

Abstract
Heart failure is one of the most serious diseases worldwide, and can be caused by many factors, among them hyperhomocysteinemia can increase the risk for development of heart failure. In this study, we treated rats with high methionine diet (HMD), which can be conversed to homocysteine in human body, to induce a novel model of heart failure. We proved the successful establishment of this model by echocardiography and pathological evaluation at the termination of treatment. Ejection fraction and fractional shortening were significantly deceased after HMD treatment, while left ventricular volume in systole was increased. HMD treatment caused hypertrophy of cardiomyocytes, disarrangement of myofibers, and infiltration of inflammatory cells, as well as abundant apoptotic cells appeared after HMD treatment. Plasmatic homocysteine level was elevated after HMD treatment. Furthermore, through electrophoretic mobility shift assay and chromatin immunoprecipitation, the activity of NF-κB in nuclear extract was also significantly elevated, showing evidence of positive relationship between hyperhomocysteinemia and activation of NF-κB in HMD-induced heart failure. The successful development and validation of this model have made it a new tool for translational medical research of metabolic disorders-related cardiovascular disease.
Keywords: Heart failure, hyperhomocysteinemia, high methionine diet, NF-kappaB, rat model
Introduction
Heart failure (HF) occurs when the heart muscle is weakened and cannot pump enough blood to meet the body’s needs for blood and oxygen [1]. It is a major public health issue causing considerable morbidity and mortality, and affecting nearly 5 million patients with about 500,000 newly diagnosed cases every year in the U.S. In developed countries, around 2% of adults have HF and in those over the age of 65, this increases to 6-10% [1]. Many factors, including hypertension, diabetes, dyslipidemia, hyperhomocysteinemia and ischemic heart disease, can increase the risk for development of HF [2].
Homocysteine (HCY) is a sulphur-containing amino acid in human body produced by conversion of methionine, an essential amino acid present in foods regularly consumed within the diet [3]. Basic and clinical research has disclosed that an increase in plasma HCY is an independent and graded risk factor for cardiovascular disease. McCully published the first paper to propose such a relationship with postmortem evidence of atherosclerosis in patients with hyperhomocysteinemia (HHCY) [4]. The following studies have linked elevated plasma concentrations of HCY to heart failure [5,6], myocardial infarction [7], peripheral vascular disease [8], and stroke [9], but the mechanism of HHCY-induced heart disease remains unclear.
Nuclear factor kappa-B (NF-κB) is a transcription factor that regulates transcription of genes involved in stress, growth, and inflammatory responses [10]. NF-κB also influences cell survival and can induce either pro- or anti-apoptotic genes depending on the cell type and stimulus [11]. HCY treatment caused an activation of NF-κB, leading to increased chemokine expression in vascular smooth muscle cells, endothelial cells and macrophages [12-14]. However, for now, the association between HHCY and activation of NF-κB in animal models, particularly of heart failure, was still unclear.
In this study, we treated rats with high methionine diet (HMD) to induce a novel model of heart failure, which was proved by echocardiography and pathological evaluation. Besides, we also tried to investigate possible mechanisms involved in this disease, especially the association between HHCY and activation of NF-κB.
Materials and methods
Animals
Male Wistar rats (5 weeks) were purchased from SLAC (Shanghai, China). Animals were housed in a temperature-controlled room (22°C) with 12-h-light/12-h-dark cycling, and had free access to food and water. All experimental procedures related to the animals complied with the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH) of the United States and were approved by Institutional Animal Care and Use Committee of The Fourth People’s Hospital of Jinan.
Modeling and in-life experiments
Rats were administered with eitherhigh methionine diet (HDTZW-005-2006; Feed Research Institute Chinese Academy of Agriculture Sciences, Beijing, China) which contains 1-3% methionine or control diet (normal chow; SLAC, Shanghai, China) from age of 6 weeks for another 6 weeks. Body weight (BW) was recorded every week. Heart failure would be diagnosed by echocardiography and pathological evaluation at the termination. After performed with echocardiography, rats were sacrificed by CO2 euthanasia. Blood was collected by heart puncture and the EDTA-treated plasma samples were saved in freezer. Heart was excised immediately, and weighted for left ventricular weight (LVW) and right ventricular weight (RVW). Part of heart was fixed in 4% parafor maldehyde for histological analysis, and the left part was homogenized for electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP).
Echocardiography
At the termination, rats were anesthetized with pentobarbital sodium (30 mg/kg) and placed on a heating pad. Echocardiography was performed to dynamically evaluate cardiac function of the rats using Vevo770 (Visual Sonics Inc., Toronto, Canada). Spatial resolutions for ventricular structure were provided by 17.5-MHz transducers. Left ventricular internal dimension in systole (LVIDs), left ventricular anterior wall in systole (LVAWs), left ventricular posterior wall in systole (LVPWs) were obtained from the M-mode tracings, and left ventricular volume in systole (LVs), ejection fraction (EF) and fractional shortening (FS) were derived automatically by the High-Resolution Electrocardiograph system.
Pathological evaluation
The fixed heart sample was embedded in paraffin, and cut into sections with 6 μm thickness. The sections were stained using Hematoxylin and Eosin (H&E) Staining Kit (Beyotime, Shanghai, China) or Terminal-deoxynucleoitidyl Transferase Mediated Nick End Labeling (TUNEL) Apoptosis Detection Kit (Beyotime) according to the manufacturer’s instruction, and photographed by digital camera conjugated with microscopy (Zeiss, Oberkochen, Germany).
Determination of plasmatic homocysteine level
Plasmatic homocysteine level was determined by commercially available Homocysteine ELISA Kit (Cell Biolabs, San Diego, CA) according to the manufacturer’s instruction.
Electrophoretic mobility shift assay
A LightShift Chemiluminescent EMSA Kit (Pierce, Rockford, IL) was used to prepare nuclear extracts from tissue homogenate and perform EMSA according to the manufacturer’s instructions. The sequence of biotin-labeled double-stranded DNA probe was listed below [15]: 5’-GGCTGGGGATTCCCCATCT-3’; 3’-CCGACCCCTAAGGGGTAGA-5’.
Chromatin immunoprecipitation
An Agarose ChIP Kit (Pierce) was used to prepare nuclear extracts from tissue homogenate and perform ChIP according to the manufacturer’s instructions. A ChIP-gradeprimary antibody against NF-κB p50 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Immunoprecipitated DNA was purified withDNA Clean-Up Column (Tiangen, Beijing, China) and then quantitated byreal-time polymerase chain reaction (RT-PCR) using PrimeScript RT-PCR-Kit (TAKARA, Dalian, China). The primers used were as follows [16-19]: IL-2: 5’-GAGGGATTTCACCTACATCCA-3’; 5’-TGCAATGCAAGACAGGAGTT-3’. IL-6: 5’-CAAGACATGCCAAAGTGCTG-3’; 5’-TTGAGACTCATGGGAAAATCC-3’. IL-8: 5’-AACAGTGGCTGAACCAGAG-3’; 5’-AGGAGGGCTTCAATAGAGG-3’. Bax: 5’-CCCGGGAATTCCAGACTGCAG-3’; 5’-GAGCTCTCCCCAGCGCAGAAG-3’. Bcl-XL: 5’-GCACCACCTACATTCAAATCC-3’; 5’-CGATGGAGGAGGAAGCAAGC-3’.
Statistical analysis
Data were presented as Mean ± SD. Significance of difference between groups was analyzed by performing two-way RM ANOVA for time course study, or one-way ANOVA with Dunnett’s multiple comparison test for other studies. P value less than 0.05 was considered statistically significant. Data were analyzed and graphed by Prism 6.0 (GraphPad Software, La Jolla, CA).
Results
We treated Wistar rats with HMD (control diet supplemented with 1, 2, or 3% methionine) for 6 weeks to induce a novel model of heart failure, which would be diagnosed by echocardiography and pathological evaluation at the termination. The BW of rats treated with 1% HMD was significantly decreased from Week 12 compared with that in naïve group, while BW in 2% and 3% HMD groups was significantly decreased from Week 11 (Figure 1). At the termination, we analyzed the LVW and RVW to disclose significant increases (dose-dependent) of LVW and LVW/BW in HMD-treated groups compared with that in naïve group (Figure 1). However, there was no significance in RVW change, and the change in RVW/BW was little.
Figure 1.

Body weight and ventricle weight. Rats were administered with either high methionine diet which contains 1-3% methionine or control diet from age of 6 weeks for another 6 weeks. A. Body weight (BW) was recorded every week. &P < 0.05, 1% HMD vs naïve group; &&P < 0.01, 1% HMD vs naïve group; &&&P < 0.001, 1% HMD vs naïve group; ##P < 0.01, 2% HMD vs naïve group; ###P < 0.001, 2% HMD vs naïve group; ***P < 0.001, 3% HMD vs naïve group. B-E. Left ventricular weight (LVW) and right ventricular weight (RVW) were recorded at the termination. *P < 0.05 vs naïve group, ***P < 0.001 vs naïve group. N=8.
At the end of treatment, we performed echocardiography and pathological evaluation to prove the success of model establishment. Heart failure was diagnosed by significantly lower EF and FS in rats treated by HMD compared with those in naïve group (Figure 2), suggesting injured cardiac function. The decreases of EF (71.6 ± 4.2%, 61.0 ± 2.5%, and 36.2 ± 1.1% vs. 95.4 ± 1.4%; P < 0.001) and FS (42.2 ± 4.4%, 33.6 ± 2.2%, and 16.2 ± 0.5% vs. 73.7 ± 4.2%; P < 0.001) weredose-dependent. Besides, HMD treatment dose-dependently in-creased LVs (86.1 ± 22.2 μL, 117.2 ± 21.1 μL, and 316.8 ± 26.4 μL vs. 6.3 ± 3.2 μL; P < 0.001) and LVIDs (4.5 ± 0.5 mm, 5.1 ± 0.4 mm, and 7.6 ± 0.3 mm vs. 1.6 ± 0.3 mm; P < 0.001), while deceased LVAWs (2.2 ± 0.3 mm, 1.5 ± 0.2 mm, and 1.1 ± 0.3 mm vs. 3.6 ± 0.2 mm; P < 0.001). However, there was nearly no significant change on LVPWs. After echocardiography, H&E and TUNEL staining were performed to evaluate pathological changes. HMD treatment caused hypertrophy of cardiomyocytes, disarrangement of myofibers, and infiltration of inflammatory cells (Figure 3). In addition, TUNEL staining disclosed that abundant apoptotic cells appeared after HMD treatment (Figure 3). Taken together, we treated rats with HMD to induce a novel animal model of heart failure.
Figure 2.

Cardiac function. At the termination, rats were anesthetized with pentobarbital sodium (30 mg/kg), and echocardiography was performed to dynamically evaluate cardiac function of the rats. Left ventricular internal dimension in systole (LVIDs), left ventricular anterior wall in systole (LVAWs), left ventricular posterior wall in systole (LVPWs), left ventricular volume in systole (LVs), ejection fraction (EF), fractional shortening (FS). *P < 0.05 vs naïve group, ***P < 0.001 vs naïve group. N=4.
Figure 3.

H&E and TUNEL staining. After performed with echocardiography, rats were sacrificed by CO2 euthanasia. The fixed heart sample was embedded in paraffin, and cut into sections with 6 μm thickness. The sections were stained for H&E and TUNEL analysis using commercially available kits. Magnification: 400.
To investigate the mechanism involved in HMD-induced heart failure, we determined the plasmatic HCY level to find that HMD significantly elevated HCY level in dose-dependent manner to induce HHCY (Figure 4). More importantly, the activity of NF-κB in nuclear extract was significantly elevated after HMD treatment (Figure 5), showing evidence of positive relationship between HHCY and activation of NF-κB in HMD-induced heart failure. Considering NF-κB is a vital transcriptional factor in regulations of genes involved in inflammation and cell survival, we performed ChIP to investigate the NF-κB-activated gene promoters. The promoters of IL-2, IL-6, IL-8 and Bax were significantly activated after HMD treatment in a dose-dependent manner (Figure 5), and their activation might be directly responsible for HMD-induced heart failure.
Figure 4.

Plasmatic homocysteine (HCY) level. At the termination, blood was collected by heart puncture and the EDTA-treated plasma samples were used to determine HCY level using commercially available kit. *P < 0.05 vs naïve group, ***P < 0.001 vs naïve group. N=8.
Figure 5.
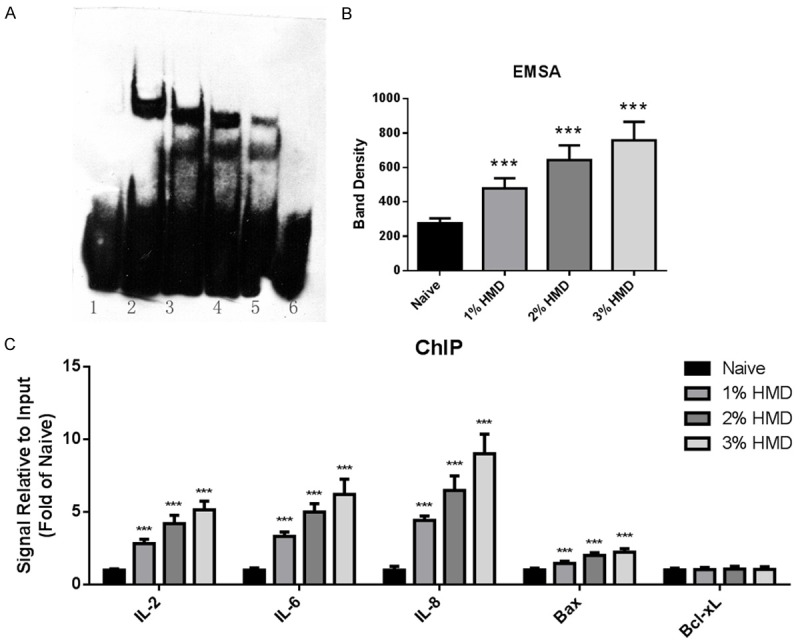
NF-κB activity. A, B. Nuclear extract was collected from heart tissue homogenate, and NF-κB/DNA binding activity was determined by electrophoretic mobility shift assay (EMSA) using commercially available kit. 1, Biotin-probe + nuclear extract (naive) + 200-fold molar excess of unlabeled probe; 2, biotin-probe + nuclear extract (3% HMD); 3, biotin-probe + nuclear extract (2% HMD); 4, biotin-probe + nuclear extract (1% HMD); 5, biotin-probe + nuclear extract (naive); 6, biotin-probe. ***P < 0.001 vs naïve group. C. Nuclear extract was collected from heart tissue homogenate, and NF-κB-regulated transcriptional activity was determined by Chromatin Immunoprecipitation (ChIP) using commercially available kit. ***P < 0.001 vs naïve group. N=8.
Discussion
Common causes of heart failure include a previous myocardial infarction (heart attack), hypertension, atrial fibrillation, valvular heart disease, excess alcohol use, smoke, infection, and metabolic disease. One well-established and widely used rat model of heart failure is coronary artery ligation model, which mimics the process of myocardial infarction to heart failure [20]. In addition, rat aortic banding, Dahl salt-sensitive rats, and spontaneous hypertensive rats (SHR) are also well-known models for translational medical research of heart failure [20-22]. Metabolic disease is another risk factor of heart failure [23,24], however, to now, there is still no established animal model under this strategy. So we tried to develop and validate a rat model through dietary intervention here, and investigate the possible involved mechanism.
In developed countries, high intake of methionine, which is abundant in animal proteins, always causes elevation of HCY level. Elevated plasmatic level of HCY has proatherothrombotic mechanisms including endothelial dysfunction and death, smooth muscle cell proliferation, inflammation, increased oxidative stress, and plaque formation [3,25]. Therefore, plasmatic HCY is a risk factor for vascular disease [26,27]. In a community-based prospective cohort study, an increased plasmatic HCY level independently predicted risk of the development of congestive heart failure in adults without prior myocardial infarction [5]. Considering the relationship between HHCY and heart failure, we treated rats with HMD to mimic the disease progress to induce heart failure. And at the termination of treatment, the condition was diagnosed in all rats by echocardiography and pathological evaluation, suggesting the model was successful.
In this rat model, we observed elevation of plasmatic HCY level after HMD treatment. More importantly, we disclosed a positive relationship between HHCY and activation of NF-κB in HMD-induced heart failure. NF-κB activation has been proved to play a vital role in the initiation and development of atherosclerosis, ischemic heart disease, and heart failure [28-30]. Several lines of evidence here indicated that NF-κB was activated in heart tissue after HMD treatment. First, results from EMSA demonstrated that HMD treatment caused a significant increase in the NF-κB/DNA binding activity. Second, results from ChIP demonstrated an enhanced NF-κB-regulated transcriptional activity. It has been well known several genes, which encode molecules involved in inflammation, apoptosis, and cell proliferation, can be regulated by NF-κB [31,32]. Here, we reported dose-dependently elevated activity of promoters of IL-2, IL-6, IL-8 and Bax, which might be directly responsible for HMD-induced heart failure. And in fact, we indeed observed phenotype changes, including inflammatory infiltration and cell apoptosis, through H&E and TUNEL staining.
Taken together, we first established a novel model of heart failure by HMD treatment, and disclosed association between HHCY and activation of NF-κB. The successful development and validation of this model have made it a new tool for translational medical research of metabolic disorders-related cardiovascular disease. In the future work, more involved mechanisms, especially those on lipid and amino acid metabolism, need to be further clarified.
Acknowledgements
This work was supported by Science and Technology Development Project of Jinan (200905036).
Disclosure of conflict of interest
None.
References
- 1.Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation. 2000;102:IV14–23. doi: 10.1161/01.cir.102.suppl_4.iv-14. [DOI] [PubMed] [Google Scholar]
- 2.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. doi: 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vizzardi E, Bonadei I, Zanini G, Frattini S, Claudia C, Raddino R, Cas LD. Homocysteine and heart failure: an overview. Recent Pat Cardiovasc Drug Discov. 2009;4:15–21. doi: 10.2174/157489009787259991. [DOI] [PubMed] [Google Scholar]
- 4.McCully KS, Wilson RB. Homocysteine theory of arteriosclerosis. Atherosclerosis. 1975;22:215–227. doi: 10.1016/0021-9150(75)90004-0. [DOI] [PubMed] [Google Scholar]
- 5.Vasan RS, Beiser A, D’Agostino RB, Levy D, Selhub J, Jacques PF, Rosenberg IH, Wilson PW. Plasma homocysteine and risk for congestive heart failure in adults without prior myocardial infarction. JAMA. 2003;289:1251–1257. doi: 10.1001/jama.289.10.1251. [DOI] [PubMed] [Google Scholar]
- 6.Herrmann M, Kindermann I, Müller S, Georg T, Kindermann M, Böhm M, Herrmann W. Relationship of plasma homocysteine with the severity of chronic heart failure. Clin Chem. 2005;51:1512–1515. doi: 10.1373/clinchem.2005.049841. [DOI] [PubMed] [Google Scholar]
- 7.Stampfer MJ, Malinow MR, Willett WC, Newcomer LM, Upson B, Ullmann D, Tishler PV, Hennekens CH. A prospective study of plasma homocyst (e) ine and risk of myocardial infarction in US physicians. JAMA. 1992;268:877–881. [PubMed] [Google Scholar]
- 8.Cheng SW, Ting AC, Wong J. Fasting total plasma homocysteine and atherosclerotic peripheral vascular disease. Ann Vasc Surg. 1997;11:217–223. doi: 10.1007/s100169900037. [DOI] [PubMed] [Google Scholar]
- 9.Parnetti L, Caso V, Santucci A, Corea F, Lanari A, Floridi A, Conte C, Bottiglieri T. Mild hyperhomocysteinemia is a risk-factor in all etiological subtypes of stroke. Neurol Sci. 2004;25:13–17. doi: 10.1007/s10072-004-0219-5. [DOI] [PubMed] [Google Scholar]
- 10.Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 11.Luo JL, Kamata H, Karin M. IKK/NF-κB signaling: balancing life and death-a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Au-Yeung KK, Woo CW, Sung FL, Yip JC, Siow YL, Karmin O. Hyperhomocysteinemia activates nuclear factor-κB in endothelial cells via oxidative stress. Circ Res. 2004;94:28–36. doi: 10.1161/01.RES.0000108264.67601.2C. [DOI] [PubMed] [Google Scholar]
- 13.Wang G, Siow Y. Homocysteine stimulates nuclear factor κB activity and monocyte chemoattractant protein-1 expression in vascular smooth-muscle cells: a possible role for protein kinase C. Biochem J. 2000;352:817–826. [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G, Siow YL, Karmin O. Homocysteine induces monocyte chemoattractant protein-1 expression by activating NF-κB in THP-1 macrophages. Am J Physiol Heart Circ Physiol. 2001;280:H2840–H2847. doi: 10.1152/ajpheart.2001.280.6.H2840. [DOI] [PubMed] [Google Scholar]
- 15.Haller D, Holt L, Kim SC, Schwabe RF, Sartor RB, Jobin C. Transforming growth factor-β1 inhibits non-pathogenic gramnegative bacteria-induced NF-κB recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J Biol Chem. 2003;278:23851–23860. doi: 10.1074/jbc.M300075200. [DOI] [PubMed] [Google Scholar]
- 16.Ashburner BP, Westerheide SD, Baldwin AS. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cianfrocca R, Muscolini M, Marzano V, Annibaldi A, Marinari B, Levrero M, Costanzo A, Tuosto L. RelA/NF-κB recruitment on the bax gene promoter antagonizes p73-dependent apoptosis in costimulated T cells. Cell Death Differ. 2008;15:354–363. doi: 10.1038/sj.cdd.4402264. [DOI] [PubMed] [Google Scholar]
- 18.Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L. CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-κB subunits on IL-8 and Bcl-xL gene promoters. Proc Natl Acad Sci U S A. 2004;101:6098–6103. doi: 10.1073/pnas.0308688101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang CH, Chiu YC, Tan TW, Yang RS, Fu WM. Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-κB pathway. J Immunol. 2007;179:5483–5492. doi: 10.4049/jimmunol.179.8.5483. [DOI] [PubMed] [Google Scholar]
- 20.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 21.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA. Animal Models of Heart Failure A Scientific Statement From the American Heart Association. Circ Res. 2012;111:131–150. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- 22.Patten RD, Hall-Porter MR. Small animal models of heart failure development of novel therapies, past and present. Circ Heart Fail. 2009;2:138–144. doi: 10.1161/CIRCHEARTFAILURE.108.839761. [DOI] [PubMed] [Google Scholar]
- 23.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–313. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 24.Malik S, Wong ND, Franklin SS, Kamath TV, Gilbert J, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 25.Steed MM, Tyagi SC. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal. 2011;15:1927–1943. doi: 10.1089/ars.2010.3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease: probable benefits of increasing folic acid intakes. JAMA. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 27.Graham IM, Daly LE, Refsum HM, Robinson K, Brattström LE, Ueland PM, Palma-Reis RJ, Boers GH, Sheahan RG, Israelsson B. Plasma homocysteine as a risk factor for vascular disease: the European Concerted Action Project. JAMA. 1997;277:1775–1781. doi: 10.1001/jama.1997.03540460039030. [DOI] [PubMed] [Google Scholar]
- 28.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-κB subunit p50 improves heart failure after myocardial infarction. FASEB J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 29.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Browder W, Kao RL. Early activation of transcription factor NF-κB during ischemia in perfused rat heart. Am J Physiol. 1999;276:H543–H552. doi: 10.1152/ajpheart.1999.276.2.H543. [DOI] [PubMed] [Google Scholar]
- 31.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roman-Blas J, Jimenez S. NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage. 2006;14:839–848. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]