

Abstract
Change in tumor size estimated using longitudinal tumor growth inhibition (TGI) modeling is an early predictive biomarker of clinical outcomes for multiple cancer types. We present the application of TGI modeling for subjects with multiple myeloma (MM). Longitudinal time course changes in M‐protein data from relapsed and/or refractory MM subjects who received single‐agent carfilzomib in phase II studies (n = 456) were fit to a TGI model. The tumor growth rate estimate was similar to that of other anti‐myeloma agents, indicating that the model is robust and treatment‐independent. An overall survival model was subsequently developed, which showed that early change in tumor size (ECTS) at week 4, Eastern Cooperative Oncology Group performance status (ECOG PS), hemoglobin, sex, percent bone marrow cell involvement, and number of prior regimens were significant independent predictors for overall survival (P < 0.001). ECTS based on M‐protein modeling could be an early biomarker for survival in MM following exposure to single‐agent carfilzomib.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ An early sensitive biomarker for predicting important clinical endpoints, such as overall survival in subjects with multiple myeloma, is lacking; its discovery would be beneficial for the clinical development of new agents. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ We evaluated whether tumor growth inhibition modeling based on longitudinal M‐protein data can be used to predict overall survival in subjects with multiple myeloma following exposure to single‐agent carfilzomib. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ This is the first full report to demonstrate the potential for longitudinal M‐protein data in predicting overall survival in subjects with multiple myeloma. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ We demonstrate two key points from our analysis: 1) a model to integrate data across various clinical studies for the purpose of predicting important clinical endpoints can be developed using longitudinal M‐protein data for multiple myeloma, and 2) prior clinical study data can be leveraged to assist in future clinical development; a model‐based approach, such as the work here, should be considered prior to the initiation of clinical studies. Importantly, overall survival is an important clinical endpoint in multiple myeloma clinical research. A robust model to predict overall survival as shown here could encourage the multiple myeloma field to adopt this model‐based approach to impact trial design and increase the success of trial outcome.
Multiple myeloma (MM) is the second most common hematologic malignancy.1 Carfilzomib (Kyprolis, Onyx Pharmaceuticals, South San Francisco, CA), a second‐generation proteasome inhibitor, has been investigated in subjects with MM, other hematologic malignancies, and solid tumors. In 2012, carfilzomib received accelerated approval from the US Food and Drug Administration for the treatment of subjects with relapsed and refractory MM.2
Carfilzomib is a tetrapeptide epoxyketone‐based irreversible proteasome inhibitor. Proteasomes are part of a major mechanism by which cells regulate the concentration of particular proteins and degrade misfolded proteins. Proteins are tagged for degradation with a small protein called ubiquitin. The result is a polyubiquitin chain bound by the proteasome, allowing it to degrade the tagged protein.3 Proteasome inhibition leads to the accumulation of polyubiquitinated protein substrates within cells and induces apoptosis. Carfilzomib is active in bortezomib‐resistant tumor cell lines,4, 5 and, as opposed to bortezomib, is highly specific for inhibiting proteasome activity.6 The improved selectivity of carfilzomib vs. bortezomib may correlate with the reduced levels of myelosuppression and peripheral neuropathy that were observed in animal toxicology and clinical studies.7
Myeloma is a malignancy of the plasma cell which produces immunoglobulins (antibodies). A myeloma protein (M‐protein) is an abnormal immunoglobulin fragment or immunoglobulin light chain produced in excess by an abnormal clonal proliferation of plasma cells, typically in MM. This increase in M‐protein concentration is a marker of tumor burden8 and has several deleterious effects on the body, including impaired immune function, abnormally high viscosity (“thickness”) of the blood, and kidney damage. In subjects with MM, blood serum M‐protein levels are part of the criteria used to assess response according to the International Myeloma Working Group Uniform Response Criteria for MM.8 Response classification is based on categorical criteria, defined by aggregate data, and does not make optimal use of available longitudinal information for predicting ultimate clinical benefits. Thus, alternative approaches that take into account early and longitudinal dynamics of M‐protein (as a marker of tumor burden) in subjects with MM may represent early biomarkers to predict clinical benefit.
In the past few years, efforts have been made to develop longitudinal tumor size (TS) models to assess the value of tumor growth inhibition (TGI) as a biomarker to quantitate drug effect. These models have been used to estimate TGI metrics that could be used as endpoints to inform early clinical decisions. A TGI model that makes use of all of the longitudinal TS data has been successfully applied to predict expected clinical responses and overall survival (OS) rates in cancer patients from a variety of clinical settings.9, 10, 11, 12, 13, 14, 15, 16, 17 The predictive performance of this model‐based approach has been assessed with prospective simulations of clinical studies in colorectal cancer10 and non‐small cell lung cancer.11, 12 Whether this TGI modeling approach can be used to simulate clinical study outcomes in MM needs to be further assessed.
Herein, we show for the first time the application of a previously described TGI model to a surrogate measurement of tumor growth for MM (M‐protein) using data from four phase II studies of single‐agent carfilzomib. We previously reported preliminary work in subjects with MM.18, 19, 20, 21, 22 A TGI model based on longitudinal M‐protein data in subjects with relapsed or refractory MM who received high‐dose (HD) dexamethasone18 was used to estimate M‐protein tumor burden at week 8 in subjects treated with pomalidomide and low‐dose dexamethasone, which was used as a basis for OS and progression‐free survival (PFS) simulation.19 The objective of this study was to assess whether changes in M‐protein over time could be used to predict clinical endpoints such as OS after carfilzomib treatment. A model for predicting long‐term outcomes based on routine short‐term M‐protein measurements may be useful in providing an early estimate of long‐term benefit for MM subjects treated with carfilzomib or with other investigational treatments.
METHODS
Trials and data
Data were obtained from four phase II studies (PX‐171‐003‐A0—Part 1, PX‐171‐003‐A1—Part 2, PX‐171‐004, and PX‐171‐005) of single‐agent carfilzomib in subjects with relapsed or relapsed and refractory MM. All studies were approved by the review boards of participating institutions. All subjects provided written informed consent.
PX‐171‐003 was a single‐arm, phase I/II trial in subjects with relapsed and refractory MM (n = 302). In PX‐171‐003‐A0—Part 1,23 carfilzomib was given as a 2‐minute intravenous (i.v.) infusion at 20 mg/m2, twice weekly, during 3 weeks in a 28‐day cycle. Premedication dexamethasone (4 mg) was given prior to all carfilzomib doses during the first cycle. Study PX‐171‐003—Part 2 (A1) was the pivotal study that was the basis for the accelerated regulatory approval in the US. This study was an open‐label, single‐arm phase II study of single‐agent carfilzomib in subjects with relapsed and refractory MM with ≥2 prior treatments. Subjects received carfilzomib (i.v. over 2 to 10 minutes) 20/27 mg/m2 on days 1, 2, 8, 9, 15, and 16 of 28‐day cycles (20 mg/m2 for all doses in cycle 1 only) for up to 12 cycles.24 Dexamethasone 4 mg was given prior to all carfilzomib doses during the first cycle and the first dose escalation cycle. A total of 274 subjects from PX‐171‐003 was included in the M‐protein TGI modeling.
PX‐171‐004 was an open‐label, single‐arm, phase II study of single‐agent carfilzomib in subjects with relapsed and/or refractory MM with between one and three prior lines of therapy. This study enrolled two cohorts: bortezomib‐treated and bortezomib‐naive subjects. Bortezomib‐treated subjects received carfilzomib (i.v. infusion over 2–10 minutes) at the 20‐mg/m2 dose (with no step up to 27 mg/m2) on days 1, 2, 8, 9, 15, and 16 of 28‐day cycles.25 Bortezomib‐naive subjects received carfilzomib (i.v. over 2–10 minutes) 20 mg/m2 or 20/27 mg/m2 on days 1, 2, 8, 9, 15, and 16 of 28‐day cycles (20 mg/m2 in all doses for cycle 1 only) for up to 12 cycles.26 In both cohorts, premedication dexamethasone 4 mg was given prior to all carfilzomib doses during the first cycle and the first dose escalation cycle.25, 26 A total of 146 subjects from PX‐171‐003 was included in the M‐protein TGI modeling.
PX‐171‐005 was a phase II, open‐label, single‐arm, multicenter study designed to assess the effect of baseline renal impairment on the pharmacokinetics (PK) of carfilzomib in subjects with relapsed or refractory MM after at least two prior treatment regimens.27 Carfilzomib was administered at 15 mg/m2 i.v. on days 1, 2, 8, 9, 15, and 16 of each 28‐day cycle for up to 12 cycles. Dose escalation to 20 mg/m2 at cycle 2 and then to 27 mg/m2 at cycle 3 occurred as appeared to be tolerated. Premedication dexamethasone 4 mg was administered prior to each carfilzomib dose during cycle 1, and could be increased to 40 mg/week in subjects who failed to achieve at least a partial response after cycle 2 or a complete response after cycle 4. Overall, 28 subjects received premedication dexamethasone 40 mg/week at varying timepoints after cycle 2. A total of 36 subjects from PX‐171‐003 was included in the M‐protein TGI modeling.
Per protocol, M‐protein was to be measured at baseline, on day 15 of cycle 1, day 1 of subsequent cycles, and at the end of the study. Pooled data from all studies were used to develop TGI and OS models.
TGI model for M‐protein data
We developed a model that accounts for the dynamics of tumor growth, exposure‐driven antitumor drug effect (Eq. (1)), and development of resistance to drug effect with time (Eq. (2)) based on a previously published TGI model.18 Since PK data were not available from all subjects who had M‐protein data, a virtual biophase modeling approach was used with dose over time as the input function (Eq. (3)).28 The TGI model is described by the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where y(t) is the M‐protein concentration at time t with the value y0 at baseline (i.e., the M‐protein level at first measurement, typically a couple of weeks before start of treatment), KL is the M‐protein growth rate, KD,CFZ is the M‐protein shrinkage rate due to carfilzomib exposure, and decreases with time (from KD,CFZ,0 at time 0) with a rate constant of λCFZ. DCFZ(t) is the amount (dose) of carfilzomib at the site of action with initial value 0. KP,CFZ is the elimination rate constant from the virtual biophase compartment for carfilzomib, and UCFZ(t) is the input function of carfilzomib dose over time.
The TGI model was used to estimate early change in tumor size (ECTS) at the start of week 4 (commencement of the second cycle) relative to the predicted M‐protein concentration immediately before the first dose (y[tfd]), as shown in Eq. (6):
| (6) |
ECTS based on week 4 was chosen because the goal of this work was to assess an early marker to predict OS benefit. In addition, there was minimal dropout at week 4, so the model was able to include data from the majority of subjects. Subject‐level log‐normal distributed random effects were allowed on all of the model parameters to account for intersubject variability. At baseline, M‐protein did not follow a log‐normal distribution (Supplementary Figure S1 online). The observed baselines with added residual error were used according to the “B2” method,29 rather than by estimating the individual baselines. No covariance between the random effects was initially considered (diagonal covariance matrix), and this model assumption was subsequently checked. Residual variability was modeled using an additive plus exponential error model. Model parameters were estimated using nonlinear mixed‐effects modeling program and first‐order conditional estimation with interaction (NONMEM, v. 7.1.0; ICON Development Solutions, Ellicott City, MD).
Potential covariates were explored graphically using individual (post hoc) random effects plotted against covariates. In a second step, covariates were tested one‐by‐one using the stepwise covariate model building feature in Perl speaks NONMEM.30 Forward inclusion/backward exclusion was employed, using an inclusion P value of 0.01 and an exclusion P value of 0.001.
Trends were detected in the exploratory analysis for the following covariates, which were tested on either M‐protein growth rate (KL) or drug potency (expressed as shrinkage rate [KD,CFZ]):
Baseline albumin on KL (<3.5 g/dL taken as the limit of normal)
Creatinine clearance (mL/min) on KD,CFZ
Body weight (kg) on KD,CFZ
ECOG PS (>0) on KL
Percent plasma cell involvement on KL
Number of prior regimens (>1, >2) on KL and KD,CFZ
Number of drugs on KD,CFZ
Bortezomib in last prior regimen (yes/no) on KL
Lenalidomide in last prior regimen (yes/no) on KL
Sex on KD,CFZ
Platelet count (109/L) on KL
The predictive performance of the model was evaluated using a posterior predictive check (PPC), which uses the model and the study design to simulate statistics of interest (ECTS at week 4) of many hypothetical trial replicates (n = 500, number of replicates limited by computation time with this model) across parameter uncertainty (for different replicates), interindividual variability (within replicates), and residual error. Recorded dosing histories and matching observed predose M‐protein levels were sampled as inputs to the model. The simulated distribution of all percentiles were recorded and compared with those observed in the studies. Observed percentiles were compared with the posterior predictive distributions by the model.
Model for OS
A parametric model for OS was developed. The model describes the survival time distribution as a function of covariates. The probability density function that best described the observed survival times was selected among normal, log‐normal, Weibull, logistic, log‐logistic, exponential and extreme, using differences in Akaike Information Criteria and goodness‐of‐fit plots of the alternative models. Model parameter estimation was performed using the software R (v. 2.15.2). The survival model can be considered a drug‐independent model that relates a biomarker response (ECTS) and prognostic factors (covariates) to a clinical endpoint (OS time).
The following baseline covariates were tested as potential prognostic factors in addition to ECTS week 4 to capture treatment effect:
Creatinine clearance (mL/min)
>1 prior regimen (yes/no)
>2 prior regimens (yes/no)
Predicted M‐protein concentration (g/L) at time of first dose, y(tfd) as shown in Eq. (6)
Prior bortezomib treatment (yes/no)
Number of previous treatments
Platelet count (109 cells/L)
Lymphocytes (109 cells/L)
Hemoglobin (g/dL)
Albumin (g/dL)
Sex
Percent cell involvement
ECOG PS >0
Covariate effects were first assessed with a Cox proportional hazard regression model using the coxph function in R v. 2.15.2. In a second step, all candidate covariates were included in a multivariate fashion using the parametric model. All covariates were then subjected to a backward stepwise elimination. At each elimination step, the relative influence of each remaining covariate on the model was reevaluated by removing it from the reduced model on an individual basis using a cutoff at P ≤ 0.01. Study effect was evaluated in the final model after adjustment for covariate effects. The obtained model was the “final” model.
The survival model was evaluated using a PPC. Survival times for the same number of subjects as in the pooled dataset were simulated 1,000 times. Parameter values for the survival model were sampled from the estimated mean values and variance‐covariance matrix (uncertainty in parameter estimates). Observed survival distributions were compared with the posterior predictive distributions by the model (95% prediction interval [PI]).
RESULTS
Longitudinal TGI model for M‐protein
The characteristics of the carfilzomib clinical trials from which the data came are summarized in Tables 1 and 2. The median number of prior regimens were 5, 1, and 4 in studies PX‐171‐003, PX‐171‐004, and PX‐171‐005, respectively.23, 24, 25, 26, 27
Table 1.
Carfilzomib phase II clinical trial characteristics
| Study | MM status | Prior therapy | Carfilzomib dosinga | N (total) | N (used in model development) | N (observations) | Reference |
|---|---|---|---|---|---|---|---|
| PX‐171‐003‐A0 | Relapsed and refractory | ≥2 regimens; responded to first‐line and refractory to most recent | 20 mg/m2 | 43 | 39 | 152 | 21 |
| PX‐171‐003‐A1b | Relapsed and refractory | ≥2 regimens; responded to ≥1 and refractory to most recent | 20/27c mg/m2 | 259 | 235 | 1393 | 22 |
| PX‐171‐004 | Relapsed and/or refractory | Responded to first‐line; relapsed or refractory to ≥1 but ≤3 regimens | 20 or 20/27c mg/m2 | 162 | 146 | 1088 | 23,24 |
| PX‐171‐005 | Relapsed and/or refractory with various levels of renal insufficiency | ≥2 regimens; achieved ≥MR to ≥1 | 15, 20, 27d mg/m2 | 49 | 36 | 102 | 25 |
| Total | 513 | 456 | 2735 |
MM, multiple myeloma; MR, minimal response; N, number of subjects or observations.
Days 1, 2, 8, 9, 15, and 16 of a 28‐day cycle.
Study PX‐171‐003‐A1 is the pivotal trial that provided response data that supported an accelerated approval of the US Food and Drug Administration for carfilzomib in the United States.
20 mg/m2 in cycle 1, and then 27 mg/m2 thereafter.
Increased each cycle as tolerated.
Table 2.
Covariate distribution by study
| Covariate | PX‐171‐003 (N = 274) | PX‐171‐004 (N = 146) | PX‐171‐005 (N = 36) |
|---|---|---|---|
| Race (Caucasian/African American/other) | 194/56/24 | 109/22/15 | 25/8/3 |
| Mean age, years (SD) | 64 (9.4) | 65 (9.6) | 66 (9.1) |
| Mean weight, kg (SD) | 79 (19) | 82 (19) | 84 (19) |
| Mean CrCl, mL/min (SD) | 77 (32) | 78 (36) | 58 (38) |
| Mean creatinine, mg/dL (SD) | 1.1 (0.39) | 1.1 (0.30) | 2.0 (1.5) |
| ECOG PS, n (0/1/2/missing) | 70/167/36/1 | 56/81/8/1 | 6/22/8/0 |
| Mean percent plasma cell involvement (SD) | 43 (29) | 31 (26) | 49 (30) |
| Mean serum β2‐microglobulin (SD) | 17 (65) | 9 (37) | 11 (16) |
| ISS stage (1/2/3/missing) | 67/84/81/42 | 62/44/30/10 | 7/9/20/0 |
| Bone marrow transplant, n (no/yes) | 64/210 | 35/111 | 13/23 |
| Median number of prior regimens | 5.0 | 1.0 | 4.0 |
| Mean number of bortezomib regimens (SD) | 2.0 (1.5) | 0.21 (0.71) | 2.1 (1.5) |
| Bortezomib in last regimen, n (no/yes) | 135/139 | 134/12 | 14/22 |
| Lenalidomide in last regimen, n (no/yes) | 159/115 | 83/63 | 26/10 |
| Thalidomide in last regimen, n (no/yes) | 241/33 | 118/28 | 23/13 |
| 132/142 | 57/89 | 16/20 | |
| Mean absolute neutrophil count, 10−3/µL (SD) | 4.0 (8.4) | 3.1 (5.1) | 5.8 (9.5) |
| Sex, n (male/female) | 162/112 | 86/60 | 20/16 |
| Mean albumin, g/dL (SD) | 3.7 (0.64) | 3.9 (0.49) | 3.4 (0.73) |
| Mean ALT, U/L (SD) | 26 (24) | 26 (15) | 23 (15) |
| Mean AST, U/L (SD) | 30 (16) | 31 (15) | 28 (13) |
| Mean bilirubin, mg/dL (SD) | 0.43 (0.26) | 0.43 (0.24) | 0.58 (0.23) |
| Mean hemoglobin, g/dL (SD) | 10 (1.5) | 11 (1.7) | 10 (1.8) |
| Mean lymphocytes, 10−3/µL(SD) | 7.0 (7.7) | 2.2 (5.5) | 4.5 (9.6) |
| Mean platelets, 10−3/µL (SD) | 160 (72) | 186 (77) | 166 (85) |
| Mean white blood cells, 10−3/µL (SD) | 4.3 (1.7) | 4.9 (2.0) | 5.0 (1.8) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CrCl, creatinine clearance; ECOG PS, Eastern Cooperative Oncology Group performance status; ISS, International Staging System; SD, standard deviation.
Among 513 subjects with M‐protein data at any timepoint, 456 (87%) had data that could be used to develop the longitudinal model for M‐protein following carfilzomib exposure; there were 2,735 total observations (median of 5.0 observations per subject). The remaining subjects had missing predose/baseline data (n = 21) or were nonsecretory at the time of the first dose (n = 36).
A large range of baseline M‐protein values (0.40–98 g/L) were observed, together with a variety of M‐protein profiles during treatment (a few typical profiles taken at random are illustrated in Figure 1).
Figure 1.
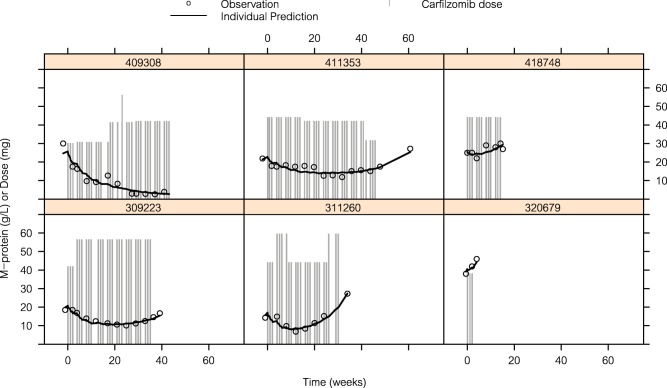
Illustration of model fit to M‐protein data in a random selection of subjects.
The parameters of the final model are presented in Table 3. The model includes two covariates: the effect of baseline platelet count on the M‐protein growth rate (KL) and the effect of number of prior regimens on the M‐protein shrinkage rate for carfilzomib (KD,CFZ). The M‐protein growth parameter KL increases with decreasing baseline platelet count. The effect is linear and corresponds to an 80% reduction in growth rate at maximum platelet count in the dataset and an increased growth rate of 30% at the lowest platelet count. The elimination rate of drugs from the virtual biophase (KP,CFZ) was estimated to be 9.94 weeks−1 when estimated with the basic model without any covariates. However, its relative standard error was 160%. As the parameter could not be estimated with precision, it was fixed to the estimate in subsequent runs. It may be noted that in the previous preliminary model on which the present TGI model18 is based, KP was fixed to a value found by likelihood profiling. KD,CFZ was reduced by ∼40% among subjects with more than one prior regimen. However, this effect is uncertain, with a relative standard error of 40%. Correlations between the estimates of the random effects were <0.64 and since this model was not intended to be used in simulations but rather to estimate ECTS, a diagonal covariance matrix was kept for the final model. The final NONMEM control file is given in the Supplementary Information online. The model was evaluated using standard goodness‐of‐fit plots.
Table 3.
Parameter estimates of the final TGI model for carfilzomib
| Estimate | RSE (%) | IIV | RSE (%) | Shrinkage (%) | |
|---|---|---|---|---|---|
| KL at median platelet count (week−1) | 0.0283 | 5.7 | 0.994 | 19 | 22 |
| KD,CFZ (1 prior regimen or less) (mg−1·week−1) | 0.0137 | 13 | 0.949 | 15 | 24 |
| λCFZ (week−1) | 0.107 | 12 | 0.814 | 21 | 38 |
| KP,CFZ (week−1) | 9.94 | FIXED | NA | NA | NA |
| Effect of platelet count on KL (mm3/week) | −0.00265 | 9.1 | |||
| Fraction of KD,CFZ among subjects with >1 prior regimen | 0.607 | 40 | |||
| σ additive (g/L) | 1.02 | 18 | NA | NA | 17 |
| σ exponential | 0.115 | 9.3 | NA | NA | 17 |
IIV, intraindividual variability; KD,CFZ, rate of M‐protein decrease induced by carfilzomib; KL, rate of M‐protein increase; KP,CFZ, rate of elimination of carfilzomib from virtual biophase compartment; NA, not available; RSE, relative standard error; SD, standard deviation of intersubject variability; TGI, tumor growth inhibition; λCFZ, rate constant of disappearance of carfilzomib effect. σ, residual error additive and exponential terms.
The dexamethasone effect is negligible at doses <20 mg, and there were too few subjects who received HD to estimate it.
The TGI model was used to estimate ECTS at the start of week 4. Week 4 was chosen, as 396 of 456 subjects (87%) were on study and sampled at that time. In contrast, at weeks 2, 6, and 8 there were data for 349 (77%), 337 (74%), and 322 (71%) subjects, respectively.
The results of the PPC are shown in Figure 2. The observed distribution (blue line) is within the 95% prediction interval (blue envelope) for most of the quantiles.
Figure 2.
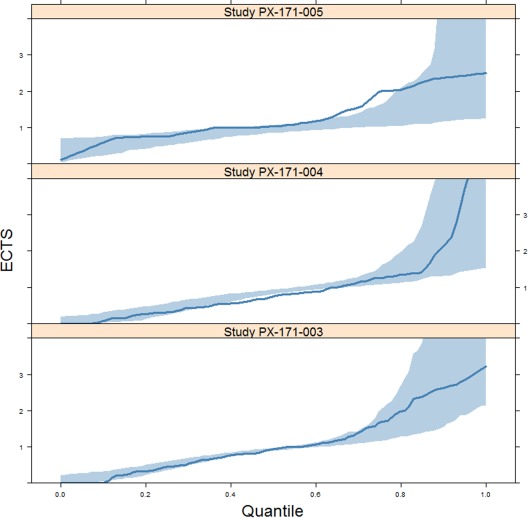
Posterior predictive check of the final tumor growth inhibition model in studies PX‐171‐003, PX‐171‐004, and PX‐171‐005. Blue lines and shading represent the observed distribution and 95% prediction intervals, respectively. ECTS, early change in tumor size.
Model for OS
A parametric model for time to death was developed based on the 456 subjects used in the TGI model development. Of those subjects, 157 (34%) died during the study. Estimated median survival was 15.8 months in study PX‐171‐003, while it was not reached in the two other studies. A log‐normal distribution best described the survival data, as previously observed in other cancer types.10, 11 The parameter estimates for the final multivariate model for OS are shown in Table 4.
Table 4.
Parameter estimates of the final OS model
| Covariates | Estimatesa | RSE (%) | 95% CI | P value |
|---|---|---|---|---|
| Intercept | 0.7277 | 94 | (−0.0609; 2.06) | 0.029 |
| ECTS week 4 | −1.160 | 16 | (−1.52; −0.801) | <0.001 |
| Hemoglobin (g/dL) | 0.3007 | 19 | (0.188; 0.414) | <0.001 |
| Female sex | 0.7441 | 23 | (0.403; 1.09) | <0.001 |
| ECOG performance status = 0 | 0.7343 | 28 | (0.327; 1.14) | <0.001 |
| Percent cell involvement | −0.01070 | 28 | (−0.0165; −0.00488) | <0.001 |
| <3 prior regimens | 0.678 | 30 | (0.280; 1.08) | <0.001 |
| Log (scale) | 0.3060 | 20 | (0.188; 0.424) | <0.001 |
CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; ECTS, early change in tumor size; OS, overall survival; RSE, relative standard error.
Positive/negative values indicate increase/decrease of survival probability.
Estimates correspond to survival times in months.
Several statistically significant baseline prognostic factors for OS were identified. Survival decreased as percent plasma cell involvement increased; survival increased with a higher hemoglobin level or when the subject was female. Survival was also longer in subjects with an ECOG PS of 0 and among subjects with fewer than three prior regimens. Finally, the probability of survival increased in subjects who demonstrated more tumor shrinkage at week 4 (decrease in ECTS) and this effect was independent of baseline prognostic factors. Study effect was highly significant in univariate analysis (P < 0.0001), but no longer in the final model after adjustment of the covariate effects (P ≥ 0.02).
Model evaluation (PPC) indicated that the model had good performance in simulating the survival distributions. Observed distributions of each study are within the 95% PI, with the only exception of the tail of study PX‐171‐003, despite large differences across studies that are accounted for by the covariates in the model (Figure 3). Studies PX‐171‐004 and PX‐171‐005 are not mature and there is censoring at the end of the distribution, but overall there were a fair number of events in the pooled dataset.
Figure 3.
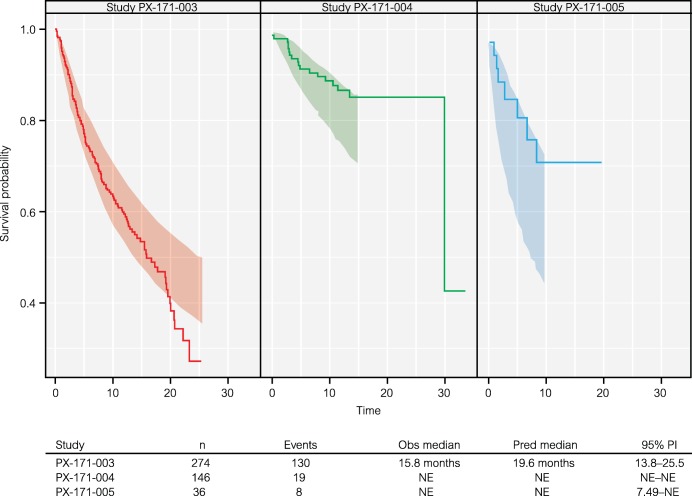
Kaplan–Meier plot of overall survival (OS) data (solid line) and 95% prediction intervals (shaded areas, 1,000 simulations) of the multivariate OS model by carfilzomib study. n, number of subjects; NE, not estimable; Obs, observed; PI, prediction interval; Pred, predicted.
DISCUSSION
Model‐based approaches can be used to integrate early clinical data to enhance learning and reduce the risk of large and costly confirmatory trials.9, 10, 11, 12, 13, 14, 15, 16, 17 Models predictive of clinical outcome measures (e.g., OS) can be used to support phase II study design, decisions at the end of phase II, and phase III planning and execution.15, 16, 17 In MM, this is important since the OS endpoint usually takes a longer time to mature, and information prior to the OS data readout will significantly facilitate efficient design of the clinical programs. The current work describes the first application of TGI and OS modeling to M‐protein in MM model simulations based on ECTS as early as week 4 (based on M‐protein time course) in conjunction with other baseline prognostic factors including ECOG PS, hemoglobin, sex, percent bone marrow cell involvement, and number of prior regimens, and shows good agreement to the observed OS data following carfilzomib treatment. This approach, which could leverage prior knowledge and early or interim clinical data to support decisions through simulations of late clinical endpoints (e.g., OS),15, 16, 17 has potential to substantially improve the efficiency of drug development for MM. In this work, we used a sequential approach for model development. We developed the tumor size model first (TGI model), estimated ECTS, and then developed the OS model. Joint modeling is theoretically better, as it is estimating model parameters from a joint likelihood that combines uncertainty in parameter estimates.14, 31 In this work, we therefore did not take into account the uncertainty in ECTS prediction in the OS model. Tumor size and OS models were nevertheless qualified for their respective intended use (i.e., to estimate ECTS and simulate OS). Further research is warranted to assess the effect of these respective approaches on model performance based on both simulated and real data. In addition, a block covariance matrix for the random effects should be considered if the TGI model is to be used in simulations.
We show that ECTS can be predictive of OS as early as 4 weeks following treatment. The link between ECTS and OS has been shown in previous studies for MM and other oncology agents,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 32 based on estimated ECTS at a later timepoint (week 8). The average time to response with carfilzomib treatment ranges from 1 month (in combination with lenalidomide) to 2 months (carfilzomib monotherapy),23, 24, 25, 26, 27 so ECTS at week 4 is consistent with the time to response and mechanism of carfilzomib on disease progression in MM subjects. In addition to ECTS, other metrics have been used in the TGI model, including time to tumor (re)growth or tumor growth rate15 as well as the full time profile of TGI or other biomarker responses.14 These longer‐term metrics capture the whole duration of drug action rather than just the early shrinkage. Our findings regarding ECTS at 4 weeks merit further exploration in MM studies. Furthermore, a comparison of predictability with other TGI metrics using early or longer‐term dynamic changes in tumor burden may be considered.
The M‐protein growth rate estimate (expressed by KL) in this study was similar to the one reported for other anti‐MM agents in earlier preliminary work,18 indicating that this disease‐specific parameter in the TGI model for MM is robust and treatment‐independent for relapsed/refractory subjects. This is very encouraging; the results support the concept of a disease model for which disease‐specific parameters, such as those related to the progression of MM, can be identified and are consistent, regardless of the anti‐myeloma treatment. As seen with the case study in which historical lenalidomide phase III data was successfully used in a TGI model to predict PFS and OS in a phase II study of pomalidomide,19 our work corroborates the previous model results, which showed that a compound‐independent disease model can be developed using the framework of the TGI model to leverage prior data to predict clinical endpoints for MM. Such a disease model framework can be used to predict clinical benefits from monotherapy vs. combination therapy. Additional systematic meta‐analyses for other anti‐myeloma agents are needed for the comprehensive development of such disease‐specific progression models for MM. The development of this kind of disease model, which requires clinical datasets from multiple agents, will require a close collaboration between industry and regulatory agencies.
The current work used drug dose (full dosing history) as the exposure metric to drive drug effect, rather than the PK data, since PK data were not collected in all subjects. It would be of value to incorporate systemic exposure (i.e., full PK profile, area under the curve) into the model, to understand the contribution of PK variability to subjects' responses in longitudinal M‐protein dynamics with the potential to optimize the dose regimen for anti‐myeloma agents. An important additional consideration is that the half‐life of M‐protein in serum is about 3 weeks. Therefore, there may be a lag time between changes in M‐protein and changes in tumor burden in subjects with MM. To capture the early treatment effect, a model to describe early dynamic change in serum‐free light chain (sFLC) can be considered.21 sFLC has a much shorter half‐life (2 to 3 hours) and can be routinely measured using existing assays in subjects with MM. In addition, a rapid reduction in FLC at day 15 following carfilzomib treatment was associated with an increased depth of response and longer PFS in patients with relapsed and relapsed and/or refractory MM.33
The clinical outcome in MM is also dependent on subject baseline characteristics, prior lines of therapy, and prognostic factors. In addition to an increase of M‐protein at week 4, which indicated a lack of drug effect, this integrated meta‐analysis of multiple studies with carfilzomib identified several baseline characteristics as significant independent prognostic factors for OS, including the number of prior regimens, baseline hemoglobin level, percent cell involvement, and good ECOG PS. Low hemoglobin is a marker of MM severity and is included in the Durie–Salmon staging system.34 Likewise, cell involvement is included in the definition of MM response criteria for a complete response. Good ECOG PS has been known to be beneficial to survival in several other cancer types.11, 13 While the covariates of OS identified in this work have yet to be confirmed by additional studies, ECOG PS and hemoglobin level were also found to be prognostic of OS in our preliminary work for a different MM agent.19 The analysis that was based on the limited phase I/II dataset also showed that females tended to have a longer OS compared to males, regardless of drug treatment. However, the limited phase I/II data may have been confounded by other factors and has not been confirmed with larger phase III datasets. With all of the potential covariates affecting OS identified in subjects with MM, a multivariate OS model and corresponding simulations adjusted for patient characteristics could be a useful tool prior to the start of the pivotal study to increase the probability of success of clinical studies.
The current work represents a first step in using a TGI model as an early biomarker to quantify the effect of carfilzomib and to ultimately predict an important clinical endpoint (OS) in subjects with MM. The proposed modeling framework assumes that the longitudinal M‐protein data will provide additional insight and granularity for predicting the ultimate benefits of carfilzomib to subjects compared with the traditional response category. ECTS at week 4 based on M‐protein modeling has the potential to be an early biomarker for survival prediction in MM following exposure to single‐agent carfilzomib. Future application of this modeling approach warrants further investigation for other agents or combination regimens for MM.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The first two authors contributed equally to this work. This study was funded by Onyx Pharmaceuticals, Inc., an Amgen subsidiary. Editorial assistance was provided by BlueMomentum, a division of KnowledgePoint360 Group, an Ashfield business. This work was presented, in part, in abstract form at the 2014 Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics (Jonsson, F. et al. A tumor growth inhibition model based on M‐protein levels in patients with relapsed/refractory multiple myeloma following single‐agent carfilzomib use [abstract PII‐044]. Clin. Pharmacol. Ther. 95 [Suppl 1], S75 [2014]).
Author Contributions
F.J., Y.O., S.A., L.C., D.S., S.J., R.V., A.B., and R.B. wrote the article; F.J., R.B., S.J., R.V., A.B., and L.C. designed the research; F.J. analyzed the data.
Conflict of Interest
F.J., R.B., and L.C. are employees of Pharsight and contractors to Onyx at the time of this work. S.A. and Y.O. are employees of Onyx.
References
- 1. Siegel, R. , Naishadham, D. & Jemal, A. Cancer statistics, 2013. C.A. Cancer. J. Clin. 63, 11–30 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Krypolis [prescribing information]. South San Francisco, CA: Onyx Pharmaceuticals; 2012. [Google Scholar]
- 3. Lodish, H. et al 3. In Molecular Cell Biology 5th edn. 66–72 (W.H. Freeman, New York, 2004). [Google Scholar]
- 4. Demo, S.D. et al Biochemical and cellular characterization of the novel proteasome inhibitor PR‐171 [abstract 1588]. Blood 106 (2005). [Google Scholar]
- 5. Kuhn, D.J. et al Potent activity of carfilzomib, a novel irreversible inhibitor of the ubiquitin‐proteasome pathway, against preclinical models of multiple myeloma. Blood 110, 3281–3290 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arastu‐Kapur, S. et al Nonproteasomal targets of proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin. Can. Res. 17, 2734–2743 (2011). [DOI] [PubMed] [Google Scholar]
- 7. Martin, T.G. Peripheral neuropathy experience in patients with relapsed and/or refractory multiple myeloma treated with carfilzomib. Oncology 27 (Suppl. 3), 4–10 (2013). [PubMed] [Google Scholar]
- 8. Durie, B. G. et al International uniform response criteria for multiple myeloma. Leukemia 20, 1467–1473 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Claret, L. et al Model‐based predictions of expected anti‐tumor response and survival in phase III studies based on phase II data of an investigational agent [abstract 2530]. J. Clin. Oncol. 24 (suppl), 307s (2006). [Google Scholar]
- 10. Claret, L. et al Model‐based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J. Clin. Oncol. 27, 4103–4108 (2009). [DOI] [PubMed] [Google Scholar]
- 11. Wang, Y. et al Elucidation of relationship between tumor size and survival in non‐small‐cell lung cancer patients can aid early decision making in clinical drug development. Clin. Pharmacol. Ther. 86, 167–174 (2009). [DOI] [PubMed] [Google Scholar]
- 12. Claret, L. , Lu, J.F. , Bruno, R. , Hsu, C.P. , Hei, Y.J. & Sun, Y.N. Simulations using a drug‐disease modeling framework and phase II data predict phase III survival outcome in first‐line non‐small‐cell lung cancer. Clin. Pharmacol. Ther. 92, 631–634, (2012). [DOI] [PubMed] [Google Scholar]
- 13. Claret, L. et al Evaluation of tumor‐size response metrics to predict overall survival in Western and Chinese patients with first‐line metastatic colorectal cancer. J. Clin. Oncol. 31, 2110–2114 (2013). [DOI] [PubMed] [Google Scholar]
- 14. Hansson, E.K. et al PKPD modeling of VEGF, sVEGFR‐2, −3 and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. Clin. Pharmacol. Ther. Pharmacometrics Syst. Pharmacol. 2, e84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruno, R. , Mercier, F. & Claret, L. Evaluation of tumor size response metrics to predict survival in oncology clinical trials. Clin. Pharmacol. Ther. 95, 386–393 (2014). [DOI] [PubMed] [Google Scholar]
- 16. Ribba, B. et al A review of mixed‐effects models of tumor growth and effects of anticancer drug treatment used in population analysis. Clin. Pharmacol. Ther. Pharmacometrics Syst. Pharmacol. 3, e113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Venkatakrishnan, K. et al Optimizing oncology therapeutics through quantitative clinical pharmacology: challenges and opportunities. Clin. Pharmacol. Ther. 97, 37–54 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Jonsson, F. et al A longitudinal tumor growth inhibition model based on serum M‐protein levels in patients with multiple myeloma treated by dexamethasone. 19, Abstract 1705. <www.page-meeting.org/?abstract=1705> (2010).
- 19. Bruno, R. et al Simulation of clinical outcome for pomalidomide plus low‐dose dexamethasone in patients with refractory multiple myeloma based on week 8 M‐protein response. Blood. 118, 1881 (2011). [Google Scholar]
- 20. Chanu, P. et al PK/PD relationship of the monoclonal anti‐BAFF antibody tabalumab in combination with bortezomib in patients with previously treated multiple myeloma: comparison of serum M‐protein and serum free light chains as predictor of progression free survival. 22 Abstract 2732. <www.page-meeting.org/?abstract=2732> (2013).
- 21. Marchand, M. , Claret, L. , Losic, N. , Puchalski, T.A. & Bruno, R. Population pharmacokinetics and exposure‐response analyses to support dose selection of daratumumab in multiple myeloma patients. 22 Abstract 2668. <www.page-meeting.org/?abstract=2668> (2013).
- 22. Chanu, P. , Marchand, M. , Claret, L. , Losic, N. , Puchalski, T.A. & Bruno, R. Population pharmacokinetic/pharmacodynamic models to support dose selection of daratumumab in multiple myeloma patients. 23 Abstract 3281. <www.page-meeting.org/?abstract=3281> (2014).
- 23. Jagannath, S. et al An open‐label single‐arm pilot phase II study (PX‐171‐003‐A0) of low‐dose, single‐agent carfilzomib in patients with relapsed and refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 12, 310–318 (2012). [DOI] [PubMed] [Google Scholar]
- 24. Siegel, D.S. et al A phase 2 study of single‐agent carfilzomib (PX‐171‐003‐A1) in patients with relapsed and refractory multiple myeloma. Blood. 120, 2817–2825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vij, R. et al An open‐label, single‐arm, phase 2 (PX‐171‐004) study of single‐agent carfilzomib in bortezomib‐naive patients with relapsed and/or refractory multiple myeloma. Blood. 119, 5661–5670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vij, R. et al An open‐label, single‐arm, phase 2 study of single‐agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br. J. Haematol. 158, 739–748 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Badros, A.Z. et al Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia. 27, 1707–1714 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacqmin, P. et al Modelling response time profiles in the absence of drug concentrations: definition and performance evaluation of the K‐PD model. J. Pharmacokinet. Pharmacodyn. 34, 57–85 (2007). [DOI] [PubMed] [Google Scholar]
- 29. Dansirikul, C. , Silber, H.E. & Karlsson, M.O. Approaches to handling pharmacodynamic baseline responses. J. Pharmacokinet. Pharmacodyn. 35, 269–283 (2008). [DOI] [PubMed] [Google Scholar]
- 30. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)—a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Desmee, S. , Mentré, F. , Veyrat‐Follet, C. & Guedj, J. Nonlinear mixed‐effect models for prostate‐specific antigen kinetics and link with survival in the context of metastatic prostate cancer: a comparison by simulation of two‐stage and joint approaches. AAPS J. 17, 691–699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruno, R. & Claret, L. On the use of change in tumor size to predict survival in clinical oncology studies: toward a new paradigm to design and evaluate phase II studies. Clin. Pharmacol. Ther. 86, 136–138 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Vij, R. et al Serum free light chain reduction correlates with response and progression‐free survival following carfilzomib therapy in relapsed/refractory multiple myeloma. Leuk. Lymphoma; e‐pub ahead of print 2015. doi:10.3109/10428194.2015.1020801. [DOI] [PubMed] [Google Scholar]
- 34. Durie, B.G. & Salmon, S.E. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 36, 842–854 (1975). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information