10.1016/S0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 7.Lim S., Naisbitt S., Yoon J., Hwang J.I., Suh P-G., Sheng M., Kim E. Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 1999;274(41):29510–29518. doi: 10.1074/jbc.274.41.29510. [DOI] [PubMed] [Google Scholar]
- 8.Sheng M., Kim E. The Shank family of scaffold proteins. J. Cell Sci. 2000;113(Pt 11):1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- 9.Bockmann J., Kreutz M.R., Gundelfinger E.D., Böckers T.M. ProSAP/Shank postsynaptic density proteins interact with insulin receptor tyrosine kinase substrate IRSp53. J. Neurochem. 2002;83(4):1013–1017. doi: 10.1046/j.1471-4159.2002.01204.x. [DOI] [PubMed] [Google Scholar]
- 10.Böckers T.M., Segger-Junius M., Iglauer P., Bockmann J., Gundelfinger E.D., Kreutz M.R., Richter D., Kindler S., Kreienkamp H-J. Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol. Cell. Neurosci. 2004;26(1):182–190. doi: 10.1016/j.mcn.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Uchino S., Wada H., Honda S., Nakamura Y., Ondo Y., Uchiyama T., Tsutsumi M., Suzuki E., Hirasawa T., Kohsaka S. Direct interaction of post-synaptic density-95/Dlg/ZO-1 domain-containing synaptic molecule Shank3 with GluR1 α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor. J. Neurochem. 2006;97(4):1203–1214. doi: 10.1111/j.1471-4159.2006.03831.x. [DOI] [PubMed] [Google Scholar]
- 12.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsäter H., Sponheim E., Goubran-Botros H., Delorme R., Chabane N., Mouren-Simeoni M-C., de Mas P., Bieth E., Rogé B., Héron D., Burglen L., Gillberg C., Leboyer M., Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39(1):25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moessner R., Marshall C.R., Sutcliffe J.S., Skaug J., Pinto D., Vincent J., Zwaigenbaum L., Fernandez B., Roberts W., Szatmari P., Scherer S.W. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007;81(6):1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gauthier J., Spiegelman D., Piton A., Lafrenière R.G., Laurent S., St-Onge J., Lapointe L., Hamdan F.F., Cossette P., Mottron L., Fombonne É., Joober R., Marineau C., Drapeau P., Rouleau G.A. Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. Part B. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- 15.Waga C., Okamoto N., Ondo Y., Fukumura-Kato R., Goto Y., Kohsaka S., Uchino S. Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr. Genet. 2011;21(4):208–211. doi: 10.1097/YPG.0b013e328341e069. [DOI] [PubMed] [Google Scholar]
- 16.Boccuto L., Lauri M., Sarasua S.M., Skinner C.D., Buccella D., Dwivedi A., Orteschi D., Collins J.S., Zollino M., Visconti P., Dupont B., Tiziano D., Schroer R.J., Neri G., Stevenson R.E., Gurrieri F., Schwartz C.E. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 2013;21(3):310–316. doi: 10.1038/ejhg.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemirovsky S.I., Córdoba M., Zaiat J.J., Completa S.P., Vega P.A., González-Morón D., Medina N.M., Fabbro M., Romero S., Brun B., Revale S., Ogara M.F., Pecci A., Marti M., Vazquez M., Turjanski A., Kauffman M.A. Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PLoS One, [Online] 2015;10 doi: 10.1371/journal.pone.0116358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uchino S., Waga C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev. 2013;35(2):106–110. doi: 10.1016/j.braindev.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y-H., Ehlers M.D. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waga C., Asano H., Sanagi T., Suzuki E., Nakamura Y., Tsuchiya A., Itoh M., Goto Y., Kohsaka S., Uchino S. Identification of two novel Shank3 transcripts in the developing mouse neocortex. J. Neurochem. 2014;128(2):280–293. doi: 10.1111/jnc.12505. [DOI] [PubMed] [Google Scholar]
- 21.Wang X., Bey A.L., Chung L., Krystal A.D., Jiang Y-H. Therapeutic approaches for shankopathies. Dev. Neurobiol. 2014;74(2):123–135. doi: 10.1002/dneu.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bozdagi O., Sakurai T., Papapetrou D., Wang X., Dickstein D.L., Takahashi N., Kajiwara Y., Yang M., Katz A.M., Scattoni M.L., Harris M.J., Saxena R., Silverman J.L., Crawley J.N., Zhou Q., Hof P.R., Buxbaum J.D. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism, [Online] (http://www.molecularautism.com/content/1/1/15. ) 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X., McCoy P.A., Rodriguiz R.M., Pan Y., Je H.S., Roberts A.C., Kim C.J., Berrios J., Colvin J.S., Bousquet-Moore D., Lorenzo I., Wu G., Weinberg R.J., Ehlers M.D., Philpot B.D., Beaudet A.L., Wetsel W.C., Jiang Y.H. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011;20(15):3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peça J., Feliciano C., Ting J.T., Wang W., Wells M.F., Venkatraman T.N., Lascola C.D., Fu Z., Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472(7344):437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Kusky J.R., Ye P., D’Ercole A.J. Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J. Neurosci. 2000;20(22):8435–8442. doi: 10.1523/JNEUROSCI.20-22-08435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bondy C.A., Cheng C.M. Signaling by insulin-like growth factor 1 in brain. Eur. J. Pharmacol. 2004;490(1-3):25–31. doi: 10.1016/j.ejphar.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez A.M., Torres-Alemán I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012;13(4):225–239. doi: 10.1038/nrn3209. [DOI] [PubMed] [Google Scholar]
- 28.Pini G., Scusa M.F., Congiu L., Benincasa A., Morescalchi P., Bottiglioni I., Marco P.D., Borelli P., Bonuccelli U., Della-Chiesa A., Prina-Mello A., Tropea D. IGF1 as apotential treatment for Rett syndrome: Safety assessment in six Rett patients. Autism Res. Treat., [Online] 2012. 2012. [DOI] [PMC free article] [PubMed]
- 29.Khwaja O.S., Ho E., Barnes K.V., O’Leary H.M., Pereira L.M., Finkelstein Y., Nelson C.A., III, Vogel-Farley V., DeGregorio G., Holm I.A., Khatwa U., Kapur K., Alexander M.E., Finnegan D.M., Cantwell N.G., Walco A.C., Rappaport L., Gregas M., Fichorova R.N., Shannon M.W., Sur M., Kaufmann W.E. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. USA. 2014;111(12):4596–4601. doi: 10.1073/pnas.1311141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bozdagi O., Tavassoli T., Buxbaum J.D. Insulin-like growth favtor-1 rescues synaptic and motor deficits in mouse model of autism and developmental delay. 2013. [DOI] [PMC free article] [PubMed]
- 31.Shcheglovitov A., Shcheglovitova O., Yazawa M., Portmann T., Shu R., Sebastiano V., Krawisz A., Froehlich W., Bernstein J.A., Hallmayer J.F., Dolmetsch R.E. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perea G., Navarrete M., Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32(8):421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Perea G., Araque A. GLIA modulates synaptic transmission. 2010. [DOI] [PubMed]
- 34.Santello M., Calì C., Bezzi P. Gliotransmission and the tripartite synapse. Adv. Exp. Med. Biol. 2012;970:307–331. doi: 10.1007/978-3-7091-0932-8_14. [DOI] [PubMed] [Google Scholar]
- 35.Chung W-S., Clarke L.E., Wang G.X., Stafford B.K., Sher A., Chakraborty C., Joung J., Foo L.C., Thompson A., Chen C., Smith S.J., Barres B.A. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L., Ragozzino D., Gross C.T. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 37.Kettenmann H., Kirchhoff F., Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77(1):10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 38.Miyamoto A., Wake H., Moorthouse A.J., Nabekura J. Microglia and synapse interactions: fine tuning neural circuits and candidate molecules. 2013. [DOI] [PMC free article] [PubMed]
- 39.Morgan J.T., Chana G., Pardo C.A., Achim C., Semendeferi K., Buckwalter J., Courchesne E., Everall I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 40.Pardo C.A., Vargas D.L., Zimmerman A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry. 2005;17(6):485–495. doi: 10.1080/02646830500381930. [DOI] [PubMed] [Google Scholar]
- 41.Enstrom A.M., Lit L., Onore C.E, Gregg J.P., Hansen R., Pessah I.N., Hertz-Picciotto I., Van de Water J.A., Sharp F.R., Ashwood P. Altered gene expression and function of peripheral blood natural killer cells in children with autism. 2009. [DOI] [PMC free article] [PubMed]
- 42.Deverman B.E., Patterson P.H. Cytokines and CNS development. 2009. [DOI] [PubMed]
- 43.Zhan Y., Paolicelli R.C., Sforazzini F., Weinhard L., Bolasco G., Pagani F., Vyssotski A.L., Bifone A., Gozzi A., Ragozzino D., Gross C.T. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014;17(3):400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- 44.Verhoeven W.M., Egger J.I., Willemsen M.H., de Leijer G.J., Kleefstra T. Phelan-McDermid syndrome in two adult brothers: atypical bipolar disorder as its psychopathological phenotype? Neuropsychiatr. Dis. Treat. 2012;8:175–179. doi: 10.2147/NDT.S30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vucurovic K., Landais E., Delahaigue C., Eutrope J., Schneider A., Leroy C., Kabbaj H., Motte J., Gaillard D., Rolland A-C., Doco-Fenzy M. Bipolar affective disorder and early dementia onset in a male patient with SHANK3 deletion. Eur. J. Med. Genet. 2012;55(11):625–629. doi: 10.1016/j.ejmg.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 46.Awadalla P., Gauthier J., Myers R.A., Casals F., Hamdan F.F., Griffing A.R., Côté M., Henrion E., Spiegelman D., Tarabeux J., Piton A., Yang Y., Boyko A., Bustamante C., Xiong L., Rapoport J.L., Addington A.M., DeLisi J.L., Krebs M.O., Joober R., Millet B., Fombonne E., Mottron L., Zilversmit M., Keebler J., Daoud H., Marineau C., Roy-Gagnon M.H., Dubé M.P., Eyre-Walker A., Drapeau P., Stone E.A., Lafrenière R.G., Rouleau G.A. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet. 2010;87(3):316–324. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauthier J., Champagne N., Lafrenière R.G., Xiong L., Spiegelman D., Brustein E., Lapointe M., Peng H., Côté M., Noreau A., Hamdan F.F., Addington A.M., Rapoport J.L., Delisi L.E., Krebs M.O., Joober R., Fathalli F., Mouaffak F., Haghighi A.P., Néri C., Dubé M.P., Samuels M.E., Marineau C., Stone E.A., Awadalla P., Barker P.A., Carbonetto S., Drapeau P., Rouleau G.A., S2D Team De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA. 2010;107(17):7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grabrucker S., Proepper C., Mangus K., Eckert M., Chhabra R., Schmeisser M.J., Boeckers T.M., Grabrucker A.M. The PSD protein ProSAP2/Shank3 displays synapto-nuclear shuttling which is deregulated in a schizophrenia-associated mutation. Exp. Neurol. 2014;253:126–137. doi: 10.1016/j.expneurol.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 49.Okamoto N., Kubota T., Nakamura Y., Murakami R., Nishikubo T., Tanaka I., Takahashi Y., Hayashi S., Imoto I., Inazawa J., Hosokai N., Kohsaka S., Uchino S. 22q13 Microduplication in two patients with common clinical manifestations: a recognizable syndrome? Am. J. Med. Genet. A. 2007;143A(23):2804–2809. doi: 10.1002/ajmg.a.31771. [DOI] [PubMed] [Google Scholar]
- 50.Föcking M., Lopez L.M., English J.A., Dicker P., Wolff A., Brindley E., Wynne K., Cagney G., Cotter D.R. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. 2014. [DOI] [PubMed]
- 51.Failla P., Romano C., Alberti A., Vasta A., Buono S., Castiglia L., Luciano D., Di Benedetto D., Fichera M., Galesi O. Schizophrenia in a patient with subtelomeric duplication of chromosome 22q. Clin. Genet. 2007;71(6):599–601. doi: 10.1111/j.1399-0004.2007.00819.x. [DOI] [PubMed] [Google Scholar]
- 52.Han K., Holder J.L., Jr, Schaaf C.P., Lu H., Chen H., Kang H., Tang J., Wu Z., Hao S., Cheung S.W., Yu P., Sun H., Breman A.M., Patel A., Lu H-C., Zoghbi H.Y. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503(7474):72–77. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]