

Abstract
To understand brain function, it is essential that we discover how cellular signaling specifies normal and pathological brain function. In this regard, chemogenetic technologies represent valuable platforms for manipulating neuronal and non-neuronal signal transduction in a cell-type-specific fashion in freely moving animals. Designer Receptors Exclusively Activated by Designer Drugs (DREADD)-based chemogenetic tools are now commonly used by neuroscientists to identify the circuitry and cellular signals that specify behavior, perceptions, emotions, innate drives, and motor functions in species ranging from flies to nonhuman primates. Here I provide a primer on DREADDs highlighting key technical and conceptual considerations and identify challenges for chemogenetics going forward.
Chemogenetics has been defined as a method by which proteins are engineered to interact with previously unrecognized small molecule chemical actuators (Forkmann and Dangelmayr, 1980; Sternson and Roth, 2014; Strobel, 1998). Over the past two decades, a large number of chemogenetic (also known as “chemical genetic”; (Bishop et al., 1998; Strader et al., 1991; Chen et al., 2005; Sternson and Roth, 2014) platforms have been invented that have been useful for biologists in general and most especially for neuroscientists.
A number of protein classes (Table 1) have been chemogenetically engineered including kinases (Bishop et al., 1998; Bishop et al., 2000; Chen et al., 2005; Cohen et al., 2005; Dar et al., 2012; Liu et al., 1998), non-kinase enzymes (Collot et al., 2003; Häring and Distefano, 2001; Klein et al., 2005; Strobel, 1998), G protein-coupled receptors (GPCRs) (Alexander et al., 2009; Armbruster and Roth, 2005; Armbruster et al., 2007; Redfern et al., 1999; Redfern et al., 2000; Vardy et al., 2015), and ligand-gated ion channels (Arenkiel et al., 2008; Lerchner et al., 2007; Magnus et al., 2011; Zemelman et al., 2003) (for recent review, see Sternson and Roth, 2014). Of these various classes of chemogenetically engineered proteins, the most widely used to date have been Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) (Armbruster and Roth, 2005; Armbruster et al., 2007), and this Neuron Primer is devoted to them.
Table 1.
Representative Chemogenetic Technologies
| Name | Protein(s) | Ligand | Reference |
|---|---|---|---|
| Representative kinases | |||
| Allele-specific kinase inhibitors | v-I388G | Compound 3g | Liu et al., 1998 |
| Analogue-sensitive kinases | v-Src (I338G, v-Src-as1), c-Fyn (T339G, c-Fyn-as1), c-Abl (T315A, c-Abl-as2), CAMK IIα (F89G, CAMK IIα-as1) and CDK2 (F80G, CDK2-as1) | K252a and PPI analogues | Bishop et al., 1998 |
| Rapamycin-insensitive TOR complex 2 | TORC2 V2227L | BEZ235 | Bishop et al., 2000 |
| ATP-binding pocket mutations in EphB1, EphB2 and EphB3 | Ephb1T697G, Ephb2T699A, and Ephb3T706A | PP1 analogues | Soskis et al., 2012 |
| ATP-binding pocket mutations of TrkA, TrkB and TrkC | TrkAF592A, TrkBF616A, and TrkCF617A | 1NMPP1 and 1NaPP1 | Chen et al., 2005 |
| Representative Enzymes | |||
| Metalloenzymes | Achiral biotinylated rhodium-diphosphine complexes | Collot et al., 2003 | |
| Engineered transaminases | Chemically conjugating a pyridoxamine moiety within the large cavity of intestinal fatty acid binding protein | Enhanced activity | Häring and Distefano, 2001 |
| Representative GPCRs | |||
| Allele-specific GPCRs | β2-adrenergic receptor, D113S | 1-(3′,4′-dihydroxyphenyl)-3-methyl-L-butanone (L-185,870) | Strader et al., 1991 |
| RASSL-Gi (receptors activated solely by synthetic ligands) | κ-opioid chimeric receptor | Spiradoline | Coward et al., 1998 |
| Engineered GPCRs | 5-HT2A serotonin receptor F340→L340 | Ketanserin analogues | Westkaemper et al., 1999 |
| Gi-DREADD | M2- and M4 mutant muscarinic receptors | Clozapine-N-Oxide | Armbruster and Roth, 2005; Armbruster et al., 2007 |
| Gq-DREADD | M1, M3, and M5- mutant muscarinic receptors | Clozapine-N-oxide | Armbruster and Roth, 2005; Armbruster et al., 2007 |
| Gs-DREADD | Chimeric M3-frog Adrenergic receptor | Clozapine-N-oxide | Guettier et al., 2009 |
| Arrestin-DREADD | M3Dq R165L | Clozapine-N-oxide | Nakajima and Wess, 2012 |
| Axonally-targeted silencing | hM4D-neurexin variant | Clozapine-N-oxide | Stachniak et al., 2014 |
| KORD | κ-opioid receptor D138N mutant | Salvinorin B | Vardy et al., 2015 |
| Representative Channels | |||
| GluCl | Insect Glutmate chloride channel; Y182F mutation | Ivermectin | Lerchner et al., 2007 |
| TrpV1 | TrpV1 in TrpV1 KO mice | capsaicin | Arenkiel et al., 2008 |
| PSAM | Chimeric channels PSAMQ79G,L141S | PSEM9S | Magnus et al., 2011 |
| PSEM | PSAM-GlyR fusions | PSEM89S; PSSEM22S | Magnus et al., 2011 |
How an Understanding of GPCR Molecular Pharmacology Facilitates the Appropriate Use of DREADD Technology
Before discussing DREADDs in detail, I will first summarize essential foundational concepts of GPCR molecular pharmacology and signaling. This background information is essential for all readers so that they may understand how DREADDs may be most effectively used. According to classical models of GPCR action GPCRs exist in multiple ligand-dependent and -independent states. These multiple GPCR states range from “fully inactive” to “partially active” to “fully active” to “signaling complexes” (Roth and Marshall, 2012; Samama et al., 1993). As depicted in Figure 1, GPCRs (R) are modulated by ligands (L) and can interact with both hetereotrimeric G proteins (G) and β-arrestins (βArr). According to the most recent findings, multiple inactive (e.g., “ground”) states exist that can be stabilized by ligands (R1L, R2L, and so on) or can even occur in the absence of ligands (R). Sodium ions stabilize the ground state by exerting a negative allosteric modulation via a highly conserved allosteric site (Fenalti et al., 2014; Katritch et al., 2014). Drugs that stabilize the R1L, R2L ground states function as inverse agonists (Samama et al., 1993, 1994). Inverse agonists are also known as “antagonists with negative intrinsic activity” (Costa and Herz, 1989). The evidence for multiple GPCR states is supported by classical molecular pharmacological (Samama et al., 1993, 1994), biophysical (Gether et al., 1995), and structural studies (Manglik et al., 2015).
Figure 1. A Modified and Extended Ternary Complex Model of GPCR Action.
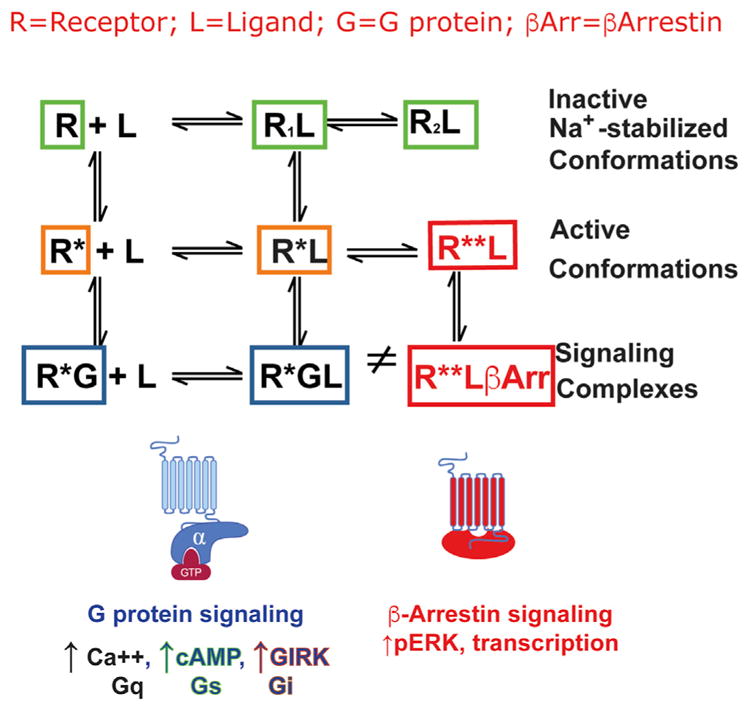
As shown in the top panel GPCRs (R) may interact with ligands (L), hetereotrimeric G proteins (G), and arrestins (βArr) and thereby form a variety of inactive (green boxes), active (orange and red boxes), and signaling complexes (blue and red boxes). The bottom panel shows a cartoon of the various signaling complexes for canonical G protein signaling (L) and β-Arrestin signaling (R).
Both full and partial agonists stabilize the active state (R*L) and promote the formation of a signaling complex (e.g., the “ternary complex”) consisting of (1) the active receptor, (2) an agonist, and (3) the heterotrimeric G protein (R*LG) (De Lean et al., 1980; Samama et al., 1993). In addition to the ligand-induced activation and inactivation of GPCRs, GPCRs can also spontaneously isomerize to an active state (R*) in the absence of ligand. Further, this active state can spontaneously interact with G proteins to yield a binary signaling complex in the absence of ligand (R*G) (Samama et al., 1993). This active state in the absence of ligand is termed “constitutive activity.”
GPCRs (R) also interact with arrestins (βArr) to form alternative signaling complexes (R**L and R**L βArr) (Luttrell et al., 1999; Wacker et al., 2013; Kroeze et al., 2015). GPCRs with high levels of basal (e.g., constitutive) activity can spontaneously interact with βArr to form an R**βArr complex in the absence of agonist (Marion et al., 2004; Kroeze et al., 2015). Based on high-resolution crystal structures of GPCR-arrestin complexes, the R*L βArr state appears to sterically occlude G-protein interactions with the receptor thereby abolishing G-protein signaling (Shukla et al., 2014; Kang et al., 2015). Accordingly, the interaction of GPCRs with βArr also represents a “desensitized” or inactive G-protein state of the complex. At the single molecule level, when GPCRs are activated by agonists, they can couple to either G-proteins or arrestins, but not both. At the cellular level conformational ensembles of all of the states identified above exist. Biasing for one particular state is dependent upon both the cellular context and the available ligand (Vardy and Roth, 2013; Wacker et al., 2013).
A clear understanding of the implications of this extended and modified ternary complex model—for which there is now compelling biochemical (Strachan et al., 2014), biophysical (Sounier et al., 2015; Nygaard et al., 2013), pharmacological (Weiss et al., 2013; Fenalti et al., 2014), and structural evidence (Fenalti et al., 2014; Manglik et al., 2015; Rasmussen et al., 2011; Wacker et al., 2013)—is crucial for understanding how GPCR-based chemogenetic technologies can be harnessed in neuroscience. Thus, for instance, a major concern for chemogenetic technologies is the possibility that high levels of expression of an engineered protein might have effects in the absence of chemical activation (Conklin et al., 2008). Indeed, many of the second-generation chemogenetic GPCRs (e.g., receptors activated solely by synthetic ligands [RASSLs]) had high basal levels of activity leading to phenotypes in the absence of chemical actuators (Hsiao et al., 2008; Sweger et al., 2007).
As depicted in Figure 1, a GPCR with constitutive activity would be more likely to exist in the R* state and thus to spontaneously interact with G proteins to yield a signaling complex in the absence of ligand (R*G). As shown in Figure 2A, high levels of expression of a GPCR with constitutive activity leads to signaling in the absence of ligand. Although no studies to date have demonstrated a basal phenotype for any of the known DREADDs, it is important to express DREADDS at the lowest level consistent with experimental design. For hM3Dq (Alexander et al., 2009) and hM4Di (Zhu et al., 2014), life-long and extremely high levels of expression were attained using a genetically encoded tetracycline-sensitive induction system without basal electrophysiological, behavioral, or anatomical abnormalities being observed. More modest life-long expression of the Gs-DREADD (GsD) also was attained without any detectible electrophysiological, behavioral, or anatomical phenotype (Farrell et al., 2013). High levels of virally mediated expression of various DREADDs have yet not been reported to yield any significant basal phenotypes (Urban et al., 2015; Vardy et al., 2015; Denis et al., 2015; Isosaka et al., 2015; Hayashi et al., 2015). Of course, the absence of reports of basal activity does not imply the absence of basal activity. Going forward, if basal activity is observed, it would be prudent to simply lower the level of DREADD expression using (1) a lower titer of virus, (2) a weaker promotor, or (3) modifying post-transcriptional expression (e.g., deleting a woodchuck hepatitis virus [WPRE] element from the 3′ end of the construct). Thus, based on the law of mass action, decreasing [R] decreases the probability of [R]→[R*]→[R*G] (e.g., inactive, active, and signaling state) transitions.
Figure 2. How Receptor Reserve and Constitutive Activity may Modify DREADD Actions In Vitro and In Vivo.

(A) Simulations of receptor activity using a standard four-parameter logistic equation for GPCR activation, and variable receptor expression (DeLean et al., 1978) was used to simulate the effects of over-expression of a DREADD with constitutive activity (red circles); high receptor reserve, minimal constitutive activity (blue circles); high receptor reserve + desensitization (green circles); low expression and no receptor reserve (purple circles); and low expression, no receptor reserve, and desensitization (orange circles).
(B and C) Potential pharmacokinetic parameters of CNO following high (B) and lower (C) doses. The dotted red line indicates the threshold concentration required for activation of the DREADD in situ.
An additional concern with DREADD technologies relates to issues of desensitization and subsequent receptor downregulation. Thus, following repeated dosing with a DREADD chemical actuator, one might observe diminished responses due to receptor desensitization and downregulation. This diminished response might be predicted because it is well known that GPCRs can be desensitized and subsequently internalized and downregulated following agonist-induced activation (DeWire et al., 2007).
As depicted in Figure 2A, the degree of desensitization depends greatly on the extent to which receptors are over-expressed and the subsequent amount of “receptor reserve.” “Receptor reserve” is a pharmacological term that describes the phenomenon whereby a maximum agonist response can be achieved with less than full occupancy of all of the receptors by agonists (Ruffolo, 1982). From a practical perspective, the concept of receptor reserve predicts that when DREADD expression is quite high, lower concentrations of the chemical actuator are needed to achieve a maximal response (Figure 2A). Additionally, when receptors are desensitized or downregulated, there may be no change in the maximum response elicited by the agonist, but there may be a shift in the dose-response curve to the right because of receptor reserve (2A). Thus, when DREADDs are expressed at high levels relative to native GPCRs via viral or transgenic approaches, the cellular and behavioral responses will be less sensitive to repeated dosing than when they are expressed at lower levels. This phenomenon might explain why no significant desensitization was seen when DREADDs were virally or transgenically expressed (Alexander et al., 2009; Krashes et al., 2011)
Another conceptual issue specific to DREADD technology relates to whether the effects observed regarding neuronal output and behavior occur due to canonical or non-canonical GPCR signaling. As shown in Figure 1, agonists may activate multiple downstream effector pathways, and it is likely that actions other than simply enhancing or silencing neural activity can result when DREADDs are activated. Specifically, one might be concerned about conditions in which βArr signaling is activated. To date, there have been no reports suggesting that the actions of the silencing (e.g., Gi-based DREADDs) or activating (e.g., Gq-based DREADDs) DREADDs on neuronal activity and subsequent physiological readouts could be explained by any mechanism other than altered neuronal firing. Pertinent to this issue, many studies have used DREADD and optogenetic technologies on the same neuronal populations. These studies have invariably identified essentially equivalent effects in terms of both the valence and magnitude of the effect on the physiological readout, although the duration is typically longer with DREADDs (Table 2 for representative examples). Indeed, many investigators now use both DREADD and optogenetic technologies to provide independent and converging lines of evidence in terms of both sufficiency and necessity when deconstructing neural circuits.
Table 2.
Examples for Apparent Equivalency of Chemo- and Optogenetic Modulation
| Cell Type | DREADD | Opsin | Electrophysiology | Behavior |
|---|---|---|---|---|
| AgRP Neurons | hM3Dq (Krashes et al., 2011) | ChR2 (Aponte et al., 2011) | Increased firing | Enhanced feeding |
| ETV-1 subfornical area neurons | hM3Dq (Betley et al., 2015) | ChR2 (Betley et al., 2015) | Increased firing | Enhanced drinking |
| Medial entorhinal cortex cells | hM4Di (Miao et al., 2015) | Arch (Miao et al., 2015) | Decreased firing | Remapping place cells |
| PBN CGRP Projection neurons | hM3Dq (Cai et al., 2014) | ChR2 (Cai et al., 2014) | Increased firing | Diminished feeding |
| Orexin neurons | hM4Di (Sasaki et al., 2011) | Halorhodopsin (Tsunematsu et al., 2011) | Decreased firing | Decreased wakefulness |
| Hippocampus | hM4Di (Zhu et al., 2014) | Arch (Sakaguchi et al., 2015) | Decreased firing | Suppression of contextual fear conditioning |
| Raphe serotonergic neurons | hM3Dq (Urban et al., 2015) | ChR2 (Ohmura et al., 2014) | Increased firing | Anxiogenic |
Current DREADDs
As shown in Table 1 (for recent reviews, see Sternson and Roth, 2014;Urban and Roth, 2015; English and Roth, 2015), there now exist many GPCR-based chemogenetic tools. These include first- (“Alelle-specific GPCRs”; Strader et al., 1991), second-(RASSLs and “Engineered GPCRs”; Coward et al., 1998; Westkaemper et al., 1999), and third-generation (DREADDs; Armbruster and Roth, 2005; Armbruster et al., 2007) platforms. Currently, DREADDs are the most widely used chemogenetic tool; the available DREADDs are shown in Figure 3A.
Figure 3. Available DREADDs and Chemical Actuators.

(A) The available DREADDs, their current accepted nomenclature, and the potential downstream neuronal effects of activation.
(B and C) (B) Shows the structures of currently available chemical actuators for CNO-based DREADDs, while (C) shows the structure of the KORD ligand salvinorin B.
Gq-DREADDS, CNO Analogues, and Basal Activity
For enhancing neuronal firing and activating Gq signaling in neuronal and non-neuronal cells, the hM3Dq DREADD is typically used (Alexander et al., 2009; Armbruster et al., 2007). hM3Dq can be activated by clozapine-N-oxide (CNO)—a pharmacologically inert metabolite of the atypical antipsychotic drug clozapine (Armbruster et al., 2007; Roth et al., 1994). When the original DREADDs were invented, three Gq-coupled DREADDs were created, each of which was based on a different human muscarinic receptor: hM1Dq, hM3Dq, and hM5Dq (Armbruster et al., 2007). All three Gq-DREADDs are activated by low nM concentrations of CNO and mobilize intracellular calcium (Armbruster et al., 2007). All three Gq-DREADDs could be used as excitatory DREADDs, although hM3Dq appears to be most frequently used.
CNO (Figure 3B) represents the prototypical chemical actuator for Gq-DREADDs. Based on many reports, CNO appears to be pharmacologically and behaviorally inert in mice (Alexander et al., 2009; Krashes et al., 2011; Farrell et al., 2013; Guettier et al., 2009; Urban et al., 2015; Zhu et al., 2014) and rats (Ferguson et al., 2011, 2013) when administered at the recommended doses (generally 0.1–3 mg/kg). CNO may be metabolized via back-transformation to clozapine—especially in guinea pigs, humans (Jann et al., 1994), and nonhuman primates (unpublished obsrevations). Although the amount of back-metabolism to clozapine is low even in humans (10% or less by mass; Jann et al., 1994), care should be taken to ensure that clozapine-like side-effects (e.g., hypotension, sedation, and anticholinergic syndrome) do not occur by keeping the dose as small as possible and by always performing appropriate controls (e.g., administering CNO to animals expressing GFP or similarly irrelevant protein).
CNO has excellent drug-like properties with rapid CNS penetration and distribution in mice (Bender et al., 1994). CNO appears to have at least a 60 min residence in vivo in mice following intraperitoneal administration (Bender et al., 1994). Given the long residence time of CNO, it is not surprising that the in vivo effects of CNO-mediated activation of hM3Dq can be both robust and prolonged (Alexander et al., 2009; Krashes et al., 2013). Clearly, unless long-term activation of Gq signaling and neuronal firing is needed, it is recommended that the lowest effective dose of CNO be administered so that only peak CNO concentrations activate the DREADD (Figures 2B and 2C). As can be seen in Figure 2B, when a large dose of CNO is administered, the effects of CNO may be greatly prolonged because brain levels of CNO remain higher than the threshold for activation of the DREADD receptor. By contrast, lower systemic doses of CNO (Figure 2C) would result in transient peak activation and then a relatively rapid decay of activity.
Because of the potential for back-metabolism of CNO to clozapine and other clozapine metabolites in non-rodent species—including the pharmacologically diverse compound N-desmethyl-clozapine (NDMC) (Davies et al., 2005)—we have developed new non-CNO chemical actuators (Chen et al., 2015). The first of these—Compound 21 (Figure 3B)—has minimal off-target activity and exquisite selectivity for activating hM3Dq versus muscarinic and other GPCRs (Chen et al., 2015). Preliminary studies indicate that Compound 21 has equivalent potency in studies in vivo when compared with CNO (unpublished data). Compound 21 likely cannot be metabolized via normal routes to clozapine or any related compound and thus represents an alternative to CNO for studies in which back metabolism of CNO to clozapine is problematic.
An additional compound especially suited for translational studies is perlapine (Figure 3B), a drug that is approved for use in Japan for insomnia. Perlapine has >10,000-fold selectivity for activating hM3Dq versus muscarinic receptors with an EC50 at hM3Dq of 2.8 nM (Chen et al., 2015). Given perlapine’s modest affinity for some biogenic amine receptors (e.g., 5-HT2A, 5-HT6, 5-HT7, and D4) (Davies et al., 2005; Roth et al., 1992, 1994, 1995), it is essential to test perlapine at the lowest possible dose in animals not expressing DREADDs before embarking on studies involving DREADDs. These preliminary studies would ensure that off-target actions of perlapine do not interfere with the phenomena studied. Perlapine will likely find its greatest utility in translational studies of DREADDs in primates and, perhaps, in humans given that it is approved for use in humans. It is likely that further investigation of the scaffolds for compound 21 and perlapine will yield even more effective, potent, and selective chemical actuators for muscarinic-based DREADDs.
The first report that CNO-induced activation of hM3Dq depolarized and excited genetically defined neurons appeared in 2009 (Alexander et al 2009). Since then, hM3Dq has been widely used to enhance neuronal firing, and I here cite only representative examples in which feeding (Krashes et al., 2011; Atasoy et al., 2012), energy expenditure (Kong et al., 2012), locomotion and striatal synaptogenesis (Kozorovitskiy et al., 2012), memory (Garner et al., 2012), and social behaviors (Peñagarikano et al., 2015) have been modulated by hM3Dq in vivo. Because hM3Dq activation induces intracellular calcium release, it has also been used to “activate” astrocytes (Agulhon et al., 2013; Bull et al., 2014; Scofield et al., 2015), hepatocytes (Li et al., 2013), pancreatic βcells (Guettier et al., 2009; Jain et al., 2013), vascular smooth muscle cells (Armbruster et al., 2007), and iPS-derived neuroblasts (Dell’Anno et al., 2014).
Multiple options are currently available for expressing hM3Dq in genetically specified cells. Thus, genetically engineered mice are now available for expressing hM3Dq under control of tetracycline (tet-off) promotor (Alexander et al., 2009; Garner et al., 2012) and via Cre-mediated recombination (Teissier et al., 2015) (Table 3); and some of these are available from Jackson Labs (Table 3). Many labs are using the FLEX switch (Schnütgen et al., 2003) as adapted by the Sternson lab for AAV (Atasoy et al., 2008) that allows for Cre-mediated cell-type-specific expression in any cell type for which there is a Cre-driver line available (Figure 4A). A key innovation for the development of AAV- and lentiviral-based FLEX switch vectors (also known as double-floxed inverse open reading frame [DIO]) (Gradinaru et al., 2010) was the use of separate antiparallel loxP-type recombination sites (especially loxP and lox2272) that allow for homotypic but not heterotypic recombination (Lee and Saito, 1998). In addition, a growing number of promotors have been characterized that allow for cell-type-specific expression using many viral vectors, including modified herpes simplex viruses (HSVs) (Ferguson et al., 2010), AAV (Zhu et al., 2014; Scofield et al., 2015), and lentivirus (Mahler et al., 2014; Vazey and Aston-Jones, 2014). Finally, the use of canine adenovirus (CAV) expressing Cre-recombinase (CAV-Cre) allows for the projection-specific expression of DREADDs. Projection-specific expression of DREADD is possible because CAV-Cre is preferentially retrogradely transported to neuronal somas. In the neuronal cell bodies, recombination of AAV-FLEX-DREADD constructs can occur to allow expression of DREADDs in a projection-specific fashion (Boender et al., 2014) (Figure 4B). The use of CAV-Cre and FLEX-DREADD constructs has been dubbed the “Retro-DREADD” technique (Marchant et al., 2016) and in theory could be used for intersectional and multiplexed applications.
Table 3.
Mice Engineered to Express DREADDs
| Mouse Name Genotype | Expression Type | First Publication | Availability |
|---|---|---|---|
| TRE-hM3Dq Tg(tetO-CHRM3*)1Blr/J | Conditional Tet-off/Tet-on driven expression of hM3Dq | Alexander et al., 2009 | JAX Mice; https://www.jax.org/strain/014093 |
| TRE-hM4Di B6.Cg-Tg(tetO-CHRM4*)2Blr/J | Conditional Tet-off/Tet-on driven expression of hM4Di | Zhu et al., 2014 | JAX Mice; https://www.jax.org/strain/024114 |
| Adora2A-rM3Ds B6.Cg-Tg(Adora2a-Chrm3*, -mCherry)AD6Blr/j | Selective expression in Adora2a-expressing D2 neurons in striatum | Farrell et al., 2013 | JAX Mice; https://www.jax.org/strain/017863 |
| FLOXED-Gi-DREADD mice B6N.129-Gt(ROSA) 26Sortm1(CAG-CHRM4*,-mCitrine)Ute/J | Conditional Cre-mediated expression of hM4Di | Unpublished resource | JAX Mice; https://www.jax.org/strain/026219 |
| FLOXED-Gq-DREADD mice Gt(ROSA) 26Sortm2(CAG-CHRM3*,-mCitrine)Ute/J | Conditional Cre-mediated expression of hM3Dq | Unpublished resource | JAX Mice; https://www.jax.org/strain/026220 |
| FLOXED-Gs-DREADD mice B6N;129-Gt(ROSA) 26Sortm3(CAG-Chrm3*,-mCitrine)Ute/J | Conditional Cre-mediated expression of GsDREADD | Unpublished resource | JAX Mice; https://www.jax.org/strain/026261 |
| FLOXED/FLP conditional Gi-DREADD mice; RC::FPDi; RC::PDi; RC::FDi | Conditional and intersectional Cre- and FLX-mediated expression of Gi-DREADD | Ray et al., 2011; Brust et al., 2014 | Dymecki Lab; http://genepath.med.harvard.edu/~dymecki/requests.html |
| β-cell-specific GqDRADD; β-R-q | Pancreatic β –cell-specific M3Gq DREADD | Guettier et al., 2009 | Jurgen Wess lab; jurgenw@helx.nih.gov |
| β-cell-specific GsDREADD; β-R-S | Pancreatic β –cell-specific Gs DREADD | Guettier et al., 2009 | Jurgen Wess lab; jurgenw@helx.nih.gov |
Figure 4. Potential Approaches for Cell- and Projection-Specific Modulation of Neuronal Activity Using DREADDs.

(A) The standard approach whereby DREADDs are expressed in a cell-type-specific manner (either virally or transgenically) and then activated by systemic administration of chemical actuator.
(B) How a combination of cell-type-specific expression (e.g., localized injection of AAV-FLEX-hSyn-DREADD) and projection-specific infusion of CAV-Cre allows for the projection-specific expression and activation of DREADDs.
(C) How local infusion of a chemical actuator provides for projection-specific effects with a limited area of activation.
(D) How distinct DREADDs may be expressed in a cell-type-specific fashion to afford multiplexed chemogenetic modulation of neural activity and physiology.
Gi-DREADDs
To date there are three Gi-DREADDs: hM2Di, hM4Di, and KORD. Of these, hM2Di and hM4Di can be activated by CNO (Armbruster et al., 2007), compound 21, and perlapine (B.L. Roth, unpublished data). Currently, hM4Di is the most commonly used inhibitory DREADD (for review, see Urban and Roth, 2015). Many labs have reported successful neuronal silencing with hM4Di, and only representative reports are cited wherein DREADDs silenced neurons (Armbruster et al., 2007), modulated amphetamine sensitization (Ferguson et al., 2011) and synaptic plasticity (Kozorovitskiy et al., 2012), regulated breathing (Ray et al., 2011), feeding (Carter et al., 2013), itching (Bourane et al., 2015), and emotional(Teissier et al., 2015) behaviors.
The κ-opioid-derived DREADD (KORD) is a new chemogenetic GPCR that is activated by the pharmacologically inert compound. Thus, salvinorin B has no activity at any other tested molecular target (>350 GPCRs, ion channels, transporters, and enzymes evaluated) and thus has no apparent off-target activity (Figure 3C) (Chavkin et al., 2004; Vardy et al., 2015). Salvinorin B does retain modest affinity for KOR (>100 nM) so that investigators using the KORD should use the lowest dose possible and verify no effects of salvinorin B in the absence of KORD. Several labs have reported successful inhibition of neural activity with KORD (Marchant et al., 2016; Vardy et al., 2015; Denis et al., 2015).
Both hM4Di and KORD appear to inhibit neuronal activity via two mechanisms: (a) induction of hyperpolarization by Gβ/γ-mediated activation of G-protein inwardly rectifying potassium channels (GIRKs) (Armbruster et al., 2007; Vardy et al., 2015) and (b) via inhibition of the presynaptic release of neurotransmitters (e.g., synaptic silencing) (Stachniak et al., 2014; Vardy et al., 2015). Thus, unlike bacterial opsins, which silence neurons via a strong hyperpolarization and with millisecond precision, DREADDs induce a modest hyperpolarization and an apparently strong inhibition of axonal release of neurotransmitter (Stachniak et al., 2014; Vardy et al., 2015) in the s-min-hr time frame. Because of the dependence upon Gβ/γ-mediated activation of GIRKs for inducing hyperpolarization, it is possible that hM4Di and KORD might not hyperpolarize all neurons. To date, there have been no reports that hM4Di or KORD fail to silence or inhibit neuronal activity.
Given that the Gi-coupled DREADDs have effects on terminal release the possibility that CNO (or an analogue) or SalB (or analogue) could micro-infused to locally suppress neural activity has been tested (Figure 4C). Thus, at least two groups (Stachniak et al., 2014; Mahler et al., 2014) have successfully silenced terminal projections via local infusion of CNO. Terminal axons have also been activated by local CNO infusion into rats expression hM3Dq (Vazey and Aston-Jones, 2014). For selective axonal silencing, an hM4Di variant has been created (Figure 3A; hM4Dnrxn) that is targeted preferentially to axons and axon terminals (Stachniak et al., 2014).
The availability of a new inhibitory DREADD—KORD—activated by a ligand orthogonal to CNO now allows for the multiplexed and bidirectional chemogenetic modulation of neural activity and behavior (Vardy et al., 2015). Thus, we recently demonstrated that KORD may be expressed simultaneously with hM3Dq to allow for the sequential chemogenetic activation (with hM3Dq and CNO) and inhibition (with SalB and KORD) (Vardy et al., 2015) of neuronal activity (Figure 4D). It is likely that KORD and hM3Dq could be combined in a combinatorial fashion with various opsins and other chemogenetic tools (e.g., PSEM and PSAMs) to afford highly multiplexed control of neuronal activity with millisecond precision (e.g., with opsins) and for long periods of time for behavioral studies (e.g., with DREADDs or PSAMs).
Gs- and β-Arrestin-DREADDs
The only DREADD coupled to Gs was created by swapping the intracellular regions of the turkey erythrocyte β adrenergic receptor for equivalent regions of a rat M3 DREADD to create a rat Gs-DREADD (Guettier et al., 2009) (Figure 3A). Unlike the current Gq and Gi-DREADDs, the Gs-DREADD (GsD) has a small degree of constitutive activity in transfected cells (Guettier et al., 2009) leading to a modest basal phenotype in pancreatic β cells (Guettier et al., 2009; Jain et al., 2013). Given that Gαolf is the major Gs-like Gα protein in some brain regions (Zhuang et al., 2000), it was critical to determine if GsD can also activate Gαolf. Importantly, we demonstrated that GsD efficiently couples to Gαolf in vitro and in vivo (Farrell et al., 2013) and that GsD had minimal constitutive activity for Gαolf-mediated signaling in vitro and in vivo (Farrell et al., 2013). GsD has been used by several groups to probe the role(s) of Gs-like signaling for a number of behaviors including ethanol consumption (Pleil et al., 2015), reward (Ferguson et al., 2013), locomotor sensitization (Farrell et al., 2013), and circadian rythmns (Brancaccio et al., 2013).
Finally, a DREADD that apparently signals exclusively via β-arrestin has been developed (Nakajima and Wess, 2012) (Figure 3A). This β-arrestin-specific DREADD (Rq(R165L) has not yet been used in vivo but has the potential to illuminate specific behaviors downstream of β-arrestin signaling (for discussion, see Allen and Roth, 2011; Allen et al., 2011).
Areas for Enhancement of DREADD Technologies
Chemogenetic technologies are now widely used neuroscientists with publications appearing at the rate of one to two per day. To date, hM4Di (for silencing) and hM3Dq (for activating) neurons have been the most frequent used DREADDs. DREADDs have been used to modulate neural activity and behavior in flies (Becnel et al., 2013), mice (Alexander et al., 2009), rats (Ferguson et al., 2011), and nonhuman primates (Eldridge et al., 2016). Although DREADD technology has clearly been useful, there are several ways in which the technological platform could be enhanced.
Outlook for New DREADDs and Chemical Actuators
In terms of creating new DREADDs, we have described a generic platform wherein human GPCRs can be expressed in yeast with engineered selectable markers and chimeric G proteins (Armbruster et al., 2007; Dong et al., 2010) and have used this platform to express dozens of human GPCRs (Huang et al., 2015b). In theory it should be possible to create new DREADDs by directed molecular evolution of human GPCRs using the prior yeast-based platforms (Armbruster et al., 2007; Dong et al., 2010; Huang et al 2015c).
An alternative approach is to use structure-guided docking of drug-like and pharmacologically inert small molecules against GPCRs for which there are available structures. This structure-guided approach was used by us to create KORD (Vardy et al., 2015). Currently there are many high-resolution structures of GPCRs including a 1.8 Å structure of the human δ-opioid receptor (Katritch et al., 2014), two serotonin receptors in partially active states (Wang et al., 2013), active and coupled states of the μ-opioid (Huang et al., 2015a), M1-muscarinic (Kruse et al., 2013), β2-adrenergic receptors (Rasmussen et al., 2011), and many other inactive state structures (Katritch et al., 2014). Additionally, my lab and collaborators have used these structures for the successful structure-guided discovery of novel small molecule modulators (Weiss et al., 2013; Carlsson et al., 2011; Shoichet and Kobilka, 2012; Huang et al., 2015c). It is thus possible that new DREADDs could be created using these sorts of approaches.
With regard to new small molecule actuators, it would be useful to identify other CNO- and salvinorin B analogues with (a) good drug-like properties, (b) excellent CNS penetrability, (c) clean off-target pharmacology, and (d) favorable pharmacokinetic and toxicological features (Arrowsmith et al., 2015). Additionally the availability of salvinorin B analogues which are water soluble—as salvinorin B is typically dissolved in dimethylsulfoxide—would also be useful. The development of these sorts of tool compounds could be achieved via conventional medicinal chemistry approaches (Chen et al., 2015) and by new technologies developed by my lab and my collaborators. These new chemical biology technologies allow for the design and validation of novel drug-like molecules using a combination of in silicio and in vitro approaches (Keiser et al., 2009; Besnard et al., 2012; Huang et al., 2015c; Kroeze et al., 2015). Additionally, new chemical biology platforms that allow for the unbiased assessment of on- and off-target pharmacologies of chemical actuators across the GPCR-ome (Kroeze et al., 2015), kinome (Elkins et al., 2015), and other targets (Arrowsmith et al., 2015) are key to validating the specificity of DREADD ligands.
Other areas of enhancement for DREADD technology would include the design of DREADDs with differential neuronal subdomain-specific targeting. Thus, in addition to the axonal targeting DREADDs previously reported (Stachniak et al., 2014), cell body, dendritically targeted, and spine-specifically targeted DREADDs would all be broadly useful. Enhancing the diversity of signaling cascades to include G12/13, Go, Golf, and so on would also be highly valuable to the community. Here, structure-based approaches might be especially valuable. Finally, improving the temporal resolution via photo-caging DREADDs or via creation of DREADD antagonists would also be potentially highly useful—particularly given the large number of laboratories currently using DREADD technology.
Potential Therapeutic Applications for Chemogenetics
Many therapeutic applications of DREADD-based therapeutics have been suggested, including diabetes (Jain et al., 2013), metabolic disorders (Li et al., 2013), Parkinson’s Disease (Dell’Anno et al., 2014), psychostimulant (Ferguson et al., 2011) and ethanol (Pleil et al., 2015) abuse, depression (Urban et al., 2015), post-traumatic stress disorder (Zhu et al., 2014), intractable seizures (Kätzel et al., 2014), inflammatory disorders (Park et al., 2014), autism (Peñagarikano et al., 2015), and many other disorders (English and Roth, 2015). DREADDs have been successfully expressed in nonhuman primates without apparent toxicity, and an exciting new report demonstrates that CNO-DREADDs can modulate circuitry, electrophysiology, and behavior in nonhuman primates (Eldridge et al., 2016). As AAV is a commonly used gene delivery platform in humans, the most likely viral vector for human studies would be AAV. In terms of the chemical actuator, given the fact that perlapine is an approved medication with a long history of safety in humans, it would be the most likely DREADD ligand for activating CNO-based DREADDs in humans. Although CNO has been given to humans without ill-effects, given its propensity for back-metabolism to clozapine and NDMC in humans, it might not be the first choice for translational studies. Salvinorin B has not been administered to humans, although salvinorin A—it’s precursor—has been used in many human studies without any apparent toxicity. Going forward it would be valuable to identify additional drugs that are approved for use in humans to accelerate translation of DREADD technology to humans.
In summary, DREADDs have transformed basic and translational neuroscience research. The availability of multiple DREADDs activated by chemically and pharmacologically distinct actuators will continue to facilitate the multiplexed, chemogenetic interrogation of circuits and cell types involved in behavior, cognition, emotion, memory, and perception.
Acknowledgments
Work involving chemogenetics in my lab is supported by an NIH BRAIN Initiative Award (UO1MH105892), the National Institute of Mental Health Psychoactive Drug Screening Program (271201300017C-1-0-1), and the Michael Hooker Distinguished Professorship in Protein Therapeutics and Translational Proteomics.
References
- Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy KD. Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol. 2013;591:5599–5609. doi: 10.1113/jphysiol.2013.261289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011;51:117–144. doi: 10.1146/annurev-pharmtox-010510-100553. [DOI] [PubMed] [Google Scholar]
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, et al. Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci USA. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14:351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenkiel BR, Klein ME, Davison IG, Katz LC, Ehlers MD. Genetic control of neuronal activity in mice conditionally expressing TRPV1. Nat Methods. 2008;5:299–302. doi: 10.1038/nmeth.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster B, Roth B. Creation of Designer Biogenic Amine Receptors via Directed Molecular Evolution. Neuropsychopharmacology. 2005;30:S265. [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488:172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becnel J, Johnson O, Majeed ZR, Tran V, Yu B, Roth BL, Cooper RL, Kerut EK, Nichols CD. DREADDs in Drosophila: a pharmacogenetic approach for controlling behavior, neuronal signaling, and physiology in the fly. Cell Rep. 2013;4:1049–1059. doi: 10.1016/j.celrep.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender D, Holschbach M, Stöcklin G. Synthesis of n.c.a. carbon-11 labelled clozapine and its major metabolite clozapine-N-oxide and comparison of their biodistribution in mice. Nucl Med Biol. 1994;21:921–925. doi: 10.1016/0969-8051(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Xu S, Cao ZF, Gong R, Magnus CJ, Yu Y, Sternson SM. Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature. 2015;521:180–185. doi: 10.1038/nature14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Shah K, Liu Y, Witucki L, Kung C, Shokat KM. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol. 1998;8:257–266. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Boender AJ, de Jong JW, Boekhoudt L, Luijendijk MC, van der Plasse G, Adan RA. Combined use of the canine adenovirus-2 and DREADD-technology to activate specific neural pathways in vivo. PLoS ONE. 2014;9:e95392. doi: 10.1371/journal.pone.0095392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourane S, Duan B, Koch SC, Dalet A, Britz O, Garcia-Campmany L, Kim E, Cheng L, Ghosh A, Ma Q, Goulding M. Gate control of mechanical itch by a subpopulation of spinal cord interneurons. Science. 2015;350:550–554. doi: 10.1126/science.aac8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M, Maywood ES, Chesham JE, Loudon AS, Hastings MH. A Gq-Ca2+ axis controls circuit-level encoding of circadian time in the suprachiasmatic nucleus. Neuron. 2013;78:714–728. doi: 10.1016/j.neuron.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust RD, Corcoran AE, Richerson GB, Nattie E, Dymecki SM. Functional and developmental identification of a molecular subtype of brain serotonergic neuron specialized to regulate breathing dynamics. Cell Rep. 2014;9:2152–2165. doi: 10.1016/j.celrep.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull C, Freitas KC, Zou S, Poland RS, Syed WA, Urban DJ, Minter SC, Shelton KL, Hauser KF, Negus SS, et al. Rat nucleus accumbens core astrocytes modulate reward and the motivation to self-administer ethanol after abstinence. Neuropsychopharmacology. 2014;39:2835–2845. doi: 10.1038/npp.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Haubensak W, Anthony TE, Anderson DJ. Central amygdala PKC-δ(+) neurons mediate the influence of multiple anorexigenic signals. Nat Neurosci. 2014;17:1240–1248. doi: 10.1038/nn.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Soden ME, Zweifel LS, Palmiter RD. Genetic identification of a neural circuit that suppresses appetite. Nature. 2013;503:111–114. doi: 10.1038/nature12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. Salvinorin A, an active component of the hallucinogenic sage salvia divinorum is a highly efficacious kappa-opioid receptor agonist: structural and functional considerations. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Chen X, Choo H, Huang XP, Yang X, Stone O, Roth BL, Jin J. The first structure-activity relationship studies for designer receptors exclusively activated by designer drugs. ACS Chem Neurosci. 2015;6:476–484. doi: 10.1021/cn500325v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collot J, Gradinaru J, Humbert N, Skander M, Zocchi A, Ward TR. Artificial metalloenzymes for enantioselective catalysis based on biotin-avidin. J Am Chem Soc. 2003;125:9030–9031. doi: 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]
- Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier JM, Chang WC, Pei Y, McCarthy KD, et al. Engineering GPCR signaling pathways with RASSLs. Nat Methods. 2008;5:673–678. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward P, Wada HG, Falk MS, Chan SD, Meng F, Akil H, Conklin BR. Controlling signaling with a specifically designed Gi-coupled receptor. Proc Natl Acad Sci USA. 1998;95:352–357. doi: 10.1073/pnas.95.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MA, Compton-Toth BA, Hufeisen SJ, Meltzer HY, Roth BL. The highly efficacious actions of N-desmethylclozapine at muscarinic receptors are unique and not a common property of either typical or atypical antipsychotic drugs: is M1 agonism a pre-requisite for mimicking clozapine’s actions? Psychopharmacology (Berl) 2005;178:451–460. doi: 10.1007/s00213-004-2017-1. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- Dell’Anno MT, Caiazzo M, Leo D, Dvoretskova E, Medrihan L, Colasante G, Giannelli S, Theka I, Russo G, Mus L, et al. Remote control of induced dopaminergic neurons in parkinsonian rats. J Clin Invest. 2014;124:3215–3229. doi: 10.1172/JCI74664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis RG, Joly-Amado A, Webber E, Langlet F, Schaeffer M, Padilla SL, Cansell C, Dehouck B, Castel J, Delbès AS, et al. Palatability Can Drive Feeding Independent of AgRP Neurons. Cell Metab. 2015;22:646–657. doi: 10.1016/j.cmet.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Dong S, Rogan SC, Roth BL. Directed molecular evolution of DREADDs: a generic approach to creating next-generation RASSLs. Nat Protoc. 2010;5:561–573. doi: 10.1038/nprot.2009.239. [DOI] [PubMed] [Google Scholar]
- Eldridge MAG, Lerchner W, Saunders RC, Kaneko H, Krausz KW, Gonzalez FJ, Ji B, Higuchi M, Minamimoto T, Richmond BJ. Chemogenetic disconnection of monkey orbitofrontal and rhinal cortex reversibly disrupts reward value. Nat Neurosci. 2016;19:37–39. doi: 10.1038/nn.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Fedele V, Szklarz M, Abdul Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP, Roth BL, et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol. 2015;34:95–103. doi: 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- English JG, Roth BL. Chemogenetics-A Transformational and Translational Platform. JAMA Neurol. 2015;72:1361–1366. doi: 10.1001/jamaneurol.2015.1921. [DOI] [PubMed] [Google Scholar]
- Farrell MS, Pei Y, Wan Y, Yadav PN, Daigle TL, Urban DJ, Lee HM, Sciaky N, Simmons A, Nonneman RJ, et al. A Gαs DREADD mouse for selective modulation of cAMP production in striatopallidal neurons. Neuropsychopharmacology. 2013;38:854–862. doi: 10.1038/npp.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, Neumaier JF. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Phillips PE, Roth BL, Wess J, Neumaier JF. Direct-pathway striatal neurons regulate the retention of decision-making strategies. J Neurosci. 2013;33:11668–11676. doi: 10.1523/JNEUROSCI.4783-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forkmann G, Dangelmayr B. Genetic control of chalcone isomerase activity in flowers of Dianthus caryophyllus. Biochem Genet. 1980;18:519–527. doi: 10.1007/BF00484399. [DOI] [PubMed] [Google Scholar]
- Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C, Mayford M. Generation of a synthetic memory trace. Science. 2012;335:1513–1516. doi: 10.1126/science.1214985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gether U, Lin S, Kobilka BK. Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand-specific conformational changes. J Biol Chem. 1995;270:28268–28275. doi: 10.1074/jbc.270.47.28268. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guettier JM, Gautam D, Scarselli M, Ruiz de Azua I, Li JH, Rosemond E, Ma X, Gonzalez FJ, Armbruster BN, Lu H, et al. A chemical-genetic approach to study G protein regulation of beta cell function in vivo. Proc Natl Acad Sci USA. 2009;106:19197–19202. doi: 10.1073/pnas.0906593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häring D, Distefano MD. Enzymes by design: chemogenetic assembly of transamination active sites containing lysine residues for covalent catalysis. Bioconjug Chem. 2001;12:385–390. doi: 10.1021/bc000117c. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Kashiwagi M, Yasuda K, Ando R, Kanuka M, Sakai K, Itohara S. Cells of a common developmental origin regulate REM/non-REM sleep and wakefulness in mice. Science. 2015;350:957–961. doi: 10.1126/science.aad1023. [DOI] [PubMed] [Google Scholar]
- Hsiao EC, Boudignon BM, Chang WC, Bencsik M, Peng J, Nguyen TD, Manalac C, Halloran BP, Conklin BR, Nissenson RA. Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc Natl Acad Sci USA. 2008;105:1209–1214. doi: 10.1073/pnas.0707457105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, et al. Structural insights into μ-opioid receptor activation. Nature. 2015a;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015b;527:477–483. doi: 10.1038/nature15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isosaka T, Matsuo T, Yamaguchi T, Funabiki K, Nakanishi S, Kobayakawa R, Kobayakawa K. Htr2a-Expressing Cells in the Central Amygdala Control the Hierarchy between Innate and Learned Fear. Cell. 2015;163:1153–1164. doi: 10.1016/j.cell.2015.10.047. [DOI] [PubMed] [Google Scholar]
- Jain S, Ruiz de Azua I, Lu H, White MF, Guettier JM, Wess J. Chronic activation of a designer G(q)-coupled receptor improves β cell function. J Clin Invest. 2013;123:1750–1762. doi: 10.1172/JCI66432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jann MW, Lam YW, Chang WH. Rapid formation of clozapine in guinea-pigs and man following clozapine-N-oxide administration. Arch Int Pharmacodyn Ther. 1994;328:243–250. [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kätzel D, Nicholson E, Schorge S, Walker MC, Kullmann DM. Chemical-genetic attenuation of focal neocortical seizures. Nat Commun. 2014;5:3847. doi: 10.1038/ncomms4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G, Humbert N, Gradinaru J, Ivanova A, Gilardoni F, Rusbandi UE, Ward TR. Tailoring the active site of chemzymes by using a chemogenetic-optimization procedure: towards substrate-specific artificial hydrogenases based on the biotin-avidin technology. Angew Chem Int Ed Engl. 2005;44:7764–7767. doi: 10.1002/anie.200502000. [DOI] [PubMed] [Google Scholar]
- Kong D, Tong Q, Ye C, Koda S, Fuller PM, Krashes MJ, Vong L, Ray RS, Olson DP, Lowell BB. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell. 2012;151:645–657. doi: 10.1016/j.cell.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Shah BP, Koda S, Lowell BB. Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab. 2013;18:588–595. doi: 10.1016/j.cmet.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguère PM, Sciaky N, Roth BL. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol. 2015;22:362–369. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Saito I. Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene. 1998;216:55–65. doi: 10.1016/s0378-1119(98)00325-4. [DOI] [PubMed] [Google Scholar]
- Lerchner W, Xiao C, Nashmi R, Slimko EM, van Trigt L, Lester HA, Anderson DJ. Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl− channel. Neuron. 2007;54:35–49. doi: 10.1016/j.neuron.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Li JH, Jain S, McMillin SM, Cui Y, Gautam D, Sakamoto W, Lu H, Jou W, McGuinness OP, Gavrilova O, Wess J. A novel experimental strategy to assess the metabolic effects of selective activation of a G(q)-coupled receptor in hepatocytes in vivo. Endocrinology. 2013;154:3539–3551. doi: 10.1210/en.2012-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shah K, Yang F, Witucki L, Shokat KM. Engineering Src family protein kinases with unnatural nucleotide specificity. Chem Biol. 1998;5:91–101. doi: 10.1016/s1074-5521(98)90143-0. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Magnus CJ, Lee PH, Atasoy D, Su HH, Looger LL, Sternson SM. Chemical and genetic engineering of selective ion channel-ligand interactions. Science. 2011;333:1292–1296. doi: 10.1126/science.1206606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, Wilson SP, Deisseroth K, Woodward JJ, Aston-Jones G. Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat Neurosci. 2014;17:577–585. doi: 10.1038/nn.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell. 2015;161:1101–1111. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NJ, Whitaker LR, Bossert JM, Harvey BK, Hope BT, Kaganovsky K, Adhikary S, Prisinzano TE, Vardy E, Roth BL, Shaham Y. Behavioral and Physiological Effects of a Novel Kappa-Opioid Receptor-Based DREADD in Rats. Neuropsychopharmacology. 2016;41:402–409. doi: 10.1038/npp.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S, Weiner DM, Caron MG. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. J Biol Chem. 2004;279:2945–2954. doi: 10.1074/jbc.M308742200. [DOI] [PubMed] [Google Scholar]
- Miao C, Cao Q, Ito HT, Yamahachi H, Witter MP, Moser MB, Moser EI. Hippocampal Remapping after Partial Inactivation of the Medial Entorhinal Cortex. Neuron. 2015;88:590–603. doi: 10.1016/j.neuron.2015.09.051. [DOI] [PubMed] [Google Scholar]
- Nakajima KI, Wess J. Design and Functional Characterization of a Novel, Arrestin-Biased Designer G Protein-Coupled Receptor. Mol Pharmacol. 2012;82:575–582. doi: 10.1124/mol.112.080358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmura Y, Tanaka KF, Tsunematsu T, Yamanaka A, Yoshioka M. Optogenetic activation of serotonergic neurons enhances anxiety-like behaviour in mice. Int J Neuropsychopharmacol. 2014;17:1777–1783. doi: 10.1017/S1461145714000637. [DOI] [PubMed] [Google Scholar]
- Park JS, Rhau B, Hermann A, McNally KA, Zhou C, Gong D, Weiner OD, Conklin BR, Onuffer J, Lim WA. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. Proc Natl Acad Sci USA. 2014;111:5896–5901. doi: 10.1073/pnas.1402087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, Lázaro MT, Lu XH, Gordon A, Dong H, Lam HA, Peles E, Maidment NT, Murphy NP, Yang XW, et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med. 2015;7:271ra8. doi: 10.1126/scitranslmed.3010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, Olson DP, Lowell BB, Grant KA, Thiele TE, Kash TL. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat Neurosci. 2015;18:545–552. doi: 10.1038/nn.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RS, Corcoran AE, Brust RD, Kim JC, Richerson GB, Nattie E, Dymecki SM. Impaired respiratory and body temperature control upon acute serotonergic neuron inhibition. Science. 2011;333:637–642. doi: 10.1126/science.1205295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern CH, Coward P, Degtyarev MY, Lee EK, Kwa AT, Hennighausen L, Bujard H, Fishman GI, Conklin BR. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat Biotechnol. 1999;17:165–169. doi: 10.1038/6165. [DOI] [PubMed] [Google Scholar]
- Redfern CH, Degtyarev MY, Kwa AT, Salomonis N, Cotte N, Nanevicz T, Fidelman N, Desai K, Vranizan K, Lee EK, et al. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci USA. 2000;97:4826–4831. doi: 10.1073/pnas.97.9.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Marshall FH. NOBEL 2012 Chemistry: Studies of a ubiquitous receptor family. Nature. 2012;492:57. doi: 10.1038/492057a. [DOI] [PubMed] [Google Scholar]
- Roth BL, Ciaranello RD, Meltzer HY. Binding of typical and atypical antipsychotic agents to transiently expressed 5-HT1C receptors. J Pharmacol Exp Ther. 1992;260:1361–1365. [PubMed] [Google Scholar]
- Roth BL, Craigo SC, Choudhary MS, Uluer A, Monsma FJ, Jr, Shen Y, Meltzer HY, Sibley DR. Binding of typical and atypical anti-psychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J Pharmacol Exp Ther. 1994;268:1403–1410. [PubMed] [Google Scholar]
- Roth BL, Tandra S, Burgess LH, Sibley DR, Meltzer HY. D4 dopamine receptor binding affinity does not distinguish between typical and atypical antipsychotic drugs. Psychopharmacology (Berl) 1995;120:365–368. doi: 10.1007/BF02311185. [DOI] [PubMed] [Google Scholar]
- Ruffolo RR., Jr Review important concepts of receptor theory. J Auton Pharmacol. 1982;2:277–295. doi: 10.1111/j.1474-8673.1982.tb00520.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Kim K, Yu LM, Hashikawa Y, Sekine Y, Okumura Y, Kawano M, Hayashi M, Kumar D, Boyden ES, et al. Inhibiting the Activity of CA1 Hippocampal Neurons Prevents the Recall of Contextual Fear Memory in Inducible ArchT Transgenic Mice. PLoS ONE. 2015;10:e0130163. doi: 10.1371/journal.pone.0130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Samama P, Pei G, Costa T, Cotecchia S, Lefkowitz RJ. Negative antagonists promote an inactive conformation of the beta 2-adrenergic receptor. Mol Pharmacol. 1994;45:390–394. [PubMed] [Google Scholar]
- Sasaki K, Suzuki M, Mieda M, Tsujino N, Roth B, Sakurai T. Pharmacogenetic modulation of orexin neurons alters sleep/wakefulness states in mice. PLoS ONE. 2011;6:e20360. doi: 10.1371/journal.pone.0020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnütgen F, Doerflinger N, Calléja C, Wendling O, Chambon P, Ghyselinck NB. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat Biotechnol. 2003;21:562–565. doi: 10.1038/nbt811. [DOI] [PubMed] [Google Scholar]
- Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG, Kalivas PW. Gq-DREADD Selectively Initiates Glial Glutamate Release and Inhibits Cue-induced Cocaine Seeking. Biol Psychiatry. 2015;78:441–451. doi: 10.1016/j.biopsych.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskis MJ, Ho HY, Bloodgood BL, Robichaux MA, Malik AN, Ataman B, Rubin AA, Zieg J, Zhang C, Shokat KM, et al. A chemical genetic approach reveals distinct EphB signaling mechanisms during brain development. Nat Neurosci. 2012;15:1645–1654. doi: 10.1038/nn.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Déméné H, Granier S. Propagation of conformational changes during μ-opioid receptor activation. Nature. 2015;524:375–378. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachniak TJ, Ghosh A, Sternson SM. Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus→midbrain pathway for feeding behavior. Neuron. 2014;82:797–808. doi: 10.1016/j.neuron.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternson SM, Roth BL. Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci. 2014;37:387–407. doi: 10.1146/annurev-neuro-071013-014048. [DOI] [PubMed] [Google Scholar]
- Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, Zhu X, Kleist A, Costa T, Lefkowitz RJ. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR) J Biol Chem. 2014;289:14211–14224. doi: 10.1074/jbc.M114.548131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strader CD, Gaffney T, Sugg EE, Candelore MR, Keys R, Patchett AA, Dixon RA. Allele-specific activation of genetically engineered receptors. J Biol Chem. 1991;266:5–8. [PubMed] [Google Scholar]
- Strobel SA. Ribozyme chemogenetics. Biopolymers. 1998;48:65–81. doi: 10.1002/(SICI)1097-0282(1998)48:1<65::AID-BIP7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Sweger EJ, Casper KB, Scearce-Levie K, Conklin BR, McCarthy KD. Development of hydrocephalus in mice expressing the G(i)-coupled GPCR Ro1 RASSL receptor in astrocytes. J Neurosci. 2007;27:2309–2317. doi: 10.1523/JNEUROSCI.4565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier A, Chemiakine A, Inbar B, Bagchi S, Ray RS, Palmiter RD, Dymecki SM, Moore H, Ansorge MS. Activity of raphe serotonergic neurons controls emotional behaviors. Cell Rep. 2015;13:1965–1976. doi: 10.1016/j.celrep.2015.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu T, Kilduff TS, Boyden ES, Takahashi S, Tominaga M, Yamanaka A. Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J Neurosci. 2011;31:10529–10539. doi: 10.1523/JNEUROSCI.0784-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol. 2015;55:399–417. doi: 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- Urban DJ, Zhu H, Marcinkiewcz CA, Michaelides M, Oshibuchi H, Rhea D, Aryal DK, Farrell MS, Lowery-Gionta E, Olsen RH, et al. Elucidation of The Behavioral Program and Neuronal Network Encoded by Dorsal Raphe Serotonergic Neurons. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.293. http://dx.doi.org/10.1038/npp.2015.293. [DOI] [PMC free article] [PubMed]
- Vardy E, Roth BL. Conformational ensembles in GPCR activation. Cell. 2013;152:385–386. doi: 10.1016/j.cell.2013.01.025. [DOI] [PubMed] [Google Scholar]
- Vardy E, Robinson JE, Li C, Olsen RH, DiBerto JF, Giguere PM, Sassano FM, Huang XP, Zhu H, Urban DJ, et al. A New DREADD Facilitates the Multiplexed Chemogenetic Interrogation of Behavior. Neuron. 2015;86:936–946. doi: 10.1016/j.neuron.2015.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazey EM, Aston-Jones G. Designer receptor manipulations reveal a role of the locus coeruleus noradrenergic system in isoflurane general anesthesia. Proc Natl Acad Sci USA. 2014;111:3859–3864. doi: 10.1073/pnas.1310025111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DR, Ahn S, Sassano MF, Kleist A, Zhu X, Strachan R, Roth BL, Lefkowitz RJ, Shoichet BK. Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem Biol. 2013;8:1018–1026. doi: 10.1021/cb400103f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westkaemper R, Glennon R, Hyde E, Choudhary M, Khan N, Roth B. Engineering in a region of bulk tolerance into the 5-HT2A receptor. Eur J Med Chem. 1999;34:441–447. [Google Scholar]
- Zemelman BV, Nesnas N, Lee GA, Miesenbock G. Photochemical gating of heterologous ion channels: remote control over genetically designated populations of neurons. Proc Natl Acad Sci USA. 2003;100:1352–1357. doi: 10.1073/pnas.242738899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Pleil KE, Urban DJ, Moy SS, Kash TL, Roth BL. Chemogenetic inactivation of ventral hippocampal glutamatergic neurons disrupts consolidation of contextual fear memory. Neuropsychopharmacology. 2014;39:1880–1892. doi: 10.1038/npp.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Belluscio L, Hen R. G(olf)alpha mediates dopamine D1 receptor signaling. J Neurosci. 2000;20:RC91. doi: 10.1523/JNEUROSCI.20-16-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]