

Abstract
Osteosarcoma is the most common type of aggressive bone cancer. Current treatment strategies include surgical resection, radiation, and chemotherapy. Doxorubicin has been widely used as a chemotherapeutic drug to treat osteosarcoma. However, drug resistance has become a challenge to its use. In this study, p53-wild type U2OS and p53-null MG-63 osteosarcoma-derived cells were used to investigate the mechanism of doxorubicin-induced cytotoxicity. In cell viability assays, doxorubicin effectively induced apoptosis in U2OS cells via the p53 signaling pathway, evidenced by elevated PUMA and p21 protein levels and activated caspase 3 cleavage. In contrast, p53-null MG-63 cells were resistant to doxorubicin-induced apoptosis, while exogenous expression of p53 increased drug sensitivity in those cells. The role of TGF-β/Smad3 signaling was investigated by using TGF-β reporter luciferase assays. Doxorubicin was able to induce TGF-β signal transduction without increasing TGF-β production in the presence of p53. Knockdown of Smad3 expression by small hairpin RNA (shRNA) showed that Smad3 was required for p53-mediated TGF-β signaling in response to doxorubicin treatment in U2OS and MG-63 cells. Taken together, these data demonstrate that p53 and TGF-β/Smad3 signaling pathways are both essential for doxorubicin-induced cytotoxicity in osteosarcoma cells.
Keywords: Osteosarcoma, P53, doxorubicin, chemotherapy, TGF-beta signaling pathway
Introduction
Osteosarcoma is a highly malignant bone tumor which predominately affects adolescents and young adults. In the majority of patients, the tumor grows rapidly, behaves aggressively, and metastasizes early [1]. Osteosarcoma is usually treated with intensive chemotherapy before or after tumor resection. Doxorubicin, a DNA intercalating agent, has been widely used in the treatment of various types of cancer [2]. It is often routinely included in the treatment regimen for osteosarcoma, but therapeutic efficacy varies dramatically among individual patients. In those patients who present a stable, continuous resistance to doxorubicin, the clinical prognosis is extremely poor [3]. Clinical evidence has suggested that around 10% of osteosarcoma patients show a variable degree of resistance to doxorubicin treatment after surgery, contributing to relapse or metastasis. Therefore, it is necessary to investigate the mechanism by which doxorubicin induces apoptosis in osteosarcoma cells, in order to overcome drug resistance and to formulate adaptive therapy within the most effective time window for treating osteosarcoma.
Recent reports have suggested that doxorubicin activates murine TGF-β signaling, and as a result, promotes lung metastasis of breast cancer [4,5]. TGF-β is a cytokine that plays dual roles in various biological processes [6-8]. On one hand, TGF-β acts as an anti-proliferative factor with the function of triggering apoptosis via its downstream signaling pathway. When it binds to type I and recruits type II serine/threonine kinase receptors at the cell surface, the receptor complex is activated and propagates the signal downstream by phosphorylating the Smad complex, which subsequently translocates to the nucleus to regulate the expression of specific genes, such as the CDK inhibitor p21 (CDKN1A, WAF1), leading to apoptosis [9]. On the other hand, TGF-β contributes to malignant cell survival and invasion via both canonical and non-canonical signaling pathways [8]. Osteosarcoma patients have a high risk of developing pulmonary metastasis. Therefore, we investigated how TGF-β signaling affects osteosarcoma cells treated with doxorubicin.
It has been reported that the tumor suppressor gene p53 exerts its anticancer function by inducing cell cycle arrest and apoptosis in cancer cells. Previous investigation has demonstrated that TGF-β-induced molecular responses, including the nuclear translocation of Smads and transcriptional activation of p21, are dependent on p53 [10]. p53 has been considered a pivotal factor that determines cytotoxicity for most chemotherapeutic agents. Li-Fraumeni syndrome is caused by germline mutations or deletion of p53 and predisposes a person to development of early-onset cancer, including some osteosarcoma cases [11].
In the present study, the effects of doxorubicin treatment were compared between two types of osteosarcoma-derived cells, U2OS cells with wild-type p53 and p53-deficient MG-63 cells. The roles of both p53 and TGF-β-dependent signaling pathways on osteosarcoma-derived cell survival in doxorubicin were explored. Our study demonstrates that p53 and TGF-β/Smad3 signaling pathways are both essential for doxorubicin-induced cytotoxicity in osteosarcoma cells, with implications for treatment of osteosarcoma.
Materials and methods
Cell lines and cell culture
U2OS (derived from bone tissues of a 15-year-old osteosarcoma patient), MG-63 (derived from bone tissues of a 14-year-old osteosarcoma patient) and HEK-293T cells (human embryonic kidney-293 cells expressing the large T-antigen of simian virus 40) were obtained from the Cell Resource Center in the Chinese Academy of Sciences and Shanghai Institute for Biological Sciences (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; HyClone Laboratories, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Gibco BRL, Gaithersburg, MD, USA) at 37°C in 5% CO2.
Plasmid constructs and transfection
To make the p53 expression plasmid (pcDNA-p53), the entire open reading frame of wild-type p53 was cloned into a pcDNA3 vector. To make the TGF-β-responsive luciferase reporter, a fragment of the CAGA-lux plasmid containing three copies of the consensus Smad2/Smad3 binding site upstream of the firefly luciferase reporter gene was cloned into the pGL3-promoter vector (Promega Biosciences, San Luis Obispo, CA, USA). For the TGF-β luciferase reporter assay, the pCMV-β-galactosidase plasmid (Invitrogen, USA) was used to normalize transfection efficiency. We purchased p53-targeting shRNAs from Santa Cruz Biotechnology (Dallas, TX, USA, shRNA #1) and Cell Signaling Technology (Danvers, MA, USA, shRNA#2). To make the Smad3 shRNA targeting construct, the sense and antisense strands specific for Smad3 separated by a loop (5’-TTCAAGAGA-3’) and a polythymidine tract to terminate transcription were cloned into the pRNAT-U6.1/Hygro vector (GenScript Corporation, Piscataway, NJ, USA). A negative control that contained scrambled shRNA (GACGCTTACCGATTCAGAA) with no significant homology to mouse or human gene sequences was used to detect any nonspecific effect. HEK-293T cells were transfected using the Calcium Phosphate Cell Transfection kit, and U2OS and MG-63 cells were transfected using LipofectamineTM2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Antibodies and reagents
Polyclonal antibodies against p53, PUMA, Caspase 3, Histone-H3 and a mouse monoclonal antibody against GAPDH were obtained from Abcam Biotechnology (Cambridge, UK). A Smad3 polyclonal antibody was purchased from Santa Cruz Biotechnology. Horseradish peroxidase (HRP)-conjugated goat anti-mouse and goat anti-rabbit IgG were obtained from Jackson ImmunoResearch (West Grove, PA, USA). FITC-conjugated secondary antibody was obtained from Beyotime (Shanghai, China). DAPI (4, 6-diamidino-2-phenylindole) was purchased from Roche Diagnostics (Indianapolis, IN, USA). Doxorubicin and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] were obtained from Sigma-Aldrich (USA). The RayBio Human TGF-β ELISA kit was purchased from RayBiotech (Norcross, GA, USA). TGF-β1 and doxorubicin were purchased from Sigma Aldrich.
Development of stable RNAi cell lines
Cells were grown to 70%-80% confluence and transfected with either p53-targeting shRNA #1 (indicated as I-p53) or Smad3-targeting shRNA (indicated as I-Smad3) using the GeneTran II transfection reagent (Biomiga, San Diego, CA). After transfection, cells were incubated for another 48 h and treated with 1.5 µg/ml puromycin for selection. After two weeks, stable cells were harvested and the efficiency of gene silencing was confirmed by Western blot.
Cell viability analysis
A colorimetric MTT assay was performed to quantify the effect of doxorubicin on cell viability. Cells were seeded in 96-well plates (1000-5000 cells/well) in 100 μL of complete growth medium. After incubating for 24 h, medium was changed to DMEM containing 3% (v/v) FBS and doxorubicin at the concentration indicated in the figures. Cells were incubated for 24 or 48 h followed by addition of 20 μL MTT solution (5 mg/mL in PBS). The plates were incubated for 4 h until purple precipitate was visible. Next, 50 μL DMSO was added to each well and agitated for 10 min to dissolve the formazan crystals. Absorbance in each well was read at 570 nm using an automated microplate reader (Bio-Rad, Hercules, CA, USA).
Apoptosis analysis
Cells were harvested and washed twice with PBS at 48 h after doxorubicin treatment or post transfection. Cells were re-suspended with PBS and counted. Cells (5×105) were stained with the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA). At least 10,000 cells were analyzed using a FACS Calibur flow cytometer (BD Biosciences).
Western blot
Cells were harvested and total protein was isolated in lysis buffer containing protease inhibitor cocktail (Roche). The nuclear and cytoplasmic fractions from cells were isolated with the NE-PER Nuclear and Cytoplasmic Extraction Kit (Life Technologies, Grand Island, NY, USA) and quantified with the Pierce BCA Protein Quantification kit (Life Technologies). Equal amounts of protein were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a PVDF membrane. Each membrane was blocked with 5% non-fat dry milk in TBS-Tween-20 (TBS-T) for 1 h, followed by incubation with primary antibodies at 4°C overnight. The membrane was then washed three times and incubated with HRP-labeled secondary antibodies before visualization using an enhanced chemiluminescence kit (Abcam, USA).
Luciferase reporter assay
To perform the TGF-β-responsive luciferase reporter assay, cells were seeded (1×105 cells/well) in a 24-well plate, and transfected with 1 μg reporter plasmid plus 0.1 μg pCMV-β-galactosidase plasmid per well to normalize transfection efficiency. After 36 h, cells were switched to 500 μL serum-free medium in the presence of 20 ng TGF-β or 2 µg/mL doxorubicin for 8 h. Cells were then harvested and washed twice with phosphate-buffered saline (PBS), and lysed in 85 μL cold lysis buffer (25 mM glycylglycine [pH 7.8], 1% Triton X-100 [v/v], 15 mM MgSO4, 4 mM ethylene glycol tetra-acetic acid [EGTA], and 1 mM dithiothreitol). After gentle agitation, 100 μL of 25 mM luciferin (BD Biosciences, San Jose, CA, USA) and 5 μL assay cocktail (1 M adenosine triphosphate, 15 mM KH2PO4 [pH 7.8], 15 mM MgCl2) were added to 45 μL cell lysate. Luciferase activity was then measured using a FLUOstar OPTIMA fluorescence reader (BMG LABTECH, Offenburg, Germany). β-Galactosidase activity was measured using a β-galactosidase assay system (Promega Biosciences).
ELISA
Cells were left untreated or treated with 2 μg/mL doxorubicin for 48 h. Supernatant was collected, and concentration of TGF-β was measured using a TGF-β specific ELISA kit following the manufacturer’s protocol.
Colony formation assay
Cells were transfected as indicated and treated with 2 µg/ml doxorubicin for 48 h. Cells were then trypsinized to a single-cell suspension, counted and seeded in a 6-well plate (1000 cells/well) for at least 10 days. At the end of the incubation period, cells were fixed with 4% paraformaldehyde and stained with hematoxylin and eosin (H&E).
Xenograft model
Male BALB/c mice (6 weeks old) were purchased from the Chinese Academy of Sciences and were housed under pathogen-free conditions. Cells that stably overexpressed p53 or had Smad3 expression reduced by gene knockdown were treated with 2 µg/ml doxorubicin for 48 h. Then cells were harvested by trypsinization, washed and re-suspended in PBS at 5×107 cells/ml. The cell suspension (0.2 ml) was injected subcutaneously into athymic male mice. At the end of the experiments, mice were sacrificed and xenografts were resected. The Chinese Academy of Sciences Animal Care and Use Committee gave approval for the animal experiments.
Statistical analysis
Statistical analysis of the data was performed using the Student’s t-test. A p value less than 0.05 (P<0.05) was considered statistically significant.
Results
Doxorubicin induces cytotoxicity in U2OS but not in p53-null MG-63 cells
The tumor suppressor gene p53 has been suggested to induce apoptosis when cellular DNA is damaged. Since doxorubicin is a DNA-damaging agent that generates DNA double-strand breaks, we speculated that there might be significant difference in doxorubicin-induced cytotoxicity between U2OS and MG-63 cells. Doxorubicin was used to treat both types of cells under equivalent conditions. An MTT assay was performed to measure the cytotoxicity of doxorubicin in these cells. As shown in Figure 1A, the growth of U2OS was inhibited much more than MG-63 cells by doxorubicin at a concentration of 2 μg/mL; U2OS cell proliferation was decreased nearly 50%. Flow cytometric analysis was performed to examine whether the decreased cell viability was due to doxorubicin-induced apoptosis. As shown in Figure 1B, the percentage of U2OS cells undergoing apoptosis after doxorubicin treatment was significantly higher than that of MG-63 cells (45% vs 7%). The MTT assay demonstrated that the Half Lethal Dose (HLD) of doxorubicin was 1.74±0.22 µg/ml in U2OS cells and 9±0.61 µg/ml in MG-63 cells (Figure 1C). These results suggest that doxorubicin-induced cytotoxicity is significantly enhanced by an intact p53 pathway.
Figure 1.

Cytotoxicity of doxorubicin in osteosarcoma-derived cells. A. U2OS and MG-63 cells were seeded in growth media containing 2 µg/mL doxorubicin for 24 hours. An MTT assay was performed to measure cell viability. Untreated cells were included as controls. B. U2OS and MG-63 cells were seeded in growth media with or without 2 µg/mL doxorubicin for 24 hours. Cells were harvested and the Annexin V apoptosis assay was performed to measure the percentage of apoptotic cells. Representative images are shown in the upper panel and results from three independent experiments are summarized in the lower panel. C. U2OS and MG-63 cells were seeded in growth media containing increasing concentrations of doxorubicin for 24 hours. MTT assay was performed to measure cell viability and the IC50 of doxorubicin was determined. *Indicates P<0.05.
Difference in doxorubicin-induced cytotoxicity between U2OS and MG-63 cells is mediated by p53
In order to investigate the role of p53 in doxorubicin-induced cytotoxicity, we first examined the amount of p53 in U2OS and MG-63 cells using a Western blot. p53 protein was detected in U2OS and the control HEK-293T cells, but not in MG-63 cells (Figure 2A). This result was consistent with previous reports indicating that MG-63 cells are p53-null. Next, we examined the effect of doxorubicin on expression of p53 downstream effectors in U2OS and MG-63 cells. p53 upregulated modulator of apoptosis (PUMA) is a key regulator of p53-dependent apoptosis [12]. p21 expression is tightly controlled by p53 and mediates p53-dependent cell cycle arrest and apoptosis [13]. As shown in Figure 2B, expression of PUMA and p21 in U2OS was upregulated by treatment with doxorubicin as early as 12 h, and the protein levels increased in a time-dependent manner. In contrast, the expression of PUMA and p21 could not be detected until 48 h after doxorubicin treatment in MG-63 cells (Figure 2C). Similarly, cleavage of the central apoptotic protease Caspase 3 was detected in U2OS cells 12 h after doxorubicin treatment, but not in MG-63 cells (Figure 2B and 2C). These results strongly suggest that the difference in doxorubicin-induced cytotoxicity between U2OS and MG-63 cells is, at least partly, mediated by p53.
Figure 2.
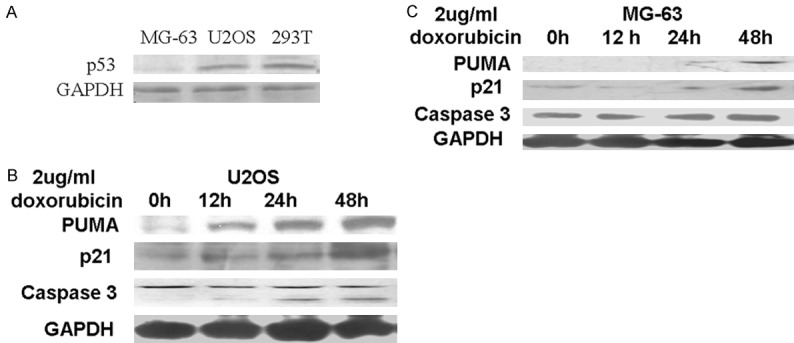
Expression of p53 and downstream effectors in U2OS and MG-63 cells after doxorubicin treatment. A. Expression of wild-type p53 was examined in MG-63, U2OS and HEK-293T cells by Western blot. B, C. U2OS and MG 63 cells were incubated with 2 µg/mL doxorubicin for 0, 12, 24 or 48 hours. Levels of PUMA, p21 and cleaved Caspase 3 were determined by Western blot. GAPDH was used as a loading control.
Exogenous expression of p53 enhances cytotoxicity of doxorubicin in MG-63 cells
In order to examine whether manipulating p53 expression could have an impact on the sensitivity of osteosarcoma cells to doxorubicin treatment, MG-63 cells were transfected with the recombinant p53 expression vector pcDNA-p53. Additionally, knockdown of p53 expression in U2OS cells was carried out by two independent p53-targeting shRNAs (I-p53): shRNA #1 and #2. The expression of p53 in each of these cell lines was confirmed by Western blot (Figure 3A). Since p53-targeting shRNA #1 and #2 exhibited similar efficacy in knocking down p53 levels in U2OS cells, we decided to use shRNA #1 in further investigations unless otherwise specified. Next, an MTT assay was performed to evaluate doxorubicin-induced growth inhibition in these cells. As predicted, viability of MG-63 cells overexpressing p53 was significantly decreased 48 h after doxorubicin treatment compared with mock transfected MG-63 cells (Figure 3B). The inhibition ratio was approximately 60%. In addition, knockdown of p53 expression by both shRNA #1 and #2 partially rescued doxorubicin-induced growth inhibition in U2OS cells. Western blotting showed that the levels of PUMA and p21 were increased and Caspase 3 cleavage was induced by 2 µg/mL doxorubicin in MG-63 cells with exogenous p53 expression (Figure 3C), suggesting that p53 is required for doxorubicin induced cell apoptosis. To our surprise, knocking down p53 did not significantly affect the anti-proliferative function of doxorubicin in U2OS cells (Figure 3B). However, when the protein level of p53 was examined in these cells, we found that doxorubicin treatment increased the endogenous p53 levels in U2OS cells, which offset the effect of shRNA-p53 (Figure 3D). These results provide further evidence suggesting that p53 is a determining factor for doxorubicin-induced cytotoxicity in osteosarcoma cells.
Figure 3.
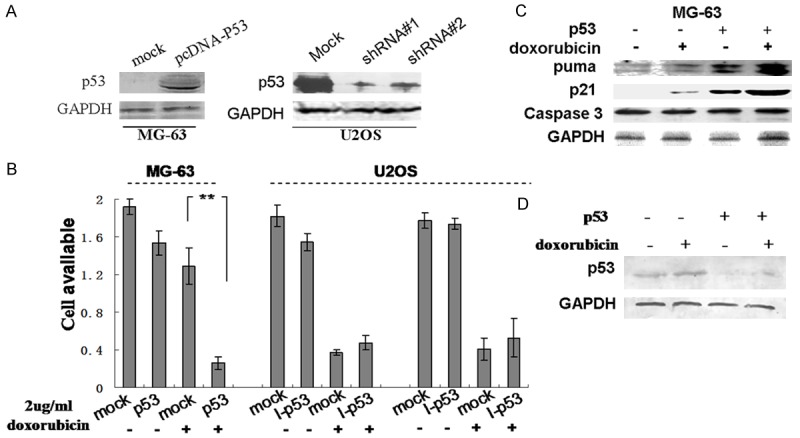
p53 is required for doxorubicin-mediated cytotoxicity. A. MG-63 and U2OS cells were transfected with a p53 expression plasmid (pcDNA-p53) or two different p53-targeting shRNAs (shRNA #1 and #2), respectively. Control cells were mock transfected. Total protein was isolated and a Western blot was performed to detect p53 protein. B. Transfected cells were incubated with or without 2 µg/mL doxorubicin for 48 hours. An MTT assay was performed to measure cell viability. **Indicates P<0.05. C. Mock or pcDNA-p53 transfected MG-63 cells were incubated with or without doxorubicin for 24 h. Protein levels of PUMA, p21, and cleaved Caspase 3 were examined by Western Blot. D. Mock or p53-targeting shRNA #1 transfected U2OS cells were incubated with or without doxorubicin for 24 h. The protein level of p53 was detected by Western blot. GAPDH was used as a loading control.
Doxorubicin functions through p53-mediated TGF-β signaling to induce apoptosis in osteosarcoma cells
A recent report has suggested that doxorubicin could activate TGF-β signaling in mammalian cells. In TGF-β-induced mammalian cell apoptosis, the transcriptional activation of p21 by nuclear translocation of Smads requires the assistance of p53. Whether doxorubicin performs its pro-apoptotic function through activation of TGF-β has not been determined. To explore this possibility, a luciferase reporter assay was performed to measure TGF-β/Smad signal transduction in osteosarcoma cells. As shown in Figure 4A, treatment with TGF-β1 in U2OS cells significantly increased the luciferase activity of the TGF-β reporter compared to the mock treatment. A similar effect was observed in doxorubicin-treated U2OS cells. When p53 expression was knocked down, the luciferase activity was only slightly reduced, which confirmed that the increased endogenous p53 levels induced by doxorubicin could contribute to the signaling activation (Figure 4A). In MG-63 cells, Smad signaling activity could be induced by TGF-β1, but not by doxorubicin. Exogenous expression of p53 compensated the loss of p53 activity in those cells and doxorubicin was then able to activate the luciferase reporter at a level similar to TGF-β1 (Figure 4A). To find out whether doxorubicin stimulated production of TGF-β, an ELISA assay was performed. In both U2OS and MG-63 cells, no significant differences in TGF-β levels were found between untreated and doxorubicin treated groups (Figure 4B), suggesting that the activation of downstream signaling was not a result of increased TGF-β production. Next, we investigated the phosphorylation and nuclear translocation of Smad3. As shown in Figure 4C, in MG-63 cells overexpressing p53, the level of Smad3 in the nucleus increased 48 h after transfection compared to the mock transfected control. After treatment with 2 μg/mL doxorubicin for 4 h, a large proportion of Smad3 was translocated into the nucleus. In untreated U2OS cells, the level of nuclear Smad3 was much higher than the cytoplasmic level (Figure 4D). p53 knockdown decreased the nuclear translocation of Smad3, and doxorubicin treatment did not significantly change the cytoplasmic-nuclear distribution of Smad3 (Figure 4D). These results indicate that doxorubicin stimulated TGF-β signaling and Smad3 activation in a p53-dependent manner.
Figure 4.
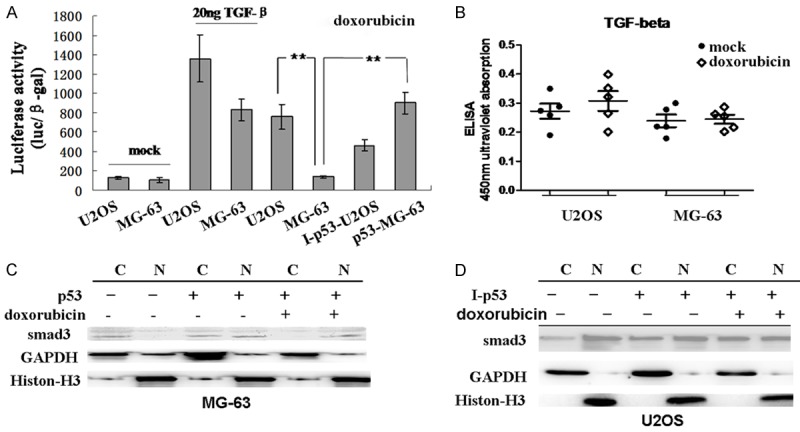
Activation of TGF-β signaling pathway is dependent on p53. A. Cells were transfected with a TGF-β luciferase reporter. After 36 h, U2OS and MG-63 cells were left untreated or treated with TGF-β or doxorubicin for 8 h. U2OS cells expressing p53-targeting shRNA (I-p53-U2OS) and MG-63 cells expressing pcDNA-p53 (p53-MG-63) were treated with doxorubicin for 8 h. Cells were lysed and a luciferase assay was performed. **Indicates P<0.05. B. U2OS and MG-63 cells were left untreated or treated with doxorubicin for 48 h. Supernatant was collected and an ELISA was performed to measure the relative level of secreted TGF-β. The experiment was repeated five times. Mean and standard deviation for each group are shown. C and D. Cytoplasmic (C) and nuclear (N) fractions were isolated from negative control, transfected (pcDNA-p53 in MG-63 and p53-targeting shRNA in U2OS cells) and doxorubicin treated transfected cells. Western blot was performed to examine the nuclear/cytoplasmic distribution of Smad3. GAPDH and Histone-H3 were used as cytoplasmic and nuclear loading controls, respectively.
Smad3 and p53 are both required for doxorubicin induced apoptosis in osteosarcoma cells
Next, we investigated whether Smad3 and p53 were both required for doxorubicin-induced apoptosis in osteosarcoma cells. First, Smad3 expression was knocked down in both U2OS and MG-63 cells (Figure 5A). Activation of TGF-β signaling in response to TGF-β or doxorubicin treatment was examined by luciferase reporter assay. As shown in Figure 5B, knockdown of Smad3 led to a significant reduction in TGF-β signaling activity induced by both TGF-β1 and doxorubicin in U2OS cells, and a complete loss of TGF-β1-induced signaling activity in MG-63 cells. These results indicated that Smad3 was required for transduction of TGF-β signaling and target gene responses. In addition, the effect of Smad3 knockdown on doxorubicin induced apoptosis in p53-positive or -negative osteosarcoma cells was examined. As shown in Figure 5C (upper panel), approximately 50% of U2OS cells with p53 knockdown were undergoing apoptosis after doxorubicin treatment, and further knockdown of Smad3 significantly suppressed doxorubicin-induced apoptosis. In p53-null MG-63 cells, exogenous expression of p53 greatly enhanced the pro-apoptotic function of doxorubicin (Figure 5C, lower panel). However, Smad3 knockdown reversed this phenotype. In addition, a colony formation assay was utilized to confirm that p53-Smad3 plays an important role in doxorubicin resistance. As shown in Figure 5D, U2OS cells with either reduced p53 or Smad3 expression showed enhanced viability. Simultaneous deficiency of both Smad3 and p53 significantly increased doxorubicin resistance in these cells. To extend our studies into an animal model, xenografts were employed to investigate whether p53-Smad3 signaling could regulate doxorubicin resistance in vivo. As shown in Figure 5E, for U2OS cell xenografts, tumor volume was markedly decreased after doxorubicin treatment, while knocking down Smad3 expression almost eliminated the cytotoxicity of doxorubicin. Moreover, simultaneous deficiency of p53 and Smad3 dramatically increased tumor size. For MG-63 cell xenografts, overexpression of p53 greatly enhanced sensitivity to doxorubicin and resulted in a significant reduction in tumor size; knocking down Smad3 partially rescued the p53-overexpressing phenotype (Figure 5E, lower panel). These results suggest that doxorubicin-induced apoptosis in osteosarcoma cells was mediated by both p53 and TGF-β signaling pathways.
Figure 5.

p53 and TGF-β signaling pathways are both required for doxorubicin induced apoptosis in osteosarcoma cells. A. U2OS and MG-63 cells were transfected with a Smad3 shRNA targeting vector (I-Smad3). After 48 h, total protein was isolated and a Western blot was performed to detect protein levels of Smad3. GAPDH was used as a loading control. B. Cells were transfected with a TGF-β luciferase reporter. After 48 h, U2OS and MG-63 cells were treated with PBS or TGF-β1 for 8 h. I-Smad3 co-transfected U2OS and MG-63 cells were treated with TGF-β1 or doxorubicin for 8 h. A luciferase reporter assay was performed. C. U2OS cells were transfected with a p53-targeting shRNA alone or co-transfected with I-Smad3. MG-63 cells were transfected with pcDNA-p53 alone or co-transfected with I-Smad3. Cells were then treated with doxorubicin for 48 h and harvested for flow cytometric analysis of apoptosis. Representative flow plots are shown in the upper panel and results from three independent experiments were summarized in the lower panel. D. Cells stably expressing shRNAs targeting p53 (I-p53), Smad3 (I-Smad3), or both (I-Smad3+I-p53) were treated with 2 µg/ml doxorubicin for 48 hours, and then seeded in a 6-well plate (1000 cells per well) for at least 10 days. At the end of the incubation period, cells were fixed and stained with H&E and the colonies were counted. Representative images from U2OS and MG-63 cells are shown in the upper panel and results from three independent experiments are summarized in the lower panel. E. U2OS cells with reduced expression of Smad3 (I-Smad3) alone or of Smad3 and p53 (I-Smad3+I-p53), and MG-63 cells overexpressing p53 in the presence or absence of Smad3 knockdown were treated with 2 µg/ml doxorubicin for 48 hours. Then cells were harvested, and 0.2 ml cell suspension (5×107 cells/ml) was injected subcutaneously into athymic male mice. At the end of the experiments, xenografts were resected and size measured. Representative images from U2OS and MG-63 cell xenografts are shown in the upper panel and results from three independent experiments (8 mice per group) are summarized in the lower panel. The scale bar shows 1 mm.
Discussion
Doxorubicin is an anticancer chemotherapeutic agent that interferes with DNA transcription and replication by intercalating between DNA base pairs and stabilizing topoisomerase II, thereby preventing normal transcription, inhibiting helicase-catalyzed DNA dissociation and generating free radicals [2]. Doxorubicin has become the cornerstone of cancer therapy in treating osteosarcoma [1]. Unfortunately, it has been shown that only 40% of osteosarcoma cells exhibit sensitivity to doxorubicin, and a subgroup of osteosarcoma patients eventually demonstrate resistance to doxorubicin treatment [14]. Increasing the dose of chemotherapeutic drugs increases the risk for side effects. By understanding the mechanism of doxorubicin-induced apoptosis in osteosarcoma cells, novel adaptive therapy could be developed in combination with doxorubicin to effectively treat osteosarcoma.
The tumor suppressor p53 plays a critical role in DNA damage-induced apoptosis and thus is considered as a pivotal factor for cytotoxicity of antitumor agents [15]. Multiple types of p53 mutations have been found in osteosarcoma patients, including point mutations, deletions and rearrangements that result in overexpression or loss-of-function of p53 protein [16,17]. In the present study, two different osteosarcoma-derived cell lines, p53-wild type U2OS and p53-null MG-63, were used to compare the efficacy of doxorubicin-induced cytotoxicity. We found that these two types of osteosarcoma-derived cells exhibited significant difference in sensitivity to doxorubicin treatment, which is consistent with previous investigation [18]. These results indicate that p53 and its related signaling pathway might play an essential role in doxorubicin-induced cytotoxicity. As expected, doxorubicin treatment effectively induced apoptosis in U2OS cells via a p53-related pathway. The protein level of PUMA was significantly increased following doxorubicin treatment, and cleavage of Caspase 3 was subsequently activated. Furthermore, the expression of downstream TGF-β target p21 was increased as well. Since p21 transcription has been shown to be cooperatively mediated by Smads and p53 [19], we speculated that doxorubicin might function through a TGF-β-mediated signaling pathway to induce apoptosis. In p53-null MG-63 cells, the pro-apoptotic effect of doxorubicin was profoundly downregulated. When p53 was overexpressed in these cells, the sensitivity to doxorubicin was restored, accompanied with elevated p21 and PUMA transcription, suggesting that p53 is required for doxorubicin-induced apoptosis in osteosarcoma cells. Interestingly, as shown in a previous report [20], we found that doxorubicin treatment could upregulate endogenous p53 expression in U2OS cells, hence knocking down p53 expression in U2OS cells did not affect the response to doxorubicin.
Previous investigation has suggested that doxorubicin could upregulate the expression of key players in the TGF-β signaling pathway, which consequently induces cardiotoxicity in rats [5]. In our study, treatment with TGF-β1 or doxorubicin independently activated Smad-driven reporter activity in U2OS cells. However, in contrast to previous reports, activation of TGF-β signaling was not due to increased TGFB1 expression. Compared to p53-wild type U2OS cells, the p53-null MG-63 line showed less reporter activity in response to TGF-β1 stimulation, suggesting that TGF-β signal transduction is partially dependent on p53. In addition, TGF-β target gene activity could not be induced by direct treatment with doxorubicin in mock-transfected MG-63 cells, while a high level of induction was achieved after exogenous expression of p53. Based on these results, we conclude that the doxorubicin-induced TGF-β signaling we observed in these osteosarcoma-derived cells was mediated by p53.
Smad3 belongs to the Smad family and functions as the key factor in the TGF-β signaling pathway. It has been shown to cooperate with p53 to synergistically regulate target gene transcription [10]. Physical interactions of p53 and Smad family members have been reported, with evidence for nuclear translocation of the resulting complex [10,19]. In our study, Smad3 was found to be a crucial factor for doxorubicin-induced apoptosis in both U2OS and MG-63 cells. In the presence of Smad3, doxorubicin could mimic the effect of TGF-β in promoting Smad3 nuclear translocation, activating target gene expression and inducing apoptosis. Knocking down Smad3 expression led to a complete loss of TGF-β-like signaling activity triggered by doxorubicin. Furthermore, doxorubicin-induced apoptosis in U2OS and in p53-overexpressing MG-63 cells was greatly reduced when Smad3 was knocked down in these cells. Integrity of the TGF-β/Smad signaling pathway is essential for doxorubicin-induced cytotoxicity, which leads us to hypothesize that patients with Smad3 deficiency may demonstrate a certain degree of resistance to doxorubicin treatment.
The results of this study indicate that doxorubicin-induced apoptosis in osteosarcoma-derived cells is dependent on p53 and is at least partially mediated through the TGF-β signaling pathway. When treating osteosarcoma patients with chemotherapy, p53 status may be an important indicator for doxorubicin efficacy. Deficiency or abnormalities of p53 in patients could be investigated before doxorubicin treatment to determine each patient’s sensitivity to doxorubicin treatment and inform prognosis. In addition, it has been previously shown that doxorubicin could increase TGF-β signaling in animal models, which in turn promotes the generation of stem cell-like cancer cells and enhances metastasis of breast cancer cells to the lung [4,8]. If this phenomenon also occurs in osteosarcoma, chemotherapy with doxorubicin may increase the risk of metastasis in a subgroup of patients and contribute to drug resistance. In clinical practice, the dosage of doxorubicin could be carefully determined and TGF-β signaling closely monitored to achieve the optimal efficacy of chemotherapy and eliminate potential risk.
Acknowledgements
The authors would like to express their gratitude to all the physicians participating in this work. This work was supported by Jilin Provincial Education Department Project (14397703D). The funder had no role in study design, data collection and analysis, decision to publish and preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment-where do we stand? A state of the art review. Cancer Treat Rev. 2014;40:523–532. doi: 10.1016/j.ctrv.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 3.Chou AJ, Gorlick R. Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther. 2006;6:1075–1085. doi: 10.1586/14737140.6.7.1075. [DOI] [PubMed] [Google Scholar]
- 4.Bandyopadhyay A, Wang L, Agyin J, Tang Y, Lin S, Yeh IT, De K, Sun LZ. Doxorubicin in combination with a small TGFbeta inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLoS One. 2010;5:e10365. doi: 10.1371/journal.pone.0010365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Shabanah OA, Aleisa AM, Hafez MM, Al-Rejaie SS, Al-Yahya AA, Bakheet SA, Al-Harbi MM, Sayed-Ahmed MM. Desferrioxamine attenuates doxorubicin-induced acute cardiotoxicity through TFG-beta/Smad p53 pathway in rat model. Oxid Med Cell Longev. 2012;2012:619185. doi: 10.1155/2012/619185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong D, Qu H, Meng X, Jiang Y, Liu D, Ye S, Chen H, Jin Y, Fu S, Geng J. S-allylmercaptocysteine promotes MAPK inhibitor-induced apoptosis by activating the TGF-beta signaling pathway in cancer cells. Oncol Rep. 2014;32:1124–1132. doi: 10.3892/or.2014.3295. [DOI] [PubMed] [Google Scholar]
- 7.Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002;307:1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 8.Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, Freeman ML, Arteaga CL. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–1313. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin X, Wu Y. Berbamine enhances the antineoplastic activity of gemcitabine in pancreatic cancer cells by activating transforming growth factor-beta/Smad signaling. Anat Rec (Hoboken) 2014;297:802–809. doi: 10.1002/ar.22897. [DOI] [PubMed] [Google Scholar]
- 10.Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 11.Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313–320. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- 12.Spender LC, Carter MJ, O’Brien DI, Clark LJ, Yu J, Michalak EM, Happo L, Cragg MS, Inman GJ. Transforming growth factor-beta directly induces p53-up-regulated modulator of apoptosis (PUMA) during the rapid induction of apoptosis in myc-driven B-cell lymphomas. J Biol Chem. 2013;288:5198–5209. doi: 10.1074/jbc.M112.410274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenzie PP, Guichard SM, Middlemas DS, Ashmun RA, Danks MK, Harris LC. Wildtype p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. Clin Cancer Res. 1999;5:4199–4207. [PubMed] [Google Scholar]
- 14.Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. Int Orthop. 2006;30:445–451. doi: 10.1007/s00264-006-0192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 16.Guo W, Wang X, Feng C. P53 gene abnormalities in osteosarcoma. Chin Med J (Engl) 1996;109:752–755. [PubMed] [Google Scholar]
- 17.Overholtzer M, Rao PH, Favis R, Lu XY, Elowitz MB, Barany F, Ladanyi M, Gorlick R, Levine AJ. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci U S A. 2003;100:11547–11552. doi: 10.1073/pnas.1934852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spina A, Sorvillo L, Di Maiolo F, Esposito A, D’Auria R, Di Gesto D, Chiosi E, Naviglio S. Inorganic phosphate enhances sensitivity of human osteosarcoma U2OS cells to doxorubicin via a p53-dependent pathway. J Cell Physiol. 2013;228:198–206. doi: 10.1002/jcp.24124. [DOI] [PubMed] [Google Scholar]
- 19.Elston R, Inman GJ. Crosstalk between p53 and TGF-beta Signalling. J Signal Transduct. 2012;2012:294097. doi: 10.1155/2012/294097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nair P, Somasundaram K, Krishna S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. J Virol. 2003;77:7106–7112. doi: 10.1128/JVI.77.12.7106-7112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]