

Abstract
Schizophrenia is considered a neurodevelopmental and neurodegenerative disorder. Cognitive impairment is a core symptom in patients with the illness, and has been suggested a major predictor of functional outcomes. Reduction of parvalbumin (PV)-positive γ-aminobutyric acid (GABA) interneurons has been associated with the pathophysiology of schizophrenia, in view of the link between the abnormality of GABA neurons and cognitive impairments of the disease. It is assumed that an imbalance of excitatory and inhibitory (E-I) activity induced by low activity of glutamatergic projections and PV-positive GABA interneurons in the prefrontal cortex resulted in sustained neural firing and gamma oscillation, leading to impaired cognitive function. Therefore, it is important to develop novel pharmacotherapy targeting GABA neurons and their activities. Clinical evidence suggests serotonin (5-HT) 1A receptor agonist improves cognitive disturbances of schizophrenia, consistent with results from preclinical studies, through mechanism that corrects E-I imbalance via the suppression of GABA neural function. On the other hand, T-817MA, a novel neurotrophic agent, ameliorated loss of PV-positive GABA neurons in the medial prefrontal cortex and reduction of gamma-band activity, as well as cognitive dysfunction in animal model of schizophrenia. In conclusion, a pharmacotherapy to alleviate abnormalities in GABA neurons through 5-HT1A agonists and T-817MA is expected to prevent the onset and/or progression of schizophrenia.
Keywords: cognitive dysfunction, GABA, glutamate, 5-HT1A agonist, neuroprotection, schizophrenia, T-817MA.
1. INTRODUCTION
Schizophrenia is a relatively common and often debilitating neuropsychiatric disorder. The illness typically starts in late adolescence or early adulthood, and in most cases, persists throughout life. Patients manifest positive and negative symptoms, as well as impairments in cognitive function. Long-term disability and social decline is most closely associated with deficits in cognition, which also determines treatment outcome [1-3]. Currently, the disease is treated mainly with antipsychotic drugs through the actions on various neurotransmitter receptors, such as dopamine (DA) receptors. However, the effect of the majority of established pharmacological therapies is mainly limited to the reduction of positive symptoms, while negative symptoms and cognitive impairments may often persist throughout life, in spite of treatment with existing antipsychotics [4, 5]. These lines of evidence lead to the concept that brain morphological changes induced by response to neurotoxic stress is a primary pathophysiology of schizophrenia.
Schizophrenia is considered as both a “neuro-degenerative” [5, 6] and “neurodevelopmental” [7, 8] disorder. While these two concepts may appear contradictory, they are, in fact, neural processes to explain the patho-physiology of the illness, if coupled with the temporal factor. Each process may predominate at different stages of schizophrenia, which may contribute to the variety of symptoms [9]. Given accumulating neuroimaging evidence for progressive volume changes occurring early in psychosis, neurodegeneration is likely to be associated with neuro-chemical dysregulation that can lead to the onset of the illness [5, 10]. Progressive cortical atrophy has been considered to be related to histological abnormalities, such as a smaller somal volume and decreased spine density, dendritic length and terminals, without gliotic reactions. It is postulated that these findings represent neurodegeneration induced by apoptosis, not necrosis [11]. The apoptotic processes, such as synaptic apoptosis and layer-specific neuronal and/or glial apoptosis, occur during the prodromal phase of schizophrenia [9, 11, 12].
Dysfunction of γ-aminobutyricacid (GABA) interneurons, particularly those containing the calcium-binding protein parvalbumin (PV), has been suggested to be associated with the pathophysiology of schizophrenia, as the result of an imbalance between excitation and inhibition in the cerebral cortex [13-15]. In fact, levels of glutamate decarboxylase (GAD) 67, the enzyme that synthesizes GABA, mRNA and protein have been found consistently to be lower in the frontal cortex of subjects with schizophrenia [16-18]. There is histological evidence for the reduction of PV-positive GABA interneuron density in the frontal cortex [19, 20]. It is suggested that a developmental deficit of inhibitory GABA interneurons may underlie neuro-degeneration through the exaggerated activation of gulutamatergic neurons [21]. Therefore, efforts to identify more effective approaches, for example, the application of neuroprotective and/or neuroregenerative compounds, as well as neuromodulators, are needed.
2. COGNITIVE DYSFUNCTION IN SCHIZOPHRENIA
2.1. Behavioral Symptoms
Schizophrenia has been characterized by positive symptoms (e.g., delusions, hallucination, thought disorder) and negative symptoms (e.g., psychomotor retardation, blunted affect, social withdrawal). Patients with the illness also demonstrated about a 1-2.5 SD decline in neuro-psychological performance, e.g. several types of memory, executive function, attention/information processing, verbal fluency, and motor function [22]. For example, working memory impairment is thought to be a core feature of schizophrenia, because it consists of several processes including storage buffers for different domains of information and a central executive component for the manipulation of information within the storage buffers [23]. It is considered that cognitive dysfunction is a major determinant of outcome, including quality of life and social function [24].
2.2. Relation to the Abnormality of GABA Neurons
Cognitive performance relies on coordinated activities of several brain regions, including the dorsolateral prefrontal cortex (DLPFC) [25]. Gamma frequency (30-80 Hz) oscillations in DLPFC neural networks are thought to provide a key neural substrate for cognition. Fast-spiking PV-positive GABAergic interneurons and pyramidal glutamatergic neurons seem to act as a basic unit of synchronous oscillatory activity, and this high frequency gamma band activity may underlie basal cognitive processes, especially working memory [26].
It is assumed that an imbalance of excitatory and inhibitory (E-I) activity induced by low activity of glutamatergic projections and PV-positive GABA interneurons in the prefrontal cortex resulted in sustained neural firing and gamma oscillation, leading to impaired working memory in schizophrenia [15, 27-29]. An upstream GAD 67 deficit in PV-positive GABA interneurons has been reported in the PFC of rats treated with MK-801, which is related to N-methyl-D-aspartate (NMDA) receptor hypofunction in these neurons, leading to disinhibition of pyramidal cells [30]. Postmortem studies report reductions in the number of PV-positive GABA interneurons in the PFC of patients with schizophrenia [13]. Moreover, the concentration of cortical GABA and the activity of GAD67 has been shown to be decreased [31, 32]. These observations led to the “GABA hypofunction” theory [33].
3. STRATEGY FOR THE DEVELOPING NOVEL PHARMACOTHERAPY IN SCHIZOPHRENIA
The first antipsychotic drugs were not prospectively designed but rather discovered serendipitously [21]. Since chlorpromazine was found in the early 1950s to alleviate agitation and hallucinations, a large amount of research has been directed to the molecular mechanisms (usually neurotransmitter receptors) underlying the effects of first-generation antipsychotic (FGA) or psychotomimetic drugs [34]. The pathophysiology of schizophrenia remains unclear; however, some promising hypotheses have been postulated. One convincing hypothesis is dysregulation of dopaminergic neurotransmissions in limbic brain regions [35], because all antipsychotic drugs are dopamine D2 receptor antagonists [36]. Specifically, there is a linear relationship between clinical potencies of antipsychotic drugs and their affinity for D2 receptors in the brain [37-39]. However, FGAs have relatively little influence on cognitive and negative symptoms, and elicit adverse side effects, such as extrapyramidal motor symptoms, tardive dyskinesia, weight gain, and sedation [1, 2, 40].
Recently, second-generation antipsychotics drugs (SGAs) have been used as first-line medication to treat patients with schizophrenia. It has been indicated that SGAs elicit a partial effect on improving cognitive dysfunction, which may be related to their relatively high affinity for serotonin (5-HT)-5HT2A receptors compared with DA-D2 receptors [41, 42]. However, about one-third of patients are resistant to treatment with existing SGA [4].
As a next step to develop novel psychotrophic compounds to treat negative symptoms and cognitive deficits, correcting the “E-I imbalance” (low activity of glutamatergic projections and PV-positive GABA interneurons) may be important to enhance cognitive performance in schizophrenia [29, 43, 44]. Several pharmacological approaches in this line are currently under study; these include agonists at the glycine site of NMDA receptor, DA-D1 receptor, metabotropic glutamate receptor (mGluR; type-2/3 or 5), or 5-HT1A receptors [45, 46].
On the other hand, there is evidence for a progressive volume loss of gray matter and reduced growth of white matter in schizophrenia, particularly in cases with multiple exacerbations [47]. Histological evidence demonstrated the reduction of PV-positive GABA interneuron density in the frontal cortex [19, 20]. These volumetric changes may be a challenge for pharmacological treatments [45]. Therefore, it is important to develop a novel paradigm targeting those morphological abnormalities Fig. (1).
Fig. (1).
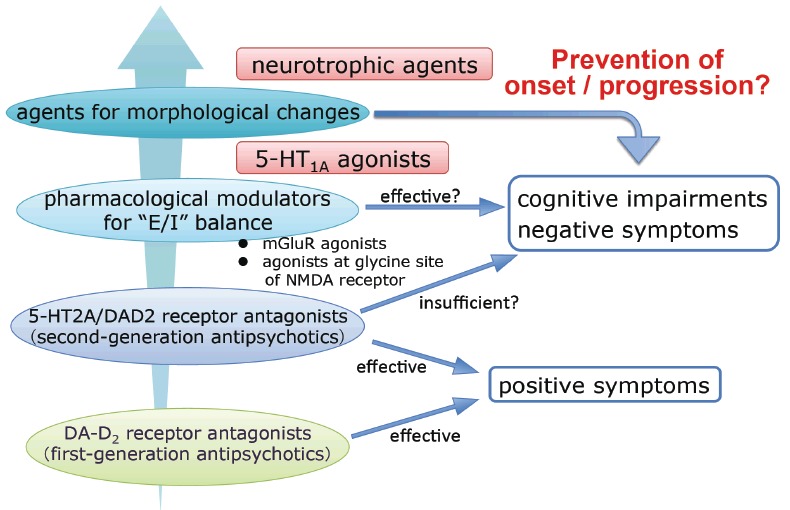
The strategy for developing new pharmacotherapy in schizophrenia.
In the following sections, we will discuss data on the effects of 5-HT1A agonists and of T-817MA, a novel neurotropic agent, on cognitive deficits and changes of PV-positive GABA interneurons.
4. 5-HT1A AGONISTS IN THE TREATMENT OF COGNITIVE DYSFUNCTION
4.1. Clinical Evidence for the Role for 5-HT1A Receptors in Cognition
Disturbances of cognitive function, evaluated by psychological and neurophysiological methods, have been shown to predict social outcome in patients with schizophrenia [3, 42]. Particularly, interests are given to the role of psychotropic compounds acting on 5-HT receptors in alleviating cognitive impairment of the disease. Among the 5-HT receptor subtypes, the 5-HT1A receptor has been a focus as a potential target for enhancing cognition [48-51]. We have carried out a series of clinical trials to determine whether 5-HT1A agonists improve cognitive function in patients with schizophrenia [52-54]. The addition of tandospirone (30 mg/kg) to typical antipsychotic drugs taken (mainly haloperidol) for 4-6 weeks, was found to improve verbal memory, memory organization, and executive function [52, 54]. Furthermore, in a randomized, double-blind, placebo-controlled study, the addition of buspirone (30 mg/kg) outperformed the placebo in improving attention/information processing in patients treated with SGAs, such as olanzapine and risperidone [53]. These findings provided the basis for the ability of 5-HT1A receptor stimulation to enhance cognition, a therapeutic approach that has promoted the development of novel antipsychotic drugs [48-50, 55].
4.2. Preclinical Evidence
The advantage of 5-HT1Aagonism for cognitive function is also suggested by animal studies. Thus, treatment with perospirone [56] or aripiprazole [57], 5-HT1A partial agonists [58, 59], dose-dependently alleviated phencyclidine (PCP)-induced cognitive deficits in mice. This effect was abolished by pretreatment with the selective 5-HT1Aantagonist WAY-100635. Moreover, tandospirone, a partial 5-HT1A receptor agonist, dose-dependently improves impaired memory performance as evaluated by the Novel Objects Recognition Test in PCP-induced rat models. This effect was also abolished by pretreatment with WAY 100635 [60]. These findings provide further support for the concept that cognitive disturbances of schizophrenia are ameliorated by stimulation of 5-HT1A receptors [50, 60].
We have examined the effects of tandospirone on extracellular lactate concentration (eLAC) as an indicator of brain energy metabolism using a microdialysis technique [61]. Lactate production was hypothesized to supply energy substrates and reflect neural activity, whose metabolism depends on glutamatergic activity [61]. Specifically, activation of postsynaptic NMDA receptors or glutamate transporters on astrocyte enhance lactate production [61, 62]. Our investigation revealed that the acute administration of tandospirone increased eLAC in the medial prefrontal cortex (mPFC) of naïve adult rats [63], while chronic treatment ameliorated abnormal lactate metabolism in the mPFC of a rat model of schizophrenia [64, 65]. These data on brain energy metabolism support the hypothesis that 5-HT1A agonism can improve cognitive deficits of schizophrenia [55, 66].
4.3. Mechanisms Underlying Cognitive Enhancement by 5-HT1A Receptors
Sumiyoshi et al. postulated a putative neural network involving glutamate, GABA, 5-HT and DA associated with the ability of 5-HT1A agonists to enhance cognition in schizophrenia [50, 51, 55] Fig. (2). Systemic administration of 8-OH-DPAT, a 5-HT1A agonist, decreases action potentials of GABA neurons. This leads to disinhibition of glutamate neurons [50, 67] and activation of meso-cortical dopamine neurons [68]. For example, administration of clozapine increases extracellular DA concentrations in the prefrontal cortex through activation of 5-HT1A receptors [68]. Findings from electrophysiological studies are consistent with the concept that cognitive benefits of 5-HT1Aagonism are mediated by glutamate and GABA neurons [67, 69].
Fig. (2).
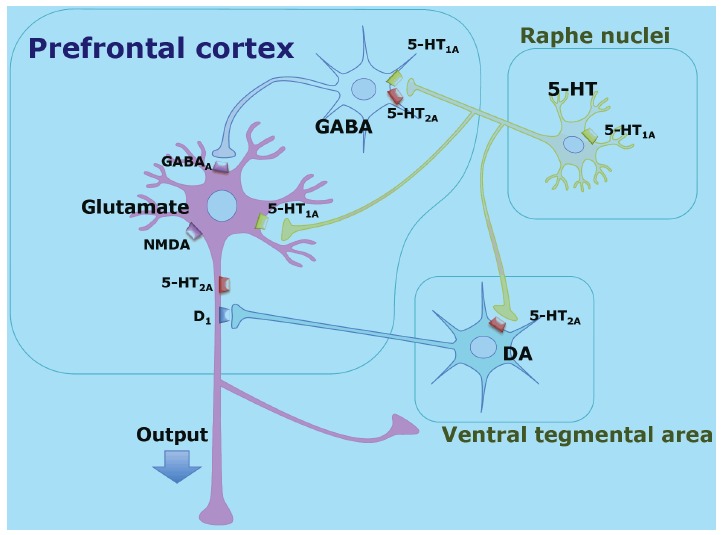
The putative mechanisms underlying cognitive enhancement by 5-HT1A receptors and Neural network in the prefrontal cortex involving glutamate, GABA, 5-HT and DA neurons. Part of the effect of 5-HT1A agonists on cognition and negative symptoms is thought to be mediated by 5-HT1A receptors located on GABAergic interneurons regulating glutamatergic pyramidal neurons [50, 51, 55].
Altogether, clinical and animal studies provide evidence for the benefits of 5-HT1Aagonism on cognitive function in schizophrenia and the mechanism is possibly involved in resetting the E-I imbalance.
It is noteworthy that activation of 5-HT1A receptors exerts a neuroprotective effect in animal models of ischemia. 5-HT1A agonists have been shown to moderate cerebral damage induced by focal cerebral ischemia in rats and mice [70]. Moreover, stimulation of 5-HT1A receptor induces neuroprotection against NMDA-induced apoptotic cell death in cell cultures [71]. Although the cellular mechanisms underlying the neuroprotective effect of 5-HT1A receptor activation remain unclear, it is assumed that 5-HT1A receptor stimulation can modulate NMDA-receptor-induced Ca2+ influx and facilitate the antiapoptotic effect of neurotrophin brain-derived neurotrophic factor (BDNF) [70]. Further studies are warranted to determine the ability of 5-HT1A receptor agonism to ameliorate morphological abnormalities in schizophrenia.
5. NEUROTROPHIC AGENTS AND MORPHOLOGICAL ABNORMALITIES
While some debate exists as to whether schizophrenia is entirely neurodevelopmental in nature, there is evidence supporting a progressive and neurodegenerative disease process as well. The pathophysiological processes possibly begin in the prodromal stage of the illness, and continue after the onset of the illness [5, 9, 10]. The underlying mechanisms may include apoptosis (a.k.a. programmed cell death). Progressive volume reduction of the brain, especially the prefrontal cortex, is found in individuals at high genetic risk of schizophrenia who later develop schizophrenia [72]. The vulnerability of neurons to pro-apoptotic insults (pro-apoptotic triggers) would lead to selective dendritic and synaptic losses observed in subjects with schizophrenia [11]. The number of spines on the dendrites of pyramidal neurons in the frontal association cortex is largely reduced in schizophrenia, while neurons themselves are reduced in size and packed more densely without any change in number [73, 74]. In the promotion of apoptosis, several stimuli have also been suggested to play a role, including 1) glutamatergic excitotoxicity, 2) excess synaptic calcium flux, 3) oxidative stress, and 4) reduced neurotrophin levels (for example, BDNF, neurotrophin-3; NT-3) [11, 75].
5.1. T-817MA as a Novel Neurotrophic Agent
Neuroprotection refers to the relative preservation of the structural integrity and normal functioning of the central nervous system against a pathological process and consequent neurobiological stress [75]. T-817MA 1-{3-2-(1-benzothiophen-5-yl) ethoxypropyl} azetidin-3- ol maleate is a newly synthesized agent that was developed for the treatment of neurodegenerative disorders, such as Alzheimer’s disease (Fig. 3). It (1) exerts neuroprotective effects against neurotoxicity caused by intracerebroventricular infusion of amyloid-β(Aβ) [76, 77], (2) facilitates neurogenesis, such as neuron proliferation, neurite outgrowth, and synaptogenesis [78], and (3) improves cognitive impairment in rats receiving intracerebroventricular infusion of Aβ [76, 77] or expressing FTDP17 human P301L mutant tau [79]. In particular, this agent elicits neuroprotective effects against H2O2-induced neurotoxicity [78] through attenuation of reactive oxygen species (ROS) production in mitochondria [80]. These data suggest that T-817MA exerts neurotrophic potency on the central nervous system, but precise mechanism of action remains unclear, for example, whether this agent itself has neurotrophic effects or leads to enhance neurotrophic factors.
Fig. (3).
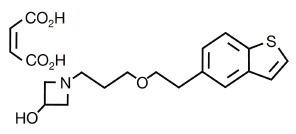
Chemical structure of T-817MA [1-{3-[2-(1-benzothiophen- 5-yl) ethoxy]propyl} azetidin-3-ol maleate] [78].
5.2. Effect of T-817MA on the Abnormality of GABA Neurons in an Animal Model of Schizophrenia
Rats administered with MK-801, a non-competitive NMDA receptor antagonist, on postnatal days (PDs) 7-10 provide an animal model of schizophrenia. These animals have been shown to elicit (1) impaired set-shifting, a measure of PFC function, in early adulthood [81], (2) disruption of prepulse inhibition (PPI), a measure of sensorimotor gating [82, 83], and (3) enhancement of methamphetamine-induced locomotor activity after puberty [83], (4) suppression of brain energy metabolism in the mPFC but not in the striatum [64], and (5) reduction of mRNA expression of mGluR3 receptors in the mPFC [84]. Moreover, the same model animals demonstrated a decreased number of PV-positive GABA interneurons in the mPFC and hippocampus [85]. These findings suggest that neonatal treatment with NMDA antagonists produces behavioral and histochemical abnormalities mimicking some features of schizophrenia.
Using rats administered with MK-801 on PD 7-10, we examined the effect of T-817MA on the number of PV-positive GABA neurons in the mPFC and hippocampus [85]. In these experiments, rats received T-817MA (10 or 20 mg/kg/day) orally. Treatment with T-817MA, at 20 mg/kg, for 14 days (from PD49 to PD62) ameliorated PPI deficits and prevented the decrease in the number of PV-positive GABAergic interneurons in the mPFC and hippocampus (mainly CA2/3 subfield) in early adulthood (PD63), while haloperidol (1.0 mg/kg/day) or risperidone (1.0 mg/kg/day) did not cause any effect Fig. (4).
Fig. (4).
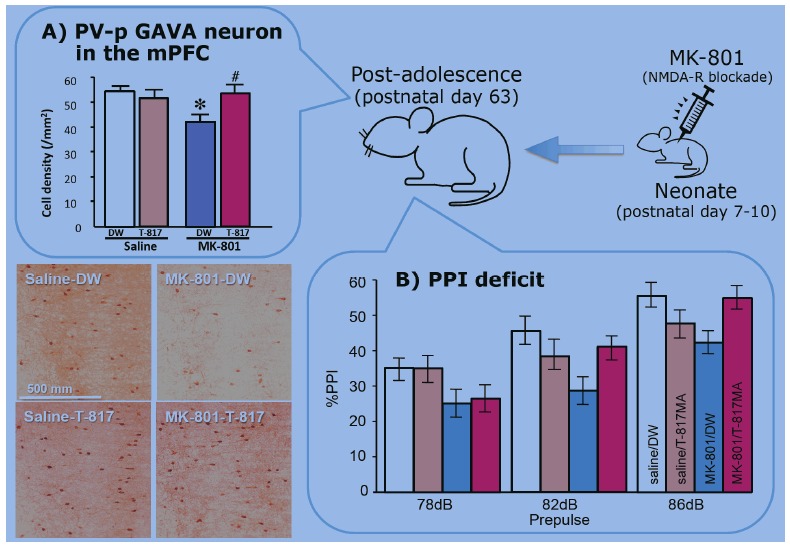
The schema of effects of T-817MA on (A) decreased number of parvalbumin positive GABA interneurons (PV-p GAVA neuron) in the medial prefrontal cortex (mPFC) and (B) prepulseinhibiton (PPI) deficit in animal model of schizophrenia. Rats were received MK-801 (0.2 mg/kg/day, s.c.) neonatally (postnatal days 7-10) [85].
The same effect of T-817MA has been confirmed with another rodent model of schizophrenia [86]. Mice with knockout of the platelet-drived growth factor (PDGF) receptor ß (PDGFR-ß) gene were considered as an animal model of schizophrenia [86, 87]. These animals have been shown to elicit PPI deficits, impeded social interaction, and disturbed spatial learning, indicated by the food search test, as well as reduction of gamma-band activity and the number of PV-positive GABA neurons in the mPFC [86, 87]. Nakamura et al. [86] have demonstrated that T-817MA (10 mg/kg/day, p.o.) for 4 weeks ameliorated these abnormalities. Importantly, sensory-evoked gamma oscillations were significantly correlated with the density of PV-positive GABA neurons in the mPFC. These results suggest that T-817MA can improve cognitive deficits in animal models of schizophrenia through ameliorating gamma oscillation generated by PV-positive GABA interneurons in the mPFC.
These results suggest that T-817MA modifies the “E-I balance” through recovery of histochemical abnormalities. The precise mechanisms underlying the effect of T-817MA on improving the cognitive dysfunction of these animal models of schizophrenia are unclear. However, this neurotrophic agent may also have the ability to facilitate neurogenesis and neuroprotective effects against apoptosis in vitro as mentioned above. Taken together, T-817MA may be able to restore some morphological changes in the brain of patients with schizophrenia Fig. (5).
Fig. (5).
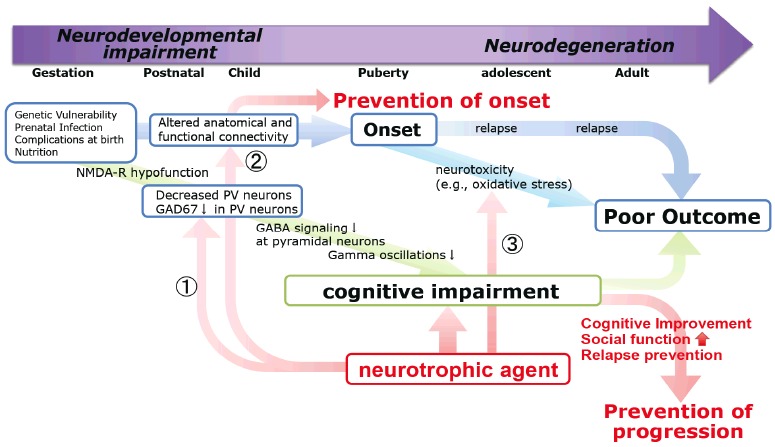
The schema of T-817MA efficacy to prevent the onset or progression of schizophrenia. T-817MA have shown the ability in vivo or animal models of schizophrenia to ①reverse the decrease in the number of parvalbumin (PV) -positive GABAergic interneurons in the mPFC and hippocampus, ②facilitate neurogenesis, such as neuron proliferation, neurite outgrowth, and synaptogenesis, through the increase of neurotrophic factors and ③elicit neuroprotective effects against H2O2-induced neurotoxicity through attenuation of reactive oxygen species (ROS) production in mitochondria.
6. CONCLUSIONS
We presented the hypothesis that 5-HT1A agonists improve cognitive dysfunction in patients with schizophrenia, through modulation of the activity of GABA neurons, leading to disinhibition of glutamate neurons in the PFC. Stimulation of 5-HT1A receptors may also protect neurodegeneration. T-817MA, a novel neurotrophicagent, also ameliorated cognitive impairments and gamma oscillation in mPFC of some animal models of schizophrenia, as well as recovering the number of PV-positive GABA interneurons in the mPFC. Further research is needed to confirm the efficacy of this agent in clinical settings. Data from preclinical research suggest the effects of the 5-HT1A receptor hyperfunction for the neurodevelopmental disability and T-817MA for the neurodegenerative disability of schizophrenia. A phar-macotherapy to alleviate abnormalities in GABA neurons through 5-HT1A agonists and T-817MA is expected to prevent the onset and/or progression of schizophrenia.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the insightful comments and criticism by Prof. Y. Kawasaki in the Department of Neuropsychiatry, Kanazawa Medical University. This study was funded by grants from the Smoking Research Foundation to Takashi Uehara, as well as the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research No. 26461761 and Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (27-1) to Tomiki Sumiyoshi.
CONFLICTS OF INTEREST
The authors confirm that this article content has no conflicts of interest.
References
- 1.Conn P.J., Tamminga C., Schoepp D.D., Lindsley C. Schizophrenia: moving beyond monoamine antagonists. Mol. Interv. 2008;8(2):99–107. doi: 10.1124/mi.8.2.7. [DOI] [PubMed] [Google Scholar]
- 2.Meltzer H.Y. Treatment of schizophrenia and spectrum disorders: pharmacotherapy, psychosocial treatments, and neurotransmitter interactions. Biol. Psychiatry. 1999;46(10):1321–1327. doi: 10.1016/S0006-3223(99)00255-3. [DOI] [PubMed] [Google Scholar]
- 3.McGurk S.R., Meltzer H.Y. The role of cognition in vocational functioning in schizophrenia. Schizophr. Res. 2000;45(3):175–184. doi: 10.1016/S0920-9964(99)00198-X. [DOI] [PubMed] [Google Scholar]
- 4.Freedman R. Schizophrenia. N. Engl. J. Med. 2003;349(18):1738–1749. doi: 10.1056/NEJMra035458. [DOI] [PubMed] [Google Scholar]
- 5.Lieberman J.A. Is schizophrenia a neurodegenerative disorder? A clinical and neurobiological perspective. Biol. Psychiatry. 1999;46(6):729–739. doi: 10.1016/S0006-3223(99)00147-X. [DOI] [PubMed] [Google Scholar]
- 6.Lewis D.A., Lieberman J.A. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28(2):325–334. doi: 10.1016/S0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- 7.Weinberger D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry. 1987;44(7):660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 8.Weinberger D.R. Neurodevelopmental perspectives on schizophrenia. In: Bloom F.E., Kupfer D.J., editors. Psychopharmacology; The Forth Generation of Progress. New York: Raven Press; 1995. pp. 1171–1183. [Google Scholar]
- 9.Lieberman J.A., Jarskog L.F., Malaspina D. Preventing clinical deterioration in the course of schizophrenia: the potential for neuroprotection. J. Clin. Psychiatry. 2006;67(6):983–990. doi: 10.4088/JCP.v67n0616. [DOI] [PubMed] [Google Scholar]
- 10.Lieberman J.A., Perkins D., Belger A., Chakos M., Jarskog F., Boteva K., Gilmore J. The early stages of schizophrenia: speculations on pathogenesis, pathophysiology, and therapeutic approaches. Biol. Psychiatry. 2001;50(11):884–897. doi: 10.1016/S0006-3223(01)01303-8. [DOI] [PubMed] [Google Scholar]
- 11.Jarskog L.F., Glantz L.A., Gilmore J.H., Lieberman J.A. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2005;29(5):846–858. doi: 10.1016/j.pnpbp.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Glantz L.A., Gilmore J.H., Lieberman J.A., Jarskog L.F. Apoptotic mechanisms and the synaptic pathology of schizophrenia. 2006. [DOI] [PubMed]
- 13.Lewis D.A., Hashimoto T., Volk D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005;6(4):312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 14.Benes F.M., Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. 2001. [DOI] [PubMed]
- 15.Lewis D.A., Curley A.A., Glausier J.R., Volk D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35(1):57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Burgos G., Hashimoto T., Lewis D.A. Alterations of cortical GABA neurons and network oscillations in schizophrenia. 2010. [DOI] [PMC free article] [PubMed]
- 17.Guidotti A., Auta J., Davis J.M., Di-Giorgi-Gerevini V., Dwivedi Y., Grayson D.R., Impagnatiello F., Pandey G., Pesold C., Sharma R., Uzunov D., Costa E. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry. 2000;57(11):1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 18.Curley A.A., Arion D., Volk D.W., Asafu-Adjei J.K., Sampson A.R., Fish K.N., Lewis D.A. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am. J. Psychiatry. 2011;168(9):921–929. doi: 10.1176/appi.ajp.2011.11010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beasley C.L., Reynolds G.P. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr. Res. 1997;24(3):349–355. doi: 10.1016/S0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- 20.Beasley C.L., Zhang Z.J., Patten I., Reynolds G.P. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol. Psychiatry. 2002;52(7):708–715. doi: 10.1016/S0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- 21.Olney J.W., Newcomer J.W., Farber N.B. NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 1999;33(6):523–533. doi: 10.1016/S0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 22.Keefe R.S., Goldberg T.E., Harvey P.D., Gold J.M., Poe M.P., Coughenour L. The Brief Assessment of Cognition in Schizophrenia: reliability, sensitivity, and comparison with a standard neurocognitive battery. Schizophr. Res. 2004;68(2-3):283–297. doi: 10.1016/j.schres.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 23.Baddeley A. Working memory. 1992. [DOI]
- 24.Green M.F. What are the functional consequences of neurocognitive deficits in schizophrenia? Am. J. Psychiatry. 1996;153(3):321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- 25.Miller E.K., Cohen J.D. An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- 26.Roopun A.K., Cunningham M.O., Racca C., Alter K., Traub R.D., Whittington M.A. Region-specific changes in gamma and beta2 rhythms in NMDA receptor dysfunction models of schizophrenia. Schizophr. Bull. 2008;34(5):962–973. doi: 10.1093/schbul/sbn059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inan M., Petros T.J., Anderson S.A. Losing your inhibition: linking cortical GABAergic interneurons to schizophrenia. Neurobiol. Dis. 2013;53:36–48. doi: 10.1016/j.nbd.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis D.A., Gonzalez-Burgos G. Pathophysiologically based treatment interventions in schizophrenia. Nat. Med. 2006;12(9):1016–1022. doi: 10.1038/nm1478. [DOI] [PubMed] [Google Scholar]
- 29.Genius J., Giegling I., Benninghoff J., Rujescu D. Disturbed function of GABAergic interneurons in schizophrenia: relevance for medical treatment? Curr. Pharm. Biotechnol. 2012;13(8):1549–1556. doi: 10.2174/138920112800784943. [DOI] [PubMed] [Google Scholar]
- 30.Homayoun H., Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007;27(43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bird J.M. Computed tomographic brain studies and treatment response in schizophrenia. Can. J. Psychiatry. 1985;30(4):251–254. doi: 10.1177/070674378503000407. [DOI] [PubMed] [Google Scholar]
- 32.Lisman J.E., Coyle J.T., Green R.W., Javitt D.C., Benes F.M., Heckers S., Grace A.A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31(5):234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kegeles L.S., Mao X., Stanford A.D., Girgis R., Ojeil N., Xu X., Gil R., Slifstein M., Abi-Dargham A., Lisanby S.H., Shungu D.C. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry, 2012;69(5):449–459. doi: 10.1001/archgenpsychiatry.2011.1519. [DOI] [PubMed] [Google Scholar]
- 34.Moore H. The role of rodent models in the discovery of new treatments for schizophrenia: updating our strategy. 2010. [DOI] [PMC free article] [PubMed]
- 35.Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1988;1(3):179–186. doi: 10.1016/0893-133X(88)90012-7. [DOI] [PubMed] [Google Scholar]
- 36.Sumiyoshi T. Possible dose-side effect relationship of antipsychotic drugs: relevance to cognitive function in schizophrenia. Expert Rev. Clin. Pharmacol. 2008;1(6):791–802. doi: 10.1586/17512433.1.6.791. [DOI] [PubMed] [Google Scholar]
- 37.Seeman P. Targeting the dopamine D2 receptor in schizophrenia. Expert Opin. Ther. Targets. 2006;10(4):515–531. doi: 10.1517/14728222.10.4.515. [DOI] [PubMed] [Google Scholar]
- 38.Seeman P., Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188(4194):1217–1219. doi: 10.1126/science.1145194. [DOI] [PubMed] [Google Scholar]
- 39.Seeman P., Lee T., Chau-Wong M., Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261(5562):717–719. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- 40.Nikam S.S., Awasthi A.K. Evolution of schizophrenia drugs: a focus on dopaminergic systems. Curr. Opin. Investig. Drugs. 2008;9(1):37–46. [PubMed] [Google Scholar]
- 41.Meltzer H.Y., Huang M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Prog. Brain Res., . 2008;172 :177–197. doi: 10.1016/S0079-6123(08)00909-6. [DOI] [PubMed] [Google Scholar]
- 42.Meltzer H.Y., McGurk S.R. The effects of clozapine, risperidone, and olanzapine on cognitive function in schizophrenia. 1999. [DOI] [PubMed]
- 43.Lewis D.A., Volk D.W., Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology (Berl.) 2004;174(1):143–150. doi: 10.1007/s00213-003-1673-x. [DOI] [PubMed] [Google Scholar]
- 44.Coyle J.T., Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl.) 2004;174(1):32–38. doi: 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- 45.Harvey P.D. Pharmacological cognitive enhancement in schizophrenia. 2009. [DOI] [PubMed]
- 46.Tandon R., Nasrallah H.A., Keshavan M.S. Schizophrenia, "just the facts" 5.Treatment and prevention.Past, present, and future. Schizophr. Res., . 2010;122 (1-3):1–23. doi: 10.1016/j.schres.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 47.Cahn W., Rais M., Stigter F.P., van Haren N.E., Caspers E., Hulshoff Pol H.E., Xu Z., Schnack H.G., Kahn R.S. Psychosis and brain volume changes during the first five years of schizophrenia. Eur. Neuropsychopharmacol. 2009;19(2):147–151. doi: 10.1016/j.euroneuro.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 48.Ohno Y., Tatara A., Shimizu S., Sasa M. Management of cognitive impairments in schizophrenia: the therapeutic role of 5-HT receptors. In: Sumiyoshi T., editor. Schizophrenia Research: Recent Advances. New York: Nova Science Publishers; 2012. pp. 321–335. [Google Scholar]
- 49.Newman-Tancredi A., Albert P.R. Gene polymorphism at serotonin 5-HT1A receptors: moving towards personalized medicine for psychosis and mood deficits? In: Sumiyoshi T., editor. Schizophrenia Research: Recent Advances. New York: Nova Science Publisher; 2012. pp. 337–358. [Google Scholar]
- 50.Sumiyoshi T., Higuchi Y. Facilitative effect of serotonin(1A) receptor agonists on cognition in patients with schizophrenia. Curr. Med. Chem. 2013;20(3):357–362. [PubMed] [Google Scholar]
- 51.Sumiyoshi T., Higuchi Y., Uehara T. Neural basis for the ability of atypical antipsychotic drugs to improve cognition in schizophrenia. Front. Behav. Neurosci. 2013;7:140. doi: 10.3389/fnbeh.2013.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sumiyoshi T., Matsui M., Yamashita I., Nohara S., Kurachi M., Uehara T., Sumiyoshi S., Sumiyoshi C., Meltzer H.Y. The effect of tandospirone, a serotonin(1A) agonist, on memory function in schizophrenia. Biol. Psychiatry. 2001;49(10):861–868. doi: 10.1016/S0006-3223(00)01025-8. [DOI] [PubMed] [Google Scholar]
- 53.Sumiyoshi T., Park S., Jayathilake K., Roy A., Ertugrul A., Meltzer H.Y. Effect of buspirone, a serotonin1A partial agonist, on cognitive function in schizophrenia: a randomized, double-blind, placebo-controlled study. Schizophr. Res. 2007;95(1-3):158–168. doi: 10.1016/j.schres.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 54.Sumiyoshi T., Matsui M., Nohara S., Yamashita I., Kurachi M., Sumiyoshi C., Jayathilake K., Meltzer H.Y. Enhancement of cognitive performance in schizophrenia by addition of tandospirone to neuroleptic treatment. Am. J. Psychiatry. 2001;158(10):1722–1725. doi: 10.1176/appi.ajp.158.10.1722. [DOI] [PubMed] [Google Scholar]
- 55.Sumiyoshi T., Uehara T. Serotonin-1A Receptors and Cognitive Enhancement in Schizophrenia: Role for Brain Energy Metabolism. In: In Tech o., editor. Neuropsychiatric Disorders, Burne, T.H.J. Rijeka, Croatia: 2012. pp. 127–140. [DOI] [Google Scholar]
- 56.Hagiwara H., Fujita Y., Ishima T., Kunitachi S., Shirayama Y., Iyo M., Hashimoto K. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the antipsychotic drug perospirone: role of serotonin 5-HT1A receptors. Eur. Neuropsychopharmacol. 2008;18(6):448–454. doi: 10.1016/j.euroneuro.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 57.Nagai T., Murai R., Matsui K., Kamei H., Noda Y., Furukawa H., Nabeshima T. Aripiprazole ameliorates phencyclidine-induced impairment of recognition memory through dopamine D1 and serotonin 5-HT1A receptors. Psychopharmacology (Berl.) 2009;202(1-3):315–328. doi: 10.1007/s00213-008-1240-6. [DOI] [PubMed] [Google Scholar]
- 58.Snigdha S., Neill J.C. Improvement of phencyclidine-induced social behaviour deficits in rats: involvement of 5-HT1A receptors. . Behav. Brain Res., . 2008; 191(1 ):26– 31. doi: 10.1016/j.bbr.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 59.Li Z., Ichikawa J., Dai J., Meltzer H.Y. Aripiprazole, a novel antipsychotic drug, preferentially increases dopamine release in the prefrontal cortex and hippocampus in rat brain. Eur. J. Pharmacol. 2004;493(1-3):75–83. doi: 10.1016/j.ejphar.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 60.Horiguchi M., Meltzer H.Y. The role of 5-HT1A receptors in phencyclidine (PCP)-induced novel object recognition (NOR) deficit in rats. Psychopharmacology (Berl.) 2012;221(2):205–215. doi: 10.1007/s00213-011-2561-4. [DOI] [PubMed] [Google Scholar]
- 61.Uehara T., Sumiyoshi T., Itoh H., Kurata K. Lactate production and neurotransmitters; evidence from microdialysis studies. Pharmacol. Biochem. Behav., 2008;90(2):273–281. doi: 10.1016/j.pbb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 62.Uehara T., Sumiyoshi T., Itoh H., Kurachi M. Role of glutamate transporters in the modulation of stress-induced lactate metabolism in the rat brain. Psychopharmacology (Berl.) 2007;195(2):297–302. doi: 10.1007/s00213-007-0881-1. [DOI] [PubMed] [Google Scholar]
- 63.Uehara T., Sumiyoshi T., Matsuoka T., Itoh H., Kurachi M. Role of 5-HT(1A) receptors in the modulation of stress-induced lactate metabolism in the medial prefrontal cortex and basolateral amygdala. Psychopharmacology (Berl.) 2006;186(2):218–225. doi: 10.1007/s00213-006-0370-y. [DOI] [PubMed] [Google Scholar]
- 64.Uehara T., Itoh H., Matsuoka T., Rujescu D., Genius J., Seo T., Sumiyoshi T. Effect of transient blockade of N-methyl-D-aspartate receptors at neonatal stage on stress-induced lactate metabolism in the medial prefrontal cortex of adult rats: role of 5-HT1A receptor agonism. Synapse. 2012;66(5):408–417. doi: 10.1002/syn.21529. [DOI] [PubMed] [Google Scholar]
- 65.Uehara T., Matsuoka T., Sumiyoshi T. Tandospirone, a 5-HT1A partial agonist, ameliorates aberrant lactate production in the prefrontal cortex of rats exposed to blockade of N-methy-D-aspartate receptors; Toward the therapeutics of cognitive impairment of schizophrenia. Front. Behav. Neurosci. 2014;8:291. doi: 10.3389/fnbeh.2014.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uehara T., Sumiyoshi T. 2013. Lactate metabolism as a new target for the therapeutics of schizophrenia. [DOI] [Google Scholar]
- 67.Llado-Pelfort L., Santana N., Ghisi V., Artigas F., Celada P. 5- HT1A receptor agonists enhance pyramidal cell firing in prefrontal cortex through a preferential action on GABA interneurons. Cereb. Cortex, . 2012;22(7):1487–1497. doi: 10.1093/cercor/bhr220. [DOI] [PubMed] [Google Scholar]
- 68.Bortolozzi A., Masana M., Díaz-Mataix L., Cortés R., Scorza M.C., Gingrich J.A., Toth M., Artigas F. Dopamine release induced by atypical antipsychotics in prefrontal cortex requires 5-HT(1A) receptors but not 5-HT(2A) receptors. Int. J. Neuropsychopharmacol. 2010;13(10):1299–1314. doi: 10.1017/S146114571000009X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Higuchi Y., Sumiyoshi T., Kawasaki Y., Ito T., Seo T., Suzuki M. Effect of tandospirone on mismatch negativity and cognitive performance in schizophrenia: a case report. 2010. [DOI] [PubMed]
- 70.Fiorino F., Severino B., Magli E., Ciano A., Caliendo G., Santagada V., Frecentese F., Perissutti E. 5-HT(1A) receptor: an old target as a new attractive tool in drug discovery from central nervous system to cancer. J. Med. Chem. 2014;57(11):4407–4426. doi: 10.1021/jm400533t. [DOI] [PubMed] [Google Scholar]
- 71.Madhavan L., Freed W.J., Anantharam V., Kanthasamy A.G. 5-hydroxytryptamine 1A receptor activation protects against N-methyl-D-aspartate-induced apoptotic cell death in striatal and mesencephalic cultures. J. Pharmacol. Exp. Ther. 2003;304(3):913–923. doi: 10.1124/jpet.102.044370. [DOI] [PubMed] [Google Scholar]
- 72.McIntosh A.M., Owens D.C., Moorhead W.J., Whalley H.C., Stanfield A.C., Hall J., Johnstone E.C., Lawrie S.M. Longitudinal volume reductions in people at high genetic risk of schizophrenia as they develop psychosis. Biol. Psychiatry. 2011;69(10):953–958. doi: 10.1016/j.biopsych.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 73.Selemon L.D., Kleinman J.E., Herman M.M., Goldman-Rakic P.S. Smaller frontal gray matter volume in postmortem schizophrenic brains. Am. J. Psychiatry. 2002;159(12):1983–1991. doi: 10.1176/appi.ajp.159.12.1983. [DOI] [PubMed] [Google Scholar]
- 74.Garey L. When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J. Anat. 2010;217(4):324–333. doi: 10.1111/j.1469-7580.2010.01231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lieberman J.A., Bymaster F.P., Meltzer H.Y., Deutch A.Y., Duncan G.E., Marx C.E., Aprille J.R., Dwyer D.S., Li X.M., Mahadik S.P., Duman R.S., Porter J.H., Modica-Napolitano J.S., Newton S.S., Csernansky J.G. Antipsychotic drugs: comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol. Rev. 2008;60(3):358–403. doi: 10.1124/pr.107.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen P.T., Kimura T., Ho S.A., Tran A.H., Ono T., Nishijo H. Ameliorative effects of a neuroprotective agent, T-817MA, on place learning deficits induced by continuous infusion of amyloid-beta peptide (1-40) in rats. Hippocampus. 2007;17(6):443–455. doi: 10.1002/hipo.20281. [DOI] [PubMed] [Google Scholar]
- 77.Kimura T., Hong Nguyen P.T., Ho S.A., Tran A.H., Ono T., Nishijo H. T-817MA, a neurotrophic agent, ameliorates the deficits in adult neurogenesis and spatial memory in rats infused i.c.v. with amyloid-beta peptide. Br. J. Pharmacol. 2009;157(3):451–463. doi: 10.1111/j.1476-5381.2009.00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hirata K., Yamaguchi H., Takamura Y., Takagi A., Fukushima T., Iwakami N., Saitoh A., Nakagawa M., Yamada T. A novel neurotrophic agent, T-817MA [1-3-[2-(1-benzothiophen-5-yl) ethoxy] propyl-3-azetidinol maleate], attenuates amyloid-beta-induced neurotoxicity and promotes neurite outgrowth in rat cultured central nervous system neurons. J. Pharmacol. Exp. Ther. 2005;314(1):252–259. doi: 10.1124/jpet.105.083543. [DOI] [PubMed] [Google Scholar]
- 79.Fukushima T., Nakamura A., Iwakami N., Nakada Y., Hattori H., Hoki S., Yamaguchi H., Nakagawa M., Terashima N., Narita H. T-817MA, a neuroprotective agent, attenuates the motor and cognitive impairments associated with neuronal degeneration in P301L tau transgenic mice. Biochem. Biophys. Res. Commun. 2011;407(4):730–734. doi: 10.1016/j.bbrc.2011.03.091. [DOI] [PubMed] [Google Scholar]
- 80.Fukushima T., Koide M., Ago Y., Baba A., Matsuda T. T- 817MA, a novel neurotrophic agent, improves sodium nitroprussideinduced mitochondrial dysfunction in cortical neurons. Neurochem. . 2006;48 (2):124–130 . doi: 10.1016/j.neuint.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 81.Stefani M.R., Moghaddam B. Transient N-methyl-D-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol. Psychiatry. 2005;57(4):433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 82.Uehara T., Sumiyoshi T., Seo T., Itoh H., Matsuoka T., Suzuki M., Kurachi M. Long-term effects of neonatal MK-801 treatment on prepulse inhibition in young adult rats. Psychopharmacology (Berl.) 2009;206(4):623–630. doi: 10.1007/s00213-009-1527-2. [DOI] [PubMed] [Google Scholar]
- 83.Uehara T., Sumiyoshi T., Seo T., Matsuoka T., Itoh H., Suzuki M., Kurachi M. Neonatal exposure to MK-801, an N-methyl-D-aspartate receptor antagonist, enhances methamphetamine-induced locomotion and disrupts sensorimotor gating in pre- and postpubertal rats. Brain Res. 2010;1352:223–230. doi: 10.1016/j.brainres.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 84.Uehara T., Sumiyoshi T., Rujescu D., Genius J., Matsuoka T., Takasaki I., Itoh H., Kurachi M. Neonatal exposure to MK-801 reduces mRNA expression of mGlu3 receptors in the medial prefrontal cortex of adolescent rats. Synapse. 2014;68(5):202–208. doi: 10.1002/syn.21734. [DOI] [PubMed] [Google Scholar]
- 85.Uehara T., Sumiyoshi T., Hattori H., Itoh H., Matsuoka T., Iwakami N., Suzuki M., Kurachi M. T-817MA, a novel neurotrophic agent, ameliorates loss of GABAergic parvalbumin-positive neurons and sensorimotor gating deficits in rats transiently exposed to MK-801 in the neonatal period. J. Psychiatr. Res. 2012;46(5):622–629. doi: 10.1016/j.jpsychires.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 86.Nakamura T., Matsumoto J., Takamura Y., Ishii Y., Sasahara M., Ono T., Nishijo H. Relationships among parvalbumin-immunoreactive neuron density, phase-locked gamma oscillations, and autistic/schizophrenic symptoms in PDGFR-β knock-out and control mice. PLoS One. 2015;10(3):e0119258. doi: 10.1371/journal.pone.0119258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nguyen P.T., Nakamura T., Hori E., Urakawa S., Uwano T., Zhao J., Li R., Bac N.D., Hamashima T., Ishii Y., Matsushima T., Ono T., Sasahara M., Nishijo H. Cognitive and socio-emotional deficits in platelet-derived growth factor receptor-β gene knockout mice. PLoS One. 2011;6(3):e18004. doi: 10.1371/journal.pone.0018004. [DOI] [PMC free article] [PubMed] [Google Scholar]