

Abstract
Epidermal growth factor receptor (EGFR) is associated with progression of many epithelial malignancies and represents a significant therapeutic target. Although clear cell renal cell carcinoma (CCRCC) has been widely investigated for EGFR molecular alterations, genetic evidences of EGFR gene activating mutations and/or gene amplification have been rarely confirmed in the literature. Therefore, until now EGFR-targeted therapies in clinical trials have been demonstrated unsuccessful. New evidence has been given about the interactions between EGFR and the sodium glucose co-transporter-1 (SGLT1) in maintaining the glucose basal intracellular level to favour cancer cell growth and survival; thus a new functional role may be attributed to EGFR, regardless of its kinase activity. To define the role of EGFR in CCRCC an extensive investigation of genetic changes and functional kinase activities was performed in a series of tumors by analyzing the EGFR mutational status and expression profile, together with the protein expression of downstream signaling pathways members. Furthermore, we investigated the co-expression of EGFR and SGLT1 proteins and their relationships with clinic-pathological features in CCRCC. EGFR protein expression was identified in 98.4% of CCRCC. Furthermore, it was described for the first time that SGLT1 is overexpressed in CCRCC (80.9%), and that co-expression with EGFR is appreciable in 79.4% of the tumours. Moreover, the activation of downstream EGFR pathways was found in about 79.4% of SGLT1-positive CCRCCs. The mutational status analysis of EGFR failed to demonstrate mutations on exons 18 to 24 and the presence of EGFR-variantIII (EGFRvIII) in all CCRCCs analyzed. FISH analysis revealed absence of EGFR amplification, and high polysomy of chromosome 7. Finally, the EGFR gene expression profile showed gene overexpression in 38.2% of CCRCCs. Our study contributes to define the complexity of EGFR role in CCRCC, identifying its bivalent kinase-dependent and kinase-independent functions, both potentially involved in CCRCC progression. These results might have important implications on therapeutic approaches to CCRCC, since the disruption of the interaction between EGFR/SGLT1, mediated by anti-EGFR antibodies and/or SGLT1 inhibitors, might constitute a novel therapeutic target for CCRCC treatment, and new clinical trials should be evaluated on the basis of this therapeutic proposal.
Keywords: Clear cell renal cell carcinoma, EGFR, SGLT1, kinase-dependent EGFR function, kinase-independent EGFR function, pAKT, p-p44/42 MAPK, p-STAT3, EGFR-variantIII, FISH analysis
Introduction
Clear cell renal cell carcinoma has been widely investigated for EGFR protein expression, and previous studies on wide series of CCRCC demonstrated that EGFR immunoreactivity is a common occurrence in CCRCCs, ranging from 50% to 90% among different series [1-6].
However, EGFR-targeted molecular therapies, namely tyrosine-kinase inhibitors, are not effective for CCRCC treatment [7-9]. In fact, genetic abnormalities such as EGFR gene activating mutations and/or gene amplification, known to be related with EGFR-targeted therapy responsiveness, have been rarely confirmed in the literature for CCRCC [10-12].
Although recent studies claimed for EGFR potential prognostic significance in CCRCC, with an apparent correlation between EGFR overexpression and higher grades and stages of the disease, this issue appears still controversial, according to previous findings [3,6,11].
Recent evidence suggests a novel potential role for EGFR in cancer progression, which seems to be unrelated to its kinase activity. SGLT1 is an integral membrane protein that mediates the active glucose transport across cellular membranes and relies on extracellular sodium concentration to transport glucose into cells, independently of glucose concentration [13]. Weihua et al. observed that EGFR maintains cellular homeostasis in neoplastic cells by a kinase-independent function; specifically, EGFR physically associates with and stabilizes SGLT1 maintaining basal intracellular glucose levels, thus promoting cancer cell survival and avoiding autophagic cancer cell death [14].
The overexpression of SGLT1 has been described in various types of cancers including colon-rectal carcinoma, lung carcinoma, head and neck carcinoma, pancreatic carcinoma and ovarian carcinoma.
SGLT1 expression in CCRCC has not previously been reported in the literature, despite of its natural location at the brush border of renal proximal tubules cells, from which the CCRCC is supposed to originate [15-20].
The aim of the present study was to perform an extensive investigation of EGFR genetic abnormalities and to evaluate its functional kinase activities in a series of CCRCCs; in addition to this, the expression of EGFR and SGLT1 in CCRCCs was analyzed and correlations between their protein expression levels and clinic-pathological features were assessed.
Material and methods
Selection of patients
Ethical approval and informed consent for this study was unnecessary, according to the Italian legislation concerning the guidelines for the performance of observational studies (G.U. n. 76. 31-3-2008); however, CCRCC samples were fully anonymized prior of any authors’ access. Consecutive 63 CCRCC were selected from the Histopathology Departments archives of Cagliari and Sassari (Italy). All cases were reviewed by at least two experienced pathologists, and categorized according to the current classification and staging systems [21,22].
From representative formalin-fixed, paraffin-embedded (FFPE) specimens, 3 µm-thick tissue sections were cut for haematoxylin and eosin stains (H&E), immunohistochemical and Fluorescence in situ hybridization (FISH) analysis. Additional consecutive sections were also obtained for genetic analysis.
Immunohistochemistry
Immunohistochemistry was performed using specific antibodies against EGFR, SGLT1, phospho-akt murine thymoma viral oncogene (p-AKT), phospho-signal transducer and activator of transcription 3 (p-STAT3) and phospho-mitogen-activated protein kinase (p-p44/42 MAPK). Sources, dilutions, and antigen retrieval conditions are summarized in Table 1.
Table 1.
Antibodies for immunohistochemical analyses
| Primary Antibodies | Type | Dilution | Antigen retrieval | Control tissue | Source |
|---|---|---|---|---|---|
| EGFR | Mouse Clone 2-18C9 | Prediluted | Proteinase K, room temperature, 5 min. | HT-29 cell line | DakoCytomation-EGFRPharmDx, Glostrup, Denmark |
| p-AKT (Ser473) | Mouse Monoclonal | 1:75 | Sodium citrate buffer pH 6, 10 mM, 99°C, 20 min. | Skin | Novocastra, Dublin, OH, USA |
| p-p44/42 MAPK (Erk1/2) (Thr202/204) | Rabbit Monoclonal | 1:100 | Sodium citrate buffer pH 6, 10 mM, 99°C, 10 min. | Colon cancer | Cell Signaling Technology, Boston, MA, USA |
| p-STAT3 (Tyr705) | Rabbit Monoclonal | 1:200 | Sodium citrate buffer pH 6, 10 mM, 99°C, 15 min. | Breast cancer | Cell Signaling Technology, Boston, MA, USA |
| SGLT1 | Rabbit Polyclonal | 1:200 | Sodium citrate buffer pH 6, 10 mM, 99°C, 20 min. | Kidney | Novus Biological, Littleton, CO, USA |
Immunoreactions were obtained by incubating sections with specific primary antibodies for 15 minutes. Immunodetection was performed using a non-biotin highly sensitive system (EnVision Peroxidase Detection System, Dako, Glostrup, Denmark), preventing possible false-positive staining due to endogenous biotin present in the tissue. The slides were then incubated with substrate chromogen solution diaminobenzidine (DAB) for 10 minutes and counterstained with haematoxylin. Specifically, EGFR immunoreaction was executed using EGFR pharmDx™ Kit (DakoCytomation), according to manufacturer’s instructions.
Results were scored semi-quantitatively including intensity (0, negative; 1+, weak; 2+, moderate; 3+, strong) and estimated percentages of labeled cells, with subcellular localization of immunostaining also being assessed for each positive case.
Fluorescence in situ hybridization
The slides were deparaffinized with two washes of xylene, 15 minutes each, and subsequently washed twice with absolute ethanol, 10 minutes each, and then air-dried in the hood. Then, the slides were treated in 0.1 mM citric acid (pH 6.0; Zymed, CA, USA) at 95°C for 10 minutes and rinsed in distilled water for 3 minutes, followed by a wash of 2× Standard Saline Citrate (SSC) for 5 minutes. Digestion of the tissue was performed by applying 0.4 ml of pepsin (5 mg/ml in 0.9% NaCl, pH 1.5; Sigma, St Louis, MO, USA) at 37°C for 40 minutes. The slides were rinsed with distilled water for 3 minutes, then washed with 2× SSC for 5 minutes, and air-dried.
Dual-color FISH was performed by using admixture of a spectrum green-labeled Centromeric α-satellite (CEP7) DNA probe and a spectrum orange-labeled locus-specific DNA probe for EGFR gene. Both probes were from Vysis (Vysis, Downers Grove, IL, USA) and were diluted with tDenHyb2 (Insitus, Alburquerque, NM, USA) in a ratio of 1:20.
Five µl of diluted probe were applied to each slide in reduced light. The slides were then covered with a 22×22 mm coverslip and sealed with rubber cement. Denaturation was achieved by incubating the slides at 80°C for 10 minutes in a humidified chamber and then hybridized at 37°C overnight. The coverslips were removed and the slides were washed extensively with two washes at 45°C with 0.1× SSC/1.5M urea, 20 minutes for each, followed by a wash with 2× SSC for 20 minutes and with 2× SSC/0.1% NP40 for 10 minutes at 45°C. The slides were further washed with 2× SSC for 5 minutes at room temperature. The slides were air-dried and counterstained with 10 ml DAPI (Insitus, Albuquerque, NM, USA), covered with coverslips and sealed with nail polish.
The slides were examined using an Olympus BX61 fluorescence microscope equipped with selective filters for the fluorochromes used. The images were acquired with a CCD camera and analyzed with Olympus DP-Softimage analysis software.
From each slide, 100 nuclei were scored for signals from LSI EGFR gene (orange) and CEP7 (green) under the fluorescence microscope with 1000x magnification and the ratio between orange and green signals was subsequently calculated; a tumor was considered amplified if the EGFR/CEP7 ratio was ≥ 2.0.
Definition of chromosomal gain was based on the Gaussian model and related to normal renal parenchyma controls, as previously described [23]. Briefly, for each slide, at least 100 nuclei were scored for green signals from centromeric probes, under the fluorescence microscope with 1000× magnification, in both tumors and adjacent non-neoplastic renal parenchyma. The cut-off value for chromosome 7 gain definition was set at mean value plus three standard deviations (SD) of the percentages of nuclei with three or more signals in normal renal parenchyma.
DNA and RNA extraction
Five 10 µm-thick consecutive sections from CCRCC specimens were prepared, and tumors were macro-dissected with a scalpel blade under sterile conditions, using corresponding H&E stained sections as a guide. Genomic DNA was obtained from neoplastic tissue blocks, and total RNA was obtained from the same neoplastic and not neoplastic specimens. Nucleic acids were extracted with a commercially available extraction kit (QIAamp DNA FFPE Tissue Kit and Rneasy FFPE Kit, Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions.
RNase A (USB Corp., Cleveland Ohio, USA) or DNase (Roche Diagnostics GmbH, Mannheim, Germany) were applied directly to the silica membrane to digest contaminating RNA or DNA, respectively. We assessed the quantity and the quality of nucleic acids spectrophotometrically (260 nm, 260/280 and 260/230 ratios, spectrum 220-320 nm) using Nanodrop ND1000 (EuroClone, Milan, Italy).
Reverse transcriptase reactions
Three µg of total RNA were reverse transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystem, Foster City, CA, USA), complying with the manufacturer’s instructions. The quality of the reverse transcription synthesis was tested by amplifying cDNA with primers of housekeeping genes (beta-actin and TATA Box Binding Protein) producing fragments of different lengths.
Quantitative real-time PCR
Primers for EGFR (Hs01076078_m1, 60 bp), and 18S rRNA (Hs99999901_S1, 187 bp) human genes were chosen using Assays-on-DemandTM-Products (Applied Biosystems). Neoplastic and non-neoplastic tissues from each patient were analyzed by quantitative real-time PCR (qRT-PCR), which was performed using TaqMan PCR chemistry and the ABI 7900HT Sequence Detection System (Applied Biosystems). A reaction volume of 50 μl containing 300 nM of primers, 200 nM of probe and 54 ng of reverse-transcribed RNA was run using the TaqMan Universal PCR Master Mix (Applied Biosystems). Cycling conditions were: 10 minutes of denaturation at 95°C, 40 cycles at 95°C for 15 seconds and at 60°C for 1 minute. 18S rRNA was used as reference gene for normalizing EGFR gene expression in qRT-PCR. Triplicate reactions were performed for each cDNA sample and the relative mRNA expression level was analyzed according to Applied Biosystem User Bulletin N°2. The calculation 2-ΔΔCt (Fold Change, FC) was chosen to represent the level of expression, with a FC of more than 2 being considered as overexpression. Moreover, data were expressed as medians and interquartile range according to a non-normal distribution of the variables.
Mutation analysis
EGFR gene mutation analysis was performed on exons 2 to 7, and on exons 18 to 24 coding for the receptor tyrosine kinase domain, which are known to harbour the most frequent and significant mutations for this gene (Table 2). Gene sequencing analysis was executed as previously reported [24].
Table 2.
Selected primers for gene amplification and sequencing
| Primers | Sequence | Annealing Temperature | Base pair |
|---|---|---|---|
| EGFR F exon 2 | CCAAGGCACGAGTAACAAGCT | 56°C | 132 |
| EGFR R exon 2 | TCATAATTCCTCTGCACATAGG | ||
| EGFR F exon 3 | TAGACCTTGAGTTCTTGAGTTC | 56°C | 155 |
| EGFR R exon 3 | CATATTTCCTCTGATGATCTGC | ||
| EGFR F exon 4 | AGTGCTCACCGCAGTTCCAT | 55°C | 228 |
| EGFR R exon 4 | CAGTGCTGTAGAGCTGTCC | ||
| EGFR F exon 5 | TATTGAATGTGCTTAACTCAGG | 58°C | 189 |
| EGFR R exon 5 | CATGGGTCTGAGGCTGTTC | ||
| EGFR F exon 6 | CTTCAGCTCACAGGGAACCTT | 57°C | 236 |
| EGFR R exon 6 | CACAGGAAGTCTTCTGTCCTG | ||
| EGFR F exon 7 | GAGTGTACTTACCTCACTTGC | 58°C | 168 |
| EGFR R exon 7 | GTGGCACCAAAGCTGTATTTG | ||
| EGFR F exon 18 | GCTTGCAAGGACTCTGGGCT | 62°C | 360 |
| EGFR R exon 18 | CCAAACACTCAGTGAAACAAAGAG | ||
| EGFR F exon 19 | GTGCATCGCTGGTAACATCCA | 55°C | 306 |
| EGFR R exon 19 | CATTTAGGATGTGGAGATGAGC | ||
| EGFR F exon 20 | GAAACTCAAGATCGCATTCATGC | 60°C | 379 |
| EGFR R exon 20 | GCAAACTCTTGCTATCCCAGGAG | ||
| EGFR F exon 21 | CTAACGTTCGCCAGCCATAAGTCC | 57°C | 370 |
| EGFR R exon 21 | GCTCACCCAGAATGTCTGGA | ||
| EGFR F exon 22 | CACTCGTAATTAGGTCCAGAG | 55°C | 295 |
| EGFR R exon 22 | CTCAGTACAATAGATAGACAGCAATG | ||
| EGFR F exon 23 | CAAGACTACAGAAATGTAGGTTTC | 63°C | 373 |
| EGFR R exon 23 | GTGATGACATTTCTCCAGGGATGC | ||
| EGFR F exon 24 | CATCACCAATGCCTTCTTTAAGC | 59°C | 310 |
| EGFR R exon 24 | GCTGGAGGGTTTAATAATGCGATC |
Additionally, to detect EGFRvIII mutation, involving the deletion of exons 2 to 7, coding for the extracellular domain of the receptor, a reverse transcription PCR (RT-PCR) was developed. cDNAs were used to amplify the deletion region by primers in exon 1 (5’-GGGCTCTGGAGGAAAAGAAA-3’) and exon 8 (5’-CCTCCATCTCATGCTGTCG-3’), producing a 91 bp or a 892 bp PCR product for EGFRvIII and EGFR wild-type, respectively.
PCR was performed in a volume of 20 µl, including 60 ng of cDNA, 0.5 μM of each primer, 0.2 mM of each dATP, dCTP, dGTP, dTTP (Invitrogen, The Netherlands) 160 mM (NH4)2SO4, 670 mM Tris-HCl pH 8.8, 1.5 mM MgCl2, 5% DMSO, 2U Super Taq (AB Analitica, Padova, Italy). PCR cycling conditions were: 5 minutes at 94°C, 30 seconds at 95°C, 30 seconds at 60°C, 1 minute at 72°C for 35 cycles, then 5 minutes at 72°C. Cloned wild-type EGFR and EGFRvIII cDNA were used as controls. Ten μl of PCR products were analyzed on a 2% agarose gel, stained with ethidium bromide.
Statistical analysis
Statistical analysis was carried out using STATA®13 (StataCorp, College Station, TX, USA). Frequencies were used to summarize qualitative variables. Spearman’s correlations between EGFR and SGLT1 expression levels were performed. Statistical differences for qualitative variables were evaluated using Chi2 or Fisher’s Exact Test, when appropriate. Logistic regression analysis, both univariate and multivariate, was carried out to assess the association between CCRCC patients with no evidence of disease compared to patients with progression of disease at 5 years follow-up, and the epidemiological, clinical, and molecular variables. The statistical significance was set-up at < 0.05.
Results
Clinic-pathological data
Sixty-three patients diagnosed with CCRCC were included in the study. Patients’ median age at the time of diagnosis was 59 years (range 30-79), with predominance of males (43 patients) compared to females (20 patients). Tumor size varied between 2 and 15 cm (median: 7 cm). Follow-up data were available for 62 out of 63 patients. Five-year follow-up data showed no evidence of disease (NED) in 33 patients, whereas 29 patients showed progression of the disease during the follow-up period: 26 developed distant metastases, and were alive with disease (AWD), while 3 patients with distant metastases died of disease (DOD). Additional clinic-pathological features are summarized on Table 3, including the Mayo Clinic stage, size, grade and necrosis (SSIGN) score [25].
Table 3.
Clinical and pathological features detected in patients with CCRCC
| CCRCC n (%) | |
|---|---|
| Pathologic Tumor classification | |
| pT1a | 12 (19.0) |
| pT1b | 11 (17.5) |
| pT2a | 8 (12.7) |
| pT2b | 3 (4.7) |
| pT3a | 17 (26.9) |
| pT3b | 11 (17.5) |
| pT3c | 1 (1.7) |
| Regional lymph nodes involvement | |
| pNx | 23 (36.5) |
| pN0 | 33 (52.4) |
| pN1 | 4 (6.4) |
| pN2 | 3 (4.7) |
| Distant metastasis | |
| M0 | 35 (55.6) |
| M1 | 28 (44.4) |
| pTNM Stage | |
| I | 14 (24.5) |
| II | 5 (8.8) |
| III | 15 (26.3) |
| IV | 23 (40.4) |
| Nuclear grading according to Fuhrman | |
| 1 | 1 (1.6) |
| 2 | 27 (42.9) |
| 3 | 26 (41.2) |
| 4 | 9 (14.3) |
| Coagulative tumor necrosis | |
| Present | 21 (33.3) |
| Absent | 42 (66.7) |
| SSIGN score | |
| 0-2 | 10 (15.9) |
| 3-4 | 9 (14.2) |
| 5-6 | 17 (27.0) |
| 7-9 | 10 (15.9) |
| ≥ 10 | 17 (27.0) |
n: number of patients. Absolute and relative frequencies (i.e., percentages) were computed.
Immunohistochemical analysis
EGFR immunostaining was reported as membranous and/or membranous-cytoplasmic, SGLT1 showed membranous-cytoplasmic immunoreactivity; p-AKT, and p-STAT3 showed nuclear staining and p-p44/42 MAPK displayed nuclear and/or cytoplasmic staining, as shown in Figure 1.
Figure 1.
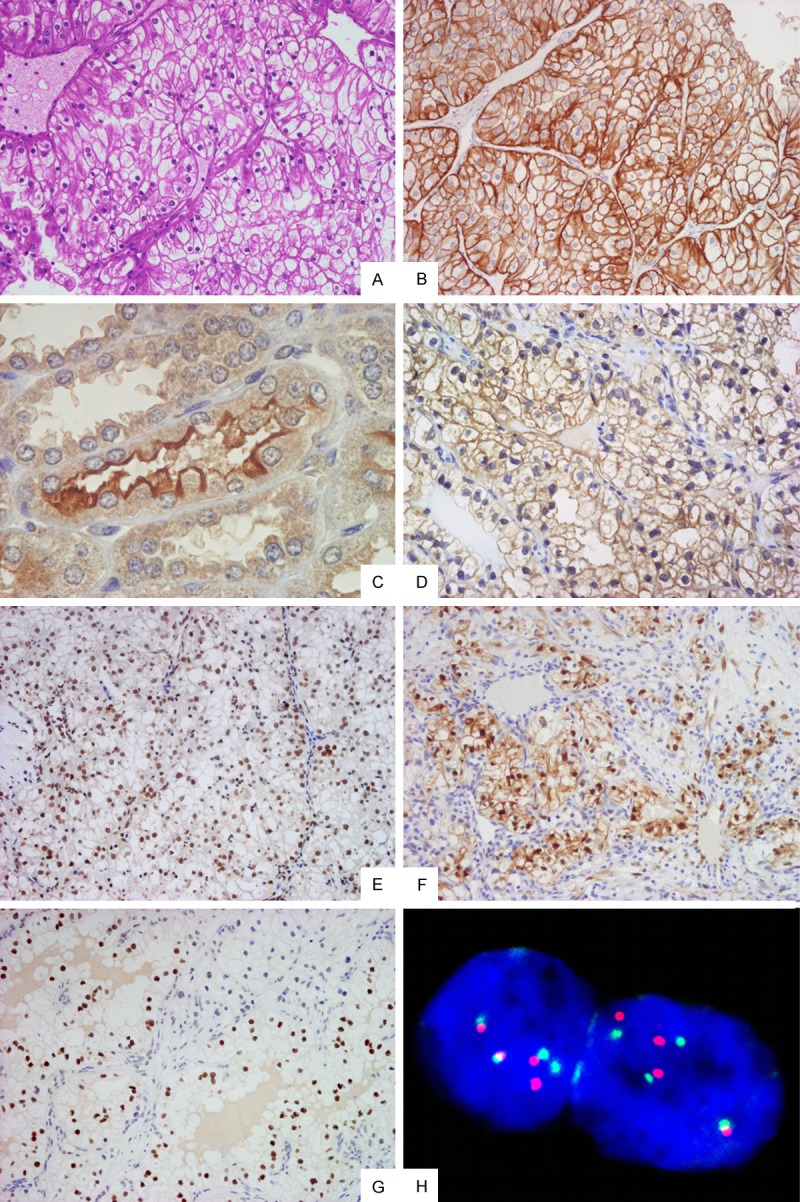
Morphologic and immunohistochemical features of Clear Cell renal Cell Carcinoma. A. Haematoxylin & Eosin stain shows typical CCRCC morphologic features (original magnification 100×); B. Immunohistochemistry for EGFR displaying diffuse and intense membranous and membranous-cytoplasmic immunoreactivity (original magnification 100×); C. Immunohistochemistry for SGLT1 on non-neoplastic renal parenchyma showing intense immunostaining on the luminal brush border of proximal renal tubules (original magnification 400×); D. Immunohistochemistry for SGLT1 on CCRCC showing diffuse and intense, predominantly membranous immunoreactivity (original magnification 200×); E. Immunohistochemistry for p-AKT showing diffuse and intense nuclear immunoreactivity (original magnification 100×); F. Immunohistochemistry for p-p44/42 MAPK displaying focal and intense nuclear-cytoplasmic immunoreactivity (original magnification 100×); G. Immunostaining for p-STAT3 displaying moderate nuclear immunoreactivity (original magnification 100×); H. FISH analysis with centromeric probe for chromosome 7 (green hybridization signals) and locus-specific EGFR gene (orange hybridization signals) showing 4 and 4 hybridization signals, respectively, which excludes gene amplification (ratio: 1), confirming chromosome 7 polysomy (original magnification 1000×).
EGFR expression was appreciable in 98.4% of the tumors, with staining intensity ranging from 1+ to 3+, and percentages of positive cells varying from 20% to 100%.
SGLT1 expression was identified in 80.9% of the tumors, with staining intensity ranging from 1+ to 3+, and percentages of positive cells varying from 15% to 90%. Co-expression of EGFR and SGLT1 was appreciable in 79.4% of cases. As positive control, renal parenchyma adjacent to neoplastic proliferation was utilized, namely proximal renal tubules, which demonstrated intense SGLT1 immunostaining on the luminal brush border (Figure 1).
p-AKT expression was detected in 58.8% of the tumors, with a 1+ to 3+ staining intensity and the range of positive cells being 20%-80%.
p-p44/42 MAPK expression was identified in 52.9% of the tumors with a staining intensity of 1+ to 3+, and positive cell percentages between 15% and 90%.
p-STAT3 expression was recognizable in 23.5% of the tumors with the staining intensity ranging from 1+ to 3+, and positive cells varying from 20% to 70%.
p-AKT, p-p44/42 MAPK and p-STAT3 expression was absent in 14.7% of CCRCCs, while 73.5% of tumors showed expression of at least 1 of the 3 downstream signaling pathway members. The 11.7% of tumors displayed concurrent immunohistochemical expression of all three pathway components.
Fluorescence in situ hybridization analysis
FISH analysis showed EGFR gene locus-specific DNA probe/CEP7 DNA probe ratios constantly below the cut-off value, ranging from 0.99 to 1.13 (mean: 1.03), thus evaluated as notamplified.
Using criteria for chromosome gain as described in the Materials and Methods section, the mean percentage of nuclei with three or more signals was scored as 1.5, whereas the standard deviation was assessed as 2. Therefore, the cut-off value to determine chromosomal gain in neoplastic specimens was 7.5. The 32.3% of CCRCCs showed percentages of nuclei with three or more centromeric signals below the cut-off value of 7.5%, ranging from 0% to 5% (mean: 2.9%), while the 67.7% were scored between 11% and 94% (mean: 44.9%) and had a chromosome 7 gain (polysomic).
Expression profiles analysis
Median values of EGFR gene expression levels were evaluated as 3.18 (interquartile range, 1.71-5.08) in CCRCC and 0.35 (interquartile range, 0.16-0.79) in the normal tissue. Statistical significance was found between tumor and normal tissue expression levels (P = 0.0003). qRT-PCR results are represented in Figure 2. Fold change evaluation for each tumor, compared to its normal counterpart, showed overexpression levels in 38.2% of CCRCCs with a range from 2.33 to 20.5 fold change.
Figure 2.
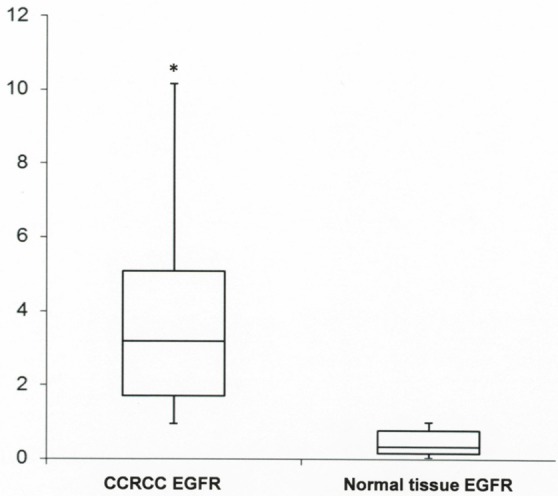
Real-time polymerase chain reaction analysis for EGFR. Box and whisker plots were used to summarize the distribution of mRNA levels in CCRCC and normal tissue controls. Statistical analysis by Mann Whitney test showed significant differences in mRNA levels between neoplastic and non-neoplastic tissues with *P-values of 0.0003.
EGFR mutational analysis
Genomic DNA sequencing of exons 2 to 7, and of exons 18 to 24 coding for the receptor tyrosine kinase domain, which are known to harbor the most frequent and significant mutations for the EGFR gene, failed to demonstrate mutations in any CCRCCs analyzed.
EGFRvIII is an oncogenic, constitutively active mutant form of EGFR. EGFRvIII is generated by in-frame genomic deletion of 801 bp from exons 2 to 7 of the coding region of the gene, which produces a truncated receptor lacking a portion of the extracellular ligand binding domain. All CCRCCs were investigated by RT-PCR to highlight this specific deletion, but no evidence of EGFRvIII deletion was found in our neoplastic series.
Statistical analysis
To analyze the relevance of EGFR and SGLT1 expression in CCRCC, the correlation between the expression of EGFR or SGLT1 with standard clinic-pathological features was computed (Table 4). The EGFR expression levels showed a significant correlation with tumor size (Ρ = 0.30; P = 0.019) and SSIGN score (Ρ = 0.29; P = 0.023), while SGLT1 had significant correlation with necrosis (Ρ = 0.29; P = 0.022). However, no significant differences were obtained comparing other variables such as age, sex, pT, pN, metastasis, grade, and stage. In addition, no correlations between the SGLT1 and EGFR expression were found for intensity (Ρ = -0.10; P = 0.420) and positive cell percentages (Ρ = -0.10; P = 0.431).
Table 4.
Correlation between EGFR and SGLT1 expression levels and clinic-pathological data
| EGFR | ||
|
| ||
| Variables | Ρ | P-value |
|
| ||
| Age | 0.16 | 0.224 |
| Sex | 0.02 | 0.877 |
| Tumor size | 0.3 | 0.019 |
| Pathologic Tumor classification | 0.23 | 0.069 |
| Regional lymph nodes involvement | 0.08 | 0.55 |
| Distant Metastasis | 0.02 | 0.847 |
| pTNM Stage | 0.11 | 0.424 |
| Nuclear grading according to Fuhrman | 0.28 | 0.27 |
| Coagulative tumor necrosis | 0.14 | 0.261 |
| SSIGN score | 0.29 | 0.023 |
|
| ||
| SGLT1 | ||
|
| ||
| Variables | Ρ | P-value |
|
| ||
| Age | -0.05 | 0.723 |
| Sex | -0.08 | 0.532 |
| Tumor size | 0.25 | 0.053 |
| Pathologic Tumor classification | -0.07 | 0.584 |
| Regional lymph nodes involvement | -0.07 | 0.608 |
| Distant Metastasis | 0.16 | 0.223 |
| pTNM Stage | -0.13 | 0.323 |
| Nuclear grading according to Fuhrman | 0.25 | 0.053 |
| Coagulative tumor necrosis | 0.29 | 0.022 |
| SSIGN score | 0.18 | 0.156 |
Spearman’s correlations were computed on the basis of the non-parametric nature of the selected variables.
A logistic regression analysis was carried out in order to assess the impact of established clinic-pathological prognostic predictors, EGFR and SGLT1 expression on CCRCC patients with no evidence of disease compared to patients with progression of disease at 5 years follow-up. Significant associations between higher grade, stage, SSIGN score and progression of disease were found in the univariate analysis, although a multivariate model confirmed only the SSIGN score as an independent prognostic factor (Table 5).
Table 5.
Association between CCRCC patients with NED and AWD or DOD and clinic-pathological and molecular variables
| Variables | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
|
| ||||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Age | 3.13 (0.87-11.24) | 0.081 | 1.59 (0.13-19.56) | 0.716 |
| Sex | 1.50 (0.51-4.42) | 0.462 | ||
| Tumor size | 1.50 (0.51-4.42) | 0.462 | ||
| Pathologic Tumor classification | 2.15 (0.78-5.97) | 0.14 | ||
| Regional lymph nodes involvement | 6.67 (0.73-60.85) | 0.093 | 1.73 (0.10-28.59) | 0.703 |
| Distant Metastasis | 1 (-) | - | ||
| pTNM Stage | 11.11 (2.71-46.61) | 0.001 | 1.16 (0.16-8.47) | 0.883 |
| Nuclear grading according to Fuhrman | 4.04 (1.38-11.81) | 0.011 | 0.54 (0.08-3.65) | 0.528 |
| Coagulative tumor necrosis | 3.02 (0.99-9.16) | 0.051 | 0.09 (0.01-1.35) | 0.081 |
| SSIGN score | 4.78 (2.31-9.90) | < 0.0001 | 11.26 (2.13-59.56) | 0.004 |
| EGFR expression levels | 1.14 (0.56-2.31) | 0.713 | ||
| SGLT1 expression levels | 1.24 (0.71-2.18) | 0.453 | ||
OR: Odds Ratio; CI: Confidence Interval. Uni- and multivariate logistic regression analysis was carried out. The risk was estimated using the odds ratio and its associated 95% confidence interval.
No statistically significant differences were detected when different combinations of EGFR/SGLT1 expression levels were observed in CCRCC patients with no evidence of disease compared to patients with progression of disease at 5 years follow-up (Table 6).
Table 6.
Correlation between EGFR-SGLT1 expression levels combinations and CCRCC patients with NED and AWD or DOD
| EGFR/SGLT1 | ||||
|
| ||||
| Indicators | +/+ | +/- | -/+ | -/- |
|
| ||||
| NED, n (%) | 8 (38.1) | 21 (60.0) | 4 (80.0) | 0 (0.0) |
| AWD/DOD, n (%) | 13 (61.9) | 14 (40.0) | 1 (20.0) | 1 (100.0) |
|
| ||||
| AWD/DOD | ||||
|
| ||||
| EGFR/SGLT1 | EGFR/SGLT1 | |||
|
| ||||
| +/+ | +/- | -/+ | ||
| +/- | 0.88 | - | - | |
| -/+ | 0.11 | 0.39 | - | |
| -/- | 0.44 | 0.23 | 0.12 | |
|
| ||||
| NED | ||||
|
| ||||
| EGFR/SGLT1 | EGFR/SGLT1 | |||
|
| ||||
| +/+ | +/- | -/+ | ||
| +/- | 0.88 | - | - | |
| -/+ | 0.09 | 0.39 | - | |
| -/- | 0.44 | 0.23 | 0.12 | |
n: number of patients. Absolute and relative frequencies (i.e., percentages) were computed. Chi2 and Fisher exact tests were computed to assess differences between the selected qualitative variables. P-values are displayed in the cells.
Discussion
EGFR protein overexpression was remarkable in our CCRCC series, since we were able to identify specific immunoreactivity in 98.4% of cases. Furthermore, we described for the first time that SGLT1 is frequently overexpressed in CCRCC (80.9% of tumors), and that co-expression with EGFR is appreciable in 79.4% of tumors.
EGFR overexpression has been previously established in renal cell cancers, and the highest frequency and intensity was recognized in CCRCC variants rather than in other renal tumors subtypes, such as papillary carcinoma and chromophobe carcinoma [2,5,6,11].
Several studies demonstrated that anti-EGFR therapy effectiveness in epithelial malignancies does not correlate with EGFR protein expression levels, while peculiar genetic abnormalities, such as gene mutations and/or increased mRNA gene expression levels, determine more precisely the responsiveness to such treatment [26,27]. In CCRCC this trend is confirmed, and the anti-EGFR therapy remains largely unsuccessful, in the light of the absence of EGFR-related genetic anomalies as reported in the literature [9,28-30].
Firstly, our study aimed to investigate the genetic abnormalities which are known to sustain EGFR overexpression. In the present study the mutational analysis of EGFR exons 2 to 7, and exons 18 to 24, coding for the receptor tyrosine kinase domain, known to harbour the most frequent and activating mutations for EGFR gene, failed to demonstrate mutations in any CCRCCs analyzed. In the same way, we were unable to identify the presence of EGFRvIII, which is an oncogenic, constitutively active mutant form of EGFR. These results confirm that structural abnormalities in EGFR gene are virtually absent in CCRCCs [11,28,31,32]. By using FISH analysis, the absence of EGFR amplification was demonstrated, instead 67.7% of CCRCCs showed high polysomy of chromosome 7, which is in keeping with data from the literature [3,4,11,12]. Finally, EGFR gene expression profile showed the presence of gene overexpression in the 38.2% of CCRCC.
EGFR overexpression, in the absence of specific gene abnormalities, should be related to alterations in the post-translational regulation machinery, with anomalous protein stabilization or defective receptor downregulation, increasing its ligand-mediated activation. Similar mechanisms are certainly involved in the biology of CCRCCs, due to their genetic specificity, i.e. Von Hippel Lindau (VHL) gene function loss, resulting in a defective polyubiquitylation-mediated degradation of activated EGFR [33,34]. Moreover, loss of VHL function in tumor cells results in the stabilization of hypoxia-inducible factor-alpha (HIFα), with its consequently increased transcriptional activity. Specifically, HIF2α increases mRNA levels of transforming growth factor-alpha (TGFα), which is a main ligand for EGFR [35]. Furthermore, HIFα increase the expression of caveolin 1, which under hypoxic conditions binds and promotes dimerization and activation of EGFR in the absence of ligand [36]. Then, the EGFR protein after stabilization can be activated both by an anomalous receptor dimerization in the absence of specific ligands, and by the production of TGFα. As a consequence, this leads to activation of downstream signaling pathways, enhancing growth, survival, motility and metabolism of neoplastic cells.
To analyze the activating role of EGFR overexpression on downstream signaling pathways in our series of CCRCC, and to highlight its kinase function, immunohistochemical analysis of pAKT, p-p44/42 MAPK and p-STAT3 were performed. According to immunohistochemical results, we were able to identify downstream pathway activation in 85.2% of cases, namely 38.2% were characterized by EGFR gene overexpression, and 47% showed EGFR protein overexpression in the absence of gene overexpression; only 14.8% of tumors displayed EGFR protein overexpression, in the absence of EGFR gene overexpression and pathway activation.
Our results demonstrate that EGFR-overexpressing CCRCCs are not to be considered as a homogeneous molecular category, since biological differences are evident from among these variants, which might explain the clinical behaviour variability and targeted treatments responsiveness. These results suggest the opportunity to select CCRCCs on the basis of their specific genomic abnormalities in order to select patients who would more likely benefit from current and novel targeted therapy strategies. However, as far as our study is concerned, the ineffectiveness of anti-EGFR therapy in CCRCC has never been correlated to the complexity of EGFR genetic-molecular scenario.
Based on our findings, correlations between EGFR immunohistochemical intensity levels and main clinic-pathological features demonstrated a significant association with size of tumors and SSIGN scores (P = 0.019 and P = 0.023, respectively), suggesting an unfavorable prognostic significance of EGFR overexpression in CCRCC. These results are in keeping with previous studies reported in the literature [5,6,11]. Conversely, no statistical significance was appreciable for EGFR expression levels, compared with patients with no evidence of disease and patients with progression of disease at 5 years follow-up.
Recent evidence indicates that EGFR retains tyrosine kinase-independent functions and a link between glucose uptake performed by SGLT1, cancer cells survival, and EGFR expression has been demonstrated. Specifically, the EGFR kinase-independent function depends on EGFR domains interaction with SGLT1, which maintains basal intracellular glucose level, avoiding cancer cell death and promoting cancer cells survival [14]. Weihua et al. observed that EGFR stabilizes SGLT1 by protein-protein interaction which leads to prevention of SGLT1 from proteasomal degradation. EGFR constantly interacts with proteins, such as SGLT1, regardless of the presence of EGFR ligands and activation or inactivation of its tyrosine kinase function [14].
The potential relation between EGFR and SGLT1 was investigated in our study, showing for the first time that SGLT1 is frequently overexpressed in CCRCCs, with a SGLT1 immunoreactivity detected in 80.9% of tumors. Since the activation of downstream EGFR pathways is found in about 79.4% of SGLT1-positive CCRCC, it is conceivable that EGFR kinase and non-kinase functions can be carried out independently of each other, and both contribute to neoplastic progression.
This tyrosine kinase-independent function of EGFR might provide cancer cells with an increased survival capacity even in the presence of chemotherapeutic agents or tyrosine kinase inhibitors, since the inhibition of both EGFR kinase and non-kinase functions might be necessary to obtain therapeutic responsiveness. As a matter of fact, the deletion of the EGFR-SGLT1 interaction promotes the down-regulation of SGTL1 via the proteasome machinery, suggesting that disruption of EGFR–SGLT1 interaction in EGFR-positive cancer cells may lead to down-regulation of SGLT1 and, consequently, facilitate cancer cell death. In facts, Ren et al. demonstrated that SGLT1 inhibitors sensitize prostate cancer cells to EGFR inhibitors [14,37]. Furthermore, preclinical data examining EGFR as a molecular target showed that the combination of EGFR tyrosine kinase inhibitors plus anti-EGFR antibodies (cetuximab) results in synergistic tumor regression [38,39], whose effect might be hypothetically ascribed to antibody-mediated disruption of EGFR-SGLT1 interaction.
SGLT1 is overexpressed in different types of malignant epithelial tumors, such as colon-rectal carcinoma, lung carcinoma, head and neck carcinoma, pancreatic carcinoma and ovarian carcinoma, and EGFR co-expression has previously been reported in oral squamous carcinoma and colon carcinoma [15-20]. According to previous reports, SGLT1 prognostic significance seems to be extremely variable depending on cancer site.
In our study, no statistically significant correlations were appreciable for SGLT1 expression alone, and in combination with EGFR expression, compared to patients with no evidence of disease and patients with progression of disease at 5 years follow-up. Interestingly, correlations between SGLT1 immunohistochemical intensity levels and the most important clinic-pathological features demonstrated a significant association with necrosis (P = 0.022), which is in agreement with the role of SGLT1 in autophagic cancer cell death [11].
In conclusion, our study contributes to define the complexity of EGFR role in CCRCC, establishing that EGFR expression is a common event in these neoplasms, and demonstrating its kinase role in downstream signal transduction pathways activation, regardless of genetic abnormalities which are actually uncommon.
Moreover, for the first time the high frequency of SGLT1 expression in CCRCC was described, which is known to have the capability to interact with EGFR as a target for EGFR kinase-independent function, determining a potential increase of neoplastic growth and progression, with improvement of cancer cell metabolism.
These results might have important implications on therapeutic approaches to CCRCC; since EGFR tyrosine-kinase inhibitors alone have been demonstrated to be unsuccessful, a wider, multi-targeted therapeutical option should be provided, taking into account also EGFR kinase-independent roles. Therefore, we hypothesized that breaking the interaction between EGFR/SGLT1, either with anti-EGFR antibodies or SGLT1 inhibitors, might be a novel therapeutic target for CCRCC treatment, and new clinical trials should be proposed based on this therapeutic approach.
Acknowledgements
This work was supported by grants from Fondazione Banco di Sardegna, Italy.
Disclosure of conflict of interest
None.
References
- 1.Stumm G, Eberwein S, Rostock-Wolf S, Stein H, Pomer S, Schlegel J, Waldherr R. Concomitant overexpression of the EGFR and erbB-2 genes in renal cell carcinoma (RCC) is correlated with dedifferentiation and metastasis. Int J Cancer. 1996;69:17–22. doi: 10.1002/(SICI)1097-0215(19960220)69:1<17::AID-IJC4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 2.Moch H, Sauter G, Buchholz N, Gasser TC, Bubendorf L, Waldman FM, Mihatsch MJ. Epidermal growth factor receptor expression is associated with rapid tumor cell proliferation in renal cell carcinoma. Hum Pathol. 1997;28:1255–1259. doi: 10.1016/s0046-8177(97)90198-2. [DOI] [PubMed] [Google Scholar]
- 3.Amare Kadam PS, Varghese C, Bharde SH, Narasimhamoorthy NK, Desai S, Advani SH, Havaldar R, Kulkarni JN. Proliferating cell nuclear antigen and epidermal growth factor receptor (EGFr) status in renal cell carcinoma patients with polysomy of chromosome 7. Cancer Genet Cytogenet. 2001;125:139–146. doi: 10.1016/s0165-4608(00)00375-7. [DOI] [PubMed] [Google Scholar]
- 4.Moch H, Sauter G, Gasser TC, Bubendorf L, Richter J, Presti JC Jr, Waldman FM, Mihatsch MJ. EGF-r gene copy number changes in renal cell carcinoma detected by fluorescence in situ hybridization. J Pathol. 1998;184:424–429. doi: 10.1002/(SICI)1096-9896(199804)184:4<424::AID-PATH1223>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 5.Merseburger AS, Hennenlotter J, Simon P, Kruck S, Koch E, Horstmann M, Kuehs U, Küfer R, Stenzl A, Kuczyk MA. Membranous expression and prognostic implications of epidermal growth factor receptor protein in human renal cell cancer. Anticancer Res. 2005;25:1901–1907. [PubMed] [Google Scholar]
- 6.Cohen D, Lane B, Jin T, Magi-Galluzzi C, Finke J, Rini BI, Bukowski RM, Zhou M. The prognostic significance of epidermal growth factor receptor expression in clear-cell renal cell carcinoma: a call for standardized methods for immunohistochemical evaluation. Clin Genitourin Cancer. 2007;5:264–270. doi: 10.3816/CGC.2007.n.002. [DOI] [PubMed] [Google Scholar]
- 7.Coppin C, Kollmannsberger C, Le L, Porzsolt F, Wilt TJ. Targeted therapy for advanced renal cell cancer (RCC): a Cochrane systematic review of published randomised trials. BJU Int. 2011;108:1556–1563. doi: 10.1111/j.1464-410X.2011.10629.x. [DOI] [PubMed] [Google Scholar]
- 8.Stadler WM. Targeted agents for the treatment of advanced renal cell carcinoma. Cancer. 2005;104:2323–2333. doi: 10.1002/cncr.21453. [DOI] [PubMed] [Google Scholar]
- 9.Motzer RJ, Amato R, Todd M, Hwu WJ, Cohen R, Baselga J, Muss H, Cooper M, Yu R, Ginsberg MS, Needle M. Phase II trial of antiepidermal growth factor receptor antibody C225 in patients with advanced renal cell carcinoma. Invest New Drugs. 2003;21:99–101. doi: 10.1023/a:1022928612511. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa J, Maeda S, Umezu K, Sugiyama T, Kamidono S. Amplification and overexpression of the epidermal growth factor receptor gene in human renal-cell carcinoma. Int J Cancer. 1990;45:1018–1021. doi: 10.1002/ijc.2910450606. [DOI] [PubMed] [Google Scholar]
- 11.Minner S, Rump D, Tennstedt P, Simon R, Burandt E, Terracciano L, Moch H, Wilczak W, Bokemeyer C, Fisch M, Sauter G, Eichelberg C. Epidermal growth factor receptor protein expression and genomic alterations in renal cell carcinoma. Cancer. 2012;118:1268–1275. doi: 10.1002/cncr.26436. [DOI] [PubMed] [Google Scholar]
- 12.El-Hariry I, Powles T, Lau MR, Sternberg CN, Ravaud A, von der Maase H, Zantl N, Harper P, Rolland F, Audhuy B, Barthel F, Machiels JP, Patel P, Kreuser ED, Hawkins RE. Amplification of epidermal growth factor receptor gene in renal cell carcinoma. Eur J Cancer. 2010;46:859–862. doi: 10.1016/j.ejca.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–794. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 14.Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–93. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanabata Y, Nakajima Y, Morita K, Kayamori K, Omura K. Coexpression of SGLT1 and EGFR is associated with tumor differentiation in oral squamous cell carcinoma. Odontology. 2012;100:156–163. doi: 10.1007/s10266-011-0033-2. [DOI] [PubMed] [Google Scholar]
- 16.Guo GF, Cai YC, Zhang B, Xu RH, Qiu HJ, Xia LP, Jiang WQ, Hu PL, Chen XX, Zhou FF, Wang F. Overexpression of SGLT1 and EGFR in colorectal cancer showing a correlation with the prognosis. Med Oncol. 2011;28(Suppl 1):197–203. doi: 10.1007/s12032-010-9696-8. [DOI] [PubMed] [Google Scholar]
- 17.Casneuf VF, Fonteyne P, Van Damme N, Demetter P, Pauwels P, de Hemptinne B, De Vos M, Van de Wiele C, Peeters M. Expression of SGLT1, Bcl-2 and p53 in primary pancreatic cancer related to survival. Cancer Invest. 2008;26:852–859. doi: 10.1080/07357900801956363. [DOI] [PubMed] [Google Scholar]
- 18.Lai B, Xiao Y, Pu H, Cao Q, Jing H, Liu X. Overexpression of SGLT1 is correlated with tumor development and poor prognosis of ovarian carcinoma. Arch Gynecol Obstet. 2012;285:1455–1461. doi: 10.1007/s00404-011-2166-5. [DOI] [PubMed] [Google Scholar]
- 19.Helmke BM, Reisser C, Idzko M, Dyckhoff G, Herold-Mende C. Expression of SGLT-1 in preneoplastic and neoplastic lesions of the head and neck. Oral Oncol. 2004;40:28–35. doi: 10.1016/s1368-8375(03)00129-5. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa N, Oguri T, Isobe T, Fujitaka K, Kohno N. SGLT gene expression in primary lung cancers and their metastatic lesions. Jpn J Cancer Res. 2001;92:874–879. doi: 10.1111/j.1349-7006.2001.tb01175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eble JN, Togashi K, Pisani P. Tumours of the Kidney. In: Eble JN, Sauter G, Epstein JI, Sesterhenn IA, editors. World Health Organization Classification of Tumours Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. 3rd edition. Lyon: IARC Press; 2004. pp. 9–87. [Google Scholar]
- 22.American Joint Committee on Cancer. AJCC Cancer Staging Manual. New York: Springer-Verlag; 2010. [Google Scholar]
- 23.Cossu-Rocca P, Eble JN, Zhang S, Martignoni G, Brunelli M, Cheng L. Acquired cystic disease-associated renal tumors: an immunohistochemical and fluorescence in situ hybridization study. Mod Pathol. 2006;19:780–787. doi: 10.1038/modpathol.3800604. [DOI] [PubMed] [Google Scholar]
- 24.De Miglio MR, Mura A, Uras MG, Manca A, Contini M, Murgia L, Zinellu A, Sotgia S, Carru C, Massarelli G, Cossu-Rocca P. High sensitivity of reverse-hybridization methodology in the detection of KRAS mutations from formalin-fixed paraffin-embedded colorectal cancer samples. Diagn Mol Pathol. 2010;19:201–208. doi: 10.1097/PDM.0b013e3181db67d5. [DOI] [PubMed] [Google Scholar]
- 25.Ficarra V, Martignoni G, Lohse C, Novara G, Pea M, Cavalleri S, Artibani W. External validation of the Mayo Clinic Stage, Size, Grade and Necrosis (SSIGN) score to predict cancer specific survival using a European series of conventional renal cell carcinoma. J Urol. 2006;175:1235–1239. doi: 10.1016/S0022-5347(05)00684-1. [DOI] [PubMed] [Google Scholar]
- 26.Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J. Clin. Oncol. 2005;23:2445–2459. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- 27.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 28.Iyevleva AG, Novik AV, Moiseyenko VM, Imyanitov EN. EGFR mutation in kidney carcinoma confers sensitivity to gefitinib treatment: a case report. Urol Oncol. 2009;27:548–550. doi: 10.1016/j.urolonc.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 29.Shek D, Longmate J, Quinn DI, Margolin KA, Twardowski P, Gandara DR, Frankel P, Pan CX, Lara PN Jr. A phase II trial of gefitinib and pegylated IFNalpha in previously treated renal cell carcinoma. Int J Clin Oncol. 2011;16:494–499. doi: 10.1007/s10147-011-0212-8. [DOI] [PubMed] [Google Scholar]
- 30.Motzer RJ, Hudes GR, Ginsberg MS, Baum MS, Harmon CS, Kim ST, Chen I, Redman BG. Phase I/II trial of sunitinib plus gefitinib in patients with metastatic renal cell carcinoma. Am J Clin Oncol. 2010;33:614–618. doi: 10.1097/COC.0b013e3181c4454d. [DOI] [PubMed] [Google Scholar]
- 31.Sakaeda T, Okamura N, Gotoh A, Shirakawa T, Terao S, Morioka M, Tokui K, Tanaka H, Nakamura T, Yagi M, Nishimura Y, Yokoyama M, Okumura K. EGFR mRNA is upregulated, but somatic mutations of the gene are hardly found in renal cell carcinoma in Japanese patients. Pharm Res. 2005;22:1757–1761. doi: 10.1007/s11095-005-7094-2. [DOI] [PubMed] [Google Scholar]
- 32.Szymańska K, Moore LE, Rothman N, Chow WH, Waldman F, Jaeger E, Waterboer T, Foretova L, Navratilova M, Janout V, Kollarova H, Zaridze D, Matveev V, Mates D, Szeszenia-Dabrowska N, Holcatova I, Bencko V, Le Calvez-Kelm F, Villar S, Pawlita M, Boffetta P, Hainaut P, Brennan P. TP53, EGFR, and KRAS mutations in relation to VHL inactivation and lifestyle risk factors in renal-cell carcinoma from central and eastern Europe. Cancer Lett. 2010;293:92–98. doi: 10.1016/j.canlet.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 33.Zhou L, Yang H. The von Hippel-Lindau tumor suppressor protein promotes c-Cbl-independent poly-ubiquitylation and degradation of the activated EGFR. PLoS One. 2011;6:e23936. doi: 10.1371/journal.pone.0023936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Yang H. The Roles of VHL-Dependent Ubiquitination in Signaling and Cancer. Front Oncol. 2012;2:35–41. doi: 10.3389/fonc.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Paulsen N, Brychzy A, Fournier MC, Klausner RD, Gnarra JR, Pause A, Lee S. Role of transforming growth factor-alpha in von Hippel--Lindau (VHL) (-/-) clear cell renal carcinoma cell proliferation: a possible mechanism coupling VHL tumor suppressor inactivation and tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:1387–1392. doi: 10.1073/pnas.031587498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, Wilson BC, Teh BT, Park M, Irwin MS, Ohh M. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factormediated upregulation of caveolin-1. Proc Natl Acad Sci U S A. 2012;109:4892–4897. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren J, Bollu LR, Su F, Gao G, Xu L, Huang WC, Hung MC, Weihua Z. EGFR-SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate. 2013;73:1453–1461. doi: 10.1002/pros.22692. [DOI] [PubMed] [Google Scholar]
- 38.Matar P, Rojo F, Cassia R, Moreno-Bueno G, Di Cosimo S, Tabernero J, Guzmán M, Rodriguez S, Arribas J, Palacios J, Baselga J. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin Cancer Res. 2004;10:6487–6501. doi: 10.1158/1078-0432.CCR-04-0870. [DOI] [PubMed] [Google Scholar]
- 39.Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, Miller VA, Pao W. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4:1036–1045. doi: 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]