

Abstract
The p38 mitogen‐activated protein kinase (p38) is a key signaling pathway involved in regulation of inflammatory cytokines. Unexpectedly, several clinical studies using p38 inhibitors found no convincing clinical efficacy in the treatment of chronic inflammation. It was the objective of this study to characterize the population pharmacokinetics (PK) of BCT197 in healthy volunteers and to examine the relationship between BCT197 exposure and pharmacodynamics (PD) measured as inhibition of ex vivo lipopolysaccharide (LPS)‐induced tumor necrosis factor alpha (TNFα), a downstream marker of p38 activity. PK was characterized using a two‐compartment model with mixed‐order absorption and limited‐capacity tissue binding. The PK‐PD relationship revealed that suppression of TNFα was partly offset over time, despite continuous drug exposure. This may indicate a mechanism by which the inflammatory response acquires the ability to bypass p38. Simulations of posology dependence in drug effect suggest that an intermittent regimen may offer clinical benefit over continuous dosing and limit the impact of tolerance development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Short‐term efficacy in phase II trials has raised concerns for the future potential of therapeutic inhibition of p38 mitogen‐activated protein kinases, a key signaling pathway involved in regulation of the proinflammatory cytokines. • WHAT QUESTIONS DID THIS STUDY ADDRESS? ☑ An increased understanding of BCT197's antiinflammatory activity was sought from a PK‐PD model that subsumes the observed biomarker escape (TNFα) under the hypothesis of a tolerance mechanism. The model was developed to optimize drug response in relation to drug exposure and dosing schedule. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ Tolerance development to chronic p38 inhibition is likely to occur in man. The investigation of schedule dependence in the drug effect revealed that moving from a continuous to an intermittent regimen may offer clinical advantage and limit the impact of tolerance development. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ This study illustrates that mechanistic PK‐PD modeling of relevant downstream markers of p38 inhibition adds insights that BCT197 might be more efficacious in treatment of acute as opposed to chronic inflammation disorders.
Chronic obstructive pulmonary disease (COPD) is characterized by chronic and progressive inflammation in the lungs that leads to airflow resistance or loss of gas exchange units.1, 2, 3 The chronic and progressive course of COPD is frequently aggravated by exacerbations—periods of increased cough, dyspnea, and production of sputum.4 The chronic inflammation in COPD is orchestrated by immune cells that are activated and recruited to the site of inflammation in response to cytokines and chemotactic factors.4, 5 The current standard of care aims at decreasing airway smooth‐muscle tone by bronchodilator drugs and modulating pulmonary inflammation with inhaled corticosteroids or the phosphodiesterase inhibitor roflumilast.4 Although these therapies can improve lung function, disease‐modifying treatments are needed to reduce the number and severity of exacerbations, and ultimately mortality.6, 7
The p38 mitogen‐activated protein kinase (p38) is a key signaling node that conveys responses to multiple cellular stressors by phosphorylating downstream substrates that are involved in regulation of the biosynthesis and actions of inflammatory cytokines such as tumor necrosis factor alpha (TNFα), interleukin (IL)‐1β, and IL‐6.8 p38 also mediates activation of matrix metalloproteinase and COX‐2 that are involved in inflammation and tissue destruction.9 Increased phosphorylation of p38 has been demonstrated in the lungs of COPD patients,10, 11 and activation of p38 correlates with the degree of lung function impairment and neutrophil airway infiltration.11, 12 Reduced cytokine production by different lung and blood cells was noted following p38 inhibition,10, 13 indicating that p38 activation may contribute to both local and systemic inflammation.
BCT197 is an oral low‐molecular‐weight p38 inhibitor currently in development for the treatment of several inflammatory conditions, including COPD.14 Intermittent short‐term dosing of BCT197 (75 mg on days 1 and 6) showed a marked improvement in lung function (FEV1) in COPD patients.15 Encouraging results were also seen for other p38 inhibitors in development for the treatment of COPD,16, 17 acute inflammation, and pain.18, 19 In contrast, several small phase II studies using continuous dosing regimens (12 weeks) in patients with rheumatoid arthritis (RA) found no convincing evidence for adequate dampening of chronic inflammation as measured by gold‐standard clinical composite scores and the acute phase protein C‐reactive protein (CRP).20, 21 The different outcome in RA compared to COPD may suggest that a biologic mechanism by which the inflammatory response acquires the ability to bypass chronic p38 inhibition cannot be ruled out. In agreement with this, an unexpected finding from RA studies was that the initial reduction of CRP was reversed by week 12 in spite of continuous and adequate drug exposure.20, 21, 22 Opportunities to obtain clinical efficacy with p38 inhibitors may remain limited in the absence of a better understanding of the relationship between drug exposure and response.
It was the objective of this study to characterize the population pharmacokinetics (PK) of BCT197 in healthy volunteers and to examine the relationship between drug exposure and pharmacodynamics (PD) measured as inhibition of ex vivo lipopolysaccharide (LPS)‐induced TNFα, a downstream surrogate marker of p38. The PK‐PD model was used to gain insight into whether efficacy in continuous treatment may possibly be compromised by a compensatory mechanism in human. Model‐based simulations were used to assess drug response in relation to drug exposure and dosing schedule.
METHODS
Study population
Clinical study CBCT197A2101 was approved by the Ethics Committees of participating centers and conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All subjects gave written informed consent before participation. Healthy male and female subjects of nonchildbearing potential aged 18–50 years with a body mass index 18–30 kg/m2 and body weight ≥50 kg were eligible to enroll. Subjects were nonsmokers in good health as determined by past medical history, physical examination, vital signs, electrocardiogram, and laboratory tests having no significance at screening. Key exclusion criteria included history of significant medical or surgical disease; significant illness within 14 days prior to initial dosing; known history of cardiovascular disease or ECG abnormality; positive urine drug screen; history of any clinically important drug allergy; or use of any investigational drug within 4 weeks prior to initial dosing. Use of any prescription, over‐the‐counter drugs or herbal medications, as well as consumption of grapefruit was not permitted from 14 days prior to dosing until 7 days following the last dose.
Study protocol
Part 1 of study CBCT197A2101 was a randomized, double‐blind, placebo‐controlled, ascending single‐dose study to evaluate safety, tolerability, PK and PD of oral BCT197 in healthy subjects. Part 2 was a 14‐day, randomized, double‐blind, placebo‐controlled, ascending multiple dose study evaluating the PK and PD of oral BCT197. PD effect of BCT197 in Parts 1 and 2 was assessed by measuring TNFα levels in ex vivo LPS‐challenged blood samples. Details of the PK and PD sampling regimen, ex vivo LPS challenge, and bioanalysis of BCT197 and TNFα are provided in the Supplementary Methods. Part 3 of the study determined the effect of a single oral administration of BCT197 on serum TNFα levels after in vivo intravenous LPS challenge. Only PK data of this part were used, as it did not measure ex vivo LPS‐induced TNFα.
Subjects fasted for 10 hours prior to BCT197 administration and continued to fast for 4 hours postdosing. No fluid intake apart from the fluid given at the time of drug intake was allowed from 2 hours before until 2 hours after dosing. Drug administrations were oral solutions with doses ranging from 0.1 to 3 mg, and tablets at doses of 5 mg and higher.
Population PK‐PD analysis
A nonlinear mixed‐effects model approach with first‐order conditional estimation (Lindström Bates) in Phoenix 6.4 NLME V1.2 (Pharsight, USA) was used to estimate population parameters and their between‐subject variability (BSV). Final model selection was guided by reduction in the objective function value (OFV), precision in parameter estimates, examination of goodness‐of‐fit, reductions in the magnitude of BSV and residual error, as well as shrinkage in random variability parameters and robust model parameter estimation. Statistical significance between nested models was based on ΔOFV (≥3.85), applying the likelihood ratio test. Model stability and performance were assessed by means of a nonparametric bootstrap with resampling and replacement. A simulation‐based diagnostic was performed by visual predictive checks.
Several combinations of absorption and distribution models were assessed: zero‐order, first‐order, mixed zero‐ and first‐order (simultaneous and sequential), with or without lag time, and transit compartments.23 Multiple‐fraction absorption kinetics24 and a shunt model were evaluated to describe the second absorption peak. Distribution models tested included open 1‐, 2‐, and 3‐compartment, and nonlinear tissue distribution from central and/or peripheral compartment. Model reductions were investigated by traditional model‐fitting criteria. BSV was modeled as lognormal and was tested on all model parameters, followed by stepwise elimination when their estimation was not adequately supported by the data. Additive, proportional, and mixed error models were tested to describe unexplained residual variability. A summary of the PK model‐building process is provided in Supplementary Table S2 online. Final PK model equations are provided in the Supplementary Methods.
A Bayesian approach conditioned on the population characteristics was used to estimate the individual PK model parameters, which were fixed and used as an input function to the PD model. Raw LPS‐induced TNFα secretion data of both BCT197 and placebo‐treated subjects was modeled simultaneously. Direct and indirect response models, with or without negative feedback, were explored. A summary of the model‐building process is provided in Supplementary Table S3. The final description of the PD data was an indirect response model with negative feedback on the system's outflow rate and nonstationarity in production of response (Eqs. (1), (2), (3)). The following expression was used to describe the oscillatory behavior of the baseline
| (1) |
where t is time, Kin is the average input rate, and t0 the shifted peak in response. The ω parameter scales the frequency of oscillations to the physical frequency 2π/cycle, where cycle equals 24 hours. Parameter A is the fraction of Kin that corresponds to the amplitude. The differential equation for ex vivo LPS‐induced TNFα secretion (E) employed Eq. 2:
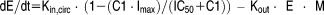 |
(2) |
where Kout is the first‐order degradation rate constant, C1 the central drug concentration, Imax the maximum inhibition, and IC50 the drug concentration at half maximum inhibition. Eq. 2 was initialized as E(0)=Kin,circ/Kout. E is counterbalanced by the ability of molecular mechanisms, denoted by M, that may regulate cellular adaptation processes to control the magnitude and duration of LPS‐induced p38 activation. Production of M is governed by relative E to baseline (E/E0) and the first‐order rate constant ktol (Eq. 3).
| (3) |
In the absence of any drug, relative drug response (E/E0) and M are unity at baseline. In response to drug effect, the first‐order decline of M is governed by ktol.
Model simulation
Stochastic model simulations in Phoenix NLME V1.2 were used to assess the drug effect in relation to drug exposure and dosing schedule. Simulations in healthy subjects employed the final PK‐PD model parameters of Table 1. Simulated individual response–time data were exported to SAS and analyzed in terms of maximum inhibition from baseline, time to inhibition nadir time above 50% inhibition, and average inhibition.
Table 1.
Population pharmacokinetic and pharmacodynamic parameters of BCT197 and bootstrap validation
| Population mean | Between‐subject variability | |||||
|---|---|---|---|---|---|---|
| Parameter | Final estimate | RSEc (%) | Bootstrap median and 90% CId | Final estimate (%CV)e | RSEc (%) | Bootstrap median and 90% CId |
| Population PKa | ||||||
| CL/F (L/h) | 1.76 | 10 | 1.77 [1.47;2.05] | 25.5 | 36 | 26.1 [20.9;37.7] |
| V1/F (L) | 71.9 | 16 | 71.5 [58.1;98.7] | 42.4 | 43 | 43.6 [34.3;67.9] |
| V2/F (L)f | 25.5 | 14 | [25.3] [19.3;31.1] | — | — | — |
| Bmax (µg) | 500 | 30 | 507 [308;806] | — | — | — |
| Kd (µg) | 367 | 35 | 371 [228;660] | — | — | — |
| CLd/F (L/h) | 7.47 | 15 | 7.46 [5.70;9.42] | — | — | — |
| Kt=Kint1 (1/h) | 1.12 | 14 | 1.13 [0.934;1.42] | 59.6 | 35 | 58.7 [43.8;81.0] |
| Ks (1/h) | 5 (fixed) | — | — | 172 | 29 | 167 [110;263] |
| Tlag (h) | 0.232 (fixed) | — | — | 44.0 | 14 | 44.1 [38.8;49.7] |
| fc (−)g | 0.344 | 9.1 | 0.342 [0.291;0.392] | — | — | — |
| Rate (µg/h) | 2300 | 5.3 | 2310 [2170;2530] | — | — | — |
| K1b (1/h) | 0.0106 | 15 | 0.0105 [0.00791;0.0132] | — | — | — |
| Db (h) | 0.1 (fixed) | — | — | — | — | — |
| Tpump (h) | 18 (fixed) | — | — | — | — | — |
| Residual error (%) | 13.4 | 3.7 | 13.3 [12.5;14.2] | — | — | — |
| Population PDb | ||||||
| Imax (−) | 0.663 | 6.4 | 0.668 [0.6121;0.752] | — | — | — |
| IC50 (µg/L)h | 44.3 | 24 | 44.3 [30.6;65.5] | — | ||
| Kin (ng/mL) | 1830 | 32 | 1850 [977;3020] | — | — | — |
| Ktol (1/h) | 0.00276 | 44 | 0.00281 [0.00173;0.00529] | — | — | — |
| Kout (1/h) | 0.348 | 35 | 0.353 [0.170;0.592] | 32.5 | 18 | 31.9 [27.1;37.4] |
| t0 (h) | 13.3 | 16 | 13.3 [11.9;14.3] | — | — | — |
| A (ng/mL) | 0.207 | 21 | 0.208 [0.161;0.317] | — | ||
| Residual error (%) | 21.2 | 3.9 | 21.1 [19.8;22.5] | — | — | — |
Bmax, total binding capacity; CL/F, oral drug clearance; CLd/F, intercompartmental distribution clearance; Db, duration of zero‐order drug release from the shunt compartment (Ashunt) into the intestinal compartment (Aint); fc, fraction of oral dose (tablet) that is absorbed in a zero‐order fashion; K1b, first order transfer rate from central compartment into the shunt compartment (Ashunt); Kd, equilibrium dissociation constant; Kint1, first order absorption rate constant of shunted drug into central compartment; Kt/Ks, first‐order oral absorption rate constant of tablet/solution; Rate, zero order oral absorption rate (tablet); Tpump, shunt compartment (Ashunt) emptying lag time; V1/F, volume of central compartment; V2/F, volume of peripheral compartment.
A, amplitude of the oscillatory baseline; IC50, concentration at half maximum inhibition; Imax, maximum inhibition; Kin, zero order production rate of drug response; Kout, first order degradation rate of drug response; Ktol, first order production rate of negative feedback; t0, shifted peak of oscillating baseline.
RSE, relative standard error calculated as 100 x (standard error/mean value) from 500 iterations of a nonparametric bootstrap.
The 90% CI is displayed as the 5th−95th percentiles of 500 iterations of a nonparametric bootstrap.
The CV% between‐subject variability is presented as 100x(EXP(mean estimate)−1)0.5.
Vss/F ranged 97.4 L (V1/F+V2/F) when dose approaches infinity to 132 L (V1/F + V2/F•(1+Bmax/Kd)) when dose approaches zero.
Fraction of dose modeled in logit domain and back transformed.
IC50 was modeled as fraction of predose baseline value (α•E(0)), where α is the fraction of baseline and E(0) the predose baseline response. Population median α and 90% CI was 10.5 [7.22;15.5].
RESULTS
Population PK model
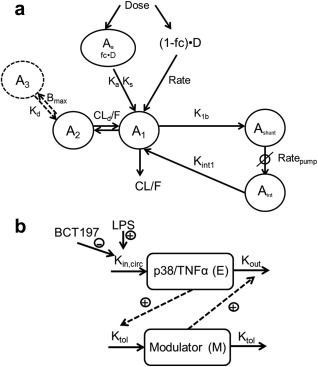
A summary of subject demographics and data used for model building is presented in Supplementary Table S1 online. Demographic and baseline characteristics were well balanced across the single and multiple dose groups. Median plasma concentration of BCT197 after single‐dose administration and once‐daily dosing for 2 weeks are depicted in Figure 1. A summary of the model building is presented in Supplementary Table S2. The final PK model is depicted in Figure 2 a. Basic goodness‐of‐fit plots of the final model did not show any relevant trends of model misspecification (Figure 3 a–d). Simulation‐based diagnostics also indicated good performance of the model (Supplementary Figures S1 and S2). η‐Shrinkage was low‐to‐moderate (<30%) for all parameters, indicating post‐hoc estimates could be obtained with confidence.
Figure 1.
Median plasma concentration time profile of BCT197 after (a) a single dose administration (inset: log scale view), and (b) once daily dosing for 14 days. Time course of inhibition of placebo‐corrected LPS‐induced TNFα secretion after (c) a single oral dose of BCT197 (inset: mean ± SD ex vivo LPS‐induced TNFα secretion in placebo treated subjects), and (d) 7 mg once daily dosing for 14 days (individual data). Doses administered were 75 mg (▵), 50 mg (○), 30 mg (■), 20 mg (□), 10 mg (▲), 7 mg (●), 5 mg ( ), 3 mg (
), 3 mg ( ), 1 mg (⋄), 0.3 mg (♦), 0.1 mg (✗).
), 1 mg (⋄), 0.3 mg (♦), 0.1 mg (✗).
Figure 2.
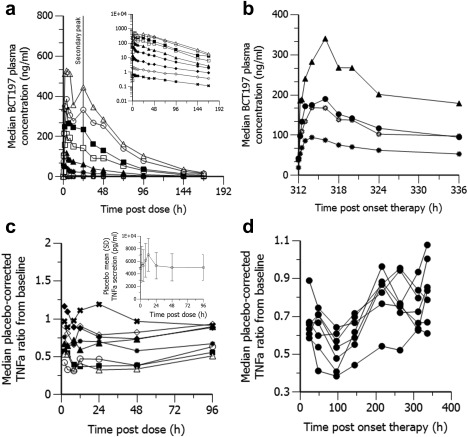
Structure of the final (a) pharmacokinetic and (b) pharmacodynamic model. For abbreviations, refer to Table 1. PK model equations are provided in the Supplementary Methods.
Figure 3.
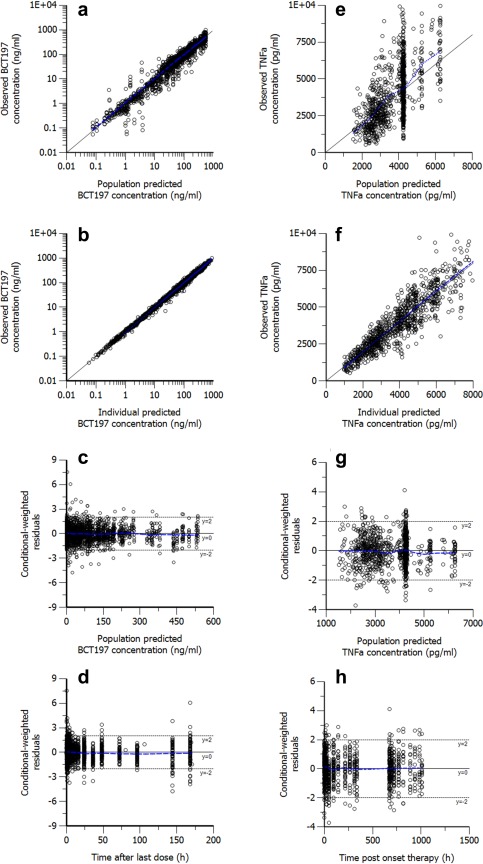
Goodness‐of‐fit plots for the final population (a–d) PK model and (e‐f) PD model. The lines of identity and local polynomial regression are represented by the solid black line and blue dashed line, respectively.
Drug administrations were oral solutions with doses up to 3 mg, and tablets at doses of 5 mg and higher. The population PK parameters along with their unexplained BSV and relative standard errors (RSE) are summarized in Table 1. BCT197 was found to be a low clearance drug (1.76 L/h), with linearity in oral drug clearance (CL/F) demonstrated over the entire dose range tested (0.1–75 mg). No relevant differences in relative bioavailability between these formulations were seen. BCT197 exhibited an apparent absorption plateau, with a tendency to less than dose‐proportional increase in peak drug concentration (Cmax). For tablets, the mixed‐order absorption model consisted of a first‐order process (Kt = 1.12 h−1), absorbing a fraction (fc) of on average 66% of dose, and a parallel zero‐order process characterized by a Rate of 2,300 µg/h (Table 1 ). A mixed‐order absorption model was significantly better than zero‐order (ΔOFV −509) or first‐order absorption (ΔOFV −403). Model parameters Rate and fc were found to be independent of dose. Addition of a random effect on Rate or fc did not improve the model fit. Oral absorption from the solution was parsimoniously described using a first‐order process (Ks). Limited data in the absorption phase impaired accurate estimation of Ks.
Population predictions from linear disposition models showed overprediction in Cmax as well as underprediction in terminal disposition half‐life, particularly at low doses, despite dose linearity in CL/F. Conversely, the quasi‐equilibrium model with negligible receptor turnover from the peripheral compartment as depicted in Figure 2 captured well the apparent nonlinearity in tissue distribution (model equations in Supplementary Methods), and dropped the OFV by 123 points (Supplementary Table S2). The dissociation constant (Kd) and maximal binding (Bmax) were estimated to be respectively 367 µg and 500 µg. Limited‐capacity binding caused steady‐state volume of distribution (Vss/F) to increase with decreasing dose, with a limiting value of 132 L when the dose approached zero (Table 1). As expected, a competing quasi‐equilibrium model with nonlinear tissue binding into the central compartment showed bias in the structural model diagnostics (not shown).
BCT197 exhibited double peak behavior at about 18 to 24 hours postdose (Figure 1). This may indicate a late absorption window or drug redistribution by a shunt, possibly enterohepatic. The addition of a shunt feature further dropped OFV by 62 points. The full shunt model, however, was overparameterized (data too sparse). A reduction in degrees of freedom was achieved by fixing the duration of drug shunting (Tpump) as well as the rate of drug transfer from Ashunt into Aint (Db). Because model fit was insensitive to Kint1, it was set equal to Kt.
Population PD model
Pharmacodynamic modeling was used to describe BCT197's ability to inhibit the ex vivo LPS‐induced TNFα secretion in peripheral blood. A summary of subject demographics and data used for model building is presented in Supplementary Table S1. Assay details are given in the Supplementary Methods.
In placebo‐treated subjects, nonstationarity in measurements of ex vivo LPS‐induced TNFα secretion was seen (Figure 2 c, inset). A circadian periodicity of shed TNFα receptors which attenuated response to LPS cannot be ruled out.25 Accordingly, drug effect was described as an inhibitory function on an oscillatory input system characterized by a physical frequency of 2π/24 hours (Eq. 1), and all available TNFα data of both BCT197 treated subjects and matching placebos were modeled simultaneously.
Exploratory plots of placebo‐corrected drug effect indicated that single‐dose BCT197 potently inhibited TNFα secretion, with a clear dose response in the dose range investigated (Figure 1 c). Remarkably, in the once‐daily regimen, placebo‐corrected nadir in drug response appeared not sustained over time (Figure 1 d). Apparent waning of drug effect was seen at about day 4 postonset of treatment, and occurred despite rising drug levels (on average 3‐fold) in reaching steady‐state. It can be speculated that continued p38 inhibition may trigger a negative feedback to response. As expected, the indirect response model with negative feedback (Figure 2 b) dropped OFV by 48 points as opposed to a turnover model without adaptation (Supplementary Table S3), and showed improved diagnostics (Supplementary Figure S3). Goodness‐of‐fit plots of the final model are depicted in Figure 3 e–h. Simulation‐based diagnostics also indicated overall good performance of the model (Supplementary Figure S4). Additional goodness‐of‐fit plots for the placebo data are provided in Supplementary Figure S5.
The population PD parameters are summarized in Table 1. BCT197 inhibited TNFα secretion with an IC50 of 44 µg/L. Predose TNFα levels correlated with IC50 and were modeled as a covariate (ΔOBJ 17). Maximum inhibition from baseline was not complete but plateaued in the typical individual at about 66% (Imax). Single‐dose BCT197 exhibited rapid onset of action with minor hysteresis in drug effect seen (t1/2,kout = 2h). In contrast, the rate constant governing tolerance was found to be relatively small (t1/2,ktol = 10.5 days), suggesting that this may reflect a slow cellular adaptation process.
Model simulations
Continuous dosing of BCT197 10 mg resulted in dose‐limiting acneiform skin rashes, whereas the drug was found to be well tolerated at single high doses up to 75 mg (data not shown). This observation, in addition to tolerance, raised the question whether BCT197 would be most efficacious in a continuous or intermittent regimen. Although the translational value of the ex vivo TNFα bioassay remains to be shown, efforts were made to assess the impact of dosing schedule on drug response through use of simulation.
Three regimens using a cumulative dose of 75 mg in a 2‐week period were considered: single dose (75 mg), an intermittent regimen (25 mg on days 1, 6, and 10), and a continuous once daily regimen (5.36 mg for 14 days). Figure 4 depicts that a single‐dose administration or an intermittent regimen may achieve BCT197 plasma concentrations exceeding IC50 (44 ng/mL) in the majority of subjects, hence may demonstrate profound pathway inhibition (Imax) and shortest time to inhibition nadir (Tmin) (Table 2). These regimens may offer clinical benefit over continuous dosing when rapid and profound inhibition is clinically indicated. Cumulative time below 50% inhibition from baseline as well as average inhibition in the first 2 days decreased with more frequent dosing, while average inhibition from baseline over the 2‐week period was similar across these regimens (Table 2).
Figure 4.
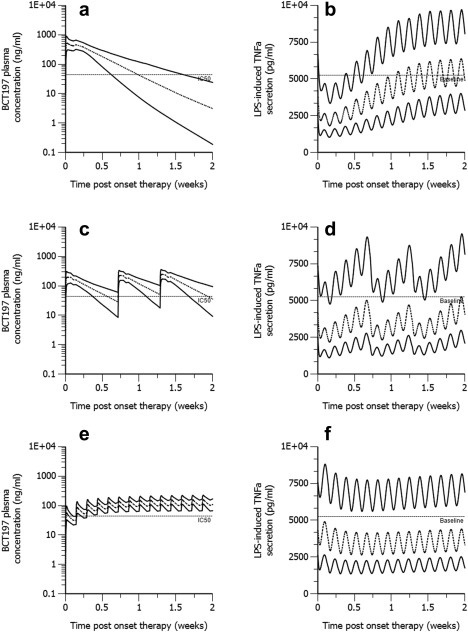
Stochastic simulation of pharmacokinetics and corresponding drug effect in naïve healthy volunteer subjects (n = 500) treated with oral BCT197 given as (a,b) 75 mg single dose, (c,d) 25 mg on days 1, 6, and 10, and (e,f) 5.36 mg once daily for 14 days. All simulations used the PK‐PD model parameters of Table 1. The lower and upper solid lines represent the 5th and 95th percentile of simulated data. The dashed line is the 50th percentile. Horizontal reference lines in the PK and PD indicate the estimated population IC50 and predose PD baseline, respectively.
Table 2.
Stochastic simulation (N = 500) of drug response (inhibition of LPS‐induced TNFα secretion) in relation to dosing schedule
| Total cumulative dose of 75 mg Single treatment period of 14 days | Total cumulative dose: 225 mg/cycle one cycle = 6 weeksa | |||||
|---|---|---|---|---|---|---|
| PD parameter | 75 mg single dose | 25 mg day 1, 6, and, 10 | 5.36 mg once daily for 14 days | 75 mg on day 1, 6, and 10 of each cycle | 37.5 mg weekly | 5.36 mg once daily |
| Imax (%) | 65.4 [59.6;68.6] | 57.7 [49.3;64.2] | 51.1 [41.2;59.0] | 65.0 [58.6;68.3] | 51.1 [43.5;56.4] | 42.9 [37.0;47.3] |
| Tmin (h) | 27 [26;28] | 27 [26;28] | 100 [75;148] | 27 [26;28] | 28 [27;28] | 3 [3;4] |
| Time above 50% inhibition from baseline in 14‐day treatment period or cycle (h) | 62 [30.1;94.0] | 36 [0;96] | 11 [0;92] | 125 [44.1;177] | 24 [0;84] | 0 [0;0] |
| Average % inhibition from baseline over 14‐day treatment period or cycle | 20.8 [12.8;33.4] | 32.9 [23.5;41.0] | 33.8 [25.3;41.3] | 15.1 [15.1;20.3] | 21.5 [14.5;29.1] | 26.4 [20.0;31.5] |
| Average % inhibition from baseline in first 2 days | 54.0 [48.0;58.2] | 44.3 [35.9;51.9] | 28.0 [19.5;37.8] | — | — | — |
| Average % inhibition from baseline in first 3 weeks of the cycle | — | — | — | 30.1 [21.3;40.6] | 21.5 [14.5;29.1] | 26.4 [20.0;31.5] |
Data are median [5th percentile, 95th percentile] of 500 simulations using model parameters of Table 1. Imax, maximum inhibition from baseline; Tmin, time to inhibition nadir.
PD data of cycle 2 (steady‐state).
Simulations involving more long‐term dosing assessed three regimens using a cumulative dose of 225 mg in a 6‐week treatment cycle: an intermittent regimen (75 mg on days 1, 6, and 10 of each cycle), a once‐weekly regimen, and a continuous regimen (5.36 mg once daily for 6 weeks). The intermittent regimen with sufficient drug holiday (>3·t1/2,ktol) between consecutive treatment cycles attenuated tolerance‐mediated erosion of Imax (Table 2). Moreover, in contrast to a continuous regimen, the intermittent regimen was more adequate in maximizing the time above 50% inhibition from baseline in each treatment cycle, with an overall improved time‐averaged drug effect in the first 3 weeks of the treatment cycle. However, neither the intermittent nor the continuous regimen conferred notable and sustained pathway inhibition over the entire treatment cycle.
DISCUSSION
Our PK‐PD work provides supportive evidence that tolerance to p38 inhibition may develop in response to chronic pathway inhibition. The rate constant governing tolerance was found to be relatively small (t1/2,ktol = 10.5 days), suggesting that this may reflect a slow cellular adaptation process. This finding is consistent with studies in RA patients showing that the initial reduction of CRP observed by week 1 was not sustained by week 4 and levels were back to baseline by week 12, despite continuous and adequate drug exposure.20, 21 Tolerance development was modeled by a negative feedback modulator, which may have some basis in mechanism, for magnitude and duration of p38 signal transduction is regulated by several phosphatases that inactivate p38.26 Chronic inhibition of p38 may downregulate dual‐specificity phosphatases, thereby upregulating p38 and JNK activity. Also, inhibition of p38 was found to stop feedback loops that suppress activity of upstream regulatory kinases,27 leading to activation of other proinflammatory MAPK pathways that are involved in TNFα transcription and expression, such as JNK or ERK.28, 29 It is therefore tempting to speculate that tolerance to p38 inhibition is likely to be a class effect. To make more informed decisions, we recommend future clinical studies explore schedule dependency in drug effect and characterize potential for tolerance development in the target population by modeling of relevant PD markers of p38.
BCT197 was found to be well tolerated at single high doses up to 75 mg, whereas continuous dosing at 10 mg resulted in dose‐limiting acneiform skin rashes. This observation, in addition to tolerance, raised the question whether BCT197 would be most efficacious in a low‐dose continuous regimen or intermittent high‐dose regimen. A single high dose or intermittent regimen with Cmax exceeding EC50 and sufficient drug washout between consecutive doses was simulated to achieve rapid and profound pathway inhibition in most subjects for several days (Figure 4). However, at the same total cumulative dose, time below 50% inhibition from baseline as well as average inhibition might decrease with more frequent dosing regimens and increasing duration of treatment (Table 2). We may therefore speculate that BCT197 could possibly have a better therapeutic window in the treatment of acute as opposed to chronic inflammation (e.g., to reduce the frequency and severity of acute exacerbations in COPD).
We acknowledge that the translational value of the ex vivo TNFα bioassay in predicting clinical outcome in the target population remains to be shown. Also, neither the extent nor duration of p38 inhibition required for therapeutic benefit has been established. Simulation‐based interrogation of posology from the bioassay data should therefore be interpreted with caution. Nonetheless, the in vivo relevance of the assay is supported, at least qualitatively, by our findings in Part 3 of the study where a single oral administration of 20 mg or 75 mg BCT197 was found to inhibit by respectively 96% and 97% the serum levels of TNFα following in vivo intravenous LPS challenge (data not shown). Noteworthy, using the same bioassay the improvement of lung function in patients treated with rofluminast was accompanied by a significant suppression of TNFα levels ex vivo.30 Single‐ and multiple‐dose BCT197 potently inhibited ex vivo LPS‐induced TNFα secretion with an IC50 of 44 ng/mL (115 nM), corresponding to a free drug IC50 of 17 nM (fup = 0.15). In‐house studies found the IC50 of BCT197 on p38α to be 12 nM (enzyme‐linked immunosorbent assay (ELISA)) and 37 nM (radiometric). In close agreement with this, BCT197 inhibited LPS‐stimulated TNFα release in the human whole blood assay with a similar free IC50 of 24 nM (total drug 51 nM). In comparison, losmapimod and PH‐797804 inhibited TNFα release in the same assay, with IC50 values of 25 nM and 85 nM, while several other p38 inhibitors appear to have lower potency.20 Inhibition of TNFα secretion was partial, which was also observed for other p38 inhibitors,31, 32 likely as a result of redundancy in MAPK signaling.
BCT197 demonstrated nonlinearity in drug distribution but not oral clearance, with an apparent increase in volume with decreasing dose observed. General approaches to modeling nonlinearity in drug distribution from a central drug compartment are found in the literature.33, 34 However such models could not describe BCT197's tendency toward less than a dose‐proportional increase in Cmax. Structural model diagnostics improved when a distribution delay to a capacity‐limited compartment was inferred. In rats, BCT197 equilibrated rapidly with peripheral tissues, but remained much longer detectable in the skin and eye of the pigmented but not albino animal, suggesting melanin‐containing structures might be a physiological correlate for this compartment (unpublished data). Nonlinear tissue binding unlikely reflects p38 binding because the distribution delay does not reconcile with the ubiquitous expression of p38,35 and drug concentration in the capacity‐limited compartment did not link with drug effect. The implementation is regarded as empirical, because absorption and disposition processes remain confounded in the absence of intravenous data.
BCT197 exhibited a larger absorption plateau with increasing dose, which was described by a mixed first‐ and zero‐order process. In theory, this flat portion might be due to zero‐order absorption or nonlinear elimination, and, unless intravenous data are available, these processes may remain confounded. Nonlinear elimination, however, is not supported by our data, with linear CL/F over the entire dose range observed. Zero‐order absorption can be encountered in the case of solubility‐limited absorption with saturated gastrointestinal drug concentrations at higher doses. This, however, is unlikely, as intestinal Cmax (dose/250 mL) would not meaningfully exceed BCT197's pH‐independent thermodynamic solubility (∼0.2 mg/mL). Moreover, the mixed‐order process in our model was parallel and dose‐independent. The initial dominant first‐order process may suggest a window‐like absorption in the upper gastrointestinal tract,24 while at more distal sites the absorption process becomes progressively affected by an interplay of various other factors. Caco‐2 model data support that intestinal uptake can be modulated by a low‐affinity efflux pump, most likely P‐glycoprotein, which was not saturated up to 0.1 mg/mL (unpublished data). Repetitive cycles of drug efflux and distal re‐uptake, with increasing P‐glycoprotein expression from upper to lower small intestine,36 may therefore be one possible mechanism to explain this absorption behavior.
Taken together, an increased understanding of BCT197's antiinflammatory activity was derived from a PK‐PD model that described the biomarker escape (TNFα) under the hypothesis of a tolerance mechanism. Our model indicates that tolerance development to continuous p38 inhibition is likely to occur in man. This may indicate a mechanism by which the inflammatory response acquires the ability to bypass p38. Simulations of schedule dependence in the drug effect suggests that a high‐dose intermittent treatment regimen of BCT197 may offer clinical advantage and limit the influence of tolerance on drug effect; hence, BCT197 might be more efficacious in treatment of acute as opposed to chronic inflammation disorders.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
This study was supported by Novartis Pharma AG and the Novartis Institute for Biomedical Research. The authors thank Fernando Romero, Helene Bon, Gaetane Woerly, for providing the plasma BCT197 and TNFα data. The authors also thank Marco Londei, Andrew Wright, Aino Launonen, Jessica Valencia, and Christoph Wanke for valuable insights and comments. We also thank Dr Scott Rasmussen, all clinical investigators, and all study coordinators for conducting the study and technicians for technical support.
Author Contributions. S.D.B. wrote the article; W.H., S.D.B., G.B., and R.W. designed the research; S.D.B., W.H., G.B., I.K., and L.M. performed the research; S.D.B., G.B., C.E., A.V., and F.S. analyzed the data; C.E., F.S., and A.V. contributed new reagents/analytical tools.
Conflict of Interest. All authors are employees of Novartis Pharma AG. The authors declare no conflict of interest directly relevant to the content of this publication.
References
- 1. Mannino, D.M. & Buist, A.S. Global burden of COPD: risk factors, prevalence, and future trends. Lancet 370, 765–773 (2007). [DOI] [PubMed] [Google Scholar]
- 2. Ngkelo, A. & Adcock, I.M. New treatments for COPD. Curr. Opin. Pharmacol. 13, 362–369 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Royce, S.G. , Moodley, Y. & Samuel, C.S. Novel therapeutic strategies for lung disorders associated with airway remodelling and fibrosis. Pharmacol. Ther. 141, 250–260 (2014). [DOI] [PubMed] [Google Scholar]
- 4. Decramer, M. , Janssens, W. & Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 379, 1341–1351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brusselle, G.G. , Joos, G.F. & Bracke, K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378, 1015–1026 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Decramer, M. & Janssens, W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir. Med. 1, 73–83 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Vestbo, J. et al Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 187, 347–365 (2013). [DOI] [PubMed] [Google Scholar]
- 8. Lee, J.C. et al A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994). [DOI] [PubMed] [Google Scholar]
- 9. Huh, Y.H. , Kim, S.H. , Kim, S.J. & Chun, J.S. Differentiation status‐dependent regulation of cyclooxygenase‐2 expression and prostaglandin E2 production by epidermal growth factor via mitogen‐activated protein kinase in articular chondrocytes. J. Biol. Chem. 278, 9691–9697 (2003). [DOI] [PubMed] [Google Scholar]
- 10. Gaffey, K. , Reynolds, S. , Plumb, J. , Kaur, M. & Singh, D. Increased phosphorylated p38 mitogen‐activated protein kinase in COPD lungs. Eur. Respir. J. 42, 28–41 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Renda, T. et al Increased activation of p38 MAPK in COPD. Eur Respir. J. 31, 62–69 (2008). [DOI] [PubMed] [Google Scholar]
- 12. Huang, C. , Xie, M. , He, X. & Gao, H. Activity of sputum p38 MAPK is correlated with airway inflammation and reduced FEV1 in COPD patients. Med. Sci. Monit. 19, 1229–1235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Armstrong, J. et al Synergistic effects of p38 mitogen‐activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 338, 732–740 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Fabian, M.A. et al A small molecule‐kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 (2005). [DOI] [PubMed] [Google Scholar]
- 15.CBCT197A2201: an exploratory, randomized, double‐blind, placebo controlled, multi‐center study to assess the efficacy, safety and tolerability of a single and a repeated dose of oral BCT197 in patients with an acute COPD exacerbation. <http://clinicaltrials.gov/show/NCT01332097>.
- 16. MacNee, W. , Allan, R.J. , Jones, I. , De Salvo, M.C. & Tan, L.F. Efficacy and safety of the oral p38 inhibitor PH‐797804 in chronic obstructive pulmonary disease: a randomised clinical trial. Thorax 68, 738–745 (2013). [DOI] [PubMed] [Google Scholar]
- 17. Watz, H. , Barnacle, H. , Hartley, B.F. & Chan, R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double‐blind, placebo‐controlled trial. Lancet Respir. Med. 2, 63–72 (2014). [DOI] [PubMed] [Google Scholar]
- 18. Anand, P. et al Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur. J. Pain. 15, 1040–1048 (2011). [DOI] [PubMed] [Google Scholar]
- 19. Tong, S.E. et al Novel p38alpha mitogen‐activated protein kinase inhibitor shows analgesic efficacy in acute postsurgical dental pain. J. Clin. Pharmacol. 52, 717–728 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Goldstein, D.M. , Kuglstatter, A. , Lou, Y. & Soth, M.J. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J. Med. Chem. 53, 2345–2353 (2010). [DOI] [PubMed] [Google Scholar]
- 21. Sweeney, S.E. The as‐yet unfulfilled promise of p38 MAPK inhibitors. Nat. Rev. Rheumatol. 5, 475–477 (2009). [DOI] [PubMed] [Google Scholar]
- 22. Millan, D.S. What is the potential for inhaled p38 inhibitors in the treatment of chronic obstructive pulmonary disease? Future Med. Chem. 3, 1635–1645 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Savic, R.M. , Jonker, D.M. , Kerbusch, T. & Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodynam. 34, 711–726 (2007). [DOI] [PubMed] [Google Scholar]
- 24. Zhou, H. Pharmacokinetic strategies in deciphering atypical drug absorption profiles. J. Clin. Pharmacol. 43, 211–227 (2003). [DOI] [PubMed] [Google Scholar]
- 25. Wipfler, P. et al Circadian rhythmicity of inflammatory serum parameters: a neglected issue in the search of biomarkers in multiple sclerosis. J. Neurol. 260, 221–227 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Cuenda, A. & Rousseau, S. p38 MAP‐kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta. 1773, 1358–1375 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Cheung, P.C. , Campbell, D.G. , Nebreda, A.R. & Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 22, 5793–5805 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muniyappa, H. & Das, K.C. Activation of c‐Jun N‐terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: a MLK‐3‐MKK7‐dependent mechanism. Cell. Signal. 20, 675–683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sabio, G. & Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Timmer, W. et al The new phosphodiesterase 4 inhibitor roflumilast is efficacious in exercise‐induced asthma and leads to suppression of LPS‐stimulated TNF‐alpha ex vivo. J. Clin. Pharmacol. 42, 297–303 (2002). [DOI] [PubMed] [Google Scholar]
- 31. Gruenbaum, L.M. et al Inhibition of pro‐inflammatory cytokine production by the dual p38/JNK2 inhibitor BIRB796 correlates with the inhibition of p38 signaling. Biochem. Pharmacol. 77, 422–432 (2009). [DOI] [PubMed] [Google Scholar]
- 32. Singh, D. , Smyth, L. , Borrill, Z. , Sweeney, L. & Tal‐Singer, R. A randomized, placebo‐controlled study of the effects of the p38 MAPK inhibitor SB‐681323 on blood biomarkers of inflammation in COPD patients. J. Clin. Pharmacol. 50, 94–100 (2010). [DOI] [PubMed] [Google Scholar]
- 33. Mager, D.E. & Jusko, W.J. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J. Pharmacokinet. Pharmacodynam. 28, 507–532 (2001). [DOI] [PubMed] [Google Scholar]
- 34. Mager, D.E. & Krzyzanski, W. Quasi‐equilibrium pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. Pharmaceut. Resh. 22, 1589–1596 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Zarubin, T. & Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 (2005). [DOI] [PubMed] [Google Scholar]
- 36. Watanabe, T. , Maeda, K. , Nakai, C. & Sugiyama, Y. Investigation of the effect of the uneven distribution of CYP3A4 and P‐glycoprotein in the intestine on the barrier function against xenobiotics: a simulation study. J. Pharmaceut. Sci. 102, 3196–3204 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information