

Abstract
Quantitative prediction of herb–drug interaction risk remains challenging. A quantitative framework to assess a potential interaction was used to evaluate a mechanism not previously tested in humans. The semipurified milk thistle product, silibinin, was selected as an exemplar herbal product inhibitor of raloxifene intestinal glucuronidation. Physiologically based pharmacokinetic (PBPK) model simulations of the silibinin–raloxifene interaction predicted up to 30% increases in raloxifene area under the curve (AUC0‐inf) and maximal concentration (Cmax). Model‐informed clinical evaluation of the silibinin–raloxifene interaction indicated minimal clinical interaction liability, with observed geometric mean raloxifene AUC0‐inf and Cmax ratios lying within the predefined no effect range (0.75–1.33). Further refinement of PBPK modeling and simulation approaches will enhance confidence in predictions and facilitate generalizability to additional herb–drug combinations. This quantitative framework can be used to develop guidances to evaluate potential herb–drug interactions prospectively, providing evidenced‐based information about the risk or safety of these interactions.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Herbal products may perpetrate untoward interactions with conventional drugs via numerous mechanisms. One potential mechanism, inhibition of intestinal glucuronidation, has been reported in preclinical models, but the clinical relevance of these observations is unexplored. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ This study addressed the application of a PBPK modeling approach to predict an herb–drug interaction mediated via inhibition of intestinal glucuronidation, an uncharted mechanism in humans. The approach was evaluated via a proof‐of‐concept clinical study using silibinin as an exemplar herbal product perpetrator and raloxifene as an intestinal UGT probe victim. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ A mechanistic PBPK interaction model accurately predicted the minimal impact of the silibinin–raloxifene interaction. This study demonstrates the utility of PBPK modeling and simulation to predict herb–drug interactions mediated via alternate mechanisms. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ These approaches can be used to establish paradigms for the prospective evaluation of herb–drug interaction potential that will provide evidence‐based information about the risk or safety of herb–drug combinations.
Herbal product usage rates continue to climb, fueled in part by the common misperception that “natural” is synonymous with “safe.” Consumers often turn to these products as a means to alleviate self‐diagnosed illnesses or supplement prescribed therapeutic regimens. As a result, herbal products are frequently taken by patients in conjunction with prescribed and over‐the‐counter medications, triggering potential unwanted herb–drug interactions.
Drug interaction liability assessment of herbal products is inherently more complicated than for conventional drugs due to their complex composition and relatively scant knowledge of individual constituents that perpetrate these interactions. A robust approach that identifies causative constituents, characterizes the pharmacokinetics of those constituents, and describes herb–drug interactions mechanistically is nonexistent. Consequently, in vitro–in vivo disconnects are more the norm than the exception when translating herb–drug interaction predictions to the clinic.
Physiologically based pharmacokinetic (PBPK) modeling and simulation has been proposed as a means to improve the quantitative prediction of herb–drug interactions.1, 2 This framework has been applied to interactions mediated via inhibition of cytochrome P450 enzymes,2 but investigation of alternate mechanisms that underlie herb–drug interactions remain relatively unexplored. Inhibition of intestinal UDP‐glucuronosyl transferases (UGTs) represents one such potential mechanism3, 4 based on in vitro and animal studies.3, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 Taken together, a PBPK modeling and simulation approach2 was expanded by applying to a an herb–drug interaction mediated via inhibition of intestinal glucuronidation, a mechanism not previously evaluated in humans.
Milk thistle (Silybum marianum (L.) Gaertn (Asteraceae)) is a popular herbal product widely used as a hepatoprotectant or cancer chemopreventive agent.16, 17 Silibinin, a semipurified milk thistle seed extract consisting of roughly a 1:1 mixture of the flavonolignans silybin A and silybin B,18 was selected as an exemplar perpetrator of herb–drug interactions due to a well‐characterized composition, sufficient quantities of individual constituents available to recover in vitro kinetic parameters, and available human pharmacokinetic data to assess model predictions.16, 17 Raloxifene, a blockbuster selective estrogen receptor modulator (SERM), was selected as a clinically relevant exemplar intestinal UGT substrate to elucidate the potential for altered drug disposition in humans via inhibition of intestinal glucuronidation. Raloxifene is extensively metabolized by intestinal UGTs, particularly UGTs 1A8 and 1A10.19, 20, 21, 22, 23 Silibinin potently inhibited these UGTs in vitro (Ki or IC50 <50 µM),13, 14, 24, 25 prompting a PBPK modeling and simulation approach to predict the interaction liability of the silibinin–raloxifene interaction prior to clinical evaluation. Two modeling platforms were used (Berkeley Madonna and SimCYP) and evaluated through a proof‐of‐concept clinical study in healthy volunteers. The results substantiated that a PBPK model‐based approach is useful to inform clinical study design and provide mechanistic insight into potential herb–drug interactions.
METHODS
PBPK model development
PBPK models were developed for raloxifene, silybin A, and silybin B using the general purpose differential equation solver, Berkeley Madonna (v. 8.3; University of California at Berkeley, Berkeley, CA), and the population‐based simulator, SimCYP (v. 13.2; SimCYP, Sheffield, UK). Two modeling and simulation platforms were assessed because standard approaches for the prediction of UGT‐mediated interactions have not been developed. The Berkeley Madonna base model structure was adapted from the literature,2 incorporating raloxifene and silibinin kinetic (Km, Vmax), (supplemental figure 2); and reversible inhibition (Ki) parameters obtained from human microsomal systems25 (Table 1) scaled to whole‐organ clearance using literature scaling factors.20, 26, 27, 28 The SimCYP model was adapted from the literature,26 incorporating kinetic and reversible inhibition parameters obtained from HEK293 cell lysates overexpressing individual UGT isoforms25 (Table 1 ) scaled to whole‐organ clearance using procedures built into the SimCYP platform for scaling of recombinant UGT systems. The SimCYP “userUGT” option was used to create duplicate UGTs 1A1, 1A8, and 1A10 needed to describe raloxifene conversion to the 4′‐glucuronide (R4G) and 6‐glucuronide (R6G) and incorporate Kis specific to each pathway (Table 1). Raloxifene partition coefficients (Kps) were predicted from physicochemical properties using SimCYP “method 2”29 and used to populate both models. The Berkeley Madonna base model structure was identical to that described in a previous report,2 and SimCYP model structures have been described previously.30 Simulated pharmacokinetic primary endpoints, raloxifene maximum concentration (Cmax) and area under the curve from time zero to infinity (AUC0‐inf), within 30% of the observed endpoints were the predefined criteria when assessing prediction accuracy.
Table 1.
Physiologically based pharmacokinetic model input parameters
| Victim compound | Perpetrator compounds | ||
|---|---|---|---|
| Parameter | Raloxifene[Link] | Silybin A[Link] | Silybin B[Link] |
| Physicochemical/Binding | |||
| Molecular weight | 510.05 | 482.44 | 482.44 |
| Fraction absorbed | 0.63 | 0.77 | 0.77 |
| ka (h−1) | 0.56 | 0.50 | 0.50 |
| Blood/plasma ratio | 1.07 | 0.58 | 0.58 |
| Unbound fraction in plasma | 0.05 | 0.04 | 0.04 |
| Metabolism | |||
| Intestinal Km (µM) | 0.81; 0.84 | 55 | 39 |
| Intestinal Vmax (pmol/min/mg) | 750; 78 | 7,600 | 94,000 |
| Hepatic Km (µM) | 3.0; 6.5 | 33 | 81 |
| Hepatic Vmax (pmol/min/mg) | 854; 310 | 20,000 | 1,40,000 |
| UGT1A1 Km (µM) | 4.3; 3.6 | – | – |
| UGT1A1 Vmax (pmol/min/mg) | 88; 92 | – | – |
| UGT1A8 Km (µM) | 5.5; 1.5 | – | – |
| UGT1A8 Vmax (pmol/min/mg) | 550; 110 | – | – |
| UGT1A10 Km (µM) | 0.87; 0.96 | – | – |
| UGT1A10 Vmax (pmol/min/mg) | 1,100; 130 | – | – |
| Inhibition | |||
| HIM Ki (µM) | – | 59; 56 | 66; 31 |
| UGT1A1 Ki (µM) | – | 5.0; 4.2 | 3.3; 2.8 |
| UGT1A8 Ki (µM) | – | 40; 67 | 47; 19 |
| UGT1A10 Ki (µM) | – | 79; 72 | 74; 65 |
See “Methods” for detailed information on model parameterization.
aParameters with two estimates correspond to the formation (Km, Vmax) or inhibition (Ki) of the raloxifene‐4′‐glucuronide (first value) or raloxifene‐6‐glucuronide (second value) pathways and were obtained from ref. 25.
ka, absorption rate constant; Ki, reversible inhibition constant.
Silibinin–raloxifene interaction model simulations
The PBPK model for perpetrator (silybin A and silybin B) and victim (raloxifene) were linked through the reversible inhibition of raloxifene intestinal glucuronidation (supplemental figure 2). Initial simulations used doses of raloxifene and silibinin products reported in previous studies. Observed concentration–time profiles were extracted from the literature using GetData Graph Digitizer (v. 2.26). Simulations were evaluated by visual inspection of predicted and observed concentration–time profiles and comparison of predicted to observed primary pharmacokinetic endpoints (AUC0‐inf, Cmax) for raloxifene (Figure 1 , Table 1). Following initial model evaluation, simulations were conducted with silibinin (480 mg Siliphos p.o. three times daily × 4 days) to predict the magnitude of interaction with raloxifene (60 mg p.o.). Pharmacokinetic outcomes (Cmax, AUC0‐inf, time to Cmax (tmax), terminal half‐life (t1/2)) from the Berkeley Madonna‐simulated concentration–time profiles were recovered via noncompartmental analysis using Phoenix WinNonlin (v. 6.3; Pharsight, Cary, NC). SimCYP default model output included the pharmacokinetic outcomes of interest. The SimCYP model considered a virtual cohort of 16 healthy volunteers (eight men, eight women), aged 18 to 65 years, with raloxifene (60 mg) and silibinin (480 mg three times daily) administered in the fasted state to mimic the proof‐of‐concept clinical study.
Figure 1.
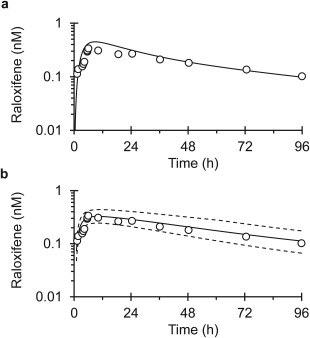
Mean concentration–time profile of raloxifene following a single 60 mg oral dose. Solid lines denote physiologically based pharmacokinetic model simulations using Berkeley Madonna (a) or SimCYP (b). Dashed lines denote upper and lower 90% confidence intervals (b). Open symbols denote observed means and were obtained from ref. 36.
Analysis of silibinin product
Siliphos capsules (n = 10) (Thorne Research, Dover, ID) were analyzed using previously described methods2 to ensure purity and content. In brief, the contents of each capsule were weighed and extracted twice with 2 mL acetone. The extract was vortex‐mixed and centrifuged (13,000g × 2 minutes). The supernatant was transferred to a clean vial for analysis of milk thistle flavonolignans using an Acquity UPLC system with an HSS‐T3 (1.8 µm, 2.1 × 100 mm) column (Waters, Milford, MA). Flavonolignan standards31 and Siliphos capsule extracts were analyzed using Empower 3 software with a gradient from 30:70 to 55:45 methanol:water (0.1% formic acid) over 6.0 minutes at a flow rate of 0.5 mL/min at 50°C; peaks were detected at 288 nm.
Proof‐of‐concept clinical study
Healthy volunteers (eight men, eight nonpregnant women) were enrolled in an open‐label, single‐dose, randomized two‐period crossover study conducted at the Washington State University (WSU) Clinical Research Unit (CRU) (Supplemental Figure 1). The WSU Institutional Review Board reviewed and approved the study protocol and consent form prior to subject enrollment. Eligibility to participate was based on screening evaluation and inclusion/exclusion criteria (Supplemental Table 1). Potential subjects provided written informed consent and Health Insurance Portability and Accountability Act authorization before undergoing screening, which consisted of the following: medical history, physical examination, liver function tests, complete blood count, and urinalysis. All women underwent a serum pregnancy test. Eligible participants were randomized to enter the control or treatment phase using a blocked design to ensure equal numbers of men and women in each sequence. The randomization schedule was created using SAS PROC PLAN (v. 9.2; SAS Institute, Cary, NC).
The control phase involved administration of 60 mg raloxifene (Teva Pharmaceuticals, Sellersville, PA) (Supplemental Figure 1). Vital signs (blood pressure, pulse, oxygen saturation) were obtained on every study day. Blood (7 mL) was collected in BD Vacutainer tubes containing EDTA (Becton, Dickinson, Franklin Lakes, NJ) through an intravenous line before and from 0.25–12 hours following drug administration. Subjects continued to fast until after the 4‐hour blood collection, when meals and snacks, devoid of fruit juices and caffeine‐containing products, were provided. Subjects returned to the CRU 24, 48, 72, and 96 hours postdrug administration for blood collection via venipuncture. Plasma (∼3 mL) was harvested by centrifugation and collected in cryogenic vials (Corning, Tewksbury, MA) (∼1 mL each) for storage at −80°C pending analysis by UHPLC‐MS/MS.
At least a seven‐day washout period separated each phase (Supplemental Figure 1). The treatment phase involved administration of 60 mg raloxifene followed immediately by 480 mg Siliphos (based on labeled content) three times daily for the entire collection interval. Each subject received his/her Siliphos in a blister pack and was asked to complete a pill diary documenting the time of administration, beverage, and approximate volume used to aid swallowing, and adverse events, if applicable. Subjects were instructed how to use the blister pack properly, and compliance was assessed at each study visit. Plasma collection times and procedures mirrored those outlined for the control phase.
Analysis of plasma for raloxifene, raloxifene glucuronides, silybin A, and silybin B
Plasma (100 µL) was treated with methanol (4 volumes with 0.1% formic acid) containing the internal standards raloxifene‐d4, raloxifene‐d4−6‐glucuronide (Rd46G), raloxifene‐d4−4′‐glucuronide (Rd44G) (Toronto Research Chemicals, Toronto, Canada), and naringin (Sigma Aldrich, St Louis, MO) and centrifuged (2,200g × 20 minutes). Chromatographic separation was achieved using an HSS T3 column (1.8 µM, 2.1 × 150 mm) with a Critical Clean precolumn filter (2.1 × 0.2 µM) (Waters) heated to 45°C and an isocratic flow rate of 0.35 mL/min (50:50 water:methanol, each with 0.1% formic acid). R6G, R4G, naringin, raloxifene, silybin A, and silybin B retention times were 1.3, 1.8, 2.0, 2.7, 4.3, and 4.9 minutes, respectively; total run time was 6.6 minutes. Samples were analyzed (3 µL injection volume) using the QTRAP 6500 UHPLC‐MS/MS system (AB Sciex, Framingham, MA) with a turbo electrospray source operated in positive (R6G, R4G, raloxifene) and negative (silybin A, silybin B, naringin) ion mode. R6G (650.0→474.0 m/z), Rd46G (654.5→478.3 m/z), R4G (650.0→474.0 m/z), Rd44G (654.5→478.3 m/z), raloxifene (474.3→112.0 m/z), raloxifene‐d4 (478.3→116.3 m/z), naringin (579.0→271.0 m/z), silybin A (481.10→125.1 m/z), and silybin B (481.10→125.1 m/z) were monitored in multiple reaction monitoring mode. Analyte concentrations were quantified using MultiQuant software (v. 2.1.1, AB Sciex) by interpolation from matrix‐matched calibration curves and quality controls with dynamic assay ranges of 0.01–5 nM (raloxifene), 0.1–200 nM (R6G), 0.3–600 nM (R4G), or 2.4–2,500 nM (silybin A and silybin B). The calibration standards and quality controls were judged for batch quality based on the 2013 US Food and Drug Administration (FDA) guidance for industry regarding bioanalytical method validation.32
Pharmacokinetic analysis
Pharmacokinetic outcomes were recovered by noncompartmental analysis using Phoenix WinNonlin. Concentrations below the limit of quantification for each analyte were excluded. The terminal elimination rate constant (λz) was estimated by linear regression using at least three data points of the terminal portion of the log‐transformed concentration–time profile. t1/2 was calculated as ln(2)/λz. Cmax, tmax, and last measured concentration (Clast) were recovered directly from the concentration–time profile. Area under the curve from time zero to Clast (AUC0‐last) was determined using the trapezoidal method with linear up/log down interpolation. AUC0‐inf was calculated as the sum of AUC0‐last and Clast/λz.
Statistical analysis
All statistical analyses were conducted using SAS (v. 9.2; SAS Institute). The sample size for the proof‐of‐concept study (n = 16 evaluable subjects) was calculated based on 80% power to detect a 25% change in the primary endpoints with a Type I error of 0.05; the primary endpoint was the test/reference ratio of log‐transformed raloxifene AUC0‐inf and Cmax, and the predefined no effect range was 0.75–1.33.33, 34 Intraindividual variability in raloxifene AUC0‐inf and Cmax were assumed to be ∼30%.35 Secondary outcomes were evaluated using a paired two‐tailed Student's t‐test on log‐transformed data (treatment vs. control). P < 0.05 was considered statistically significant.
RESULTS
Modeling and simulation
PBPK model development
Simulated raloxifene concentration–time profiles closely approximated previously published model predictions,26 observed profiles,36, 37 and reported pharmacokinetic outcomes.35 Model‐predicted primary endpoints, raloxifene AUC0‐inf and Cmax, were within the prespecified criterion (30%) for satisfactory model performance (Table 2).
Table 2.
Comparison of previously published and model‐predicted raloxifene pharmacokinetic outcomes
| Outcome | Previously published[Link] | Berkeley Madonna model‐predicted[Link] | SimCYP model‐predicted |
|---|---|---|---|
| Raloxifene (60 mg) | |||
| t½ (h) | 32 (39) | 55 | 58.5 (17) |
| tmax (h) (median (range)) | 6.0 (4.0–12) | 9.7 | 9.8 (12) |
| Cmax (nM) | 0.37 (34) | 0.45 | 0.33 (38) |
| AUC0‐inf (nM *h) | 21 (33) | 20.7 | 18.6 (40) |
aGeometric or arithmetic means and coefficients of variation (%) unless indicated otherwise from refs. 35 and 36.
bPoint estimates.
t1/2, terminal half‐life; tmax, time to maximal concentration; Cmax, maximal concentration; AUC0‐inf, area under the concentration‐time curve from time zero to infinity.
Prediction of the silibinin–raloxifene interaction
Simulations conducted using Berkeley Madonna predicted an ∼30% increase in raloxifene AUC0‐inf and Cmax with minimal change (3%) in t1/2. The SimCYP model predicted negligible changes in raloxifene pharmacokinetic outcomes (≤5%) (Table 3). Both models predicted similar overall exposure to silybin A and silybin B (Table 3 , Figure 4). The SimCYP model predicted rapid silibinin elimination (t1/2 <3 hours). The Berkeley Madonna model predicted longer half‐lives for silybin A and silybin B, consistent with previous clinical study outcomes when silibinin was administered at the same dose for 7 days.2 Maximum silibinin (silybin A + silybin B) intestinal tissue concentrations predicted by the SimCYP model (∼26 µM) were lower than those predicted by the Berkeley Madonna Model (∼61 µM). Both estimates were within reported intestinal tissue concentrations (20–310 µM).38
Table 3.
Comparison of clinical study outcomes to physiologically based pharmacokinetic model predictions
| Observed | Predicted (Berkeley Madonna) | Predicted (SimCYP) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Geometric mean (CV%) | Geometric mean | Geometric mean | ||||||||||
| Outcome | Control | Treatment | Treatment/control ratio (90% CI) | P‐value[Link] | Control | Treatment | Treatment/control ratio | Control | Treatment | Treatment/control ratio (90% CI) | ||
| Raloxifene | ||||||||||||
| t1/2 (h) | 32.6 (32) | 30.2 (47) | 0.93 (0.90–1.3) | – | 55.5 | 57.0 | 1.03 | 58.5 (17) | 56.0 (19) | 0.96 (0.94–0.99) | ||
| Cmax (nM) | 0.40 (58) | 0.40 (77) | 0.99 (0.81–1.2) | 0.96 | 0.45 | 0.58 | 1.29 | 0.33 (38) | 0.34 (38) | 1.04 (1.02–1.07) | ||
| AUC0‐inf (nM[Link]h) | 16.3 (63) | 17.9 (75) | 1.09 (0.92–1.3) | 0.35 | 20.7 | 27.9 | 1.31 | 18.6 (40) | 19.6 (41) | 1.05 (1.03–1.08) | ||
| R4G | ||||||||||||
| t1/2 (h) | 15.4 (48) | 16.8 (75) | 1.09 (0.65–1.3) | – | – | – | – | – | – | – | ||
| Cmax (nM) | 106 (61) | 114 (76) | 1.08 (0.75–1.5) | 0.63 | – | – | – | – | – | – | ||
| AUC0‐inf (nM[Link]h) | 1380 (68) | 1290 (66) | 0.93 (0.65–1.3) | 0.58 | – | – | – | – | – | – | ||
| R6G | ||||||||||||
| t1/2 (h) | 15.6 (75) | 15.8 (77) | 1.01 (0.70–1.4) | – | – | – | – | – | – | – | ||
| Cmax (nM) | 15.8 (69) | 14.5 (58) | 0.92 (0.77–1.1) | 0.45 | – | – | – | – | – | – | ||
| AUC0‐inf (nM[Link]h) | 324 (91) | 317 (80) | 0.97 (0.78–1.2) | 0.87 | – | – | – | – | – | – | ||
| Silybin A | ||||||||||||
| Cmax (nM) | – | 319 (118) | – | – | – | 243 | – | – | 252 (53) | – | ||
| t1/2 (h) | – | 1.52 (50) | – | – | – | 6.3 | – | – | 2.8 (48) | – | ||
| Silybin B | ||||||||||||
| Cmax (nM) | – | 210 (136) | – | – | – | 231 | – | – | 218 (40) | – | ||
| t1/2 (h) | – | 1.23 (50) | – | – | – | 5.3 | – | – | 1.7 (48) | – | ||
R4G, raloxifene‐4′‐glucuronide; R6G, raloxifene‐6‐glucuronide; t1/2, terminal elimination half‐life; Cmax, maximal concentration; AUC0‐inf, area under the concentration‐time curve from time zero to infinity.
*Paired two‐tailed Student's t‐test on log‐transformed data (control vs. treatment).
Figure 4.
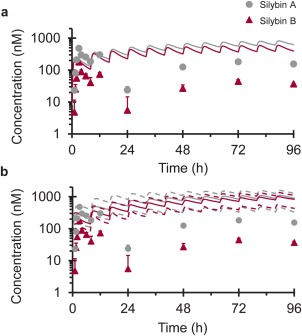
Geometric mean concentration–time profiles of silybin A (gray circles) and silybin B (crimson triangles) following oral administration of silibinin (480 mg three times daily × 4 days). Solid lines denote PBPK model simulations of mean concentration–time profiles using Berkeley Madonna (a) or SimCYP (b). Dashed lines denote upper and lower 90% confidence intervals (b). Symbols and error bars denote observed geometric means and upper limits of the 90% confidence interval, respectively.
Proof‐of‐concept clinical assessment
Verification of silibinin content in test herbal product
A single lot (#313823) of Siliphos capsules, labeled to contain 60 mg silibinin per capsule, was selected as the test herbal product. The capsules were overfilled consistently, containing 73.4 ± 2.47 mg silibinin comprised of 31.3 ± 0.84 mg silybin A and 42.1 ± 1.97 mg silybin B. The capsules also contained small amounts of the regioisomers isosilybin A (1.21 ± 0.03 mg) and isosilybin B (1.01 ± 0.06 mg).
Study participants
Prior to the first study day, two participants (one man, one woman) withdrew consent due to unforeseen scheduling conflicts. Two additional participants were screened, enrolled, and assigned to the randomization schedule of the participants they replaced. All participants who entered the study completed both phases (Supplemental Table 2). Raloxifene and Siliphos were well tolerated. One subject experienced nausea in response to venous catheter placement that was self‐limiting and not study drug‐related (occurred prior to administration). The event did not limit the subject's continued participation. No other adverse events were reported.
Effects of silibinin on raloxifene pharmacokinetics
The effects of silibinin (480 mg three times daily × 4 days) were compared to baseline oral pharmacokinetics of raloxifene (control phase). Silibinin did not alter raloxifene Cmax nor tmax, with a geometric mean Cmax of 0.40 n M occurring at a median of 6 hours in both phases (Figure 2 a, Table 3). Geometric mean AUC0‐inf of raloxifene was increased by 9% (Figure 2 a; Table 3), with that for five subjects outside the predefined no effect range (0.75–1.33) (Figure 3 b). One subject demonstrated a 2‐fold increase in raloxifene AUC0‐inf and a 3‐fold increase in Cmax (Figure 3 a,b). The 90% confidence intervals for the raloxifene primary endpoints (Cmax and AUC0‐inf) lay within the predefined no effect range (Table 3).
Figure 2.
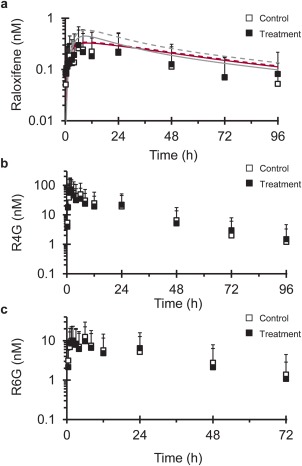
Geometric mean concentration–time profile of (a) raloxifene, (b) raloxifene‐4′‐glucuronide (R4G), and (c) raloxifene‐6‐glucuronide (R6G) following a single 60 mg oral dose of raloxifene given alone (open symbols) or after silibinin (480 mg three times daily × 4 days) treatment (solid symbols). Lines in (a) denote PBPK model simulations of raloxifene concentration–time profiles when given alone (solid line) or with silibinin (dashed line) using Berkeley Madonna (gray) or SimCYP (crimson). R6G concentrations are not reported beyond 72 hours due to concentrations being below the lower limit of quantitation (c). Symbols and error bars denote observed geometric means and upper limits of the 90% confidence interval, respectively.
Figure 3.
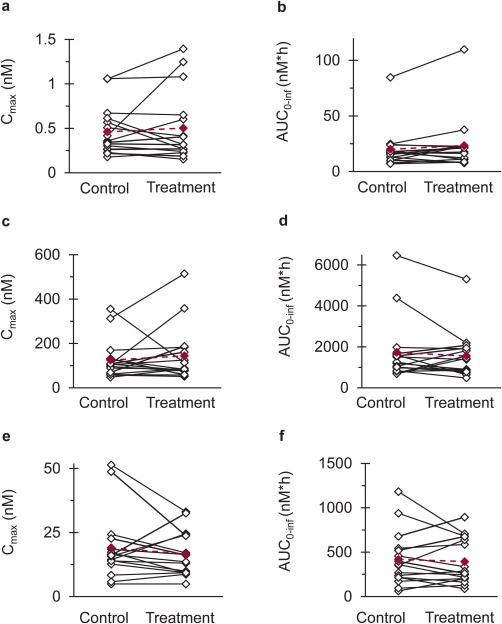
Effects of silibinin (480 mg three times daily for 4 days) on (a,c,e) Cmax and (b,d,f) AUC0‐inf of (a,b) raloxifene, (c,d) raloxifene‐4′‐glucuronide, and (e,f) raloxifene‐6‐glucuronide in 16 healthy volunteers following oral administration of raloxifene (60 mg). Open symbols connected by solid lines (black) denote individual values. Solid symbols connected by dashed lines (crimson) denote geometric means.
Pharmacokinetic outcomes for the raloxifene glucuronides were minimally altered by silibinin (<10%). Relative to control, geometric mean R4G Cmax increased by 8%, whereas R6G Cmax decreased by the same extent (Figure 3 c,e; Table 3). Geometric mean AUC0‐inf of R4G and R6G decreased by 7 and 3%, respectively (Figure 3 d,f; Table 3). Three subjects demonstrated >2‐fold reductions in the AUC0‐inf of R4G, the primary glucuronide of raloxifene (Figure 3 d). Secondary peaks observed in the concentration–time profiles of raloxifene and raloxifene glucuronides are consistent with enterohepatic recirculation (Figure 2).35, 36
The sampling strategy was not intended to capture silybin A and silybin B pharmacokinetics; as such, these outcomes are presented for qualitative purposes. Silybin A and silybin B were rapidly eliminated (geometric mean t1/2 of ∼90 minutes) following oral administration (Figure 4 ; Table 3). Silybin A Cmax exceeded that of silybin B by ∼50%. Pill diaries and detection of silybin A and silybin B in treatment phase samples indicated compliance with the herbal product regimen.
DISCUSSION
Despite considerable investigation, quantitative prediction of herb–drug interaction liability remains elusive. Regulatory guidelines in Western countries typically require premarket evaluation of drug–drug interactions, but these guidelines generally do not apply to herb–drug interactions. Approaches developed for the prediction of drug–drug interactions are often inadequate to overcome the additional hurdles unique to the evaluation of herb–drug interactions.1, 39, 40, 41 The complex composition of herbal products renders traditional static models of drug–drug interaction potential generally less useful for the prediction of herb–drug interactions. Highly variable and limited pharmacokinetic data available for herbal constituents, combined with a lack of herbal product standardization, continues to challenge the development of predictive herb–drug interaction models. Consequently, approaches to assess herb–drug interaction liability vary widely, yielding conflicting results, inconclusive translation to the clinical setting, and uncertain generalizability. The relative paucity of reliable pharmacokinetic data to describe herbal product constituents further complicates efforts to extrapolate preclinical data to predict clinical consequences.
PBPK modeling and simulation approaches have been suggested as a method to predict the consequences of drug–drug and herb–drug interactions mediated via modulation of CYP‐mediated metabolism.2, 33, 34 SimCYP, a population‐based PBPK simulation platform, is designed explicitly to predict the absorption, distribution, metabolism, and excretion (ADME) of drug molecules and their interplay with xenobiotics that can modulate one or more of these processes. The intuitive user interface of SimCYP provides a means to populate built‐in model structures and generate simulated pharmacokinetic outcomes that can be output in a standard format. However, the built‐in model structures are limited in that a maximum of two perpetrators can be accommodated. This limitation applies particularly to the prediction of herb–drug interactions, as herbal products are typically mixtures of multiple constituents that can alter ADME processes via multiple mechanisms. In contrast, Berkeley Madonna is a general‐purpose differential equation solver that is limited in terms of model structures and perpetrator count only by the user's ability to adequately describe these processes mathematically. This flexibility, however, is accompanied by a lack of functionality to easily incorporate population and parameter variability and generate standardized outputs. These limitations may require additional software platforms to generate desired outcome measures and evaluate the impact of population and parameter uncertainty. Based on these advantages and disadvantages of SimCYP and Berkeley Madonna, both platforms were used and expanded with unique approaches to evaluate the consequences of an herb–drug interaction mediated via inhibition of intestinal glucuronidation, a mechanism uncharted in humans.
Silibinin was selected as a model herbal product perpetrator based on well‐characterized composition, availability of human pharmacokinetic data, and in vitro inhibitory potency (Kis) towards intestinal UGTs at concentrations below those measured in colorectal tissue specimens obtained from cancer patients administered oral silibinin.24, 38 Raloxifene is a SERM widely used to reduce the risk of developing breast cancer and for the treatment of osteoporosis. Raloxifene was selected as a clinically relevant probe substrate for intestinal UGT activity due to the dominance of intestinal glucuronidation to raloxifene disposition, relatively high affinity for two intestinal UGT isoforms (UGT1A8 and 1A10),14, 19, 23 and excellent safety profile for administration to healthy volunteers.35, 37
Because standard approaches to evaluate herb–drug interactions mediated via inhibition of intestinal glucuronidation in humans have not been described, two different modeling platforms were used to predict the likelihood and magnitude of the silibinin–raloxifene interaction. The Berkeley Madonna model incorporated data recovered from human microsomal systems,25 which contain the full complement of UGTs present in the intestine and liver. This approach illustrates the impact of modulation of overall intestinal raloxifene glucuronidation. The SimCYP model incorporated data from HEK293 cell lysates25 to describe the UGT isoform‐specific inhibition of raloxifene glucuronidation. Both models provided an accurate bottom‐up description of raloxifene pharmacokinetics. These data are consistent with the assertion that in vitro to in vivo extrapolation accuracy for UGT substrates is improved by incorporating parameters to describing drug disposition in multiple organs with metabolic capacity.20, 42 Raloxifene modulation of silibinin disposition via competitive inhibition was not incorporated into model predictions, as quantitative data (Kis) describing these processes have not been reported. Recovery of robust parameters describing inhibition of silibinin metabolism is hindered by a lack of authentic glucuronide standards of silybin A and silybin B. In the absence of such standards, substrate depletion methods must be used that are exceedingly time‐ and resource‐intensive, particularly for Ki determinations.24
The Berkeley Madonna interaction model overpredicted, whereas the SimCYP interaction model accurately predicted, the observed magnitude of interaction. The discrepancy between the two models can be partially explained by the different in vitro parameters used to populate each model. The differing absorption models also may contribute. The Berkeley Madonna model used a relatively simple first‐order absorption model, whereas the SimCYP model used the advanced dissolution, absorption, and metabolism (ADAM) model. The ADAM model incorporates population variability of multiple component processes within a mechanistic framework to describe the passage of drug molecules along and through the gastrointestinal tract.30 The ADAM model divides the intestine into multiple transit compartments, effectively distributing any interactions between the herbal product perpetrator, silibinin, and the victim drug, raloxifene, along the entire length of the gastrointestinal tract. The net interaction potential reflects a summary of multiple processes governing the release, dissolution, permeability, metabolism, and transit of both the perpetrator and victim. In contrast, the Berkeley Madonna model assumes that the interaction occurs within a single, homogenous compartment. This assumption can result in overprediction of inhibitor concentration and residence time at the active site and may explain the overprediction of the interaction magnitude by the Berkeley Madonna model. A mechanistic static model of the silibinin–raloxifene interaction predicted up to a 5‐fold increase in raloxifene systemic exposure in the presence of silibinin.25 However, dynamic modeling and simulation more accurately predicted the observed lack of clinical interaction and provided another example of overprediction by a static model of interaction risk for rapidly cleared reversible inhibitors.2 Collectively, results from the current work provide credence to the incorporation of isoform‐specific UGT metabolism and inhibition data into a mechanistic PBPK model for the prediction of herb–drug interactions mediated via inhibition of intestinal glucuronidation.
In general, the proof‐of‐concept clinical study suggested a low interaction risk of silibinin administered with raloxifene. These data are consistent with evaluation of herbal products as inhibitors of hepatic glucuronidation in healthy volunteers43, 44, 45, 46, 47 and support the assertion that drug interaction risk for UGT substrates is generally low.48 However, multiple subjects demonstrated nearly 2‐fold increases in raloxifene exposure, suggesting that the interaction potential should not be disregarded entirely. Alternate study designs, including silibinin predosing and multiple dosing of raloxifene, could provide further insight into interaction potential. One subject demonstrated high baseline raloxifene AUC0‐inf (control phase), consistent with UGT1A8 slow metabolizer status,23 that increased by 50% in the presence of silibinin (Figure 3 b). Functional polymorphisms in UGT1A8 may be associated with these observations but suggest that impaired basal raloxifene metabolism may not explain the observed differences in interaction magnitude, which is supported further by the lack of correlation between control phase AUC0‐inf and interaction magnitude. Clinical observations will facilitate refinement and enhance the predictive power of the PBPK interaction model.
PBPK modeling is an iterative process whereby the model should be updated with knowledge gained from clinical observations to enhance confidence and better predict unknown clinical situations.49, 50 Future applications of this modeling approach include alternate combinations of herbal product perpetrators and drug victims. Design of drug molecules to avoid oxidative metabolism continues to emphasize the importance of conjugative metabolism in drug development. As a result, these modeling approaches may also be useful in the evaluation of drug–drug interaction potential of new chemical entities cleared predominantly by glucuronidation.
In summary, quantitative prediction of herb–drug interactions remains challenging. This work evaluated the application of a quantitative framework to assess the risk of an herb–drug interaction mediated via a mechanism not previously explored in humans. Model‐informed clinical evaluation of the silibinin–raloxifene interaction indicated minimal clinical interaction liability, albeit some individuals may be more sensitive for as‐yet unknown reasons. Further development and validation of PBPK modeling and simulation approaches will enhance confidence in model predictions and facilitate generalizability to alternate combinations of herbal products and conventional drugs. Ultimately, these approaches can be used to establish paradigms for the prospective evaluation of herb–drug interaction potential that will provide evidence‐based information about the risk or safety of herb–drug combinations.
Conflict of Interest/Disclosure
The authors have no disclosures and declare no personal or financial conflicts of interest.
Author Contributions
B.T.G., J.R.W., J.T.B., N.H.O., and M.F.P. wrote the article. B.T.G. and M.F.P. designed research. B.T.G., J.T.B., V.G.P., J.R.W., and M.E.L. performed research. B.T.G., N.H.O., and M.F.P. analyzed data. N.H.O. contributed new reagents/analytical tools.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors thank Judy Griffin and Yili Zhong (WSU) for their assistance in conducting the clinical study and Tyler Graf (UNCG) for verifying silibinin content in the test herbal product. The Health Sciences and Services Authority of Spokane provided financial support to purchase bioanalytical instrumentation. This work was supported by the National Institutes of Health National Institute of General Medical Sciences (Grant R01 GM077482‐S1). B.T.G. was supported by a fellowship awarded by the American Foundation for Pharmaceutical Education. B.T.G. is currently supported by the National Institute of General Medical Sciences (Grant T32 GM008425). M.F.P. dedicates this article to Dr. David P. Paine.
References
- 1. Brantley, S.J. , Argikar, A.A. , Lin, Y.S. , Nagar, S. & Paine, M.F. Herb–drug interactions: challenges and opportunities for improved predictions. Drug Metab. Dispos. 42, 301–317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brantley, S.J. et al Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb–drug interaction. CPT Pharmacometrics Syst. Pharmacol. 3, e107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mohamed, M.E. & Frye, R.F. Effects of herbal supplements on drug glucuronidation. Review of clinical, animal, and in vitro studies. Planta Med. 77, 311–321 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Ritter, J.K. Intestinal UGTs as potential modifiers of pharmacokinetics and biological responses to drugs and xenobiotics. Expert Opin. Drug Metab. Toxicol. 3, 93–107 (2007). [DOI] [PubMed] [Google Scholar]
- 5. Gurley, B.J. Pharmacokinetic herb–drug interactions (part 1): origins, mechanisms, and the impact of botanical dietary supplements. Planta Med. 78, 1478–1489 (2012). [DOI] [PubMed] [Google Scholar]
- 6. Gurley, B.J. , Fifer, E.K. & Gardner, Z. Pharmacokinetic herb–drug interactions (part 2): drug interactions involving popular botanical dietary supplements and their clinical relevance. Planta Med. 78, 1490–1514 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Kiang, T.K. , Ensom, M.H. & Chang, T.K. UDP‐glucuronosyltransferases and clinical drug‐drug interactions. Pharmacol. Ther. 106, 97–132 (2005). [DOI] [PubMed] [Google Scholar]
- 8. Li, L. , Hu, H. , Xu, S. , Zhou, Q. & Zeng, S. Roles of UDP‐glucuronosyltransferases in phytochemical metabolism of herbal medicines and the associated herb–drug interactions. Curr. Drug Metab. 13, 615–623 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Ma, G. , Wu, B. , Gao, S. , Yang, Z. , Ma, Y. & Hu, M. Mutual regioselective inhibition of human UGT1A1‐mediated glucuronidation of four flavonoids. Mol. Pharm. 10, 2891–2903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohamed, M.F. & Frye, R.F. Inhibition of intestinal and hepatic glucuronidation of mycophenolic acid by Ginkgo biloba extract and flavonoids. Drug Metab. Dispos. 38, 270–275 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Mohamed, M.F. , Tseng, T. & Frye, R.F. Inhibitory effects of commonly used herbal extracts on UGT1A1 enzyme activity. Xenobiotica 40, 663–669 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Moon, Y.J. , Wang, X. & Morris, M.E. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol. In Vitro 20, 187–210 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Sridar, C. , Goosen, T.C. , Kent, U.M. , Williams, J.A. & Hollenberg, P.F. Silybin inactivates cytochromes P450 3A4 and 2C9 and inhibits major hepatic glucuronosyltransferases. Drug Metab. Dispos. 32, 587–594 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Jeong, E.J. , Liu, Y. , Lin, H. & Hu, M. Species‐ and disposition model‐dependent metabolism of raloxifene in gut and liver: role of UGT1A10. Drug Metab. Dispos. 33, 785–794 (2005). [DOI] [PubMed] [Google Scholar]
- 15. Chen, Y. , Jia, X. , Chen, J. , Wang, J. & Hu, M. The pharmacokinetics of raloxifene and its interaction with apigenin in rat. Molecules 15, 8478–8487 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Polyak, S.J. , Oberlies, N.H. , Pecheur, E.I. , Dahari, H. , Ferenci, P. & Pawlotsky, J.M. Silymarin for HCV infection. Antivir. Ther. 18, 141–147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramasamy, K. & Agarwal, R. Multitargeted therapy of cancer by silymarin. Cancer Lett. 269, 352–362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kroll, D.J. , Shaw, H.S. & Oberlies, N.H. Milk thistle nomenclature: why it matters in cancer research and pharmacokinetic studies. Integr. Cancer Ther. 6, 110–119 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Kemp, D.C. , Fan, P.W. & Stevens, J.C. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab. Dispos. 30, 694–700 (2002). [DOI] [PubMed] [Google Scholar]
- 20. Cubitt, H.E. , Houston, J.B. & Galetin, A. Relative importance of intestinal and hepatic glucuronidation‐impact on the prediction of drug clearance. Pharm. Res. 26, 1073–1083 (2009). [DOI] [PubMed] [Google Scholar]
- 21. Dalvie, D. , Kang, P. , Zientek, M. , Xiang, C. , Zhou, S. & Obach, R.S. Effect of intestinal glucuronidation in limiting hepatic exposure and bioactivation of raloxifene in humans and rats. Chem. Res. Toxicol. 21, 2260–2271 (2008). [DOI] [PubMed] [Google Scholar]
- 22. Kosaka, K. et al Impact of intestinal glucuronidation on the pharmacokinetics of raloxifene. Drug Metab. Dispos. 39, 1495–1502 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Sun, D. , Jones, N.R. , Manni, A. & Lazarus, P. Characterization of raloxifene glucuronidation: potential role of UGT1A8 genotype on raloxifene metabolism in vivo. Cancer Prev. Res. (Phila) 6, 719–730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gufford, B.T. , Chen, G. , Lazarus, P. , Graf, T.N. , Oberlies, N.H. & Paine, M.F. Identification of diet‐derived constituents as potent inhibitors of intestinal glucuronidation. Drug Metab. Dispos. 42, 1675–1683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gufford, B.T. , Chen, G. , Vergara, A.G. , Lazarus, P. , Oberlies, N.H. & Paine, M.F. Milk thistle constituents inhibit raloxifene intestinal glucuronidation: a potential clinically relevant natural product‐drug interaction. Drug Metab. Dispos. 43, 1353–1359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zientek, M. & Dalvie, D. Use of a multistaged time‐dependent inhibition assay to assess the impact of intestinal metabolism on drug‐drug interaction potential. Drug Metab. Dispos. 40, 467–473 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Paine, M.F. et al Characterization of interintestinal and intraintestinal variations in human CYP3A‐dependent metabolism. J. Pharmacol. Exp. Ther. 283, 1552–1562 (1997). [PubMed] [Google Scholar]
- 28. Barter, Z.E. et al Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug Metab. 8, 33–45 (2007). [DOI] [PubMed] [Google Scholar]
- 29. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 30. Jamei, M. et al Population‐based mechanistic prediction of oral drug absorption. AAPS J. 11, 225–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Graf, T.N. , Wani, M.C. , Agarwal, R. , Kroll, D.J. & Oberlies, N.H. Gram‐scale purification of flavonolignan diastereoisomers from Silybum marianum (Milk Thistle) extract in support of preclinical in vivo studies for prostate cancer chemoprevention. Planta Med. 73, 1495–1501 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Food and Drug Administration Center for Drug Evaluation and Research. Bioanalytical method validation (draft guidance). (2013).
- 33. European Medicines Agency . Guideline on the Investigation of Drug Interactions (Final). (2012).
- 34.Food and Drug Administration Center for Drug Evaluation and Research. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations (draft guidance). (2012).
- 35.Eli Lilly and Company, Indianapolis, IN. Evista [package insert]. (Approved 1997; Revised 2007).
- 36. Czock, D. , Keller, F. , Heringa, M. & Rasche, F.M. Raloxifene pharmacokinetics in males with normal and impaired renal function. Br. J. Clin. Pharmacol. 59, 479–482 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Food and Drug Administration Center for Drug Evaluation and Research . Application Number 020815: Raloxifene hydrochloride Clinical Pharmacology and Biopharmaceutics Review. (1997).
- 38. Hoh, C. et al Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin. Cancer Res. 12, 2944–2950 (2006). [DOI] [PubMed] [Google Scholar]
- 39. Gufford, B.T. , Lazarus, P. , Oberlies, N.H. & Paine, M.F. Predicting pharmacokinetic herb‐drug interactions: overcoming hurdles that extend beyond drug‐drug interactions. AAPS Newsmag. 19–22 (2014). [Google Scholar]
- 40. Won, C.S. , Oberlies, N.H. & Paine, M.F. Influence of dietary substances on intestinal drug metabolism and transport. Curr. Drug Metab. 11, 778–792 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Won, C.S. , Oberlies, N.H. & Paine, M.F. Mechanisms underlying food‐drug interactions: inhibition of intestinal metabolism and transport. Pharmacol. Ther. 136, 186–201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cubitt, H.E. , Houston, J.B. & Galetin, A. Prediction of human drug clearance by multiple metabolic pathways: integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab. Dispos. 39, 864–873 (2011). [DOI] [PubMed] [Google Scholar]
- 43. Blonk, M. , Colbers, A. , Poirters, A. , Schouwenberg, B. & Burger, D. Effect of ginkgo biloba on the pharmacokinetics of raltegravir in healthy volunteers. Antimicrob. Agents Chemother. 56, 5070–5075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Volak, L.P. et al Effect of a herbal extract containing curcumin and piperine on midazolam, flurbiprofen and paracetamol (acetaminophen) pharmacokinetics in healthy volunteers. Br. J. Clin. Pharmacol. 75, 450–462 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee, L.S. , Wise, S.D. , Chan, C. , Parsons, T.L. , Flexner, C. & Lietman, P.S. Possible differential induction of phase 2 enzyme and antioxidant pathways by American ginseng, Panax quinquefolius. J. Clin. Pharmacol. 48, 599–609 (2008). [DOI] [PubMed] [Google Scholar]
- 46. van Erp, N.P. et al Effect of milk thistle (Silybum marianum) on the pharmacokinetics of irinotecan. Clin. Cancer Res. 11, 7800–7806 (2005). [DOI] [PubMed] [Google Scholar]
- 47. Gwilt, P.R. et al The effect of garlic extract on human metabolism of acetaminophen. Cancer Epidemiol. Biomarkers Prev. 3, 155–160 (1994). [PubMed] [Google Scholar]
- 48. Williams, J.A. et al Drug‐drug interactions for UDP‐glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab. Dispos. 32, 1201–1208 (2004). [DOI] [PubMed] [Google Scholar]
- 49. Zhao, P. , Rowland, M. & Huang, S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 92, 17–20 (2012). [DOI] [PubMed] [Google Scholar]
- 50. Huang, S.M. & Rowland, M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin. Pharmacol. Ther. 91, 542–549 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information