

Abstract

Two new bifunctional tris(hydroxypyridinone) (THP) chelators designed specifically for rapid labeling with 68Ga have been synthesized, each with pendant isothiocyanate groups and three 1,6-dimethyl-3-hydroxypyridin-4-one groups. Both compounds have been conjugated with the primary amine group of a cyclic integrin targeting peptide, RGD. Each conjugate can be radiolabeled and formulated by treatment with generator-produced 68Ga3+ in over 95% radiochemical yield under ambient conditions in less than 5 min, with specific activities of 60–80 MBq nmol–1. Competitive binding assays and in vivo biodistribution in mice bearing U87MG tumors demonstrate that the new 68Ga3+-labeled THP peptide conjugates retain affinity for the αvβ3 integrin receptor, clear within 1–2 h from circulation, and undergo receptor-mediated tumor uptake in vivo. We conclude that bifunctional THP chelators can be used for simple, efficient labeling of 68Ga biomolecules under mild conditions suitable for peptides and proteins.
Introduction
The generator-produced positron-emitting isotope gallium-68 (68Ga) possesses a decay profile (t1/2 = 68 min, 90% positron yield, 1.9 MeV) that makes it suitable for molecular imaging with positron emission tomography (PET) using peptide-based targeting agents.1−3 The advent of an approved pharmaceutical grade 68Ge/68Ga generator3 (68Ge t1/2 = 270 days) allows hospitals economical access to a PET isotope without expensive cyclotron facilities. Indeed, current clinical use of 68Ga tracers for neuroendocrine (68Ga-DOTATATE) and prostate (68Ga-HBED-PSMA) tumor-targeting radiopharmaceuticals has had significant impact in the management of patients.4−9
The macrocycle 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), utilized in 68Ga-DOTATATE, has been extensively used in radiolabeled 68Ga3+ PET tracers despite the fact that elevated temperatures are required to achieve quantitative radiochemical yield, and some clinical productions of 68Ga-DOTATATE require a postsynthetic purification step1,2,10−12 which adds undesirable complexity to the radiopharmaceutical preparation, creating a barrier to widespread implementation of 68Ga PET. To allow for simplification of labeling, alternative bifunctional chelators for 68Ga3+ have been designed. These include bifunctional chelators based on 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA and NODAGA),13−20 1,4,7-triazacyclononane macrocycles substituted with phosphinic acid groups at the amine (TRAP and NOPO),21−27 hexaazamacrobicycles,28 a pyridyl-substituted DOTA macrocycle (PCTA),10,29 bis(2-hydroxybenzyl)ethylenediaminediacetic acid (HBED), and related compounds (notably HBED-CC) possessing phenol, amine, and carboxyl donor groups,4,30 substituted 6-amino-perhydrodiazepines (AAZTA),31 a siderophore-derived macrocyclic chelator with hydroxamate groups (FSC),32 and an acyclic chelator based on a substituted pyridine carboxylate with an N4O2 binding mode (DEDPA).33−35 Derivatives of NOTA/NODAGA, TRAP/NOPO, HBED, FSC, AAZTA, and DEDPA conjugates have demonstrated desirable radiolabeling properties, with labeling proceeding at room temperature for all these chelators. We have described a tripodal ligand containing three 1,6-dimethyl-3-hydroxypyridin-4-one groups (H3THP-Ac) that, upon loss of three protons, coordinates 68Ga3+ via 6 O-atoms at pH 6–7 and low ligand concentrations (10 μM) in <5 min (Chart 1).36 This chelator can be radiolabeled quantitatively and faster, at lower chelator concentration and under conditions that are milder36 (ambient temperature without acidic pH) than other chelators such as DOTA and NOTA that are commonly employed for 68Ga3+ and that typically require acidic conditions (pH 2–5) and, in the case of DOTA, high temperatures (≥80 °C) to achieve quantitative labeling.10−13 A maleimide derivative of this chelator (for conjugation to thiol groups) has been prepared (H3THP-mal, Chart 1),36,3768Ga-labeled conjugates of which demonstrated high in vivo stability over 90 min with respect to metal complex integrity.
Chart 1. Structures of H3THP-Ac and H3-THP-mal.

To increase the versatility of this unique chelation system to enable labeling of noncysteine-containing biomolecules, including small proteins and antibodies, new derivatives are required. We report the synthesis of two new tris(hydroxypyridinone) chelators based on 1,6-dimethyl-3-hydroxypyridin-4-one groups that contain pendant isothiocyanates for conjugation to primary amines, and their use for labeling exemplar peptide conjugates with 68Ga for PET imaging.
Results
Synthesis and Radiolabeling of Peptide Conjugates
To synthesize H3THP-NCS (Scheme 1), (1)38 was reacted with triethylamine and carbon disulfide in ethanol, to give a precipitated dithiocarbamate intermediate upon addition of water.39 The precipitated intermediate was resuspended in a solution of carbon disulfide/ethanol and addition of di-tert-butyl dicarbonate and a catalytic amount of 4-dimethylaminopyridine resulted in formation of (2). Subsequent removal of the benzyl groups with boron trichloride in dichloromethane, followed by addition of trifluoroethanol resulted in H3THP-NCS. To synthesize H3THP-PhNCS (Scheme 1), an excess of p-phenylene diisothiocyanate and diisopropylethylamine in dimethylformamide were added to a solution of (1), followed by isolation of (3) using reverse-phase semipreparative high-performance liquid chromatography (HPLC). The benzyl groups of (3) were removed using boron trichloride in dichloromethane, followed by addition of methanol, resulting in the bifunctional chelator H3THP-PhNCS.
Scheme 1. Synthesis of Tris(hydroxpyridinone) Bifunctional Chelators Containing Isothiocyanate Groups, H3THP-NCS (top) and H3THP-PhNCS (bottom).

The αvβ3 integrin-targeting pentapeptide, cyclic(RGDfK) (RGD), was chosen as an appropriately well-characterized peptide targeting vector40 for conjugation to the new tris(hydroxypyridinone) bifunctional chelators. Both H3THP-NCS and H3THP-PhNCS were reacted with the primary amine of the lysine side chain of RGD under microwave conditions similar to those often employed for peptide synthesis (dimethyl sulfoxide containing diisopropylethylamine, 120 °C, 300 W, 30 min). Use of dimethylformamide as a solvent in place of dimethyl sulfoxide resulted in precipitation of the bifunctional chelator upon addition of base, and no reaction took place, necessitating the use of dimethyl sulfoxide as a solvent. The reaction products were purified using semipreparative HPLC. Both conjugates H3THP-NCS-RGD and H3THP-PhNCS-RGD (Figure 1a,c) were isolated in 98% purity.
Figure 1.

(a) H3THP-NCS-RGD; (b) HPLC traces (λ220) of H3THP-NCS-RGD (black) and [natGa(THP-NCS-RGD)] (blue), and radio-HPLC trace of [68Ga(THP-NCS-RGD)] (red). Inset: Mass spectral signal of [natGa(THP-NCS-RGD) + 2H]2+; (c) H3THP-PhNCS-RGD; (d) HPLC traces (λ220) of H3THP-PhNCS-RGD (black) and [natGa(THP-PhNCS-RGD)] (blue), and radio-HPLC trace of [68Ga(THP-PhNCS-RGD)] (red). Inset: Mass spectral signal of [natGa(THP-PhNCS-RGD) + 2H]2+.
Both peptide derivatives could be radiolabeled with generator-produced eluate that was added either directly from the generator, or eluate that was preconditioned to concentrate activity and remove any contaminating 68Ge. In the case of the former, addition of generator-produced 68Ga3+ (90–110 MBq, Eckert and Ziegler generator) in aqueous HCl (0.1 M, 1 mL) to the tris(hydroxypyridinone) conjugates (10–12 nmol) at ambient temperature, followed by addition of aqueous ammonium acetate (1 M, 300 μL) to obtain solutions of pH ∼ 6.5, provided 68Ga-labeled conjugate. Within 2–5 min of addition of 68Ga3+ to the conjugates, the solutions were subjected to analytical reverse-phase HPLC analysis. Each reaction mixture gave a single peak in the HPLC radiochromatogram (Figure 1b,d, red traces), and in these experiments, in which a low activity generator was utilized, the radiochemical yield measured >99%, with specific activities of 8–9 MBq nmol–1.
For radiolabeling conjugates for in vivo studies, a generator eluting higher activities was employed, and eluate was subjected to pretreatment prior to radiolabeling to remove 68Ge.41 Generator-produced 68Ga3+(800–1000 MBq, iThemba Laboratories generator) was concentrated on an AG 50W × 4 cation exchange cartridge, and eluted with 200 μL 0.9 M HCl in ethanol/water (90%/10%).41 This volume was diluted in deionized water (800 μL) and directly added to H3THP-NCS-RGD and H3THP-PhNCS-RGD (∼12 nmol) at ambient temperature, followed immediately by addition of aqueous ammonium acetate (2 M, 400 μL) and saline to obtain solutions of pH ∼ 6.5, resulting in solutions of [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)]. Within 2–5 min of addition of 68Ga3+ to the conjugates, the solutions were subjected to analytical reverse-phase HPLC and instant thin layer chromatography (ITLC) analysis. Each reaction mixture gave a single major peak in the HPLC radiochromatogram (Figure S1), and under these conditions, the radiochemical yield for all of these reactions was >95% (determined by ITLC), with specific activities of 60–80 MBq nmol–1.
To verify the identity of the radiolabeled products, the 68Ga-labeled conjugates and their nonradioactive (natGa) analogs were analyzed using analytical reverse-phase HPLC with UV and sodium iodide scintillation detection. H3THP-NCS-RGD eluted earlier than [nat/68Ga(THP-NCS-RGD)] (Figure 1b, black trace, retention time of 7.15 min). Co-elution of [natGa(THP-NCS-RGD)] (Figure 1b, blue trace, retention time of 7.57 min) with [68Ga(THP-NCS-RGD)] (red trace, retention time of 7.83 min) confirmed the identity of the radiolabeled compound, with the difference in retention time a result of the configuration of the different detectors in series. Only a single signal was observed in the liquid chromatography/mass spectrometry (LCMS) total ion count chromatogram, corresponding to [natGa(THP-NCS-RGD)] (Figure 1b, inset). The dipositive ion ([natGa(THP-NCS-RGD)+2H]2+: {C65H90N17O17SGa}2+, observed monoisotopic peak = 740.78, calculated = 740.78) was the strongest in the spectrum, but the corresponding tripositive and monopositive species were also observed. Similarly, a solution containing [natGa(THP-PhNCS-RGD)] resulted in a signal in the UV chromatogram with a retention time of 8.15 min, matching that of [68Ga(THP-PhNCS-RGD)] (8.35 min, radiochromatogram), and distinct from H3THP-PhNCS-RGD (7.80 min) (Figure 1d). A single signal was observed in the LCMS total ion count chromatogram, corresponding to [natGa(THP-PhNCS-RGD)], and the major peak in the associated mass spectrum was the dipositive ion ([natGa(THP-PhNCS-RGD) + 2H]2+: {C72H96N19O17S2Ga}2+, observed monoisotopic peak = 815.80, calculated 815.80). No Ga3+ complexes with metal/ligand stoichiometry other than 1:1 were detected in any fraction eluting from the HPLC column in either case.
Serum stability studies were undertaken to determine whether [68Ga(THP-NCS-RGD)] or [68Ga(THP-PhNCS-RGD)] release 68Ga3+ to endogenous serum proteins. Addition of generator-produced 68Ga3+ to a solution of human serum resulted in 68Ga-bound protein and small molecule adducts that possessed distinct retention times of 5.9, 9.1, and 13.1 min when applied to the size exclusion HPLC column utilized in this study (Figure S2a). Under the same conditions, the radiolabeled peptide conjugates [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] possessed retention times of 18.1 and 20.2 min, respectively (Figure S2b,d). Both [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] were incubated in fresh human serum at 37 °C, and at 1 and 4 h, aliquots of these serum solutions were applied directly to the size exclusion HPLC column. Size exclusion chromatograms acquired at 1 and 4 h time points exhibited single signals with the same retention times as the control samples of [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)], indicating that no transchelation of 68Ga3+ from peptide conjugate to serum constituents occurred in either case (Figure S2c,e).
In vivo metabolic stability studies were also performed in Balb/c mice to assess whether [68Ga(THP-NCS-RGD)] or [68Ga(THP-PhNCS-RGD)] are modified, metabolized, or degraded while in circulation. Blood samples were obtained 20 and 90 min postinjection (PI), the serum fraction was separated from erythrocytes, serum proteins precipitated, and the supernatant applied to an analytical reverse-phase HPLC column. The acquired chromatograms revealed single signals for each trace, and each signal possessed a retention time that matched that of the administered radiotracer (Figures S3a-c and S4a-c). Urine was also collected and analyzed by reverse-phase HPLC (Figures S3d and S4d). While signals corresponding to the retention time of the administered radiotracers were observed in the chromatograms for both conjugates, several other broad signals were observed. These data indicated that both [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] were metabolically stable in circulation in vivo over a period of 90 min; however, renal clearance pathways for both radiotracers involved significant metabolism of both [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)].
The half-maximal inhibitory concentration (IC50) values of [Ga(THP-NCS-RGD)], [Ga(THP-PhNCS-RGD)], and RGD were determined using a solid-phase competitive binding assay with 125I-echistatin. Binding of 125I-echistatin to αvβ3 integrin was inhibited by [Ga(THP-NCS-RGD)], [Ga(THP-PhNCS-RGD)], and RGD in a concentration-dependent manner demonstrating high binding affinity and specificity of [Ga(THP-NCS-RGD)], [Ga(THP-PhNCS-RGD)], and RGD for αvβ3 integrin (Table 1, Figure 2). The IC50 value of [Ga(THP-NCS-RGD)] (8.3 ± 1.5 nM) was comparable to that of RGD (8.7 ± 1.6 nM), indicating similar affinity for the αvβ3 integrin receptor. The IC50 value of [Ga(THP-PhNCS-RGD)] (4.8 ± 0.6 nM) was lower than that of [Ga(THP-NCS-RGD)] and RGD, suggesting a higher relative affinity for the αvβ3 integrin receptor. Importantly, IC50 values indicated that the tris(hydroxypyridinone) conjugates retain the affinity of the parent peptide for αvβ3 integrin receptors.
Table 1. IC50 Values for [Ga(THP-NCS-RGD)], [Ga(THP-PhNCS-RGD)], and RGD, Determined Using a Solid-Phase Competitive Binding Assay with 125I-Echistatin.
| compound | IC50 (nM) (±standard error) | 95% confidence interval (nM) |
|---|---|---|
| [Ga(THP-NCS-RGD)] | 8.3 ± 1.5 | 5.2–11.3 |
| [Ga(THP-PhNCS-RGD)] | 4.8 ± 0.6 | 3.6–5.9 |
| RGD | 8.7 ± 1.6 | 5.5–11.9 |
Figure 2.
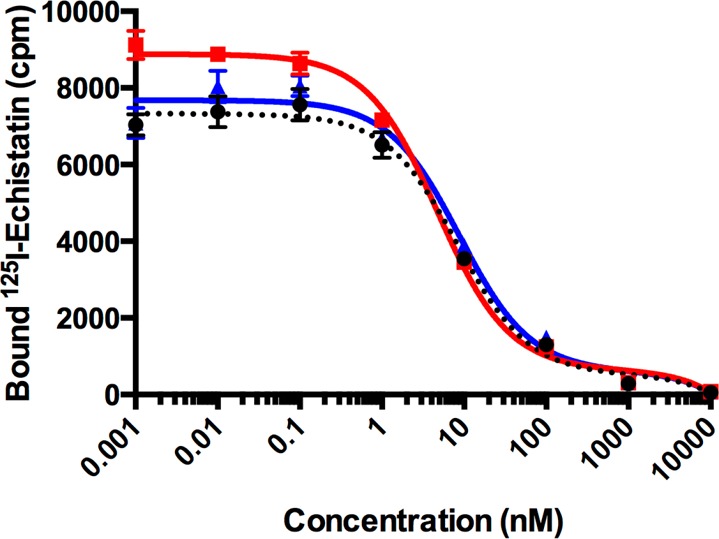
Mean concentration response curve for 125I-echistatin titrated with [Ga(THP-NCS-RGD)] (black), [Ga(THP-PhNCS-RGD)] (red), and RGD (blue) (n = 6 for each concentration; error bars correspond to standard error of the mean).
Biodistribution of 68Ga-Labeled Peptide Conjugates
The biodistribution of [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] was assessed in Balb/c nu/nu mice bearing αvβ3 integrin-positive glioblastoma U87MG tumors (n = 3). Each animal was administered 16–21 MBq of tracer (containing ∼1 μg of conjugate) and PET scanned at either 1 or 2 h PI for 10 min, followed by euthanasia and organ harvesting for ex vivo radioactivity counting. To assess specificity of the radiotracer, separate groups of animals were coadministered the tracer and RGD (0.4 mg per animal), followed by scanning, euthanasia, and ex vivo organ counting 1 h PI.
In PET scans of mice administered [68Ga(THP-NCS-RGD)] 1 h PI (Figure 3a), the tumor was visible, with a tumor to background (mediastinum) concentration ratio of 2.76 ± 0.18. The kidneys and liver of all animals were also discernible with a tumor to kidney concentration ratio of 1.03 ± 0.05, and a tumor to liver concentration ratio of 1.59 ± 0.16. There was no evidence of bone uptake being above that of surrounding muscle. Excretion was largely renal and the bladder was associated with the highest levels of activity in all PET images. Images of animals coadministered RGD demonstrated that RGD effectively blocks αvβ3 integrin receptor binding by [68Ga(THP-NCS-RGD)].
Figure 3.

PET imaging and ex vivo biodistribution of (a) [68Ga(THP-NCS-RGD)] and (b) [68Ga(THP-PhNCS-RGD)]. Representative PET maximum intensity projection of Balb/c nu/nu mice bearing a U87MG tumor on right flank at (i) 1 h PI of tracer, (ii) 1 h after coinjection of tracer and RGD, and (iii) 2 h PI of tracer, (iv) ex vivo biodistribution in mice at 1 and 2 h PI of tracer, and 1 h PI of tracer coadministered with RGD (blocked); n = 3. Error bars correspond to standard error of the mean.
The ex vivo biodistribution data of [68Ga(THP-NCS-RGD)] (Figure 3b) were consistent with PET imaging data. In animals administered solely [68Ga(THP-NCS-RGD)], there was significantly higher radioactivity concentration in the tumor than in animals co-administered [68Ga(THP-NCS-RGD)] and RGD, indicative of receptor-mediated tumor uptake (2.35 ± 0.06%ID g–1, vs 0.62 ± 0.10%ID g−1, respectively, mean difference = 1.73%ID g–1, 95% CI = 1.45 – 1.99%ID g–1, p = 6.00 × 10–5. Coadministration of RGD also significantly reduced uptake of the tracer at 1 h PI in the heart, liver, spleen, muscle, and lungs (Table S1). At both 1 and 2 h PI, the tumor, kidneys, and liver contained the highest concentration of radioactivity (Figure 3b). At 2 h PI, blood activity had cleared significantly compared to 1 h PI (0.16 ± 0.04 vs 0.84 ± 0.09%ID g–1, respectively), and in all organs and tissues, decay-corrected activity was slightly decreased compared to 1 h PI.
Similarly, PET scans of animals administered [68Ga(THP-PhNCS-RGD)] demonstrated that [68Ga(THP-PhNCS-RGD)] accumulated in the tumor, with a tumor to background concentration ratio of 2.34 ± 0.06, and a tumor to liver concentration ratio of 0.91 ± 0.03 at 1 h PI. There was no evidence of bone uptake being above that of surrounding muscle. Ex vivo biodistribution data demonstrated that there was significantly more activity associated with the tumor for animals administered solely [68Ga(THP-PhNCS-RGD)] 1 h PI (2.86 ± 0.43%ID g–1) than for animals coadministered [68Ga(THP-PhNCS-RGD)] and RGD (0.99 ± 0.12%ID g–1, mean difference = 1.87%ID g–1, 95% CI = 0.86–2.89%ID g–1, p = 6.89 × 10–3), consistent with αvβ3 integrin receptor mediated tumor uptake. Significantly reduced uptake was also observed for the spleen, liver, and muscle in animals coadministered RGD (Table S2). In contrast to [68Ga(THP-NCS-RGD)], uptake of [68Ga(THP-PhNCS-RGD)] in the tumor, spleen, and liver was slightly higher 2 h PI compared to 1 h PI, while uptake in other organs decreased (Figure 3b-iv). Blood activity at 2 h PI (0.25 ± 0.02%ID g–1) was lower than that observed at 1 h PI (0.57 ± 0.03%ID g–1). Blood activity at 1 h PI in the group coadministered RGD was significantly higher (1.29 ± 0.10%ID g–1) than in the group administered only [68Ga(THP-PhNCS-RGD)] (0.57 ± 0.03%ID g–1) (Table S2).
Tumor uptake of [68Ga(THP-PhNCS-RGD)] was higher than that of [68Ga(THP-NCS-RGD)] at 1 h PI and 2 h PI (1 h: 2.86 ± 0.43 vs 2.35 ± 0.06%ID g–1, mean difference = 0.52%ID g–1, 95% CI = −0.48–1.51%ID g–1, p = 0.22; 2 h: 3.32 ± 0.20 vs 1.90 ± 0.21%ID g–1, mean difference = 1.41%ID g–1, 95% CI = 0.75–2.08%ID g–1, p = 4.15 × 10–3, respectively). This is consistent with the lower IC50 value determined for [Ga(THP-PhNCS-RGD)] relative to [Ga(THP-NCS-RGD)]. The observed higher affinity of [Ga(THP-PhNCS-RGD)] (both in the solid-phase binding assay and in vivo) is possibly a result of the greater distance between the tris(hydroxypyridinone) complex and the RGD motif in [68Ga(THP-PhNCS-RGD)] compared to [68Ga(THP-NCS-RGD)].
Similarly, accumulation of radioactivity in the liver and spleen is higher in animals administered [68Ga(THP-PhNCS-RGD)] compared with animals administered 68Ga(THP-NCS-RGD)] at both 1 and 2 h PI. This difference in accumulation of the two tracers in the liver and spleen could be a result of two factors: (i) the increased αvβ3 integrin affinity of [68Ga(THP-PhNCS-RGD)] relative to [68Ga(THP-NCS-RGD)], and/or (ii) increased nonspecific uptake of [68Ga(THP-PhNCS-RGD)] in the liver and spleen compared to [68Ga(THP-NCS-RGD)]. There is some evidence in support of the latter—in the blockade experiments, uptake of [68Ga(THP-PhNCS-RGD)] is higher than uptake of [68Ga(THP-NCS-RGD)] in both the liver (2.68 ± 0.12 vs 1.30 ± 0.16%ID g–1) and spleen (0.82 ± 0.03 vs 0.42 ± 0.09%ID g–1).
Discussion
Simplicity and efficiency of labeling is a key to wider adoption of 68Ga PET in hospitals. The new bifunctional chelators, H3THP-NCS and H3THP-PhNCS, enable a facile, single-step route for preparation of peptide conjugates containing a tris(hydroxypyridinone) chelator. We chose to attach these new bifunctional chelators to the αvβ3 integrin-targeting peptide RGD to evaluate the radiolabeling and biodistribution profile of a simple tris(hydroxypyridinone) peptide conjugate and to establish whether such conjugates retain their receptor targeting affinity in vivo. Conjugation via the pendant isothiocyanate proceeds under microwave conditions frequently employed in peptide synthesis.42
The 68Ga-radiolabeling of H3THP-NCS-RGD and H3THP-PhNCS-RGD could be accomplished at ambient temperatures simply by addition of acidic solutions of generator-produced 68Ga3+ to solutions of the conjugates, followed by addition of ammonium acetate. High radiochemical yields (>95%) and specific activities (60–80 MBq nmol–1) were achieved, circumventing the requirement for postsynthetic purification. These uniquely efficient, simple, and mild radiolabeling properties would afford exceptionally rapid radiochemical synthesis of a 68Ga radiopharmaceutical and are conducive to translation to a one-step, kit-based preparation of a peptidic 68Ga-labeled radiotracer based on a tris(hydroxypyridinone) conjugate.
Serum and metabolic stability data indicate that both [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] are stable to 68Ga3+ transchelation to serum proteins, as well as degradation or modification of the conjugates while in circulation, respectively. Furthermore, PET scans and ex vivo biodistribution data in Balb/c nu/nu mice bearing glioblastoma U87MG αvβ3 integrin-positive tumors indicate that both [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] retain affinity for αvβ3 integrin receptors in vivo. Ex vivo biodistribution data are consistent with the expression profile of αvβ3 integrin, which is expressed in tumors including late stage glioblastomas, angiogenesis,43 and at low levels in normal vascular tissue.40
Animals coadministered RGD demonstrate that RGD effectively blocks αvβ3 integrin receptor binding by both [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] in tumors, and as such, tumor uptake of the new radiotracers is receptor-mediated. Biodistribution data also indicate significant receptor-mediated uptake in the liver and spleen for both radiotracers, and in the heart, muscle, and lungs for [68Ga(THP-NCS-RGD)]. Similar decreased uptake in blockade experiments for the liver, spleen, heart, lungs, muscles, adrenals, and intestines has been previously observed with other RGD conjugates labeled with PET isotopes 68Ga, 64Cu, 18F, and 44Sc.19,20,26,27,44,45 Tumor uptake of [68Ga(THP-NCS-RGD)] (1 h: 2.35 ± 0.06%ID g–1, 2 h: 1.90 ± 0.21%ID g–1) and [68Ga(THP-PhNCS-RGD)] (1 h: 2.86 ± 0.43, 2 h: 3.32 ± 0.20) is comparable to that of other conjugates of RGD reported in U87MG tumor bearing mice, although generally tumor to nontarget organ ratios, notably the liver, are lower for the tris(hydroxypyridinone) conjugates. 68Ga-labeled conjugates of RGD demonstrate receptor-mediated tumor uptake at 1 h PI of 5.19 ± 1.45%ID g–1 for 68Ga-NODAGA-RGD and 3.47 ± 0.78%ID g–1 for 68Ga-DOTA-RGD. Similar accumulation is observed for 64Cu-labeled analogs.20 The 18F-galacto-RGD PET tracer has an uptake of 1.16%ID g–1 1 h PI, but significantly lower nontarget organ uptake.46 The dimeric conjugate containing two αvβ3 integrin targeting cyclic(RGDyK) groups, 68Ga-NODAGA-E(RGDyK)2, demonstrates receptor mediated accumulation, with U87MG tumor uptake measuring 2.23 ± 0.08%ID g–1 1 h PI and liver uptake of 2.97 ± 0.39%ID g–1,19 values comparable to the monomeric tracers described here.
Concluding Remarks
The new bifunctional chelators, H3THP-NCS and H3THP-PhNCS, enable a facile route for preparation of conjugates containing a tris(hydroxypyridinone) chelator, and the conjugates retain the uniquely efficient radiolabeling properties reported for the maleimide derivative,36 affording exceptionally rapid radiochemical synthesis of a 68Ga radiopharmaceutical under mild conditions without the need for subsequent purification. Ga3+-complexed conjugates of RGD retain affinity for αvβ3 integrin receptors, and provide PET images that allow clear delineation of αvβ3 integrin-positive tumors. The efficiency of labeling tris(hydroxypyridinone) chelators at very low concentrations and under mild conditions brings the possibility of kit-based production of 68Ga PET tracers, including sensitive proteins, without complex automated synthesis typical of multistep PET radiochemistry. This would greatly increase 68Ga PET access to hospitals that lack expertise or facilities to implement such automated synthetic technologies but are adept in preparation of kit-based radiopharmaceuticals—notably radiopharmacy laboratories routinely producing 99mTc radiopharmaceuticals. Thus, such kit-based technologies will expand the use of the 68Ga generator for the benefit of more hospitals and patients.
Experimental Procedures
Materials and Instrumentation
Chemicals and reagents were obtained from Sigma-Aldrich (Dorset, UK) unless otherwise specified. High-performance liquid chromatography (HPLC) analysis was carried out using an Agilent 1200 LC system with in-line UV and gamma detection (Flow-Count, LabLogic). NMR spectra were acquired on a Bruker Avance 400 spectrometer (Bruker UK Limited, Coventry, UK) equipped with a 5 mm QNP probe at 400.13 MHz for 1H NMR spectra (using a zg30 pulse program) and 100.6 MHz for 13C NMR spectra (using a zgpg30 pulse program). Spectra were referenced to residual solvent signals. Mass spectra were recorded in the positive ion mode on an Agilent 6510 Q-TOF LC/MS mass spectrometer coupled to an Agilent 1200 LC system (Agilent, Palo Alto, CA). Data were acquired and reference mass-corrected via a dual-spray electrospray ionization source, using the factory-defined calibration procedure. Analytical reverse-phase LCMS and radio-LCMS traces were acquired using an Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm) with a 1 mL min–1 flow rate. Instant thin layer chromatography strips (ITLC-SG) were obtained from Varian Medical Systems UK, Ltd. (Crawley, UK), and ITLC strips were visualized using a Raytest Rita-Star TLC scanner. Semipreparative reverse-phase HPLC was conducted using an Agilent Eclipse XDB-C18 column (9.4 × 250 mm, 5 μm) coupled to an Agilent 1200 LC system, with a 3 mL min–1 flow rate and UV spectroscopic detection at 220 nm. Mobile phase A contained water with 0.2% TFA and mobile phase B contained acetonitrile with 0.2% TFA. All methods started with 100% A at 0 min. For method 1, the concentration of B increased at a rate of 1% min–1, and for method 2, the concentration of B increased at a rate of 0.5% min–1. Analytical reverse-phase HPLC and radio-HPLC traces were acquired using two different instruments: (1) an Agilent 1200 LC system and an Agilent Zorbax Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm) with a 1 mL min–1 flow rate and UV spectroscopic detection at either 214 or 220 nm. The radio-HPLC was coupled to a LabLogic Flow-Count detector with a sodium iodide probe (B-FC-3200). Aliquots (10–200 μL) of each radiolabeled sample were injected onto the column, using a flow rate of 1 mL min–1. Mobile phase A contained water with 0.1% TFA and mobile phase B contained acetonitrile with 0.1% TFA. For method 3, the concentration of B increased at a rate of 5% min–1, with 100% A at 0 min, and 100% B at 20 min. (2) An Agilent Zorbax Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm) with a 1 mL min–1 flow rate and UV spectroscopic detection at either 214 or 220 nm coupled to a Shimadzu HPLC consisting of a SCL-10AVP system controller, a SIL-10ADVP autoinjector, a LC-10 ATVP solvent delivery unit, a FCV-10AL control valve, a DGU-14A degasser, and a SPD-10AVP UV detector. This was coupled to a radiation detector consisting of an Ortec model 276 Photomultiplier Base with Preamplifier, Amplifier, BIAS supply and SCA and a Bicron 1 M 11.2 Photomultiplier Tube. For method 4, the concentration of B increased at a rate of 6.67% min–1, with 100% A at 0 min, and 80% B at 12 min. For initial radiolabeling and characterization studies that utilized <400 MBq, an Eckert and Ziegler 68Ge/68Ga generator (Berlin, Germany) was used. For biodistribution studies, and experiments that utilized >600 MBq 68Ga, an iThemba Laboratories 1.8 GBq 68Ge/68Ga generator (IDB Holland BV, Netherlands) was used. Analytical size exclusion radio-HPLC traces were acquired using an Agilent 1200 Series HPLC system and a Phenomenex Biosep 2000 (300 × 7.8 mm) size exclusion column with a phosphate buffered saline mobile phase with a flow rate of 1 mL min−1.
Synthesis
Compound 2
Compound 1 (synthesized according to previously reported procedures38) (100 mg, 96 μmol) was dissolved in ethanol (5 mL), and carbon disulfide (60 μL, 12 equiv) and triethylamine (13.4 μL, 1 equiv) were added.39 Addition of water (5 mL) resulted in formation of a white precipitate, corresponding to the dithiocarbamate intermediate. Excess carbon disulfide was removed under reduced pressure, and the remaining solution frozen and lyophilized. The dry residue was resuspended in ethanol (5 mL) containing carbon disulfide (60 μL) and triethylamine (13.4 μL). Di-tert-butyl dicarbonate (4 equiv, 21 mg) and 4-dimethylaminopyridine (2–3% molar equiv) were added,39 and the reaction stirred at room temperature for 6 h. The reaction mixture was evaporated to dryness, the residue dissolved in water/acetonitrile (60%/40%) and applied to a semipreparative reverse-phase Agilent Eclipse XDB-C18 column (9.4 × 250 mm, 5 μm) with a 3 mL min–1 flow rate and UV spectroscopic detection at 220 nm. Using HPLC method 1, 1 eluted with 40% B (40 min). Fractions containing the desired product were lyophilized. Yield = 50 mg, 48%. 1H NMR ((CD3)3SO, 400 MHz): δ 1.80 (m, 6H), 2.05 (m, 6H), 2.45 (t, J = 6.37, 2H), 2.55 (s, 9H), 3.75 (broad, 2H), 3.75 (s, 9H), 4.53 (d, J = 5.06, 6H), 5.13 (s, 6H), 7.13 (s, 3H), 7.35–7.48 (m, 15H + NH), 8.30 (t, J = 5.06, 3NH). 13C NMR ((CD3)3SO, 100 MHz): δ 20.4, 28.9, 29.6, 34.5, 35.4, 38.5, 41.1, 56.6, 74.0, 114.6, 127.5, 128.1, 128.2, 128.3, 136.1, 142.9, 146.3, 151.5, 165.0, 167.9, 171.2. ESI-MS: m/z for C59H68N8O10S + H+ calc 1081.48, found 1081.48.
H3THP-NCS
A solution of chilled boron trichloride in dichloromethane (10 mL, 1 M) was added to a sealed vial containing compound 2 (46 mg, 43 μmol), and the reaction was stirred at ambient temperature for 1 h. The reaction vial was then cooled in an ice bath, and trifluoroethanol (3 mL) was added dropwise to the flask. The reaction solution was evaporated to dryness, and the residue dissolved in water/acetonitrile (80%/20%) and filtered. The filtrate was diluted to 10 mL using a solution of 0.2% TFA in water, and applied to a semipreparative HPLC column. Using HPLC method 2, H3THP-NCS eluted with 17–18% B (37 min). Fractions containing the desired product were lyophilized. Yield of trifluoroacetate salt = 25 mg, 47% yield. 1H NMR ((CD3)3SO, 400 MHz): δ 1.82 (m, 6H), 2.08 (m, 6H), 2.45 (t, J = 6.41 2H), 2.52 (s, 9H), 3.77 (t, J = 6.41, 2H), 3.80 (s, 9H), 4.54 (d, J = 5.21, 6H), 6.97 (s, 3H), 7.43 (s, 1NH), 8.47 (t, J = 5.21, 3NH). 13C NMR ((CD3)3SO, 100 MHz): δ 20.3, 25.5, 28.8, 34.4, 35.4, 38.3, 41.1, 57.0, 112.4, 127.8, 138.6, 143.2, 148.3, 160.9, 168.2, 173.1. ESI-MS: m/z for C38H50N8O10S + H+ calc 811.34, found 811.36.
Compound 3
An excess of p-phenylene diisothiocyanate (40 mg) and diisopropylethylamine (40 μL) in DMF (0.5–1 mL) were added to a solution of 1 (40 mg, 39 μmol) in DMF (0.5–1 mL). The reaction solution was agitated, and after 5–10 min, applied to a semipreparative HPLC column. Using HPLC method 1, 3 eluted with 42% B (42 min). Fractions containing the desired product were lyophilized. Yield = 39 mg, 81% yield. 1H NMR (CD3OD, 400 MHz): δ 1.94 (m, 6H), 2.15 (m, 6H), 2.46 (t, J = 6.14, 2H), 2.60 (s, 9H), 3.79 (broad, 2H), 3.84 (s, 9H), 4.60 (s, 6H), 5.20 (s, 6H), 7.17 (d, J = 8.79, 2H), 7.33–7.46 (15H), 7.41 (d, J = 8.79, 2H). 13C NMR (CD3OD, 100 MHz): δ 21.4, 30.7, 31.0, 36.5, 36.9, 39.9, 42.1, 59.1, 76.3, 116.2, 125.7, 127.1, 128.6, 129.7, 129.9, 130.0, 136.6, 137.5, 145.0, 148.4, 154.1, 166.6, 173.6, 175.4, 182.0. ESI-MS: m/z for C66H74N10O10S2 + 2H+ calc 616.26, found 616.26.
H3THP-PhNCS
A solution of chilled boron trichloride in dichloromethane (5 mL, 1 M) was added to a sealed vial containing compound 3 (20 mg, 16 μmol), and the reaction was stirred at ambient temperature for 1 h. The reaction vial was then cooled in an ice bath, and methanol (5–10 mL) was added dropwise to the flask. The reaction solution was evaporated to near dryness under reduced pressure, and acetone (50 mL) was added to the residue, resulting in a flocculant white precipitate. This suspension was transferred to a 50 mL centrifugal tube, and the mixture centrifuged at 3000 rpm for 10 min. After this, the solution was decanted and discarded, acetone added (50 mL), the suspension agitated, and centrifuged again for 10 min. This process of washing with acetone was repeated again. Finally, the product was dissolved in water/acetonitrile (50/50), filtered, frozen and lyophilized. Yield = 10 mg, 58% yield. 1H NMR (CD3OD, 700 MHz): δ 1.98 (m, 6H), 2.22 (m, 6H), 2.49 (t, J = 6.13 2H), 2.59 (s, 9H), 3.82 (broad, 2H), 3.93 (s, 9H), 4.69 (s, 6H), 7.00 (s, 3H), 7.23 (d, J = 8.44, 2H), 7.43 (d, J = 8.44, 2H). 13C NMR (CD3OD, 175 MHz): δ 21.1, 30.6, 31.0, 36.3, 36.9, 39.5, 42.2, 59.1, 114.1, 125.8, 127.2, 128.8, 136.8, 139.4, 140.0, 145.2, 150.4, 162.2, 173.6, 176.3, 182.1. ESI-MS: m/z for C45H56N10O10S2 + H+ calc 961.37, found 961.37.
Synthesis of RGD Conjugates
The cyclic peptide cyclic(RGDfK) (RGD) was dissolved in dimethyl sulfoxide (100–300 μL) and added to a solution of either H3THP-NCS or H3THP-PhNCS in dimethyl sulfoxide (100–300 μL), and diisopropylethylamine (5–10 μL) was added. The reaction solutions were heated in a microwave (120 °C, 300 W, 30 min) and then applied to a reverse-phase HPLC column. Fractions containing the desired conjugate in sufficient purity were combined and lyophilized. Using HPLC method 2, H3THP-NCS-RGD eluted with 21% solvent B (42 min) and H3THP-PhNCS-RGD eluted with 23% solvent B (47 min). Isolated yields = 30–40%. H3THP-NCS-RGD: ESI-MS: m/z for C65H91N17O17S + 3H+ calc 472.22, found 472.22; HPLC: 220 nm, RT = 7.15 min, >98% purity (HPLC method 3). H3THP-PhNCS-RGD: ESI-MS: m/z for C72H97N19O17S2 + 3H+ calc 522.23, found 522.24; HPLC: 220 nm, RT = 7.80 min, >98% purity (HPLC method 3).
Complexing H3THP-NCS-RGD and H3THP-PhNCS-RGD with 68Ga3+ and natGa3+
Initial radiolabeling experiments utilized an Eckert & Ziegler 68Ge/68Ga generator. Aqueous HCl solution (0.1 M, 5 mL) was passed through the generator and the eluate was fractionated (5 × 1 mL). The second fraction (1 mL, containing 90–100 MBq 68Ga) was added directly to an ethanol/water solution (50%/50%, 50–100 μL) of either H3THP-NCS-RGD (22.5 μg) or H3THP-PhNCS-RGD (25 μg), immediately followed by a solution of ammonium acetate (1 M, 300 μL). This solution was immediately applied to an analytical reverse-phase C18 HPLC column. [68Ga(THP-NCS-RGD)]: radiochemical yield >99% (HPLC), HPLC: RT = 7.83 min (HPLC method 3). [68Ga(THP-PhNCS-RGD)]: radiochemical yield >99% (HPLC), HPLC: RT = 8.15 min (HPLC method 3). For both conjugates, specific activity at calibration = 8–9 MBq nmol–1 conjugate.
The nonradioactive analogues, [natGa(THP-NCS-RGD)] and [natGa(THP-PhNCS-RGD)], were also prepared. An aqueous solution of GaCl3 (2 mg mL–1, 5 μL, 50–60 nmol) was added to either H3THP-NCS-RGD (45 μg, ∼24 nmol) or H3THP-PhNCS-RGD (50 μg, ∼25 nmol) dissolved in deionized water (50 μL). The solutions were applied to an analytical reverse-phase C18 HPLC column and subjected to LCMS analysis. [natGa(THP-NCS-RGD)]: HPLC RT = 7.57 min (HPLC method 3); MS C65H88N17O17SGa + 2H+, observed monoisotopic peak = 740.78, calculated =740.78. [natGa(THP-PhNCS-RGD)]: HPLC RT = 8.15 min (HPLC method 3); MS C72H94N19O17S2Ga + 2H+, observed monoisotopic peak = 815.80, calculated 815.80.
For biodistribution studies, 68Ga eluate from an iThemba Lab generator was preconditioned as previously described.41 Briefly, a cation exchange cartridge containing AG 50W×4 resin (50 mg) was conditioned by passing through aqueous HCl solution (4 M, 1 mL) and deionized water (1 mL) sequentially. To elute the 68Ge/68Ga generator, aqueous HCl solution (0.4 M, 5 mL) was passed through the generator and transferred directly onto the cation exchange cartridge. The cartridge was dried with air (1 mL), washed with 0.15 M HCl in water/ethanol (20%/80%), and again dried with air (1 mL). A solution of 0.9 M HCl in water/ethanol (200 μL, 10%/90%) was used to elute 68Ga (800–1000 MBq), which was diluted to a volume of 1 mL with deionized water. Lyophilized peptide conjugate—H3THP-NCS-RGD (22.5 μg, trifluoroacetate salt) or H3THP-PhNCS-RGD (25 μg, trifluoroacetate salt)—dissolved in 20–40 μL of water/ethanol (50%/50%) was added to the solution containing 68Ga, immediately followed by a solution of ammonium acetate (2 M, 400 μL) and 0.9% saline (1100 μL). An aliquot for ITLC analysis was immediately applied to an ITLC-SG plate. The ITLC-SG plate was developed using an aqueous citrate buffer (0.1 M, pH 5.5) mobile phase. [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)]: Rf < 0.1; [68Ga(citrate)2]3–: Rf > 0.8. [68Ga(THP-NCS-RGD)]: radiochemical yield = 96–98% (ITLC), HPLC: RT = 9.42 min (HPLC method 4). [68Ga(THP-PhNCS-RGD)]: radiochemical yield = 97–99% (ITLC), HPLC: RT = 9.78 min (HPLC method 4). For both conjugates, specific activity at dose measurement = 60–80 MBq nmol–1 conjugate.
Serum Stability Studies
Solutions containing [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] (each 250 μL, containing 50 MBq 68Ga3+ and ∼5 μg of peptide conjugate, synthesized using eluate from an Eckert and Ziegler generator as described above) were added to 1.25 mL of fresh human female O+ serum, and incubated at 37 °C for 4 h. At 1 and 4 h, aliquots were applied to a size exclusion HPLC column. Solutions of [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] were also separately subjected to size exclusion chromatographic analysis to determine retention times. A solution of 68Ga3+ in 0.33 M ammonium acetate (10 MBq, 50 μL) was added to 250 μL of serum and incubated at 37 °C for 1 h to determine the retention time of 68Ga3+-bound serum proteins.
Metabolic Stability Studies
Solutions containing [68Ga(THP-NCS-RGD)] and [68Ga(THP-PhNCS-RGD)] (synthesized using eluate from an Eckert and Ziegler generator as described above) were diluted with physiological saline solution (15–30 MBq of each radiotracer containing 1–3 μg conjugate) and administered intravenously to seven- to nine-week-old female Balb/c mice (Harlan, UK) under isoflurane anesthesia. At 20 or 90 min PI, animals were euthanized, and serum and urine samples collected and analyzed using analytical reverse-phase HPLC (method 3).
Determination of IC50
The relative affinity of [Ga(THP-NCS-RGD)], [Ga(THP-PhNCS-RGD)], and RGD for integrin αvβ3 was determined in a solid-phase competitive binding assay45,47 with 125I-echistatin (PerkinElmer, Boston). In brief, wells of a 96 well plate were coated with integrin αvβ3 (150 ng mL–1) in coating buffer (100 μL, 25 mM Tris HCl pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2, and 1 mM MnCl2) overnight at 4 °C. Wells were then washed twice in binding buffer (coating buffer containing 0.1% bovine serum albumin (w/v) (BSA)) before being blocked for 2 h at room temperature with blocking buffer (coating buffer containing 1% BSA (w/v)). After a further two washes in binding buffer, 125I-echistatin (0.5 kBq) and [Ga(THP-NCS-RGD)], [Ga(THP-PhNCS-RGD)], or RGD were added simultaneously (to a total volume of 100 μL, and a conjugate/(RGDfK) concentration of 10 000 nM to 0.001 nM) for 1 h at room temperature, before being washed twice as before. Finally, the amount of activity bound to the wells via integrin αvβ3 was counted using a Wallac 1282 Compugamma Universal Gamma Counter. Measurements at each concentration for each compound were obtained in sextuplicate. IC50 values were calculated using a nonlinear regression model (Binding/Saturation, one site–total) in GraphPad Prism 5.04.
PET Scanning and Biodistribution
All animal experiments were performed with approval from the Peter MacCallum animal ethics committee. Six- to eight-week-old Balb/c nude mice (Animal Resources Centre, Western Australia) were implanted subcutaneously on the right flank with 4 million U87MG cells. Once the tumors reached a volume of >250 mm3 the animals were injected intravenously with 13–20 MBq [68Ga(THP-NCS-RGD)] (containing 1 μg of H3THP-NCS-RGD). For blocking studies, animals were coinjected with RGD peptide (400 μg). At 1 and 2 h, the animals were anaesthetized and imaged on a Philips MOSAIC small animal PET scanner. The images were reconstructed using a 3D RAMLA algorithm and tracer uptake determined as described previously.48 On completion of the scan animals were euthanized and tissues harvested, weighed, and radioactivity counted using a Gamma Counter (Biomedex).
Acknowledgments
M.T.M. acknowledges the support of the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement number 299009, and the Royal Society of Chemistry through a Researcher Mobility Fellowship. S.Y.A.T. was supported by a grant from Leukaemia and Lymphoma Research. J.B.-T. was supported by a grant from the Alzheimer’s Society. We thank Wayne Noonan, Kerry Ardley, and Rachael Walker for expert technical support. We thank David C. Muller (Genetic Epidemiology Group, International Agency for Research on Cancer) for his statistical advice and support. This research was supported by the Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC (WT088641/Z/09/Z), the KCL and UCL Comprehensive Cancer Imaging Centre funded by CRUK and EPSRC in association with the MRC and DoH (England), and by the NIHR Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the DoH.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.5b00335.
HPLC chromatograms, serum stability, metabolic stability and biodistribution data (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): PJB holds patents whose claims encompass the newly described chelators. All other authors declare that they have no conflict of interest.
Supplementary Material
References
- Price E. W.; Orvig C. (2014) Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 43, 260–90. 10.1039/C3CS60304K. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Houghton J. L.; Evans M. J.; Viola-Villegas N.; Lewis J. S. (2014) Underscoring the influence of inorganic chemistry on nuclear imaging with radiometals. Inorg. Chem. 53, 1880–99. 10.1021/ic401607z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikyan I. (2014) Prospective of 68Ga-radiopharmaceutical development. Theranostics 4, 47–80. 10.7150/thno.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar-Oromieh A.; Zechmann C. M.; Malcher A.; Eder M.; Eisenhut M.; Linhart H. G.; Holland-Letz T.; Hadaschik B. A.; Giesel F. L.; Debus J.; et al. (2014) Comparison of PET imaging with a 68Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 41, 11–20. 10.1007/s00259-013-2525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman M. S.; Kong G.; Neels O. C.; Eu P.; Hong E.; Hicks R. J. (2012) High management impact of Ga-68 DOTATATE (GaTate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours. J. Med. Imaging Radiat Oncol 56, 40–7. 10.1111/j.1754-9485.2011.02327.x. [DOI] [PubMed] [Google Scholar]
- Ambrosini V.; Campana D.; Tomassetti P.; Fanti S. (2012) 68Ga-labelled peptides for diagnosis of gastroenteropancreatic NET. Eur. J. Nucl. Med. Mol. Imaging 39, 52–60. 10.1007/s00259-011-1989-4. [DOI] [PubMed] [Google Scholar]
- Srirajaskanthan R.; Kayani I.; Quigley A. M.; Soh J.; Caplin M. E.; Bomanji J. (2010) The role of 68Ga-DOTATATE PET in patients with neuroendocrine tumors and negative or equivocal findings on 111In-DTPA-octreotide scintigraphy. J. Nucl. Med. 51, 875–82. 10.2967/jnumed.109.066134. [DOI] [PubMed] [Google Scholar]
- Haug A. R.; Auernhammer C. J.; Wangler B.; Schmidt G. P.; Uebleis C.; Goke B.; Cumming P.; Bartenstein P.; Tiling R.; Hacker M. (2010) 68Ga-DOTATATE PET/CT for the early prediction of response to somatostatin receptor-mediated radionuclide therapy in patients with well-differentiated neuroendocrine tumors. J. Nucl. Med. 51, 1349–56. 10.2967/jnumed.110.075002. [DOI] [PubMed] [Google Scholar]
- Conry B. G.; Papathanasiou N. D.; Prakash V.; Kayani I.; Caplin M.; Mahmood S.; Bomanji J. B. (2010) Comparison of 68Ga-DOTATATE and 18F-fluorodeoxyglucose PET/CT in the detection of recurrent medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 37, 49–57. 10.1007/s00259-009-1204-z. [DOI] [PubMed] [Google Scholar]
- Ferreira C. L.; Lamsa E.; Woods M.; Duan Y.; Fernando P.; Bensimon C.; Kordos M.; Guenther K.; Jurek P.; Kiefer G. E. (2010) Evaluation of bifunctional chelates for the development of gallium-based radiopharmaceuticals. Bioconjugate Chem. 21, 531–6. 10.1021/bc900443a. [DOI] [PubMed] [Google Scholar]
- Mueller D.; Klette I.; Baum R. P.; Gottschaldt M.; Schultz M. K.; Breeman W. A. P. (2012) Simplified NaCl based 68Ga concentration and labeling procedure for rapid synthesis of 68Ga radiopharmaceuticals in high radiochemical purity. Bioconjugate Chem. 23, 1712–7. 10.1021/bc300103t. [DOI] [PubMed] [Google Scholar]
- Zhernosekov K. P.; Filosofov D. V.; Baum R. P.; Aschoff P.; Bihl H.; Razbash A. A.; Jahn M.; Jennewein M.; Roesch F. (2007) Processing of generator-produced 68Ga for medical application. J. Nucl. Med. 48, 1741–8. 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]
- Velikyan I.; Maecke H.; Langstrom B. (2008) Convenient preparation of 68Ga-based PET-radiopharmaceuticals at room temperature. Bioconjugate Chem. 19, 569–73. 10.1021/bc700341x. [DOI] [PubMed] [Google Scholar]
- Eisenwiener K.-P.; Prata M. I. M.; Buschmann I.; Zhang H.-W.; Santos A. C.; Wenger S.; Reubi J. C.; Maecke H. R. (2002) NODAGATOC, a new chelator-coupled somatostatin analogue labeled with 67/68Ga and 111In for SPECT, PET, and targeted therapeutic applications of somatostatin receptor (hsst2) expressing tumors. Bioconjugate Chem. 13, 530–41. 10.1021/bc010074f. [DOI] [PubMed] [Google Scholar]
- Fani M.; Braun F.; Waser B.; Beetschen K.; Cescato R.; Erchegyi J.; Rivier J. E.; Weber W. A.; Maecke H. R.; Reubi J. C. (2012) Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J. Nucl. Med. 53, 1481–9. 10.2967/jnumed.112.102764. [DOI] [PubMed] [Google Scholar]
- Morfin J.-F.; Toth E. (2011) Kinetics of Ga(NOTA) formation from weak Ga-citrate complexes. Inorg. Chem. 50, 10371–8. 10.1021/ic201445e. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Niu G.; Wang F.; Chen X. (2009) 68Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur. J. Nucl. Med. Mol. Imaging 36, 1483–94. 10.1007/s00259-009-1123-z. [DOI] [PubMed] [Google Scholar]
- de Sa A.; Matias A. A.; Prata M. I. M.; Geraldes C. F. G. C.; Ferreira P. M. T.; Andre J. P. (2010) Gallium labeled NOTA-based conjugates for peptide receptor-mediated medical imaging. Bioorg. Med. Chem. Lett. 20, 7345–8. 10.1016/j.bmcl.2010.10.059. [DOI] [PubMed] [Google Scholar]
- Oxboel J.; Brandt-Larsen M.; Schjoeth-Eskesen C.; Myschetzky R.; El-Ali H. H.; Madsen J.; Kjaer A. (2014) Comparison of two new angiogenesis PET tracers 68Ga-NODAGA-E[c(RGDyK)]2 and 64Cu-NODAGA-E[c(RGDyK)]2; in vivo imaging studies in human xenograft tumors. Nucl. Med. Biol. 41, 259–67. 10.1016/j.nucmedbio.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Dumont R. A.; Deininger F.; Haubner R.; Maecke H. R.; Weber W. A.; Fani M. (2011) Novel 64Cu- and 68Ga-labeled RGD conjugates show improved PET imaging of αvβ3 integrin expression and facile radiosynthesis. J. Nucl. Med. 52, 1276–84. 10.2967/jnumed.111.087700. [DOI] [PubMed] [Google Scholar]
- Simecek J.; Schulz M.; Notni J.; Plutnar J.; Kubicek V.; Havlickova J.; Hermann P. (2012) Complexation of metal ions with TRAP (1,4,7-triazacyclononane phosphinic acid) ligands and 1,4,7-triazacyclononane-1,4,7-triacetic acid: phosphinate-containing ligands as unique chelators for trivalent gallium. Inorg. Chem. 51, 577–90. 10.1021/ic202103v. [DOI] [PubMed] [Google Scholar]
- Simecek J.; Hermann P.; Wester H.-J.; Notni J. (2013) How is 68Ga labeling of macrocyclic chelators influenced by metal ion contaminants in 68Ge/68Ga generator eluates?. ChemMedChem 8, 95–103. 10.1002/cmdc.201200471. [DOI] [PubMed] [Google Scholar]
- Notni J.; Pohle K.; Wester H.-J. (2012) Comparative gallium-68 labeling of TRAP-, NOTA-, and DOTA-peptides: practical consequences for the future of gallium-68-PET. EJNMMI Research 2 (28), 5. 10.1186/2191-219X-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notni J.; Hermann P.; Havlickova J.; Kotek J.; Kubicek V.; Plutnar J.; Loktionova N.; Riss P. J.; Rosch F.; Lukes I. (2010) A triazacyclononane-based bifunctional phosphinate ligand for the preparation of multimeric 68Ga tracers for positron emission tomography. Chem. - Eur. J. 16, 7174–85. 10.1002/chem.200903281. [DOI] [PubMed] [Google Scholar]
- Notni J.; Simecek J.; Hermann P.; Wester H.-J. (2011) TRAP, a Powerful and Versatile Framework for Gallium-68 Radiopharmaceuticals. Chem. - Eur. J. 17, 14718–22. 10.1002/chem.201103503. [DOI] [PubMed] [Google Scholar]
- Notni J.; Pohle K.; Wester H.-J. (2013) Be spoilt for choice with radiolabelled RGD peptides: Preclinical evaluation of 68Ga-TRAP(RGD)3. Nucl. Med. Biol. 40, 33–41. 10.1016/j.nucmedbio.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Simecek J.; Notni J.; Kapp T. G.; Kessler H.; Wester H.-J. (2014) Benefits of NOPO as chelator in gallium-68 peptides, exemplified by preclinical characterization of 68Ga-NOPO-c(RGDfK). Mol. Pharmaceutics 11, 1687–95. 10.1021/mp5000746. [DOI] [PubMed] [Google Scholar]
- Ma M. T.; Neels O. C.; Denoyer D.; Roselt P.; Karas J. A.; Scanlon D. B.; White J. M.; Hicks R. J.; Donnelly P. S. (2011) Gallium-68 complex of a macrobicyclic cage amine chelator tethered to two integrin-targeting peptides for diagnostic tumor imaging. Bioconjugate Chem. 22, 2093–103. 10.1021/bc200319q. [DOI] [PubMed] [Google Scholar]
- Ferreira C. L.; Yapp D. T. T.; Mandel D.; Gill R. K.; Boros E.; Wong M. Q.; Jurek P.; Kiefer G. E. (2012) 68Ga small peptide imaging: comparison of NOTA and PCTA. Bioconjugate Chem. 23, 2239–46. 10.1021/bc300348d. [DOI] [PubMed] [Google Scholar]
- Eder M.; Schaefer M.; Bauder-Wuest U.; Hull W.-E.; Waengler C.; Mier W.; Haberkorn U.; Eisenhut M. (2012) 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjugate Chem. 23, 688–97. 10.1021/bc200279b. [DOI] [PubMed] [Google Scholar]
- Waldron B. P.; Parker D.; Burchardt C.; Yufit D. S.; Zimny M.; Roesch F. (2013) Structure and stability of hexadentate complexes of ligands based on AAZTA for efficient PET labelling with gallium-68. Chem. Commun. 49, 579–81. 10.1039/C2CC37544C. [DOI] [PubMed] [Google Scholar]
- Knetsch P. A.; Zhai C.; Rangger C.; Blatzer M.; Haas H.; Kaeopookum P.; Haubner R.; Decristoforo C. (2015) [68Ga]FSC-(RGD)3 a trimeric RGD peptide for imaging αvβ3 integrin expression based on a novel siderophore derived chelating scaffold-synthesis and evaluation. Nucl. Med. Biol. 42, 115–22. 10.1016/j.nucmedbio.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros E.; Ferreira C. L.; Cawthray J. F.; Price E. W.; Patrick B. O.; Wester D. W.; Adam M. J.; Orvig C. (2010) Acyclic chelate with ideal properties for 68Ga PET imaging agent elaboration. J. Am. Chem. Soc. 132, 15726–33. 10.1021/ja106399h. [DOI] [PubMed] [Google Scholar]
- Boros E.; Ferreira C. L.; Yapp D. T. T.; Gill R. K.; Price E. W.; Adam M. J.; Orvig C. (2012) RGD conjugates of the H2dedpa scaffold: synthesis, labeling and imaging with 68Ga. Nucl. Med. Biol. 39, 785–94. 10.1016/j.nucmedbio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Ramogida C. F.; Cawthray J. F.; Boros E.; Ferreira C. L.; Patrick B. O.; Adam M. J.; Orvig C. (2015) H2CHXdedpa and H4CHXoctapa-chiral acyclic chelating ligands for 67/68Ga and 111In radiopharmaceuticals. Inorg. Chem. 54, 2017–31. 10.1021/ic502942a. [DOI] [PubMed] [Google Scholar]
- Berry D. J.; Ma Y.; Ballinger J. R.; Tavare R.; Koers A.; Sunassee K.; Zhou T.; Nawaz S.; Mullen G. E. D.; Hider R. C.; et al. (2011) Efficient bifunctional gallium-68 chelators for positron emission tomography: tris(hydroxypyridinone) ligands. Chem. Commun. 47, 7068–70. 10.1039/c1cc12123e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M. T.; Meszaros L. K.; Paterson B. M.; Berry D. J.; Cooper M. S.; Ma Y.; Hider R. C.; Blower P. J. (2015) Tripodal tris(hydroxypyridinone) ligands for immunoconjugate PET imaging with 89Zr4+: comparison with desferrioxamine-B. Dalton Trans. 44, 4884–900. 10.1039/C4DT02978J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Neubert H.; Liu D. Y.; Liu Z. D.; Ma Y. M.; Kong X. L.; Luo W.; Mark S.; Hider R. C. (2006) Iron binding dendrimers: a novel approach for the treatment of hemochromatosis. J. Med. Chem. 49, 4171–82. 10.1021/jm0600949. [DOI] [PubMed] [Google Scholar]
- Munch H.; Hansen J. S.; Pittelkow M.; Christensen J. B.; Boas U. (2008) A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 49, 3117–9. 10.1016/j.tetlet.2008.03.045. [DOI] [Google Scholar]
- Kerr J. S.; Slee A. M.; Mousa S. A. (2000) Small molecule αv integrin antagonists: novel anticancer agents. Expert Opin. Invest. Drugs 9, 1271–9. 10.1517/13543784.9.6.1271. [DOI] [PubMed] [Google Scholar]
- Eppard E.; Wuttke M.; Nicodemus P. L.; Roesch F. (2014) Ethanol-based post-processing of generator-derived 68Ga toward kit-type preparation of 68Ga-radiopharmaceuticals. J. Nucl. Med. 55, 1023–8. 10.2967/jnumed.113.133041. [DOI] [PubMed] [Google Scholar]
- Yu H. M.; Chen S. T.; Wang K. T. (1992) Enhanced coupling efficiency in solid-phase peptide synthesis by microwave irradiation. J. Org. Chem. 57, 4781–4. 10.1021/jo00044a001. [DOI] [Google Scholar]
- Terry S. Y. A.; Abiraj K.; Frielink C.; van Dijk L. K.; Bussink J.; Oyen W. J.; Boerman O. C. (2014) Imaging integrin αvβ3 on blood vessels with 111In-RGD2 in head and neck tumor xenografts. J. Nucl. Med. 55, 281–6. 10.2967/jnumed.113.129668. [DOI] [PubMed] [Google Scholar]
- Hernandez R.; Valdovinos H. F.; Yang Y.; Chakravarty R.; Hong H.; Barnhart T. E.; Cai W. (2014) 44Sc: An attractive isotope for peptide-based PET imaging. Mol. Pharmaceutics 11, 2954–61. 10.1021/mp500343j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschauer S.; Haubner R.; Kuwert T.; Prante O. (2014) 18F-Glyco-RGD peptides for PET imaging of integrin expression: efficient radiosynthesis by click chemistry and modulation of biodistribution by glycosylation. Mol. Pharmaceutics 11, 505–15. 10.1021/mp4004817. [DOI] [PubMed] [Google Scholar]
- Liu S.; Liu Z.; Chen K.; Yan Y.; Watzlowik P.; Wester H.-J.; Chin F. T.; Chen X. (2010) 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol. Imaging Biol. 12, 530–8. 10.1007/s11307-009-0284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf I.; Terry S. Y. A.; McBride W. J.; Goldenberg D. M.; Laverman P.; Franssen G. M.; Oyen W. J. G.; Boerman O. C. (2013) Imaging integrin alpha-v-beta-3 expression in tumors with an 18F-labeled dimeric RGD peptide. Contrast Media Mol. Imaging 8, 238–45. 10.1002/cmmi.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson B. M.; Roselt P.; Denoyer D.; Cullinane C.; Binns D.; Noonan W.; Jeffery C. M.; Price R. I.; White J. M.; Hicks R. J.; et al. (2014) PET imaging of tumours with a 64Cu labeled macrobicyclic cage amine ligand tethered to Tyr3-octreotate. Dalton Trans. 43, 1386–96. 10.1039/C3DT52647J. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.