

Abstract
Background
The link between inflammation and cancer has been confirmed by the use of anti-inflammatory therapies in cancer prevention and treatment. 5-aminosalicylic acid (5-ASA) was shown to decrease the growth and survival of colorectal cancer (CRC) cells. Studies also revealed that metformin induced apoptosis in several cancer cell lines.
Methods
We investigated the combinatory effect of 5-ASA and metformin on HCT-116 and Caco-2 CRC cell lines. Apoptotic markers were determined using western blotting. Expression of pro-inflammatory cytokines was determined by RT-PCR. Inflammatory transcription factors and metastatic markers were measured by ELISA.
Results
Metformin enhanced CRC cell death induced by 5-ASA through significant increase in oxidative stress and activation of apoptotic machinery. Moreover, metformin enhanced the anti-inflammatory effect of 5-ASA by decreasing the gene expression of IL-1β, IL-6, COX-2 and TNF-α and its receptors; TNF-R1 and TNF-R2. Significant inhibition of activation of NF-κB and STAT3 transcription factors, and their downstream targets was also observed. Metformin also enhanced the inhibitory effect of 5-ASA on MMP-2 and MMP-9 enzyme activity, indicating a decrease in metastasis.
Conclusion
The current data demonstrate that metformin potentiates the antitumor effect of 5-ASA on CRC cells suggesting their potential use as an adjuvant treatment in CRC.
Keywords: 5-ASA, Inflammation, CRC, NF-κB, STAT3, Metformin
Background
Colorectal cancer (CRC) is the third most common cancer with a lifetime risk of 5 %. A functional link between chronic inflammation and cancer has long been suspected but the complete underlying molecular pathways remain unknown [1]. Inflammatory bowel diseases (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD) are chronic inflammatory disorders of the gastrointestinal tract that lead to impairment of the gastrointestinal structure and function [2–4]. Patients suffering from IBD are at an increased risk of developing CRC, this depends on disease duration, as well as, the extent and severity of inflammation [4]. IBD-associated CRC accounts for 1–2 % of all CRC cases, however, IBD patients are six times more likely to die from CRC than the general population [4, 5]. Although carcinogenesis in IBD follows a different sequence of genetic alterations than that observed in sporadic CRC, patients with sporadic CRC have elevated inflammatory cytokine levels indicative of subclinical inflammatory disease [6–8].
Mesalamine [5-aminosalicylic acid (5-ASA)] is the drug of choice in IBD, mainly UC, for maintenance of remission and treatment of mild relapses. It is a weak COX and LOX inhibitor as it is structurally related to NSAIDs, but unlike these compounds it has low systemic resorption and very few side effects even at high doses for long time periods [9]. Epidemiological investigations suggested that long term 5-ASA consumption decreases the risk of developing CRC in IBD patients [10, 11]. In addition, several experimental studies showed that 5-ASA decreases growth and survival of CRC cells [12–15]. The anti-proliferative and pro-apoptotic effects of 5-ASA on several tumor-derived cell lines have been previously reported and different mechanisms have been proposed namely, inhibition of; Wnt/β-catenin pathway [16], EGFR activation [17–19], NF-κB [20], and COX-2 expression [21]. Moreover, Rousseaux et al. showed that mesalamine activates PPAR-y and enhances its expression in CRC cells [22].
Type 2 DM prevalence is estimated to be 8–18 % in newly diagnosed cancer patients, [23]. Evidence indicates that type 2 DM is positively associated with incidence and mortality of CRC with a 30 % increased relative risk compared with non-diabetic individuals [24]. The associated hyperglycemia and hyperinsulinemia result in direct stimulation of cell growth and DNA synthesis along with an increase in pro-inflammatory cytokines production [25]. Metformin, a biguanide derivative, is an oral anti-diabetic drug used as the first line pharmacological treatment in type 2 DM. It acts mainly by inhibiting hepatic glucose production and decreasing peripheral tissue resistance to insulin. This reduces the circulating glucose and insulin levels thus reducing the incidence of diabetes-related complications [26].
Metformin is a safe drug with the most frequent adverse effects being gastrointestinal symptoms but they are usually mild and transient [27]. Many studies revealed the beneficial effect of metformin in decreasing CRC risk [28, 29]. Moreover, it was found to synergistically increase apoptosis of CRC cells in-vitro when combined with other chemotherapy drugs [30, 31].
Since inflammation and hyperglycemia are associated with increased risks of cancer as well as of the major causes of cancer progression, the aim of the present study was to evaluate the effect of combining 5-ASA and metformin on CRC cell lines.
Methods
Drugs
Metformin was obtained from CID Co. (Cairo, Egypt). It was freshly dissolved in culture medium, Roswell Park Memorial Institute 1640 (RPMI-1640), as 80 mM stock solution. 5-ASA was kindly provided by Minapharm (Cairo, Egypt) and dissolved in phosphate buffered saline (PBS) just before use. It was added to the medium with the final maximum concentration of PBS 0.1 % v/v, and experiments carried out protected from light.
Chemicals and antibodies
PBS, RPMI-1640 medium, and sulphorodamine-B (SRB) were all purchased from Sigma Aldrich (St Louis, Missouri, USA). Polyclonal anti-human Bax and Bcl-2 antibodies were obtained from Invitrogen (Carlsbad, CA, USA). Monoclonal anti-human β-actin was obtained from Sigma-Aldrich. All other chemicals were of reagent grade and used without further purification.
Cell culture
The two available human colorectal cancer cell lines, Caco-2 and HCT-116, were obtained from the American Type Culture Collection (Manassas, USA). They were maintained and grown at the Egyptian National Cancer Institute (Cairo, Egypt) in RPMI-1640 supplemented with 10 % fetal bovine serum, 2 mM L-glutamine, 1.5 g/l sodium bicarbonate and 1 % penicillin/streptomycin. Cells were cultured in a humidified incubator at 37 °C in 5 % carbon dioxide (CO2). No ethical approval was required for any aspect of this study.
Cytotoxicity assay
Cytotoxicity was evaluated using the SRB assay. Briefly, exponentially growing cells were seeded in 96-well microtitre plates at an initial density of 5 × 103 /well. After 24 h, metformin and 5-ASA were added with various concentrations and incubated at 37 °C for 48 h to determine their IC50s (the concentration of the drug required to produce 50 % inhibition of cell growth). Cells were fixed with 10 % trichloroacetic acid for 1 h at 4 °C and stained with 0.4 % SRB for 30 min., wells were then washed four times with 1 % acetic acid and air-dried. The dye was solubilized with 10 mM Tris base (pH 10.5) and the optical density (O.D.) was measured spectrophotometrically at 570 nm with the microplate reader (Tecan SunriseTM, Männedorf, Switzerland). The percentage of cell survival was calculated as follows: survival fraction = O.D. (treated cells)/O.D. (control cells).
The IC50 values of the two cancer cell lines after 48 h treatment were calculated using sigmoidal dose response curve-fitting models (Graphpad Prism Software, version 5.03, Inc. Avendia de la Playa La Jolla, USA).
Caco-2 and HCT-116 cells were then treated with a combination of subIC50 concentrations of metformin and different concentrations of 5-ASA to determine the concentration at which 5-ASA would give a significant difference than metformin alone.
Oxidative stress markers
Caco-2 and HCT-116 cells were grown in 75 cm2 flasks and allowed to adhere for 24 h. Cells were treated with metformin, 5-ASA or the combination of both substances for 48 h then collected by trypsinisation. The cell pellet was washed twice with PBS. To the cell suspension 1 ml of 20 % (w/v) trichloroacetic acid (TCA; Sigma, USA) containing 0.8 % (w/v) thiobarbituric acid (TBA; Sigma, USA) was added and mixed well. MDA (decomposition product of the lipid peroxidation process) level was determined colorimetrically by measuring the pink pigment product resulting from the reaction of one molecule of MDA with two molecules of TBA at 535 nm. Protein was measured by the method of Bradford and MDA is expressed in nmol MDA per mg protein. For determination of total SH group, protein precipitation was carried out using 10 % TCA then samples were centrifuged at 3000 rpm for 10 min at 4 °C. The resultant supernatant was mixed with phosphate buffer and Ellman’s reagent (Sigma-Aldrich, Milan, Italy). The method depends on the reduction of thiol reagent; Ellman’s reagent by the sulfhydryl SH group in GSH to form the yellow chromophore; 5-thionitrobenzoic acid, measured spectrophotometrically at 412 nm. GSH is expressed in nmol GSH per mg protein.
Real-time PCR analysis
Analysis of COX-2, IL-1β, IL-6, TNF-α, TNF-R1, and TNF-R2 RNA expression was performed by real-time PCR. Caco-2 and HCT-116 cells, treated with metformin and 5-ASA, were collected by trypsynisation. Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Milan, Italy), according to the manufacturer’s instructions. Concentration and purity of the RNA was checked by A260/A280 optical density ratio. RNA (1 μg/sample) was retro-transcribed into complementary DNA (cDNA) and 1 μl of cDNA/sample was then amplified using the following conditions: denaturation 1 min at 95 °C, annealing 30s at 60 °C for COX-2, TNF-R2, IL-1β, and β-actin or 30s at 57 °C for TNF-α, TNF-R1 and IL-6, followed by 30s of extension at 72 °C. Primers sequence was as shown in Table 1 and they were obtained from Invitrogen (Milan, Italy). RT-PCR was performed using the IQ SYBER Green Supermix (Bio-Rad Laboratories, Milan, Italy). mRNA levels were calculated relative to β-actin, which was unaffected by metformin and 5-ASA treatment.
Table 1.
Primer sequences
| Gene | Primer |
|---|---|
| COX-2 | FWD: 5′-CCC TTC CTT CGA AAT GCA AT-3′ REV: 5′-CAT TTG AAT CAG GAA GCT GC-3′ |
| IL-1β | FWD: 5′-GGA CAA GCT GAG GAA GAT GC-3′ REV: 5′-TTT TTT GCT GTG AGT CCC GG-3′ |
| IL-6 | FWD: 5′-GAG ACT TGC CTG GTG AAA AT-3′ REV: 5′-CAG GGG TGG TTA TTG CAT CT-3′ |
| TNF-α | FWD: 5′ ACA AGC CTG TAG CCC ATG TT-3′ REV: 5′ AAA GTA GAC CTG CCC AGA CT-3′ |
| TNF-R1 | FWD: 5′-CGC TTC AGA AAA CCA CCT CAG AC-3′ REV: 5′-CCA AAG AAA ATG ACC AGG GGC-3′ |
| TNF-R2 | FWD: 5′-GCT CTG ACC AGG TGG AAA CTC AAG-3′ REV: 5′-GGA TGA AGT CGT GTT GGA GAA CG-3′ |
| β-actin | FWD: 5′-TCT GGC ACC ACA CCT TCT ACA ATG-3′ REV: 5′-AGC ACA GCC TGG ATA GCA ACG-3′ |
Western blot analysis
Aliquots of protein supernatants containing equal amounts of protein and sodium dodecyl sulphate (SDS) reducing buffer were boiled for 5 min. They were then electrophoresed on SDS-polyacrylamide gels and transferred to polyvinyldiene difluoride membranes. The membranes were blocked with 5 % non-fat dry milk and probed with specific primary antibodies of monoclonal anti-Bax and Bcl2 antibodies followed by incubation with peroxidase-conjugated secondary antibodies. The blots were developed with Amersham ECL western blotting kit (GE Healthcare, Amersham Place, Little Chalfont, U.K) according to the manufacturer’s instructions. The blots were quantified by ChemiDoc XRS 4.6.9 (Bio-Rad Laboratories Inc., Hercules, CA, USA.) software and protein loading was corrected for β-actin as loading control.
ELISA techniques
The different proteins were determined in both cell lines according to the kit manufacturer’s instructions. For NF-κB the Kamiya Biomedical assay kit (Seattle, USA) was used, while the RayBiotech (Georgia, USA) was used for TNF-α, IL-6, STAT3, MMP-2 and −9. The assay employs the quantitative sandwich enzyme immunoassay technique. A monoclonal antibody specific for human NF-κB, TNF-α, IL-6, STAT3, MMP-2 or −9 has been pre-coated onto a microplate. Samples were pipetted into the wells and the measured human biomarkers present in the solutions were bound by the immobilized antibody. A yellow color is developed which is proportional to the amount of NF-κB/ TNF-α/IL-6/ STAT3/MMP-2/MMP-9 bound. The intensity of the color is measured at 450 nm.
Caspase-3 activity was measured based on spectrophotometric detection of the chromophore p-nitroaniline (pNA) at 405 nm after cleavage from its labelled substrate DVD-pNA. Protein concentration of the samples was analyzed and normalized in lysis buffer to equal protein concentrations. Colorimetric assay (Caspase-3/CPP32, BioVision, Milpitas, USA) was used according to the manufacturer’s instructions.
Scratch wound healing assay
Caco-2 and HCT-116 cells were grown in 6 well plates and allowed to adhere for 24 h. Gently and slowly the monolayer was scratched in one direction with a new 1 ml pipette tip across the center of the well. The resulted gap distance therefore equals to the outer diameter of the end of the tip. The wells were then washed twice with medium to remove the detached cells. Cells were treated with metformin, 5-ASA or the combination of both substances for 48 h. Cells were washed twice with 1x PBS, then fix the cells with 3.7 % paraformaldehye for 30 min and they were stained with 1 % crystal violet in 2 % ethanol for 30 min. Photos were taken for the stained monolayer on a microscope. The gap distance was measured using the Leica Qwin-Plus software (Leica Microsystems, UK).
Statistical analysis
All the data are expressed as mean ± SD from three different experiments and comparisons between means were carried out using one way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparisons test. A probability level of less than 0.05 was accepted as being significant in all types of statistical tests. All statistical analysis was performed using GraphPad InStat, version 5.0 (GraphPad, San Diego, California, USA).
Results
Co-incubation of metformin enhanced 5-ASA-mediated inhibition of cell counts in Caco-2 and HCT-116 cells
Caco-2 cells were treated with 13 mM metformin, 2.5 mM 5-ASA, or the combination of both for 48 h. Similarly, HCT-116 cells were treated with 8 mM metformin, 3 mM 5-ASA, or the combination of both for 48 h. Significant inhibition of cell proliferation were seen in all treatment groups with the combination group showing the highest inhibition reaching 55 % of the control compared to nearly 40 % reached by the solo treatments in both cell lines Fig. 1.
Fig. 1.
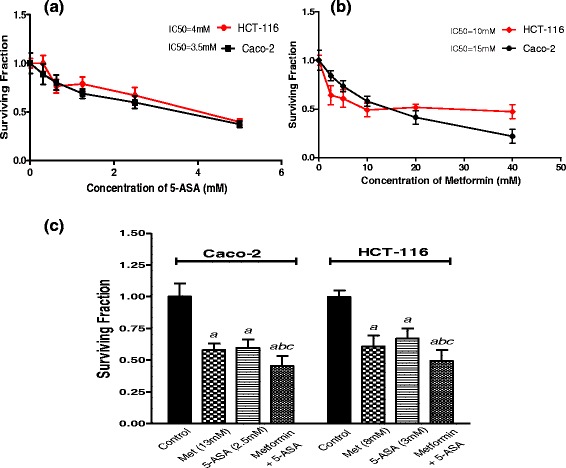
Effect of different concentrations of (a) 5-ASA and (b) Metformin on surviving fractions of Caco-2 and HCT-116 cells treated for 48 h. c Surviving fraction of Caco-2 and HCT-116 cells after treatment with subIC50 concentrations of metformin, 5-ASA and a combination of both for 48 h. All data are expressed as mean ± SD of 3 separate experiments performed in triplicates. The statistical significance of the results was analyzed using one way ANOVA followed by Tukey-Kramer multiple comparison test. a Significantly different from control, b from metformin and c from 5-ASA (P <0.05)
Exaggerated increase in oxidative stress upon combined treatment with metformin and 5-ASA
Combination of subIC50 concentrations of both drugs produced a pronounced increase in MDA level (Fig. 2a) and a greater decrease in the intracellular GSH level than each drug alone (Fig. 2b). In Caco-2 cells the combination of metformin and 5-ASA resulted in a significant increase in MDA level of 3 folds compared to, 1.9 and 1.75 folds produced by either of the treatments. In addition, a reduction of GSH exceeding 85 % was observed compared to nearly 50 % decrease produced by each drug alone. A similar effect was produced when both drugs were added to the HCT-116 cells where MDA level showed an elevation mounted to 2.5 folds compared to nearly 1.7 fold increase produced by solo treatments. Moreover, intracellular GSH depletion reached 65 %, while only a 35 % decrease in GSH levels was observed after adding metformin or 5-ASA alone.
Fig. 2.
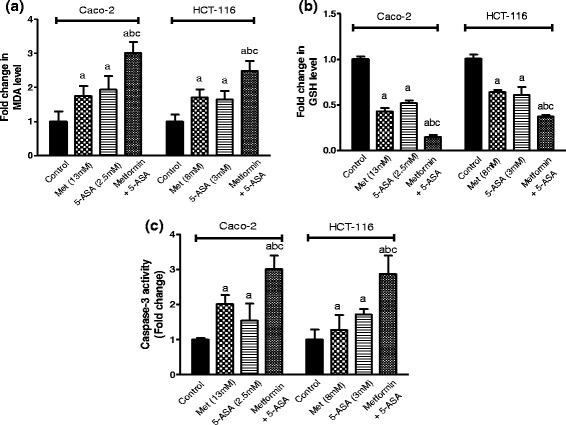
Effect of treatment of Caco-2 and HCT-116 cells for 48 h with subIC50 concentrations of metformin, 5-ASA or the combination of both drugs on oxidative stress markers measured as (a) MDA level (b) GSH level and on (c) caspase-3 activity. Data are indicative of 3 separate experiments. The statistical significance of the results was analyzed using one way ANOVA followed by Tukey-Kramer multiple comparison test. a Significantly different from control, b from metformin and c from 5-ASA (P <0.05)
Co-incubation of metformin enhances 5-ASA-induced apoptosis
The addition of metformin to 5-ASA succeeded in activating the caspase-3 enzyme more than the individual treatments in both cell lines reaching 3 fold the control group (Fig. 2c). Solo treatments in Caco-2 and HCT-116 cells increased active caspase-3 to levels ranging from 1.2–1.7 folds.
The increase in caspase-3 activity was accompanied by increased apoptotic Bax levels and decreased expression of the anti-apoptotic Bcl-2 protein in both cell lines (Fig. 3), following treatment with 5-ASA, metformin or their combination after 48 h. The combination group showed the most significant change in Bax levels, as compared to individual treatments where it produced a 16 and 13 folds increase in Caco-2 and HCT-116 cells, respectively. On the other hand, the anti-apoptotic Bcl-2 expression decreased by 85 % and 80 % in Caco-2 and HCT-116 cells after combination treatment.
Fig. 3.
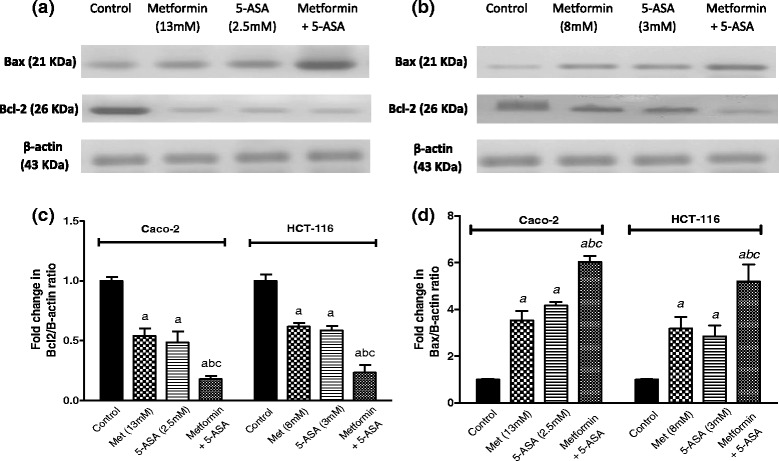
Western blot for Bcl-2 and Bax levels in (a) Caco-2 and (b) HCT-116 cells after treatment with metformin, 5-ASA or their combination. One representative western blot of three independent experiments is shown. Effect of treatment of Caco-2 and HCT-116 cells for 48 h with subIC50 concentrations of metformin, 5-ASA or the combination of both drugs on (c) Bcl-2 level (d) Bax level. All data are expressed as mean ± SD of 3 separate experiments. The statistical significance of the results was analyzed using one way ANOVA followed by Tukey-Kramer multiple comparison test. a Significantly different from control, b from metformin and c from 5-ASA (P <0.05)
Combination of metformin and 5-ASA downregulates TNF-α, TNF-α receptors (TNF-R1 and TNF-R2), and IL-1β and inhibits the activation of NF-κB
All treatment groups produced a prominent decrease in the expression of TNF-α and its receptors (Fig. 4). In Caco-2 cells, the combination group did not significantly differ from the 5-ASA group although it produced 85 % decrease in TNF-α expression while 5-ASA produced 80 % decrease. However, in HCT-116 cells the combination of both drugs downregulated TNF-α expression by 80 % compared to 60 % decrease produced by either drug. These results were in agreement with the protein levels of TNF-α that decreased significantly after the combination treatment compared to either of the drugs alone in both cell lines as shown in Fig. (4e). Moreover, following treatment of Caco-2 cells with the two drugs, there was a downregulation of both TNF-α receptors that was greater and statistically significant compared to the solo drug treatments where TNF-R1 and TNF-R2 gene expression levels decreased by 95 % and 90 % compared to the control group. In addition, the combination group in HCT-116 cells decreased TNF-R2 expression level reaching solely 10 % compared to the untreated cells.
Fig. 4.

Effect of treatment of Caco-2 and HCT-116 cells for 48 h with subIC50 concentrations of metformin, 5-ASA or the combination of both drugs on (a) TNF-α (b) TNF-R1 (c) TNF-R2 and (d) IL-1β gene expression levels and on (e) TNF-α and (f) IL-6 protein levels. All data are expressed as mean ± SD of 3 separate experiments. The statistical significance of the results was analyzed using one way ANOVA followed by Tukey-Kramer multiple comparison test. a Significantly different from control, b from metformin and c from 5-ASA (P <0.05)
Concerning IL-1β gene expression level, after treatment of Caco-2 and HCT-116 cells with a combination of metformin and 5-ASA, IL-1β levels decreased by 55 % compared to the control (Fig. 4d). This decrease was statistically significant from the metformin group but not the 5-ASA group. On the other hand, a 75 % decrease was observed in the HCT-116 cells that was significant from all treatment groups.
In parallel with the previous results, NF-κB levels (Fig. 5c) showed significant decrease to 29 % and 51 % in Caco-2 and HCT-116 cells upon the addition of metformin to 5-ASA.
Fig. 5.

Effect of treatment of Caco-2 and HCT-116 cells for 48 h with subIC50 concentrations of metformin, 5-ASA or the combination of both drugs on (a) IL-6 and (b) COX-2 gene expression levels. Fold change in levels of (c) NF-κB and (d) STAT3 transcription factors in Caco-2 and HCT-116 cells after 48 h of treatment with metformin, 5-ASA or their combination. All data are expressed as mean ± SD of 3 separate experiments. The statistical significance of the results was analyzed using one way ANOVA followed by Tukey-Kramer multiple comparison test. a Significantly different from control, b from metformin and c from 5-ASA (P <0.05)
Intensified inhibition of IL-6 gene expression and inhibition of STAT3 activation upon addition of metformin to 5-ASA
The expression of IL-6 (Fig. 5a) after exposure to metformin or 5-ASA was decreased significantly by 30 % for both drugs in the Caco-2 cell line. On HCT-116 cells, metformin and 5-ASA produced a 65 % and 50 % decrease in IL-6 expression, compared to the control. Furthermore, the combination resulted in an exaggerated decrease of 80 % in IL-6 expression compared to either drug alone in both cell lines. Confirming the gene expression results, the IL-6 protein level showed a significant decrease after treatment in both cell lines than the solo treatments as shown in Fig. (4f).
As illustrated in Fig. (5d) the greatest inhibition of STAT3 activity was observed in the combination group reaching 29 % and 50 % in Caco-2 and HCT-116 cell lines.
Metformin exaggerates the 5-ASA-induced inhibition of COX-2 enzyme gene expression
The expression of COX-2 gene, after treatment of Caco-2 cells with metformin decreased by 75 % compared to control, while treatment with the anti-inflammatory agent 5-ASA caused a further downregulation in the gene expression by 85 % (Fig. 5b). The COX-2 gene expression reached its maximum inhibition (90 %) when the Caco-2 cells were treated by both agents.
The same pattern was reflected on the HCT-116 cells, where the COX-2 gene expression was downregulated by metformin, 5-ASA and their combination in this descending order by 50 %, 60 % and 90 %, compared with the control group.
Pronounced suppression of matrix metalloproteinase-2 and −9 and inhibition of cell migration after combining metformin and 5-ASA
The addition of metformin to 5-ASA in Caco-2 cells significantly lowered MMP-2 and −9 levels to 33 % and 35 % respectively (Fig. 6). Similarly, treatment of the HCT-116 cells with a combination of both drugs decreased MMP-2 and −9 enzyme levels to 34 % and 21 % compared to the control. The results of the scratch wound healing assay were in accordance with the MMP levels as the combination of both drugs resulted in a 25 % decrease from control at zero time in Caco-2 cells and a 20 % inhibition in HCT-116 cells. Moreover the results of this assay confirm the fact that HCT-116 cells are highly tumorigenic and metastatic Fig. 7.
Fig. 6.
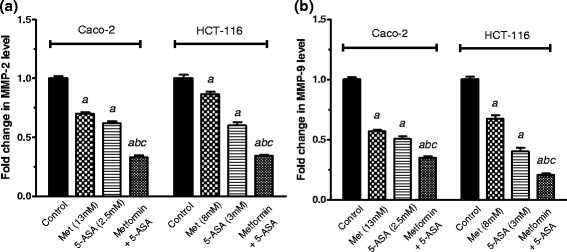
Fold change in (a) MMP-2 and (b) MMP-9 enzyme levels after 48 h of treatment of Caco-2 and HCT-116 cells with metformin, 5-ASA or their combination. All data are expressed as mean ± SD of 3 separate experiments. The statistical significance of the results was analyzed using one way ANOVA followed by Tukey-Kramer multiple comparison test. a Significantly different from control, b from metformin c from 5-ASA (P <0.05)
Fig. 7.
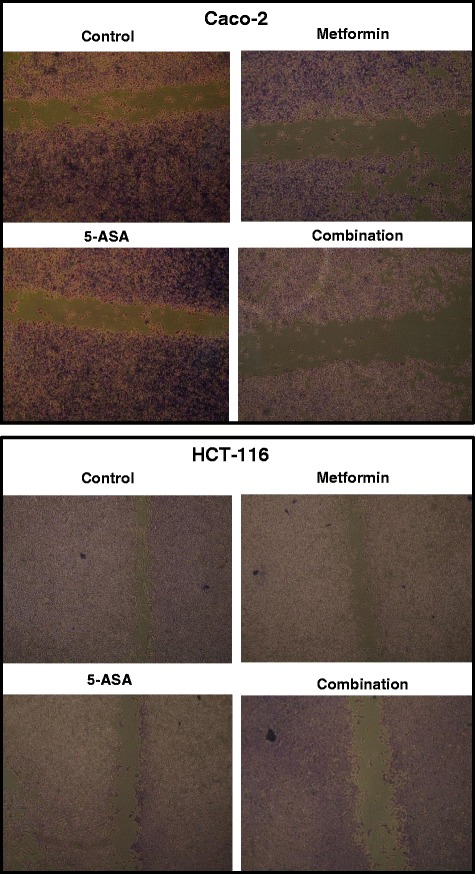
Photos of the stained monolayer of Caco-2 and HCT-116 cells without treatment and after treatment for 48 h with subIC50 concentrations of metformin, 5-ASA or the combination of both drugs
Discussion
In the current study, we used 2 colorectal cancer cell lines Caco-2 and HCT116. Caco-2 cell line is colorectal adenocarcinoma cells with intermediate characteristics, as it is spontaneously differentiating tumor in normal culture, it is tumorigenic but hasn’t a metastatic behavior in vivo.. Treatment with very low drug concentrations of metformin, 5-ASA and their combination was coupled by a significant decrease in the surviving fraction of CRC cells. The increased cell death could be explained by intracellular GSH depletion and increase in MDA level, indicating that cell death could be initiated or underwent under oxidative stress originating from modulation of the intracellular redox system. Our results are in accordance with previous studies showing that 5-ASA or metformin can increase oxidative stress in certain types of cancer thus leading to increased apoptosis [12, 32].
Another explanation for the increased apoptosis is the inhibition of inflammation. The central players involved in inflammation-mediated tumor progression include IL-1β, IL-6 and TNF-α [33]. Data from several inflammation-associated cancer models implicate these inflammatory cytokines in being the bridge between inflammation and tumorigenesis [34]. On CRC cells, addition of metformin to 5-ASA significantly reduced the gene expression of TNF-α and its receptors, TNF-R1 and TNF-R2.
Altered expression of the genes coding for TNF-α and its receptors was observed in neoplastic diseases [35]. In CRC, malignant cell derived TNF-α, enhances the growth and metastasis of the tumor as evidenced from animal models [36]. Chronic TNF-α production by malignant or host cells or both may directly contribute to oncogene activation, DNA damage and metastasis [37]. The main receptor mediating TNF-α effects is TNF-R1 which when stimulated, induces activation of NF-κB after a series of intracellular events [38]. On the other hand, the increased TNF-α level could be due to the continuous activation of NF-κB [39]. NF- κB activation has long been known to suppress apoptosis as it promotes transcription of anti-apoptotic genes, one of which is Bcl-2 [40]. The decline in TNF-α or TNF-R1 genetic expression will therefore decrease NF-κB activation and thus increase apoptosis. In our study, combination of 5-ASA and metformin resulted in the greatest inhibition in the TNF-α and TNF-R1 gene expression, and NF- κB protein level with a subsequent increase in apoptosis, evident by decreased level of Bcl-2 protein expression.
Although TNF-α mediates its action mainly through TNF-R1 activation, TNF-R2 has been linked to increased colon cancer cell growth and proliferation [41, 42]. It was found that both IL-6 and TNF-α interact to induce TNF-R2 expression and function in colon cancer cells, suggesting that a specific microenvironment of multiple cytokines is required to induce TNF-R2, as found in IBD or IBD associated CRC.
Our study revealed a pronounced down-regulation of TNF-R2 gene expression in Caco-2 and HCT-116 cell lines in all treatment groups with the greatest effect being that of the combination group. There were no previous studies conducted on 5-ASA or metformin that assessed their effect on TNF-R2 expression. In this study evidence of the effect of both drugs either alone or in combination on TNF-R2 expression levels was demonstrated.
Together with TNF-α, IL-1β also increases NF-κB transcription thus increasing tumor adhesiveness, invasion and angiogenesis [43]. Likewise, activation of NF-κB results in an increase in IL-1β [39]. Polymorphisms of IL-1β are associated with increased cancer risk [44]. Caco 2 cells expresses active COX 2 and epidermal growth factor receptor (EGFR). On the other hand HCT-116 cell line are undifferentiated colorectal carcinoma cells that don’t express COX2 and are positive for transforming growth factor beta 1 (TGF beta 1) and beta 2 (TGF beta 2) expression. In the current study, combining metformin with 5-ASA showed a remarkable decrease in IL-1β gene expression in both cell lines respectively. The decrease in NF-κB protein level, cell proliferation, increase in apoptosis and decrease in MMP-2 and −9 expression, all support the inhibition of the downstream signaling pathway of IL-1β.
Adding to the panoply of molecules, IL-6 is one of the best characterized pro-tumorigenic cytokines. IL-6 is not only produced by immune cells but also by epithelial and malignant cells. Its production is induced by a variety of stimuli one of which is NF-κB. Therefore, IL-6 production can be stimulated indirectly by TNF-α or IL-1β that activate the NF-κB or directly by PGE2 [45]. IL-6 promotes colon cancer cell proliferation, survival, migration, invasion, metastasis, angiogenesis and inflammation [46]. These effects are a result of the activation of the downstream target of IL-6, the STAT3 transcription factor. Persistent activation of STAT3 has been reported in a variety of human tumors, including the colon [47]. This persistent activation can also be the reason of increased interleukins levels [48].
The current study revealed that addition of metformin to 5-ASA resulted in a prominent decrease in IL-6 mRNA levels that was accompanied by a decrease in STAT3 level and therefore increased cell death. It is demonstrated herein that the combination group expressed the lowest Bcl-2 and highest Bax protein levels. This was associated with increased caspase-3 activity and apoptosis in both cell lines. This effect may be attributed in part to the suppression of IL-6 and STAT3.
COX-2 is another important pro-inflammatory mediator that is implicated in the process of carcinogenesis. It was shown to be upregulated early in CRC and plays a major role in its progression [49] by regulating the process of proliferation, angiogenesis and metastasis [50]. It was previously demonstrated that one of the mechanisms of 5-ASA to decrease proliferation was by inhibiting COX-2 enzyme [21]. Metformin as well, has been reported to inhibit inflammatory responses and COX-2 expression [51], however, its effect on COX-2 expression in CRC cells has not been clarified. Our data show that combination of both drugs resulted in an exaggerated inhibition of COX-2 expression than that produced by the solo treatments suggesting a synergistic effect. This explains the inhibition of MMP’s level that was observed in the combination group since COX-2 overexpression increases invasiveness of CRC cells by inducing MMP expression [52]. It was reported that COX-2 inhibitors decrease MMP-2 and −9 expression [53]. Therefore, the decrease in MMP-2 and −9 level could be due to inhibition of COX-2 and the NF-κB and STAT3 signaling pathways. Several studies showed that metformin or 5-ASA can inhibit MMP’s expression [54–56].
Conclusions
Our data (Fig. 8) clarify that treatment of CRC cells with a combination of 5-ASA and metformin increases cell death than either drug alone. This increase in apoptosis may be due to inhibition of the STAT3 and NF-κB signaling pathways. The decreased level of cytokines that stimulate those pathways (TNF-α, IL-1β and IL-6), lowered protein levels of the activated transcription factors (STAT3 and NF-κB) and decreased protein or expression levels of their target genes (TNF-α, IL-6, COX-2, MMP-2, MMP-9 and Bcl-2) confirm the inhibition of the key pathways in inflammation-mediated tumor promotion and progression.
Fig. 8.
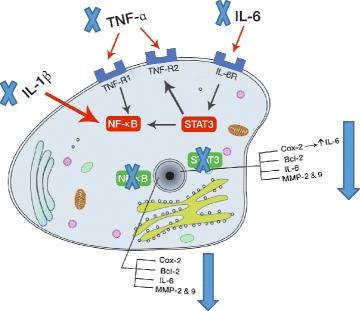
Addition of 5-ASA and metformin on CRC cells inhibit the NF-κB and STAT3 signaling pathways. Inhibition of gene expression of IL-1β and TNF-α decreases activation of NF-κB resulting in inhibition of target gene expression. Moreover, decrease in IL-6 mRNA levels reduces STAT3 activation and thus decreasing target gene transcription. The target genes for both pathways include COX-2, Bcl-2, IL-6 and MMP’s. Low levels of COX-2 and IL-6 gene expression and Bcl-2, MMP-2 and −9 protein levels confirm the inhibition of both the signaling pathways
Acknowledgements
This work was supported and in part by Pharmacology and Toxicology Department, Faculty of Pharmacy and Cancer Biology Department, National Cancer Institute, Cairo University.
Abbreviations
- COX
cyclooxygenase
- CRC
colorectal cancer
- DM
diabetes mellitus
- EGFR
Epidermal growth factor receptor
- GSH
glutathione
- IBD
inflammatory bowel disease
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- LOX
Lypooxygenase
- MDA
malondialdehyde
- MMP
matrix metalloproteinase
- NF-κB
nuclear factor kappa B
- NSAIDs
Non-steroidal anti-inflammatory drugs
- O.D.
optical density
- PPAR-y
peroxisome-proliferator-activated receptor gamma
- RPMI-1640
Roswell Park Memorial Institute 1640
- STAT3
signal transducer and activator of transcription 3
- TNF-R
Tumor necrosis factor receptor
- TNF-α
Tumor necrosis factor-α
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM carried out the practical work except western blotting and drafted the manuscript. MG did the statistical analysis, carried western blot and drafted the manuscript. AA-S participated in the design of the work and revised the manuscript. SS offered tissue culture cancer lab and shared with A A-S in designing and reviewing the manuscript. All authors read and approved the final manuscript.
Contributor Information
Mona M. Saber, Email: mona.magdy@pharma.cu.edu.eg
May A. Galal, Email: mayouya2010@yahoo.com
Afaf A. Ain-Shoka, Email: drafafainshoka@yahoo.com
Samia A. Shouman, Email: samia.shouman@nci.cu.edu.eg
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3(7):521–33. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 3.Eaden J. Review article: colorectal carcinoma and inflammatory bowel disease. Aliment Pharmacol Ther. 2004;20(Suppl 4):24–30. doi: 10.1111/j.1365-2036.2004.02046.x. [DOI] [PubMed] [Google Scholar]
- 4.Choi PM, Zelig MP. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut. 1994;35(7):950–4. doi: 10.1136/gut.35.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lennard-Jones JE. Cancer risk in ulcerative colitis: surveillance or surgery. Br J Surg. 1985;72(Suppl):S84–S86. doi: 10.1002/bjs.1800721344. [DOI] [PubMed] [Google Scholar]
- 6.Rhodes JM, Campbell BJ. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trends Mol Med. 2002;8(1):10–6. doi: 10.1016/S1471-4914(01)02194-3. [DOI] [PubMed] [Google Scholar]
- 7.Erdman SE, Poutahidis T. Roles for inflammation and regulatory T cells in colon cancer. Toxicol Pathol. 2010;38(1):76–87. doi: 10.1177/0192623309354110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15(2):79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brimblecombe R. Mesalazine: a global safety evaluation. Scand J Gastroenterol Suppl. 1990;172:66. doi: 10.3109/00365529009091915. [DOI] [PubMed] [Google Scholar]
- 10.Rubin DT, LoSavio A, Yadron N, Huo D, Hanauer SB. Aminosalicylate therapy in the prevention of dysplasia and colorectal cancer in ulcerative colitis. Clin Gastroenterol Hepatol. 2006;4(11):1346–50. doi: 10.1016/j.cgh.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Eaden J. Review article: the data supporting a role for aminosalicylates in the chemoprevention of colorectal cancer in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18(Suppl 2):15–21. doi: 10.1046/j.1365-2036.18.s2.3.x. [DOI] [PubMed] [Google Scholar]
- 12.Fina D, Franchi L, Caruso R, Peluso I, Naccari GC, Bellinvia S, Testi R, Pallone F, Monteleone G. 5-aminosalicylic acid enhances anchorage-independent colorectal cancer cell death. Eur J Cancer. 2006;42(15):2609–16. doi: 10.1016/j.ejca.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda I, Tomimoto A, Wada K, Fujisawa T, Fujita K, Yonemitsu K, Nozaki Y, Endo H, Takahashi H, Yoneda M, Inamori M, Kubota K, Saito S, Nagashima Y, Nakagama H, Nakajima A. 5-aminosalicylic acid given in the remission stage of colitis suppresses colitis-associated cancer in a mouse colitis model. Clin Cancer Res. 2007;13(21):6527–31. doi: 10.1158/1078-0432.CCR-07-1208. [DOI] [PubMed] [Google Scholar]
- 14.Clapper ML, Gary MA, Coudry RA, Litwin S, Chang WC, Devarajan K, Lubet RA, Cooper HS. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2008;14(10):1341–7. doi: 10.1002/ibd.20489. [DOI] [PubMed] [Google Scholar]
- 15.Reinacher-Schick A, Schoeneck A, Graeven U, Schwarte-Waldhoff I, Schmiegel W. Mesalazine causes a mitotic arrest and induces caspase-dependent apoptosis in colon carcinoma cells. Carcinogenesis. 2003;24(3):443–51. doi: 10.1093/carcin/24.3.443. [DOI] [PubMed] [Google Scholar]
- 16.Bos CL, Diks SH, Hardwick JC, Walburg KV, Peppelenbosch MP, Richel DJ. Protein phosphatase 2A is required for mesalazine-dependent inhibition of Wnt/beta-catenin pathway activity. Carcinogenesis. 2006;27(12):2371–82. doi: 10.1093/carcin/bgl071. [DOI] [PubMed] [Google Scholar]
- 17.Svrcek M, Cosnes J, Tiret E, Bennis M, Parc Y, Flejou JF. Expression of epidermal growth factor receptor (EGFR) is frequent in inflammatory bowel disease (IBD)-associated intestinal cancer. Virchows Arch. 2007;450(2):243–4. doi: 10.1007/s00428-006-0338-7. [DOI] [PubMed] [Google Scholar]
- 18.Spano JP, Lagorce C, Atlan D, Milano G, Domont J, Benamouzig R, Attar A, Benichou J, Martin A, Morere JF, Raphael M, Penault-Llorca F, Breau JL, Fagard R, Khayat D, Wind P. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16(1):102–8. doi: 10.1093/annonc/mdi006. [DOI] [PubMed] [Google Scholar]
- 19.Monteleone G, Franchi L, Fina D, Caruso R, Vavassori P, Monteleone I, Calabrese E, Naccari GC, Bellinvia S, Testi R, Pallone F. Silencing of SH-PTP2 defines a crucial role in the inactivation of epidermal growth factor receptor by 5-aminosalicylic acid in colon cancer cells. Cell Death Differ. 2006;13(2):202–11. doi: 10.1038/sj.cdd.4401733. [DOI] [PubMed] [Google Scholar]
- 20.Bantel H, Berg C, Vieth M, Stolte M, Kruis W, Schulze-Osthoff K. Mesalazine inhibits activation of transcription factor NF-kappaB in inflamed mucosa of patients with ulcerative colitis. Am J Gastroenterol. 2000;95(12):3452–7. doi: 10.1111/j.1572-0241.2000.03360.x. [DOI] [PubMed] [Google Scholar]
- 21.Stolfi C, Fina D, Caruso R, Caprioli F, Sarra M, Fantini MC, Rizzo A, Pallone F, Monteleone G. Cyclooxygenase-2-dependent and -independent inhibition of proliferation of colon cancer cells by 5-aminosalicylic acid. Biochem Pharmacol. 2008;75(3):668–76. doi: 10.1016/j.bcp.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, Wahli W, Desvergne B, Naccari GC, Chavatte P, Farce A, Bulois P, Cortot A, Colombel JF, Desreumaux P. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201(8):1205–15. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, Hartwick W, Hoffman B, Hood N. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002;20(1):42–51. doi: 10.1200/JCO.20.1.42. [DOI] [PubMed] [Google Scholar]
- 24.Larsson SC, Orsini N, Wolk A. Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst. 2005;97(22):1679–87. doi: 10.1093/jnci/dji375. [DOI] [PubMed] [Google Scholar]
- 25.Rinaldi S, Rohrmann S, Jenab M, Biessy C, Sieri S, Palli D, Tumino R, Mattiello A, Vineis P, Nieters A, Linseisen J, Pischon T, Boeing H, Hallmans G, Palmqvist R, Manjer J, Wirfalt E, Crowe FL, Khaw KT, Bingham S, Tjonneland A, Olsen A, Overvad K, Lund E, Skeie G, et al. Glycosylated hemoglobin and risk of colorectal cancer in men and women, the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2008;17(11):3108–15. doi: 10.1158/1055-9965.EPI-08-0495. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda T, Iwata K, Murakami H. Inhibitory effect of metformin on intestinal glucose absorption in the perfused rat intestine. Biochem Pharmacol. 2000;59(7):887–90. doi: 10.1016/S0006-2952(99)00396-2. [DOI] [PubMed] [Google Scholar]
- 27.DeFronzo RA, Goodman AM. Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. The Multicenter Metformin Study Group. N Engl J Med. 1995;333(9):541–9. doi: 10.1056/NEJM199508313330902. [DOI] [PubMed] [Google Scholar]
- 28.Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W, Zhao G, Kip KE. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: a meta-analysis. Diabetes Care. 2011;34(10):2323–8. doi: 10.2337/dc11-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng CH. Diabetes, metformin use, and colon cancer: a population-based cohort study in Taiwan. Eur J Endocrinol. 2012;167(3):409–16. doi: 10.1530/EJE-12-0369. [DOI] [PubMed] [Google Scholar]
- 30.Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E, Majumdar AP. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One. 2014;9(1):e84369. doi: 10.1371/journal.pone.0084369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Guan M, Zheng Z, Zhang Q, Gao F, Xue Y. Effects of metformin on CD133+ colorectal cancer cells in diabetic patients. PLoS One. 2013;8(11):e81264. doi: 10.1371/journal.pone.0081264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, Dekker RF, Fortes ZB, Khaper N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9(5):e98207. doi: 10.1371/journal.pone.0098207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 34.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 35.Muc-Wierzgon M, Nowakowska-Zajdel E, Kokot T, Kozowicz A, Zubelewicz B, Klakla K, Mazurek U, Cholewa K, Wilczok T, Wierzgon J, Sosada K. Genetic disregulation of gene coding tumor necrosis factor alpha receptors (TNFalpha Rs) in colorectal cancer cells. J Exp Clin Cancer Res. 2004;23(4):651–60. [PubMed] [Google Scholar]
- 36.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–71. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 37.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457(7225):102–6. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12(4):419–29. doi: 10.1016/S1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 39.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18(1):19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3(1):17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 41.Hamilton KE, Simmons JG, Ding S, Van LL, Lund PK. Cytokine induction of tumor necrosis factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol Cancer Res. 2011;9(12):1718–31. doi: 10.1158/1541-7786.MCR-10-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, Whitehead R, Polk DB. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Physiol Cell Physiol. 2003;284(4):C953–C961. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 43.Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA, Apte RN. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007;67(3):1062–71. doi: 10.1158/0008-5472.CAN-06-2956. [DOI] [PubMed] [Google Scholar]
- 44.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Jr, Rabkin CS. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 45.Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011;3:177–89. doi: 10.2147/CMR.S18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker C, Fantini MC, Wirtz S, Nikolaev A, Lehr HA, Galle PR, Rose-John S, Neurath MF. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle. 2005;4(2):217–20. doi: 10.4161/cc.4.2.1413. [DOI] [PubMed] [Google Scholar]
- 47.Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal K, Beug H, Ohlschlager P, Schutz A, Halbhuber KJ, Friedrich K. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7(6):545–55. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BK, Sethi G, Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–54. doi: 10.1016/j.bbcan.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 49.Carlson ML, Wilson ET, Prescott SM. Regulation of COX-2 transcription in a colon cancer cell line by Pontin52/TIP49a. Mol Cancer. 2003;2:42. doi: 10.1186/1476-4598-2-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu YM, Azahri NS, Yu DC, Woll PJ. Effects of COX-2 inhibition on expression of vascular endothelial growth factor and interleukin-8 in lung cancer cells. BMC Cancer. 2008;8:218. doi: 10.1186/1471-2407-8-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SA, Choi HC. Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem Biophys Res Commun. 2012;425(4):866–72. doi: 10.1016/j.bbrc.2012.07.165. [DOI] [PubMed] [Google Scholar]
- 52.Tsujii M, Kawano S, Dubois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A. 1997;94(7):3336–40. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao M, Kargman S, Lam EC, Kelly CR, Zheng Y, Luk P, Kwong E, Evans JF, Wolfe MM. Inhibition of cyclooxygenase-2 by rofecoxib attenuates the growth and metastatic potential of colorectal carcinoma in mice. Cancer Res. 2003;63(3):586–92. [PubMed] [Google Scholar]
- 54.Kim YH, Kim MH, Kim BJ, Kim JJ, Chang DK, Son HJ, Rhee PL, Rhee JC. Inhibition of cell proliferation and invasion in a human colon cancer cell line by 5-aminosalicylic acid. Dig Liver Dis. 2009;41(5):328–37. doi: 10.1016/j.dld.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 55.Hwang YP, Jeong HG. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Calpha-dependent pathway: phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. Br J Pharmacol. 2010;160(5):1195–211. doi: 10.1111/j.1476-5381.2010.00762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Esfahanian N, Shakiba Y, Nikbin B, Soraya H, Maleki-Dizaji N, Ghazi-Khansari M, Garjani A. Effect of metformin on the proliferation, migration, and MMP-2 and −9 expression of human umbilical vein endothelial cells. Mol Med Rep. 2012;5(4):1068–74. doi: 10.3892/mmr.2012.753. [DOI] [PMC free article] [PubMed] [Google Scholar]