

Abstract
Connective tissue growth factor (CTGF) plays a critical role in the hepatic stellate cells (HSCs)-mediated development of hepatic fibrosis. Nevertheless, the effects of CTGF gene promoter methylation in the pathogenesis of hepatic fibrosis remain largely unknown. In the current study, we isolated and overexpressed CTGF in primary HSCs. We analyzed the CTGF gene promoter methylation inHSCs that undergo a phenotypic change into myofibroblast-like cellsthat express α-smooth muscle actin (α-SMA) in vitro and in vivo in a CCl4-induced rat hepatic fibrosis model. We found that CTGF promoted the phenotypic changes of HSCs into myofibroblasts in vitro, while inhibition of CTGF promoter methylation augmented the process, suggesting that CTGF gene promoter methylation may negatively regulate hepatic fibrosis. In vivo, CCl4 induced hepatic fibrosis in rats, and the severity of hepatic fibrosis inversely correlated with the levels of CTGF gene promoter methylation in HSCs. Together, our data demonstrate that CTGF gene promoter methylation may prevent the development of hepatic fibrosis, and low level of CTGF gene promoter methylation in HSCs may be a predisposing factor for developing liver fibrotic disease.
Keywords: Connective tissue growth factor (CTGF), hepatic stellate cells (HSCs), hepatic fibrosis, myofibroblasts, α-smooth muscle actin (α-SMA)
Introduction
Liver fibrosis is a wound-healing process in response to chronic liver injuries associated with various etiologies [1]. The underlying pathological alterations in liver involve the activation of quiescent hepatic stellate cells (HSCs) into contractile myofibroblast-like cells, which produce and secrete excessive extracellular matrix (ECM) proteins in the liver [2-4]. Hepatic fibrosis may ultimately develop into cirrhosis or hepatocellular carcinoma (HCC). Thus, any therapeutic approaches to reverse the liver fibrosis may be critical for preventing those highly malignant and lethal diseases like cirrhosis and HCC [5,6].
A variety of animal models mimic different liver fibrosis and cirrhosis in mice, rats, and other animals [7,8]. These models use either ethanol treatment, or immunogens including plant protein concanavalin A and xenogeneic serum, or an operation of bile duct ligation, or genetic modification,or chemical compounds such as CCl4, thioacetamide and dimethylnitrosamine, which injure hepatocytesand trigger secondary inflammatory reaction to induce liver fibrosis and cirrhosis [7,8]. Due to the relative simple and reproducible approach, we used intraperitoneal administration of CCl4 in our study to induce hepatic fibrosis in rats [9].
Connective tissue growth factor (CTGF) is a highly profibrogenic molecule that overexpresses in many fibrotic lesions, including liver fibrosis [10]. CTGF is known to be transcriptionally activated by transforming growth factor-beta (TGFβ), which is the most potent and important molecular that regulate fibrogenesis [11,12]. Indeed, TGFβ has been shown to mediate activation of CTGF, and they coordinate the stimulation of connective tissue cell proliferation and synthesis of ECM in vitro [13]. In fibrotic liver, CTGF mRNA and protein are produced by fibroblasts, myofibroblasts, hepatic stellate cells (HSCs), endothelial cells and bile duct epithelial cells [13-15]. CTGF in cultured HSCs is activated upon TGFβ stimulation to promote HSC adhesion, proliferation and collagen production, as well as induction of HSC to a myofibroblast phenotype [16]. It is known that CTGF gene expression is sensitive to DNA methylation, and the methylation state of the CTGF promoter is negatively correlated with CTGF expression in ovarian cancer cells. Moreover, the epigenetic inactivation caused by hypermethylation of the CTGF promoter plays a role in ovarian tumorigenesis [17,18]. Furthermore, it has been reported that abnormal CTGF gene methylation occurred in a variety of liver cancer cell lines and primary liver cancer tissues, suggesting that CTGF methylation may be involved in liver tumorigenesis [19]. A previous study has investigated the effects of 5aza2’deoxycytidine (5azadCyd), a selective DNA methyltransferase inhibitor, on CTGF expression and methylation levels in human glomerular mesangial cells [20]. This study showed that 5azadCyd was able to induce the demethylation process of the CTGF gene promoter and to increase the expression of CTGF mRNA and protein [20]. However, so far, the methylation state of the CTGF promoter in HSCs in the setting of liver fibrosis, as well as its importance to disease progression, is ill-defined.
The current study was thus designed to use methylationspecific polymerase chain reaction (MSP) and bisulfite sequencing (BS) to investigate the methylation levels of theCTGF promoter in rat HSCs in vitro and in vivo in in a CCl4-induced rat hepatic fibrosis model.
Materials and methods
Ratand treatments
Male Sprague-Dawley rats (250-300 g) were used for in vivo experiments. The rats were obtained from the Shanghai Laboratory Animal Center (Shanghai, China) and had free access to standard laboratory water and chow. The rats were maintained under specific pathogen-free (SPF) conditions. Animal experiments were carried out in strict accordance with the regulations in the Guide for the Care and Use of Laboratory Animals issued by Shanghai Jiaotong University. The protocol was approved by the Institutional Animal Care and Use Committee of the Shanghai Jiaotong University.All the animals were randomly divided into two groups: control and carbon tetrachloride (CCl4). Liver fibrosis was induced in the CCl4 treatment group by intraperitoneal (i.p.) injection of CCl4 (Sigma-Aldrich, St. Louis, MO, USA) (dissolved CCl4 at 40% in olive oil) at 2.5 ml/kg body weight twice a week for 8 weeks. The animals in the control group were treated similarly including i.p. injection with the same volume of olive oil at the same frequency. Rats were sacrificed at the end of 8 weeks. HSCs from liver tissue samples were isolated for further study.
Isolation and culture of HSCs
HSCs were isolated from rat liver according to standard protocols, as have been previously described [21]. HSCs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), 1% L-glutamine and 1% penicillin-streptomycin (Invitrogen) in a humidified chamber with 5% CO2 at 37°C.
Plasmid and cell transfection
CTGF-overexpressingplasmids were prepared using routine methods. Briefly, the complete coding sequence for human CTGF was cloned using human cDNA as a template. A control scrambled sequence (SCR) was used as a control. These plasmids were cloned into pcDNA3.1-EGFP to generate the CTGF-overexpression plasmids and control SCR plasmids. These plasmids of 2 μg were transfected into cultured HSCs using Lipofectamine 2000, according to the manufacturer’s instructions (Invitrogen). The plasmids also contained a GFP reporter to allow determination of transfection efficiency, which was more than 95%.
Quantitative PCR (RT-qPCR)
Total RNA was extracted using RNeasy kit (Qiagen, Hilden, Germany). For cDNA synthesis, complementary DNA (cDNA) was randomly primed from 2 μg of total RNA using the Omniscript reverse transcription kit (Qiagen). RT-qPCR was subsequently performed in triplicate with a 1:4 dilution of cDNA using the Quantitect SyBr green PCR system (Qiagen). All primers were purchased from Qiagen. Data were collected and analyzed using 2-ΔΔCt method. Values of genes were first normalized against α-tubulin, and then compared to the experimental controls.
Western blot
The protein was extracted and homogenized in RIPA lysis buffer (1% NP40, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 0.5% sodium deoxycholate, in PBS) on ice. The supernatants were collected after centrifugation at 12000×g at 4°C for 20 min. Protein concentration was determined using a BCA protein assay kit (Bio-rad, China), and whole lysates were mixed with 4×SDS loading buffer (125 mmol/l Tris-HCl, 4% SDS, 20% glycerol, 100 mmol/l DTT, and 0.2% bromophenol blue) at a ratio of 1:3. Protein samples were heated at 100°C for 5 min and were separated on SDS-polyacrylamide gels. The separated proteins were then transferred to a PVDF membrane. The membrane blots were first probed with a primary antibody. After incubation with horseradish peroxidase-conjugated second antibody, autoradiograms were prepared using the enhanced chemiluminescent system to visualize the protein antigen. The signals were recorded using X-ray film. Primary antibodies for Western Blot are anti-CTGF, anti- α-smooth muscle actin (α-SMA) and α-tubulin (all from Cell Signaling, San Jose, CA, USA). α-tubulin was used as protein loading controls. Secondary antibody is HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Images shown in the figures were representative from 5 individuals. Densitometry of Western blots was quantified with NIH ImageJ software (Bethesda, MA, USA). The protein levels were first normalized to α-tubulin, and then normalized to experimental controls.
Methylationspecific polymerase chain reaction (MSP) analysis of CTGF promoter methylation
Methylation-specific primers were designed based on the promoter sequence of CTGF, with 5’-TCGTTTCGGTCGATAGTTTC-3’ as the forward primer and 5’-CGAAACCCATACTAACGACG-3’ as the reverse primer. The sequences 5’-TTGTTTTGGTTGATAGTTTT-3’ and 5’-CAA AACCCATACTAACAACA-3’ were used for the forward and reverse non-methylation-specific primers, respectively. The following thermal cycling conditions were used: initial denaturation at 95°C for 5 min; 35 cycles of denaturation at 94°C for 45 sec, annealing at 50°C for 30 sec and extension at 72°C for 45 sec; final extension at 72°C for 10 min. The 159-bp MSP product was isolated using electrophoresis in a 1.5% agarose gel and analyzed using an ultraviolet (UV) gel imaging system (ImageQuant 350; GE Healthcare Co., Little Chalfont, UK).
Bisulfite sequencing (BS) analysis of CTGF promoter methylation
BS primers for the CTGF promoter region were designed to avoid methylated CpGs. Once the DNA sample treated with sodium bisulfite was fully sulfonated, the BS product was amplified with forward 5’-GTTGAGAGGAGATAGTTAGTG-3’ and reverse 5’-GGTTGTTAGGGAGGGATT-3’ primers. The polymerase chain reaction (PCR) thermal cycling conditions were: initial denaturation at 95°C for 5 min; 35 cycles of denaturation at 94°C for 45 sec, annealing at 52°C for 30 sec and extension at 72°C for 30 sec; final extension at 72°C for 10 min. The 296-bp amplification product was isolated using electrophoresis in a 1.5% agarose gel and visualized under UV light. A 10 μl aliquot of the PCR product was subjected to further sequencing by the Beijing Genomics Institute (Beijing, China). The BS primer amplification products from the samples of the three groups were compared with completely sulfonated promoter target sequences using the JellyFish 1.3 data application software (Field Scientific, LCC, Lewisburg, PA, USA). The target sequence was known to contain 39 methylated cytosine-guanine (C-G) pairs, and any remaining methylated cytosines (mCs) in the sequence indicated a methylation site. The methylation level was calculated as: (mC/C-G) x 100%.
Histology of liver injury
A small portion of the liver was fixed in 10% formalin for 12 hours and then paraffin-embedded. All the tissue specimens were cut into 4μm sections. The changes in liver histology were examined using hematoxylin and eosin (HE) and Masson’s trichrome staining, which were observed under a light microscope. Three sections per rat liver were blindly examined independently by two experienced liver pathologists who did not know the project details. Fibrosis in the Masson’s trichrome staining sections was evaluated according to the Semiquantitative Scoring System, as has been described before [22].
Statistics
All statistical analyses were carried out using the GraphPad Prism 6.0 statistical software (GraphPad Software, Inc. La Jolla, CA, USA). All values in cell and animal studies are depicted as mean ± standard deviation and are considered significant if p<0.05. All data were statistically analyzed using one-way ANOVA with a Bonferroni correction, followed by Fisher’ Exact Test for comparison of two groups. Bivariate correlations were calculated by Spearman’s Rank Correlation Coefficients.
Results
Overexpression of CTGF in HSCs
HSCs were isolated from rat liver. In order to evaluate the promoter methylation of CTGF in HSCs, we overexpressed CTGF in HSCs by transfection of a CTGF-overexpressing plasmids, and used a plasmid expressing a scrambled sequence (SCR) as a control. The increased expression of CTGF in HSCs by CTGF transfection was confirmed by RT-qPCR (Figure 1A), and by Western blot (Figure 1B).
Figure 1.
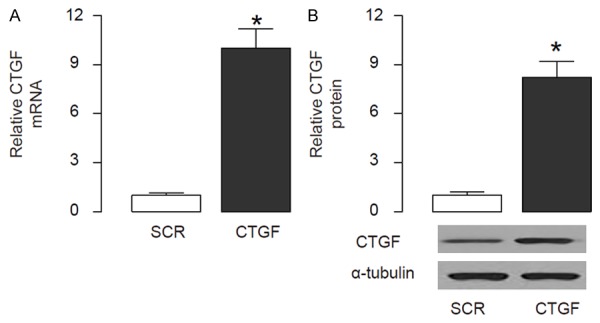
Overexpression of CTGF in HSCs. HSCs were isolated from rat liver. In order to evaluate the promoter methylation of CTGF in HSCs, we overexpressed CTGF in HSCs by transfection of a CTGF-overexpressing plasmids, and used a plasmid expressing a scrambled sequence (SCR) as a control. A. RT-qPCR for CTGF. B. Western blot for CTGF. *p<0.05. N=5.
CTGF promotes transition of HSCs into myofibroblast-like cells in vitro
The CTGF-modified HSCs were then put in culture to allow automatic transition into myofibroblast-like cells. The phenotypic changes of the cells were monitored at 3 day (early stage), 5 day (middle stage) and 7 day (activated stage) (Figure 2A). Then the cells were isolated and analyzed for expression of α-SMA, a marker for myofibroblasts. We found that expression of α-SMA increased with time in control HSCs (SCR), while significantly augmented in CTGF-overexpressing HSCs, by RT-qPCR (Figure 2B), and by Western blot (Figure 2C). These data suggest that CTGF promotes transition of HSCs into myofibroblast-like cells in vitro.
Figure 2.
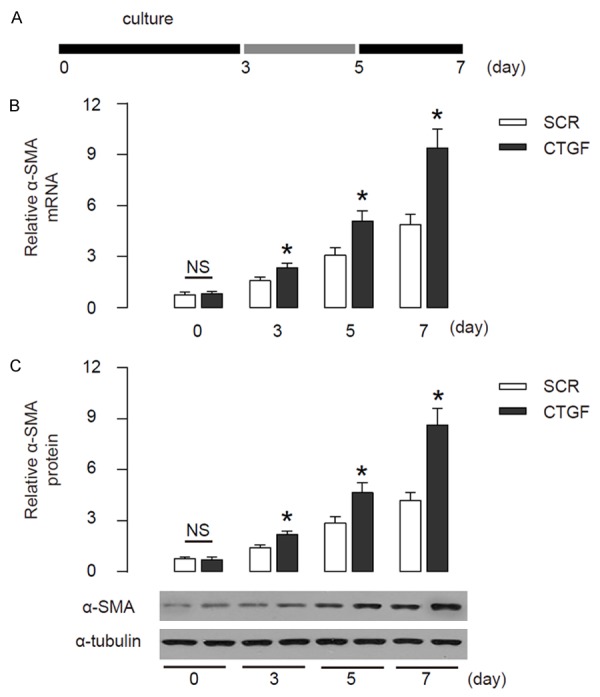
CTGF promotes transition of HSCs into myofibroblast-like cells in vitro. (A) The CTGF-modified HSCs were then put in culture to allow automatic transition into myofibroblast-like cells. The phenotypic changes of the cells were monitored at 3 day (early stage), 5 day (middle stage) and 7 day (activated stage). (B, C) The levels of α-SMA in control HSCs (SCR) andCTGF-overexpressing HSCs (CTGF), by RT-qPCR (B), and by Western blot (C). *p<0.05. NS: non-significant. N=5.
5azadCyd reduces CTGF gene promoter methylation in vitro
Next, we used 5aza2’deoxycytidine (5azadCyd), a selective DNA methyltransferase inhibitor, to treat CTGF-modified HSCs (Figure 3A). BS amplification results confirmed complete sulfonation of the DNA in HSCs. MSP analysis revealed high levels of methylation in the CTGF gene promoter in HSCs. Methylation levels in the CTGF promoter were significantly reduced by 5azadCyd (Figure 3B-C). These data suggest that 5azadCyd reduces CTGF gene promoter methylation in vitro.
Figure 3.
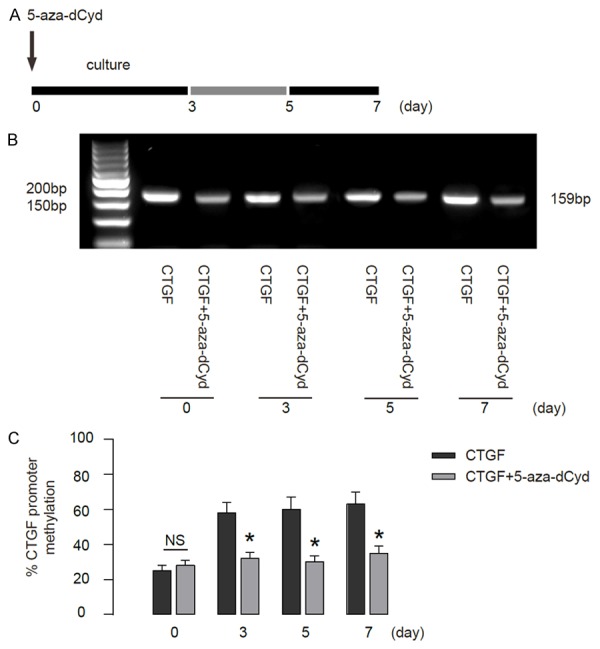
5azadCyd reduces CTGF gene promoter methylation in vitro. A. We used 5aza2’deoxycytidine (5azadCyd) to treat cultured CTGF-modified HSCs and analyzed the phenotypic changes of the cells at 3 day (early stage), 5 day (middle stage) and 7 day (activated stage). B. MSP analysis of the CTGF gene promoter methylation in HSCs. C. The quantification of CTGF gene promoter methylation by BS. *p<0.05. NS: non-significant. N=5.
Reduction in CTGF gene promoter methylation significantly attenuates HSC transition
Then we analyzed the effects of reduction in CTGF gene promoter methylation in HSC transition. We found that 5azadCyd-induced reduction in CTGF gene promoter methylation significantly attenuated the activation of α-SMA in HSCs, shown by RT-qPCR (Figure 4A), and by Western blot (Figure 4B). These data suggest that reduction in CTGF gene promoter methylation significantly attenuates HSC transition in vitro.
Figure 4.
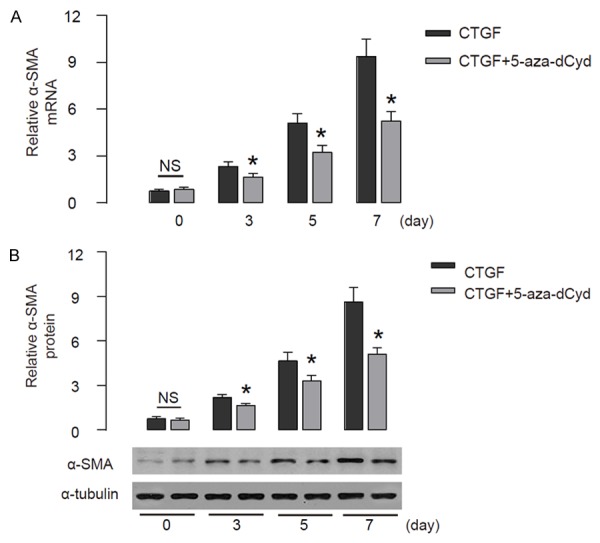
Reduction in CTGF gene promoter methylation significantly attenuates HSC transition. The effects of reduction in CTGF gene promoter methylation on HSC transition were analyzed. (A, B) The levels of α-SMA in HSCs were analyzed, by RT-qPCR (A), and by Western blot (B). *p<0.05. NS: non-significant. N=5.
Severity of hepatic fibrosis inversely correlates with CTGF gene promoter methylation in vivo
Then we analyzed the effects of reduction in CTGF gene promoter methylation in hepatic fibrosis. We used CCl4 to induced hepatic fibrosis in 12 rats, which was confirmed by histological score (Figure 5A). Next, we performed a correlation test between the severity of hepatic fibrosis and the levels of CTGF gene promoter methylation in isolated HSCs. We found that the severity of hepatic fibrosis inversely correlated with CTGF gene promoter methylation (R=-0.80, p<0.001, n=12) (Figure 5B). Thus, these data demonstrate that CTGF gene promoter methylation may prevent the development of hepatic fibrosis, and low level of CTGF gene promoter methylation may be a predisposing factor for developing liver fibrotic disease.
Figure 5.
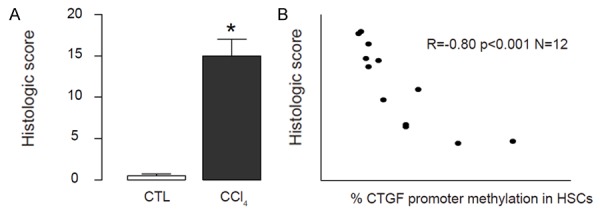
Severity of hepatic fibrosis inversely correlates with CTGF gene promoter methylation in vivo. CCl4 was used to induce hepatic fibrosis in 12 rats. A. Histological score was analyzed. B. A correlation test between the severity of hepatic fibrosis and the levels of CTGF gene promoter methylation was analyzed, showing that the severity of hepatic fibrosis inversely correlated with CTGF gene promoter methylation (R=-0.80, p<0.001, n=12). *p<0.05. N=12.
Discussion
Hepatic fibrosis and cirrhosis are a major health problem affecting millions of people worldwide [1-4]. During the chronic inflammation of the liver, normal quiescent HSCs undergo activation and transdifferentiation into myofibroblasts, which produce majority of hepatic ECM [1-4]. Thus, HSCs play a critical role in the initiation, maintenance and progression of liver fibrosis [1-4]. Therefore, suppression of activation of HSCs has been proposed as an effective therapy for patients with chronic liver injury and fibrosis.
CTGF is a highly profibrogenic molecule that plays a key role in liver fibrosis [10]. In fibrotic liver, CTGF are mainly produced by myofibroblasts and HSCs upon TGFβ stimulation [13-15]. CTGF gene expression is largely regulated by DNA methylation, which is the most common type of apparent genetic modification, and plays essential roles in immune system imbalance, oxidative stress, inflammation, insulin resistance and fibroblast activation [17,18]. Moreover, DNA methylation is critical for the formation of renal fibrosis and cardiovascular complications in patients with chronic kidney disease [23].
In the present study, we used CCl4 to establish liver fibrosis model in rats, as proved by histological evaluation. We found that in a rat hepatic fibrosis model, the severity of hepatic fibrosis inversely correlated with the levels of CTGF gene promoter methylation in HSCs. In vitro, we also provided evidence that CTGF promoted the phenotypic changes of HSCs into myofibroblasts, while inhibition of CTGF promoter methylation augmented the process, suggesting that CTGF gene promoter methylation may negatively regulate hepatic fibrosis. CTGF gene promoter existed in a relative low methylation state in rats that developed relative heavy hepatic fibrosis in rats. Furthermore, the methylation level of the CTGF promoter is an independent factor of CTGF expression. Together, these data indicate that DNA methylation is a regulatory mechanism of CTGF expression, possibly contributing to the pathogenesis of hepatic fibrosis, cirrhosis and HCC. Understanding the role of the demethylation of the CTGF promoter in the development of hepatic fibrosis may lead to the identification of novel strategies and/or additional therapeutic targets for the prevention and treatment of hepatic fibrosis.
The present study had several limitations. The current studies were performed in vitro and in vivo in rat models. The analyses on human specimens may further provide evidence of a role of CTGF promoter methylation in hepatic fibrosis. A fundus examination may be used in future studies as the screening method for patients with hepatic fibrosis, cirrhosis and HCC. In addition, despite the fact that previous studies and the current work have indicated that methylation is involved in the regulation of CTGF expression in HSCs, and that here our study revealed that mice with severe hepatic fibrosis had relative low methylation levels of CTGF, the mechanisms underlying the control of CTGF gene promoter methylation still requires further investigation.
Together, our data demonstrate that CTGF gene promoter methylation may prevent the development of hepatic fibrosis, and low level of CTGF gene promoter methylation may be a predisposing factor for developing liver fibrotic disease.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (NO: 81400631).
Disclosure of conflict of interest
The authors have declared that no competing interests exist.
References
- 1.Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. doi: 10.1038/nature12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Son MK, Ryu YL, Jung KH, Lee H, Lee HS, Yan HH, Park HJ, Ryu JK, Suh JK, Hong S, Hong SS. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci Rep. 2013;3:3470. doi: 10.1038/srep03470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu PS, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A, Mikami Y, Sugiyama K, Tomita K, Kanai T, Saito H, Hibi T. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology. 2013;58:337–350. doi: 10.1002/hep.26351. [DOI] [PubMed] [Google Scholar]
- 5.Ji D, Li B, Shao Q, Li F, Li Z, Chen G. MiR-22 Suppresses BMP7 in the Development of Cirrhosis. Cell Physiol Biochem. 2015;36:1026–1036. doi: 10.1159/000430276. [DOI] [PubMed] [Google Scholar]
- 6.Li B, Shao Q, Ji D, Li F, Chen G. Mesenchymal Stem Cells Mitigate Cirrhosis through BMP7. Cell Physiol Biochem. 2015;35:433–440. doi: 10.1159/000369708. [DOI] [PubMed] [Google Scholar]
- 7.Olivier AK, Gibson-Corley KN, Meyerholz DK. Animal models of gastrointestinal and liver diseases. Animal models of cystic fibrosis: gastrointestinal, pancreatic, and hepatobiliary disease and pathophysiology. Am J Physiol Gastrointest Liver Physiol. 2015;308:G459–471. doi: 10.1152/ajpgi.00146.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi H, Sakai T. Animal models for the study of liver fibrosis: new insights from knockout mouse models. Am J Physiol Gastrointest Liver Physiol. 2011;300:G729–738. doi: 10.1152/ajpgi.00013.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vassiliadis E, Veidal SS, Kristiansen MN, Hansen C, Jorgensen M, Leeming DJ, Karsdal M. Peptidyl arginine deiminase inhibitor effect on hepatic fibrogenesis in a CCl4 pre-clinical model of liver fibrosis. Am J Transl Res. 2013;5:465–469. [PMC free article] [PubMed] [Google Scholar]
- 10.Paradis V, Dargere D, Vidaud M, De Gouville AC, Huet S, Martinez V, Gauthier JM, Ba N, Sobesky R, Ratziu V, Bedossa P. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology. 1999;30:968–976. doi: 10.1002/hep.510300425. [DOI] [PubMed] [Google Scholar]
- 11.Massague J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, Song Z, El-Gohary Y, Prasadan K, Shiota C, Gittes GK. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci U S A. 2014;111:E1211–1220. doi: 10.1073/pnas.1321347111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakai K, Jawaid S, Sasaki T, Bou-Gharios G, Sakai T. Transforming growth factor-beta-independent role of connective tissue growth factor in the development of liver fibrosis. Am J Pathol. 2014;184:2611–2617. doi: 10.1016/j.ajpath.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao C, Xie Y, Peng M, Ma L, Zhou Y, Zhang Y, Kang W, Wang J, Bai X, Wang P, Jia Z. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin Exp Med. 2014;14:141–150. doi: 10.1007/s10238-013-0229-6. [DOI] [PubMed] [Google Scholar]
- 15.Tong Z, Chen R, Alt DS, Kemper S, Perbal B, Brigstock DR. Susceptibility to liver fibrosis in mice expressing a connective tissue growth factor transgene in hepatocytes. Hepatology. 2009;50:939–947. doi: 10.1002/hep.23102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Tan Z, Wang Y, Tang J, Jiang R, Hou J, Zhuo H, Wang X, Ji J, Qin X, Sun B. PTPROassociated hepatic stellate cell activation plays a critical role in liver fibrosis. Cell Physiol Biochem. 2015;35:885–898. doi: 10.1159/000369746. [DOI] [PubMed] [Google Scholar]
- 17.Kroening S, Neubauer E, Wullich B, Aten J, Goppelt-Struebe M. Characterization of connective tissue growth factor expression in primary cultures of human tubular epithelial cells: modulation by hypoxia. Am J Physiol Renal Physiol. 2010;298:F796–806. doi: 10.1152/ajprenal.00528.2009. [DOI] [PubMed] [Google Scholar]
- 18.Kikuchi R, Tsuda H, Kanai Y, Kasamatsu T, Sengoku K, Hirohashi S, Inazawa J, Imoto I. Promoter hypermethylation contributes to frequent inactivation of a putative conditional tumor suppressor gene connective tissue growth factor in ovarian cancer. Cancer Res. 2007;67:7095–7105. doi: 10.1158/0008-5472.CAN-06-4567. [DOI] [PubMed] [Google Scholar]
- 19.Chiba T, Yokosuka O, Fukai K, Hirasawa Y, Tada M, Mikata R, Imazeki F, Taniguchi H, Iwama A, Miyazaki M, Ochiai T, Saisho H. Identification and investigation of methylated genes in hepatoma. Eur J Cancer. 2005;41:1185–1194. doi: 10.1016/j.ejca.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Yi B, Zhang H, Zhou H, Cai X, Sun J, Liu Y. [High glucose induce the demethylation of CTGF promoter and gene expression] . Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2011;27:747–750. [PubMed] [Google Scholar]
- 21.Hendriks HF, Verhoofstad WA, Brouwer A, de Leeuw AM, Knook DL. Perisinusoidal fatstoring cells are the main vitamin A storage sites in rat liver. Exp Cell Res. 1985;160:138–149. doi: 10.1016/0014-4827(85)90243-5. [DOI] [PubMed] [Google Scholar]
- 22.Chevallier M, Guerret S, Chossegros P, Gerard F, Grimaud JA. A histological semiquantitative scoring system for evaluation of hepatic fibrosis in needle liver biopsy specimens: comparison with morphometric studies. Hepatology. 1994;20:349–355. [PubMed] [Google Scholar]
- 23.Chiba T, Yokosuka O, Arai M, Tada M, Fukai K, Imazeki F, Kato M, Seki N, Saisho H. Identification of genes up-regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J Hepatol. 2004;41:436–445. doi: 10.1016/j.jhep.2004.05.018. [DOI] [PubMed] [Google Scholar]