

Abstract
The Virtual Physiological Human (VPH) project aims to develop integrative, explanatory and predictive computational models (C-Models) as numerical investigational tools to study disease, identify and design effective therapies and provide an in silico platform for drug screening. Ultimately, these models rely on the analysis and integration of experimental data. As such, the success of VPH depends on the availability of physiologically realistic experimental models (E-Models) of human organ function that can be parametrized to test the numerical models. Here, the current state of suitable E-models, ranging from in vitro non-human cell organelles to in vivo human organ systems, is discussed. Specifically, challenges and recent progress in improving the physiological realism of E-models that may benefit the VPH project are highlighted and discussed using examples from the field of research on cardiovascular disease, musculoskeletal disorders, diabetes and Parkinson's disease.
Keywords: physiology, experimental models, human
1. Introduction
Computational modelling in science constitutes a means to capture and integrate accumulated knowledge and contemporary paradigms, and test their consistency with quantitative experimental observations [1,2]. From its very beginning, the application of this methodological tool to biology has been driven by a desire to answer focused scientific questions (e.g. metabolic control of glycolysis [3]) rather than serve any grand, broad ambition to eventually capture and predict ‘everything’ (e.g. the E-Cell project [4]). Indeed, when Chance and co-workers in the late 1950s reported the results of their pioneering application of machine-based computing to test contemporary understanding of metabolic control in aerobic glycolysis, the authors made a point of explicitly stating that biochemical completeness was not a design criterion [3].
The nature of the C-models developed within the Virtual Physiological Human (VPH) project [5–7] is in that sense somewhat different in that its objective is to develop predictive numerical tools to address medical rather than abstract scientific problems. First of all, this specific objective significantly raises the bar of admissible minimal level of C-model complexity. Suitable VPH C-models must ultimately mechanistically incorporate model output sensitivity towards, on the one hand, risk factors embedded in individual human genotypes and, on the other hand, therapeutic drug interactions [5–7]. In addition, it raises the need for species-specificity and detail of the quantitative database for VPH model development. For example, in the example of the early numerical investigation of metabolic control in glycolysis, it was deemed adequate and sufficient to parametrize the C-model using in vitro enzymatic data from yeast and subsequently test their model against experimental data obtained at non-physiological temperature and pH in suspensions of non-vascularized mouse carcinoma cells [3]. It remains to be proved that such a ‘Frankenstein’ approach to human C-model development is adequate and sufficient to accomplish the objectives of the VPH [8]. It may well turn out that it will require multi-dimensional quantitative data collected from human cells and organs, preferably including interactions with other cell types and organs, in both healthy and diseased states. Of course, this provision does not per se disqualify the use of any non-human animal E-model towards C-model parameterization, verification and validation, as will be discussed below. The specific medical objective of the VPH project does, however, impose constraints on which E-models do qualify.
Physiological realism is an essential criterion in this matter. Here, its measure is defined as the quality of reconstitution of the in vivo state in a laboratory setting. The ideal E-model requires complete functional reconstitution of the multi-scale network of genes, proteins, cells and tissues that interact in the intact living human body. Obviously, such level of completeness in a laboratory setting (or in silico, for that matter) is either practically untenable or experimentally intractable. As such, many of the same objective-guided rational and/or practical choices and compromises that must be made in computational modelling need to be made in E-model design. An exciting future perspective is the application of personalized E- and C-models for in vitro and in silico drug testing to guide personalized therapeutic interventions.
In this essay, challenges, choices and recent progress in improvement of the physiological realism of E-models ranging from isolated cell organelles to human organs in vivo that may support C-model development for the VPH are highlighted and discussed using examples from the field of research on cardiovascular disease, musculoskeletal disorders, diabetes and Parkinson's disease (PD).
2. Improving experimental models to study human organ function
2.1. Isolated mitochondria
Mitochondria are cellular organelles where carbon substrates derived from carbohydrates, amino acids (following deamidation) and fatty acids undergo terminal oxidation, and the free energy obtained from those terminal oxidations is transduced into the free energy of ATP hydrolysis reaction through oxidative phosphorylation (OxPhos), which then drives cellular functions. In other words, the net reactions occurring in mitochondria are the complete oxidation of carbon substrates to CO2 and H2O and the synthesis of ATP from ADP and inorganic phosphate (Pi) in response to cellular energy demand (i.e. hydrolysis of ATP into ADP and Pi in the extramitochondrial space) [9]. Key questions in the context of the role of mitochondria as cellular energy transducers are how mitochondria sense ATP demand, how the flux of ATP synthesis is regulated in order to achieve energetic homeostasis and how those processes are altered in pathophysiological states [8]. Isolated mitochondria-based E-models have played a crucial role in the development of C-models of OxPhos, which then provided a means to predict in vivo regulation and dysregulation of energy metabolism. This section provides a brief history of isolated mitochondria as an E-model and how E-models and C-models for cardiac and skeletal muscle mitochondria have contributed to our understanding of in vivo regulation of oxidative ATP synthesis in the mammalian heart and skeletal muscle. Potential future directions for improving the physiological realism of isolated mitochondria-based E-models are provided in conclusion.
The most essential ingredient of E-models for studying mitochondrial function in isolation is a preparation of mitochondria from primary tissue by homogenization and differential or density gradient centrifugation. Media and preparation strategies for intact mitochondria were derived from the work of Hogeboom et al. [10] and Claude [11]. Applying those strategies, Chance & Williams [12] isolated intact mitochondria from rat and guinea pig liver and showed that in the presence of excess phosphate the flux of respiration (ATP synthesis) was approximately a hyperbolic function of the concentration of phosphate acceptor ADP. Current methods for preparing intact muscle mitochondria are derived from the initial work of Chance & Hagihara [13] using bacterial proteases to soften the tissue mince prior to homogenization and differential centrifugation, which proved critical to obtaining chemi-osmotically well-coupled preparations. The criteria for purity and homogeneity of isolated mitochondrial preparations are discussed by de Duve [14]. Subsequent work in the 1970s using isolated cardiac mitochondria most notably by LaNoue et al. [15] was focused on elucidating the regulation of the Krebs cycle through substrate feedback via conserved moieties NADH and CoA. Bishop and Atkinson [16,17] further extended the realism of the cardiac mitochondrial E-model by reconstituting steady states in mitochondrial respiration in response to fluxes of net ATP hydrolysis in the extramitochondrial space at defined concentrations of Pi and total extramitochondrial adenine nucleotide pool sizes. They showed that respiration was a Hill function of ADP (also see [18]) and that Pi reduced the apparent Km of respiration towards ADP and increased the apparent Hill coefficient. They also postulated that Pi could potentially act as a feedback signal in vivo for OxPhos. Denton and co-workers showed that divalent cations Ca2+ and Mg2+ modulated the activities of TCA cycle dehydrogenases and could play a role in regulating the flux of OxPhos [19,20]. More recently, a substantial body of work by Balaban and co-workers identified multiple sites of regulation in the OxPhos system by Ca2+ [21,22] and Pi [23] in cardiac and skeletal muscle mitochondria [24] oxidizing glutamate and malate at physiological temperature and ionic conditions. In summary, the isolated mitochondria E-model showed that OxPhos could be regulated by nucleotide, Pi, carbon substrates and divalent cations.
Beard and co-workers developed C-models of mitochondrial oxidative phosphorylation [25] and TCA cycle [26] based on data from Balaban and co-workers [23] and LaNoue et al. [27], which were then successfully applied to predict in vivo measurements of myocardial phosphate energetics and oxygen consumption [28]. The mitochondrial C-model was also applied successfully towards prediction of steady-state energetics in human skeletal muscle in vivo in healthy individuals and in subjects with mitochondrial complex I and ANT deficiencies [29]. These models showed primarily that feedback from products of ATP hydrolysis may be sufficient to explain the regulation of OxPhos in vivo and that the energetic phenotype of heart failure can be explained by remodelling of nucleotide and phosphate pools without any intrinsic mitochondrial functional deficit [28,30]. Indeed, additional tests using isolated mitochondria using a physiologically oxidized substrate showed only a modest contribution of Ca2+ to the in vivo range of respiration [31,32], an observation explained by the finding cardiac function was unaffected in mouse models in which the mitochondrial calcium uniporter was genetically ablated [33,34]. However, conditional tissue-specific cardiac mitochondrial uniporter knockout models by Kwong et al. [35] led to a more nuanced picture where exercising mice required a substantial ‘warm up time’ before being able to exercise at a level similar to control mice and that those changes were associated with changes in calcium content in mitochondria due to adrenergic stimulation. The physiological realism of the Beard mitochondrial C-model was recently revisited by Vinnakota et al. [36], who showed that the near equilibrium behaviour of F1FO ATPase and the rapid transport of Pi from extramitochondrial space to the matrix governed the kinetic feedback from Pi to the TCA cycle [36]. The results did not support any direct activation of complex III by Pi contrary to the models of Beard [25] and Bose et al. [23].
The main challenge in obtaining a physiologically realistic in vitro preparation of mitochondria is to achieve a complete reconstruction of inherently homeostatic systems [37] with the same control structure observed in vivo. This is largely due to the difficulty of reconstituting mitochondria with extramitochondrial systems that can maintain non-equilibrium steady states in carbon substrates and possess the same elasticities as the in vivo system. A well-known practical problem with this particular E-model, for example, has been the reconstitution—and maintenance during the course of the experiments—of physiologically realistic pool sizes of matrix cations and metabolites (e.g. Krebs cycle substrates (E. Gnaiger 2005, personal communication)). A significant advance in the quality of the experimental preparation used by Vinnakota and co-workers has therefore been to reconstitute a family of respiratory steady states in isolated cardiac mitochondria oxidizing pyruvate while measuring respiration, NADH, inner membrane potential and steady-state ADP at specified total adenine nucleotide and phosphate pool sizes at physiological temperature and ionic conditions (figure 1) [36]. Prior numerical simulations of the experiments proved valuable to identify particular sensitivities of the preparation. Thus, despite the challenges, research in this area has indeed demonstrated that observations on in vitro E-models indeed can effectively inform C-models that explain and predict in vivo physiological function in health and disease. This successful extrapolation from in vitro to in vivo requires a judicious design of appropriate isolated mitochondrial E-models to simulate physiologically relevant conditions obtained from intact organ measurements and C-models to bridge the gap between in vitro and in vivo levels of organization.
Figure 1.

Improved characterization of the mitochondrial E-model. (a) 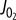 versus [ADP] plot and a fit to the expression Vmax [ADP][Pi]/(KADP+[ADP])(KPi + [Pi]) (solid line) show that a single KADP and KPi can describe the relationship between [ADP], [Pi] and
versus [ADP] plot and a fit to the expression Vmax [ADP][Pi]/(KADP+[ADP])(KPi + [Pi]) (solid line) show that a single KADP and KPi can describe the relationship between [ADP], [Pi] and 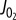 . (b) Contour plot of
. (b) Contour plot of 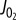 as a function of both [ADP] and [Pi] showing the [ADP], [Pi] concentrations spanned in this study and
as a function of both [ADP] and [Pi] showing the [ADP], [Pi] concentrations spanned in this study and 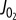 data and contours of constant
data and contours of constant 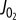 in [ADP], [Pi] space. (c)
in [ADP], [Pi] space. (c) 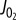 plotted against the free energy of the ATP hydrolysis (ΔrGATPase) reaction in the cytosol showing that at higher [Pi] mitochondria can synthesize ATP at a higher ΔrGATPase for a given ATP synthesis flux. (d) Normalized steady-state NADH plotted against
plotted against the free energy of the ATP hydrolysis (ΔrGATPase) reaction in the cytosol showing that at higher [Pi] mitochondria can synthesize ATP at a higher ΔrGATPase for a given ATP synthesis flux. (d) Normalized steady-state NADH plotted against 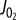 showing the oxidation of NADH with increase in
showing the oxidation of NADH with increase in 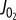 and no distinguishable difference between 5 mM [Pi]0 and 1 mM [Pi]0 experiments. The solid line shows a power law fit to the entire dataset. (e) Δψ plotted against
and no distinguishable difference between 5 mM [Pi]0 and 1 mM [Pi]0 experiments. The solid line shows a power law fit to the entire dataset. (e) Δψ plotted against 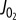 shows depolarization with increasing flux of respiration, demonstrating that the phosphorylation subsystem, which consumes the proton electrochemical gradient, is the fastest component of OxPhos. (f) CytC2+ normalized with respect to total cyt c plotted against
shows depolarization with increasing flux of respiration, demonstrating that the phosphorylation subsystem, which consumes the proton electrochemical gradient, is the fastest component of OxPhos. (f) CytC2+ normalized with respect to total cyt c plotted against 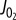 shows no difference between the curves for 1 mM [Pi]0 and 5 mM [Pi]0 experiments, contradicting prior reports in the literature on the role of [Pi] in modulating cytochrome C redox state via complex III. Data in all figures were plotted as mean ± s.e.m., where the number of biological replicates was 3–6 for
shows no difference between the curves for 1 mM [Pi]0 and 5 mM [Pi]0 experiments, contradicting prior reports in the literature on the role of [Pi] in modulating cytochrome C redox state via complex III. Data in all figures were plotted as mean ± s.e.m., where the number of biological replicates was 3–6 for 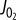 data, 2 for [ADP] data, 3–6 for NADH data, 3 for Δψ data and 2 for CytC2+ data. (Adapted from [36].) (Online version in colour.)
data, 2 for [ADP] data, 3–6 for NADH data, 3 for Δψ data and 2 for CytC2+ data. (Adapted from [36].) (Online version in colour.)
2.2. Isolated pancreatic islet cells from human donors
The pancreatic islets secrete insulin and glucagon, two of the body's most important metabolic hormones. Glucagon (secreted by the islet alpha-cells) is the body's principal hyperglycaemic hormone (i.e. increases plasma glucose), whereas insulin (secreted by the islet beta-cell) is the only hypoglycaemic (i.e. lowering plasma glucose) hormone. Much of our knowledge of the regulation of insulin and glucagon secretion comes from work on rodent pancreatic islet cells [38,39]. Here, we will focus on the control of insulin secretion in the beta-cells. Although human and rodent beta-cells share many features, they are not identical [38] and it is therefore crucial that key observations in rodents are validated in human cells. Moreover, some of the characteristics that make laboratory mice attractive experimentally (e.g. the limited genetic diversity) also represent a shortcoming that makes successful translation of results into the clinic more difficult [40]. In this section, results of comparative physiological studies of rodent versus human beta-cells are reviewed and discussed in the context of whether or not rodent E-models may be adequate and sufficient to support C-model development of human organ systems. The merit of experimental approaches to ‘humanize’ mouse models to study disease and discover drug targets [40] will also be considered.
Pancreatic beta-cells are electrically excitable and generate action potentials when exposed to glucose concentrations stimulating insulin secretion [39]. In mouse beta-cells, the glucose-induced electrical activity (that emerges at glucose concentrations 6–7 mM) is oscillatory and consists of depolarized plateaux on which action potentials are superimposed that are separated by repolarized and electrically silent intervals [41,42]. Work on isolated mouse beta-cells has identified and characterized many of the membrane currents that underlie this electrical activity. Key players include ATP-regulated K+ channels (KATP channels) and voltage-gated L-type Ca2+ channels. The KATP channels are active at low glucose concentrations. Following an increase in (plasma) glucose concentration, stimulation of glucose metabolism in the beta-cells leads to an increased cytoplasmic ATP/ADP ratio which leads to inhibition of the KATP channels that are spontaneously active at low glucose concentrations (when the intracellular ATP/ADP ratio is low). Glucose-induced closure of the KATP channels results in membrane depolarization and when it exceeds a threshold potential of −60 to −50 mV, regenerative electrical activity consisting of Ca2+-dependent action potentials is generated. Thus, beta-cell electrical activity results in stimulation of Ca2+ influx and is therefore associated with an elevation of the cytoplasmic Ca2+ concentration, which culminates in initiation of Ca2+-dependent exocytosis of insulin-containing secretory granules [39]. Insulin thus released into the islet interstitium diffuses into the islet capillary network and travels to the target organs via the circulation and there promotes the uptake and deposition of glucose [43]. The resultant reduction of plasma glucose leads to termination of insulin secretion via a reversal of the process outlined above, thus providing an elegant feedback system for the regulation of systemic glucose metabolism. Defects in this regulation (i.e. due to reduced capacity of the beta-cell to increase the cytosolic ATP/ADP ratio) leads to reduced insulin secretion and this is believed to be a cause of type-2 diabetes (adult onset diabetes) [44].
Many of the processes outlined above for mouse beta-cells have their counterparts in human beta-cells. However, there are notable differences. First, insulin secretion is stimulated at lower glucose concentrations in man than in mice (3 mM versus 6–7 mM) [45,46]. Second, glucose-induced electrical activity in human beta-cells does not appear to exhibit the membrane potential oscillations that are so conspicuous in mouse beta-cells [47]. Third, resting KATP channel activity is much lower in human than in mouse beta-cells, a feature that may contribute to the lower threshold for stimulation of insulin secretion in man [38]. Finally, the ion channel complement of human beta-cells is quite different from that of mouse beta-cells [48]. For example, voltage-gated, tetrodotoxin-sensitive Na+ channels play a more important role in human beta-cell electrical activity and insulin secretion. Human beta-cells also have a large low-threshold (T-type) Ca2+ current, absent in mouse beta-cells, that is critical to action potential initiation in human beta-cells; and P/Q-type rather than L-type Ca2+ channels are linked to insulin granule exocytosis. These differences are naturally highly significant when it comes to the interpretation of the genetic data that are now emerging. Clearly, ion channel gene variants (polymorphisms) linked to diabetes are unlikely to affect insulin secretion if the gene is not expressed in the human beta-cell. In such cases, any association may instead be secondary to neuronal appetite control and obesity.
There have been numerous mathematical models of mouse beta-cell electrical activity (see [49] for a few references to this earlier work). Surprisingly, they almost invariably reproduce the oscillatory electrical activity that can be recorded from beta-cells in freshly isolated mouse pancreatic islets although the parameters used in the modelling work are almost exclusively and deliberately [50] derived from experimental studies on isolated cells that do not generate the characteristic oscillatory electrical activity [51]. This correlates with the loss of a slowly activating Ca2+-dependent K′ current (Kslow) in isolated beta-cells. It would be interesting to construct a robust model of the isolated human beta-cell [52] based on actual functional parameters (i.e. without any tweaking) and compare that to the electrical activity that can be recorded experimentally. Such a model would be a useful base for future work and to explore the significance of differences observed between isolated beta-cells maintained in tissue culture and beta-cells within intact pancreatic islets. The parameters may then be altered to reflect differences between beta-cells in the intact organ after isolation to determine if these changes are sufficient to produce oscillatory electrical activity. Indeed, the addition of Kslow to a non-bursting model of isolated beta-cells is sufficient to give rise to bursting electrical activity, providing proof of concept for the usefulness of the approach (figure 2).
Table 1.
Overview of modified model parameter values for simulation of the electrical activity of a human pancreatic beta-cell.
| model components | parameters | original value in Cha et al. [50] | revised value |
|---|---|---|---|
| IPMCA | PPMCA (pA) | 1.56 | 3.26 |
| KPMCA (mM) | 0.00014 | 0.0002 | |
| ITRPM | PTRPM (pA mM−1) | 0.0234 | 0.01872 |
| KTRPM (mM) | 0.00076 | 0.0006 | |
| IKATP | GKATP (pA mV−1) | 2.31 | 3.7 |
| kdd (mM) | 0.01 | 0.017 | |
| INaK | Kd_MgATP (mM) | 0.6 | 0.2 |
| glycolysis and oxidative phospholylation | kATP (ms−1) | 0.000062 | 0.000155 |
| kATP,Ca (mM−1 ms−1) | 0.187 | 0.1309 | |
| kglc (ms−1) | 0.000126 | 0.000252 | |
| kβ,ox (ms−1) | 0.0000063 | 0.0000145 | |
| [ATPtot] (mM) | 4 | 5 |
Figure 2.
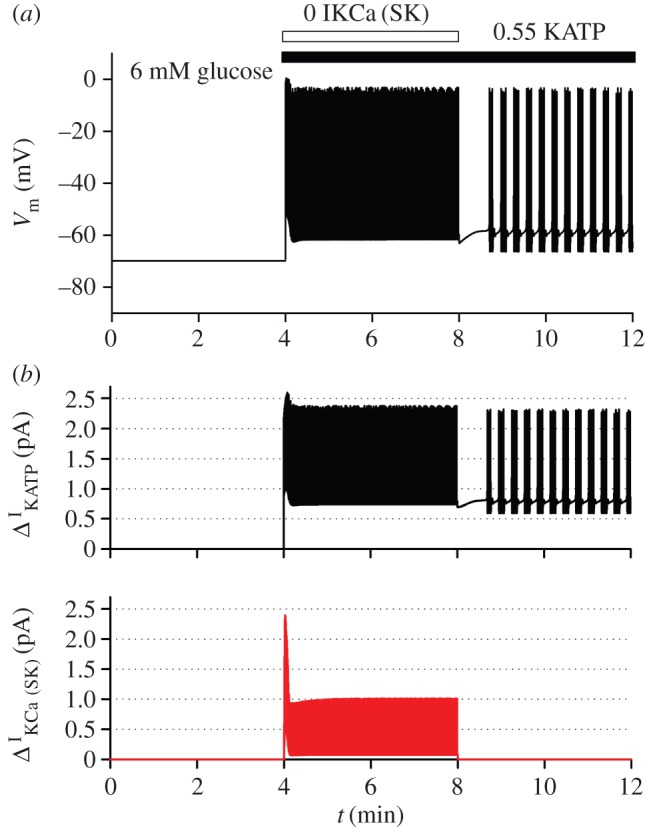
Model-based simulations of electrical activity of a human pancreatic beta-cell. To reproduce the electrical activity of pancreatic beta-cell, we used a model of Cha et al. [50] which was adjusted with some updated parameter values (table 1) [50]. Specifically, it has been reported that IKslow was experimentally a mixed current of small-conductance KCa channel current (IKCa(SK)*) [53] and KATP current (IKATP) [54]. The latter has been supposed to compose IKslow about by half [54]. Therefore, to simulate effects of the inhibition of IKslow, IKCa(SK) and IKATP were decreased to zero and 55%, respectively, from 4 min (horizontal bars above the traces). The cell was stabilized at 6 mM glucose at first, but started to excite with regular spikes by the inhibition of the two currents. To examine the role of interaction between Ca2+ and IKCa(SK) in state transition from regular spiking to bursting oscillation, IKCa(SK) was restored to its control value from 8 min. In the simulation, intracellular Na+ and K+ concentration was fixed at 5 mM and 140 mM, respectively, to mimic the effect of intracellular perfusion by a patch pipette. Each panel shows time-dependent change in Vm (a), and amplitude of decreased currents (b) (IKATP, black; IKCa(SK), red). * IKCa(SK) was marked as ‘IKslow’ in the model by Cha et al. [50], but we renamed here to avoid confusion. (Online version in colour.)
As mentioned above, membrane potential recordings from human beta-cells have provided only limited evidence for oscillatory electrical activity but it has not been conclusively established whether this represents a true difference between mouse and human islets or whether it is simply a consequence of the experimental protocols. To date, no membrane potential recordings from beta-cells within acutely isolated intact human islets have been reported. It is also noteworthy that these experiments should perhaps be conducted at lower glucose concentrations than those used in mice as suggested by the lower threshold for glucose-induced insulin secretion Furthermore, it is unknown to what extent the characteristics of beta-cell electrical activity observed in vitro are relevant to the control of insulin secretion/electrical activity in vivo. In the intact body, the islets are strongly innervated and they are—via the rich vascular supply—exposed to a plethora of circulating factors, many of which are likely to influence both insulin secretion and beta-cell electrical activity.
It is important to note that despite the significant differences between mouse and human beta-cells, this does not invalidate studies in mouse beta-cells. Starting with a good model of mouse beta-cell electrical activity, it may be possible to ‘humanize’ the mouse beta-cell in silico by addition/removal of membrane currents, study the impact on electrical activity and compare the properties of electrical activity in such humanized models of beta-cell electrical activity with that experimentally observed. Such humanized models might also be of great use to predict potential side effects of novel drugs on beta-cell electrical activity and insulin secretion. A similar approach may be used to predict the impact of variants of genes encoding ion channels on electrical activity and insulin secretion [55]. For example, variants of KCNQ1 (that encodes a voltage-gated potassium channel) has been linked to diabetes risk [56] but it is debated whether the effect is mediated by a genetic or electrophysiological mechanism [57]. However, the finding that KCNQ1 is expressed at only low levels in human beta-cells [58] and the absence of a potassium current component with the biophysical signature expected for such a current militate against the latter possibility.
Another ‘complication’ inherent to the work on human beta-cells is that they (by necessity) are obtained from dead organ donors and the islets have experienced a variable period of ischaemia prior to isolation of the islets. It is also unclear to what extent medication during the intensive care that preceded the death of the donor may have affected the function and properties of the isolated islets cells. It is possible that human beta-cells/islet-like structures derived from stem cells represents a means of better defining the functional characteristics of the ‘healthy’ beta-cell [59].
As mentioned above, defects in the stimulus-secretion coupling of the beta-cell outlined above is likely to result in type-2 diabetes. However, the exact nature of the dysfunction of the diabetic beta-cell remains poorly characterized, mainly because of very limited access to human beta-cells. A further complication is the progressive nature of diabetes. Although it is likely that the function of the beta-cell is compromised in the type-2 diabetic individual, it should be kept in mind that the beta-cell may have functioned adequately for 50–60 years prior to the onset of the disease. What triggers the deterioration of beta-cell function in those that develop diabetes is not known. Age itself is not necessarily the causal factor. In fact, there are some data suggesting that the beta-cells become hyper-responsive with age (i.e. the reverse to what may have been expected) [60]. Again, in silico analysis combining models of insulin secretion with insulin signalling in the target organs may help to resolve this apparent conundrum.
Finally, a central question in all experimental studies is how much of the function of the beta-cell in vivo can be gleaned from studies on isolated cells in vitro. Ideally, much of the work should of course be conducted on human beta-cells residing within living (healthy or diabetic) individuals but such techniques are not currently available and will probably remain so for the foreseeable future.
2.3. Parkinson's disease phenotypes of human dopaminergic neurons in culture
Availability of living human cells for physiological study from organs and tissues, like the pancreatic islet cells from human donors discussed in the previous section, is typically very limited. For many neurological diseases such as PD, cells from the affected organ (the brain) are inaccessible and are only available post-mortem. As such, any alternative to the invasive procedure of harvesting living cells from human organs to obtain suitable E-models for medical application other than blood and skin would be highly desirable. Here, a promising novel approach in PD research is discussed that exploits recent advances in cellular phenotypic reprogramming technologies and may well offer such an alternative.
Similar to type-2 diabetes, animal models of PD have been useful for elucidating disease mechanisms and identification genetic causes [61]. For example, early animal models of PD using neurotoxins that target dopaminergic neurons such as MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydopyrine) produced motor deficits that mimic those observed in human PD [62]. However, these E-models do not faithfully recapitulate the slow time-dependent progression of neurodegeneration observed in human patients nor capture the effect of genetic mutation in patients [63]. As such, it may be proposed that E-modelling the disease in the very same human neurons that die in PD will be necessary to discover therapies that will actually prevent neurodegeneration rather than merely provide relief of symptoms as is currently the case in PD [63]. But how can this be done?
Induced pluripotent stem cells (iPSC) offer an unparalleled opportunity to generate and study physiologically relevant cell types in culture [64]. Recently, this approach has been adopted to generate human cellular E-models of PD in culture [63,65]. Briefly, a small biopsy is taken from the skin of a patient and explanted so that fibroblasts may grow out from the biopsy sample in vitro. Next, a set of reprogramming factors is delivered, typically by viral transduction, to transform the cells into iPSC. The cells are then differentiated into midbrain dopaminergic neurons. Functional studies confirmed the dopaminergic nature of these neurons by demonstrating electrical activity and the production and release of dopamine (figure 3) [65]).
Figure 3.

Highly physiological dopaminergic neurons generated from human iPSC lines to study PD. (a) Differentiated dopaminergic neurons express the neuronal marker β3-tubulin (red) and the dopaminergic neuronal marker tyrosine hydroxylase (TH; green) shown by immunofluorescence. (b) Representative spontaneous pace-making firing traces of differentiated dopaminergic neurons. (c) Human iPSC-derived dopaminergic neurons exhibit the spontaneous pace-making Ca2+ signals characteristic of dopaminergic neurons of the substantia nigra pars compacta. Neurons show increasing rhythmicity over six to eight weeks of differentiation. (d) With maturation over time, dopaminergic neurons acquire the ability to fire a train of action potentials on depolarization. (adapted from [65].) (Online version in colour.)
Study of these cells is advantageous towards understanding the aetiology of PD as well as discovery of preventative therapy. First and foremost, no alternative source of living human midbrain dopaminergic neurons has been previously available. Secondly, patient-derived iPSCs carry the genetic background of the disease and should help discover genetic interactions which may lead to PD. iPSC lines have been generated from fibroblasts of patients carrying SNCA, PINK1, PARK2 or LRRK2 mutations, all of which are known PD-related mutations [63]. Thirdly, these E-models will enable studying the early changes in cellular physiology that eventually lead to pathology. The latter is key to the development of therapies that may prevent neurodegeneration in PD.
The ‘how to’ knowledge of generation and differentiation of human iPSCs into relevant cell types discussed here in the context of PD offers a generic technology platform to generate human in vitro E-models of disease. Indeed, the application of this technology to develop in vitro human cell models of other significant diseases has been rapidly expanding. Recent examples include human cell models of other neurodegenerative diseases such as Huntington's disease [66] and Alzheimer's disease [67], familial cardiomyopathy [68,69] and familial cancer [70]. In addition to enabling study of the development and progression of the diseases, these human E-models offer a versatile platform for therapy and drug testing [69]. Some limitations of these cell models remain, however. First of all, careful phenotypic benchmarking of a particular cell population under investigation is required. Secondly, living human organs typically consists of multiple interacting cell types rather than any single cell type. As such, any proper reconstitution of the in vivo steady-state in these human cell models may well require reconstituting this cellular heterogeneity using approaches from the field of ‘organoid’ cell model development [71].
2.4. The isolated perfused heart
The isolated saline perfused heart is likely one of the most popular in vitro preparations in cardiovascular research. Despite its popularity, based mostly on its ease of preparation and rapid collection of data, it has been suggested for many years that the oxygen tension is inadequate to support oxidative phosphorylation even under resting conditions. This is despite the large increase in coronary flow rates when compared to estimated coronary flows in vivo. Various studies added back red blood cells or perfluorcarbon emulsions to increase the oxygen-carrying capacity of the tissue and found profound increases in cardiac performance coupled with reductions in coronary flow, consistent with the notion that oxygenation was limiting and that a flow reserve for experimentation was now available much like under in vivo conditions [72–80]. Furthermore, others went on to demonstrate that myoglobin was significantly deoxygenated in heart perfused with ordinary saline [81], providing direct evidence that the cytosol of the myocyte was not adequately oxygenated while NADH fluorescence studies of the complex 1 redox state also suggested that the perfused heart was on the brink of mitochondrial hypoxia [82]. This is consistent with the very early work from Chance's laboratory suggesting the existence of remarkable gradients in oxygen tension within the heart [83]. Even though much of this work came from leading laboratories, the problem with insufficient oxygenation and its functional implication has remained understudied and the traditional (saline perfused) heart preparation has remained the preparation of choice in pharmaceutical and physiological studies.
Recently, methods for monitoring the redox-sensitive chromophores of the complexes of oxidative phosphorylation in isolated mitochondria and perfused hearts have been developed [84]. It was this thus possible to demonstrate that the different redox states of cytochrome oxidase can be differentiated in intact mitochondria and tissues [85]. This approach permitted the differentiation of cytochrome oxidase reduced by ATP synthesis and hypoxia. Also important for this discussion is the ability to monitor myoglobin independently from the redox species of cytochrome oxidase permitting the estimate of the cytosolic and mitochondria oxygenation levels. In preliminary studies in the saline perfused rabbit heart (figure 4; [84]) significant levels of reduced cytochrome a,a3 in its fully reduced state were observed, and any induced change in workload with pacing or beta agonists resulted in an increase in the hypoxia form of cytochrome a,a3. These results are consistent with early studies on the saline perfused rabbit heart where increases in perfusion flow with afterload resulted in an oxidation of cytochrome oxidase and myoglobin when compared to the control [86]. However, the spectral analyses of these early studies were limited and hampered by an error in using a chemically oxidized form of myoglobin as a reference. However, all these studies were consistent with the concept that oxygen restricts oxidative phosphorylation in the saline perfused heart when compared to the in vivo condition [87]. We contend that any future use of the isolated saline perfused heart should be reconsidered in favour of more physiological experimental paradigms that ensure adequate tissue oxygenation. To this end, the perfusate should be supplemented with either red cells or perfluorocarbon and the oxygen tension should be monitored. Preliminary studies support the earlier functional studies suggesting that the perfluorocarbons improves tissue oxygenation, coronary flow reserve and restores mitochondrial redox state to levels more consistent with what is observed in the heart in vivo.
Figure 4.

Spectral fitting of data acquired from isolated Langendorff perfused rabbit hearts. (a) Raw, fit and residual data of rabbit heart myocardium following perfusion with cyanide to induce reduction of mitochondrial chromophores. (b) Contribution of the reference spectra to the fit data shown in (a). Note the lack of change in myoglobin oxygenation. (c) Raw, fit and residual data of rabbit heart myocardium following global no-flow ischaemia to induce reduction of mitochondrial chromophores and myoglobin deoxygenation (as evidenced by the trough at approx. 580 nm). (d) Contribution of the reference spectra to the fit data shown in (c). (Online version in colour.)
2.5. Human organs in vivo
2.5.1. The human heart
The ideal assessment of cardiac function includes assessment throughout a range of haemodynamic stressors, just as the heart is required to respond to differing haemodynamic environments during daily life. It is also important to note that the first sign of heart disease typically occurs during exercise and thus it is critical to understand normal cardiac physiology during exercise to distinguish abnormalities in function due to disease. For example, ischaemic heart disease does not usually cause any abnormalities in cardiac function at rest but manifests as metabolic and functional derangements in those regions in which the metabolic requirements of the exercising myocardium exceed coronary blood supply. Similarly in heart failure, patients most often progress from asymptomatic or ‘sub-clinical’ cardiac dysfunction through exertional breathlessness to severe symptoms when abnormalities may be readily detectable at rest. Assessment of cardiac function during exercise provides an opportunity to identify cardiac pathology at a far earlier stage of the disease process, a time in which treatments often have greatest efficacy.
Cardiac reserve is a term used to describe the heart's capacity to respond to the increasing workload of exercise. Although there are numerous theoretical reasons as to why cardiac reserve should serve as a critical key measure in understanding cardiac symptoms and limitations, the promise has not been realized because cardiac imaging is more difficult and less accurate during exercise. Increases in heart rate and respiratory capacity represent substantial challenges for ultrasound, nuclear and magnetic resonance imaging. Thus, much of the potential increase in diagnostic yield during exercise is attenuated by the inaccuracies of our imaging techniques.
Two differing approaches have been taken to address the impasse between the potential benefits of imaging during haemodynamic stress and the difficulties of imaging in this setting. Some investigators have accepted compromises and used ‘exercise surrogates’ which are more conducive to imaging, whilst others have attempted to improve cardiac imaging such that it can accurately appraise changes in cardiac function during exercise. Exercise surrogates can replicate various aspects of exercise physiology but each has limitations. Beta-adrenergic agents (such as dobutamine) increase myocardial contractility substantially, while the chronotropic response is less profound. More importantly, beta-adrenergic agents do not affect venous return, which is normally facilitated during exercise by the substantial increase in vascular flow (cardiac output), the increase in the skeletal muscle pump and the respiratory pump resulting from increasingly negative intra-thoracic pressures during inspiration. Without these factors, the ionotropic and chronotropic stimulation is not matched by cardiac filling and so there is minimal augmentation, or even attenuation, in cardiac filling. Thus there is a loss of stroke volume augmentation due to the Frank–Starling mechanism and increases in cardiac output are modest.
An alternative strategy is to use pacing or predominantly chronotropic stimuli (such as isoprenaline) but this strategy has even less impact on cardiac output and the lack of increase in preload and contractility mean that stroke volume tends to fall, quite the opposite effect of exercise. Cardiac work can be approximated as the product of stroke volume and afterload (mean systemic or pulmonary pressure for the left ventricle and right ventricle, respectively) and increasing exercise intensity demands a progressive augmentation of cardiac work due to an increase in both stroke volume and afterload. Dobutamine tends to increase stroke volume less than exercise and afterload is minimally impacted, such that it represents a lesser stress on the ventricle than does exercise [88,89]. Figure 5 (and the accompanying online video; electronic supplementary material) compares dobutamine- versus exercise-induced stress in the same patient using real-time cardiac magnetic resonance imaging. As compared with rest, the end-systolic volumes are only minimally reduced during exercise because much of the augmentation in stroke volume comes from the increase in end-diastolic volume. This is very different from the stimulus of dobutamine where all of the augmentation comes from increases in contractility against little afterload and thus there is a far greater reduction in end-systolic volume.
Figure 5.
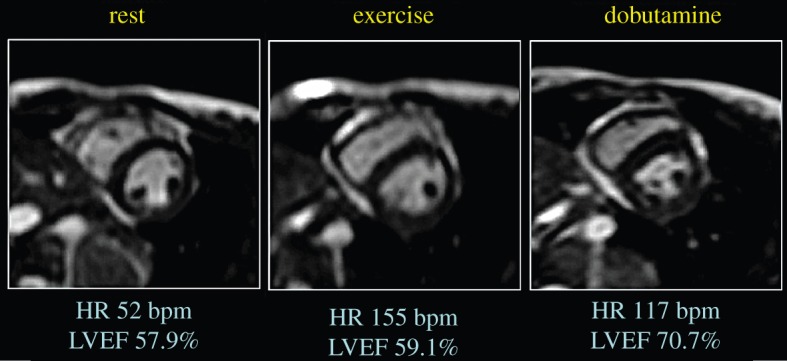
Augmentation of cardiac output during exercise as compared with dobutamine. As compared with rest, cardiac output increases substantially during exercise and modestly with dobutamine. Short-axis cine magnetic resonance images taken at end-systole demonstrate that augmentation during exercise is due to an increase in end-diastolic volumes and a reduction in end-systolic volumes. This is distinctly different from the response to dobutamine, in which the end-diastolic volume is reduced and the end-systolic volume is markedly reduced (seen as a pronounced reduction in end-systolic area in the figure). These differences are more clearly seen in the accompanying video (electronic supplementary material). (Online version in colour.)
In conclusion, there have been significant advances in cardiac imaging during exercise over recent years and we now have the capacity to assess the function of both the left and right ventricles during intense exercise with great accuracy. Real-time exercise cardiac magnetic resonance (Ex-CMR) imaging with compensation for respiratory translation enables cardiac volumetric quantification of both ventricles during free breathing and maximal exercise. Cardiac outputs determined using this technique correlate very strongly with invasive Fick measures but the reproducibility is arguably better with Ex-CMR [90]. Furthermore, although echo-cardiographic images cannot be acquired in all subjects, the accuracy of measures that can be obtained are reasonably impressive [91]. Thus, we can now move away from the limitations of pharmacological surrogates and more directly assess cardiac function during exercise in humans.
2.5.2. Human skeletal muscle
Skeletal muscle has traditionally been a popular in vitro organ E-model in biological energy transduction research (e.g. [92]) due to its large dynamical range of ATP turnover, spanning two orders of magnitude from the resting to the maximally activated states [93]. The pioneering application of 31P NMR spectroscopy to study ATP metabolism in living amphibian muscle was reported in the late 1970s [94,95]. Soon after, the first in vivo 31P MR spectra from mammalian muscle were obtained in rodents [96] and humans [97]. Using voluntary exercise as an experimental intervention, various biochemical and physiological processes in living muscle including ATP turnover and cellular energy balance, respectively, could be tracked in vivo and near real-time. Medical application of this non-invasive analytical technique soon followed, including diagnosis and evaluation of therapy efficacy in mitochondrial myopathy [98,99].
An important scientific advance was the validation of the hypothesis [100] that the phosphate potential, ΔGp, in resting muscle fibres is 20 kJ mol−1 higher than had been assumed on basis of chemical analysis of muscle biopsy samples (i.e. −65 versus −45 kJ mol−1, respectively [97])). This outcome clearly exposed the limitations of the latter destructive E-model for the investigation of human skeletal muscle biochemistry and physiology. Yet, biopsy findings that human skeletal muscle pH drops 0.6 pH units during strenuous dynamic exercise were confirmed in vivo (e.g. [101]), strengthening the evidence base for the acidification paradigm of short-term muscle fatigue that, at the time, was widely supported ([102]). In vivo MRS observations revalidated a rapid drop in muscle phosphocreatine (PCr) concentration, pH and myoglobin oxygenation in exercising human muscle. This led to the proposal that homeostatic control of myocellular energy balance involves a low-gain, closed-loop, concentration-dependent control mechanisms both with regard to cellular ATP as well as pO2 [93], as opposed to a high-gain, open loop, control paradigm proposed at the time for the special case of cardiac muscle [103].
For various reasons, including the physical dimensions of the horizontal magnet bore of early in vivo NMR spectrometers, voluntary exercise paradigms used in these studies typically involved some form of cyclic contraction and relaxation of a muscle group in a single limb (e.g. the digital flexor muscles of the forearm and the calf muscle of the lower leg) [104]. With the advent of ‘whole body’ MRI scanners in the late 1980s, the dynamic range of in-magnet bodily motion was significantly increased, enabling for the first time in vivo MRS data gathering from the quadriceps muscle [105,106]. An in vivo 31P MRS study of energy and proton balance in this muscle in healthy subjects performing alternating bipedal leg extension exercise against an adjustable hydraulic resistance observed that the concentration ratio PCr/(PCr + Pi), instead of falling abruptly at low workloads, decreased linearly over the entire work-range from rest to exhaustion [107]. Intra-sarcoplasmic pH likewise fell monotonously during exercise rather than abruptly at low workloads and attained a minimal value of a mere 6.8 [107]—i.e. significantly above the values of 6.5 or less commonly quoted in textbooks [101]. These findings were at odds with all previous results obtained either in biopsy studies or in vivo MRS studies employing single-limb exercise regimens. As such, they raised the question of what homeostatic control mechanisms of muscle PCr content and pH had previously been missed in single-limb exercise studies, and why.
The answer to this question goes right to the heart of the matter of this essay—i.e. the problem of reconstitution of the in vivo state in a laboratory setting. In skeletal muscle, the concentrations of PCr, Pi and H+ are determined not only by the magnitude of fluxes within the intra-myocellular network of metabolic pathways and membrane transporters but also by fluxes within the supra-myocellular network of tissues and organs that interact via the vascular system to supply working muscle with O2 and remove any accumulated CO2 and lactic acid, respectively (figure 6). Hence, the quality of reconstitution of the in vivo concentrations of PCr, Pi and H+ at any particular dynamic- or steady-state in any E-model of human skeletal muscle is dependent both on the level of reconstitution of this multi-scale network as well as on the maximal rates of muscle perfusion and pulmonary ventilation that may be achieved during any experimental intervention. The latter are regulated by changes in arterial pCO2 and pH during exercise [108]. Whereas the single-limb quadriceps exercise protocol was associated with a change in the arterial pCO2 but not pH [106], any form of whole body exercise is typically associated with a significant change in both variables [109] due to recruitment of a much larger muscle mass than during any form of single-limb exercise. This finding may explain the finding of severe quadriceps muscle acidification during dynamic knee-flexion exercise [106] versus robust quadriceps muscle pH homeostasis during in-magnet whole body exercise [107]. The latter finding has since been confirmed independently in studies employing an alternative mode of in-magnet whole body exercise (i.e. in-magnet bicycling exercise; [110,111]).
Figure 6.

Schematic drawing of the multi-scale network including interactions involved in quadriceps proton balance (left plane) and energy balance (right plane) during exercise. Atot: total weak acid content; CO2: total CO2 content; SID: strong ion difference. Arrows in the left plane indicate elasticities; hatched arrows indicate mass flow from the muscle (CO2 and lactate, respectively). Solid arrows in the right plane indicate metabolic mass flow during cyclic muscle contraction and relaxation. Open arrows between both planes depict interactions and their elasticities between the networks controlling muscle proton and energy balance during exercise. (Adapted from [107].)
The employment of in vivo experimental interventions that recruit homeostatic actions of the supracellular bodily network of interacting organs and tissues in addition to cellular homeostatic mechanisms also benefits the clinical investigation of multifactorial disease. For example, in juvenile dermatomyositis, an inflammatory disease of the skin and muscle with a suspected vascular component, it was found that robustness of homeostatic control of muscle CEC and pH during bicycling exercise was maintained in a patient that responded well to anti-inflammatory treatment but lost in another patient that maintained idiopathic pain complaints following exercise even after treatment [112]. Specifically, the same abrupt drop in muscle pH observed previously in healthy subjects during single leg knee-flexion exercise [106] was observed in the symptomatic patient during in-magnet bicycling exercise (figure 7). Mitochondrial function in this patient was, however, intact if not superior as evidenced by twofold faster than normal kinetics of PCr recovery post-exercise [112]. On basis of these findings in muscle, it was concluded that vascular function during exercise was compromised and that treatment in this particular case should additionally focus on normalizing the latter. This diagnosis may well have been missed if a single-limb exercise protocol and its associated normal range of CEC and pH dynamics had been used instead.
Figure 7.
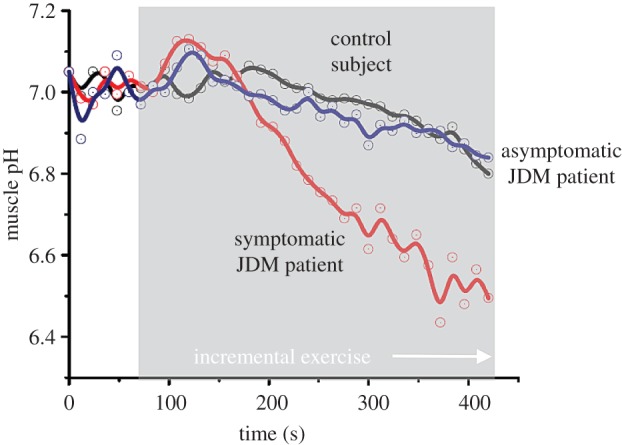
Quadriceps pH dynamics during progressive bicycling exercise to exhaustion in two paediatric patients diagnosed with juvenile dermatomyositis and an age-matched control. Patient no. 1 presented with lowered exercise tolerance and idiopathic upper leg muscle pain following exercise at the time of the MRS examination while exercise tolerance had fully recovered in patient no. 2. Both patients had received anti-inflammatory treatment. Solid lines depict the trend in the pH dynamics calculated using a spline function (adapted from [112]). (Online version in colour.)
3. Summary
Any accomplishment of the scientific objectives of the VPH project is critically dependent on the availability of physiologically realistic E-models of human cells and organs for disease studies and drug screening. The examples discussed in this review highlight some of the challenges and recent solutions in the field of medical experimental biology to improve the physiological realism of such E-models. Using the quality of reconstitution of the in vivo state in a laboratory setting as its measure, two issues in particular stand out. First, the main challenge is to avoid the pitfall of reductionist experimental design. The latter may result either from physical truncation of the multi-scale in vivo network (e.g. by isolating cell organelles or organs from the body; §§2.1 and 2.4), from studying surrogate cell models rather than human cells (§2.2), or from the use of surrogate instead of physiologically realistic experimental interventions (e.g. using ionotropic pharmeutical agents versus physical exercise to stress the human heart; §2.5.1). Secondly, the main path to E-model improvement has and will come from continuing application of technological innovation (e.g. the application of cell phenotypic reprogramming knowledge to develop patient-specific human cell models of disease; §2.3).
Competing interests
We declare we have no competing interests.
Funding
This work was supported in part by NIH grants HL072011 (JALJ and DAB) and GM094503 (DAB) and by a Wellcome Trust senior investigator award (PR).
References
- 1.Beard DA, Bassingthwaighte JB, Greene AS. 2005. Computational modeling of physiological systems. Physiol. Genomics 23, 1–3. ( 10.1152/physiolgenomics.00117.2005) [DOI] [PubMed] [Google Scholar]
- 2.Beard DA, Kushmerick MJ. 2009. Strong inference for systems biology. PLoS Comput. Biol. 5, e1000459 ( 10.1371/journal.pcbi.1000459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chance B, Garfinkel D, Higgins J, Hess B. 1960. Metabolic control mechanisms. 5. A solution for the equations representing interaction between glycolysis and respiration in ascites tumor cells. J. Biol. Chem. 235, 2426–2439. [PubMed] [Google Scholar]
- 4.Tomita M. 2001. Whole-cell simulation: a grand challenge of the 21st century. Trends Biotechnol. 19, 205–210. ( 10.1016/S0167-7799(01)01636-5) [DOI] [PubMed] [Google Scholar]
- 5.Viceconti M, Clapworthy G, Van Sint Jan S. 2008. The virtual physiological human—a European initiative for in silico human modelling. J. Physiol. Sci. 58, 441–446. ( 10.2170/physiolsci.RP009908) [DOI] [PubMed] [Google Scholar]
- 6.Hunter P, et al. 2010. A vision and strategy for the virtual physiological human in 2010 and beyond. Phil. Trans. R. Soc. A 368, 2595–2614. ( 10.1098/rsta.2010.0048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bono B, Hunter P. 2012. Integrating knowledge representation and quantitative modelling in physiology. Biotechnol. J. 7, 958–972. ( 10.1002/biot.201100304) [DOI] [PubMed] [Google Scholar]
- 8.Schmitz JP, Vanlier J, van Riel NA, Jeneson JA. 2011. Computational modeling of mitochondrial energy transduction. Crit. Rev. Biomed. Eng. 39, 363–377. ( 10.1615/CritRevBiomedEng.v39.i5.20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholls DG, Ferguson SJ. 2013. Bioenergetics, 4th edn, pxiv, 419 p. Amsterdam, The Netherlands: Academic Press. [Google Scholar]
- 10.Hogeboom GH, Schneider WC, Pallade GE. 1948. Cytochemical studies of mammalian tissues; isolation of intact mitochondria from rat liver; some biochemical properties of mitochondria and submicroscopic particulate material. J. Biol. Chem. 172, 619–635. [PubMed] [Google Scholar]
- 11.Claude A. 1946. Fractionation of mammalian liver cells by differential centrifugation: II. Experimental procedures and results. J. Exp. Med. 84, 61–89. ( 10.1084/jem.84.1.61) [DOI] [PubMed] [Google Scholar]
- 12.Chance B, Williams GR. 1955. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 217, 383–393. [PubMed] [Google Scholar]
- 13.Chance B, Hagihara B (eds). 1963. Direct spectroscopic measurements of interaction of components of the respiratory chain with ATP, ADP, phosphate and uncoupling agents. In Proc. of the Fifth Int. Congress of Biochemistry, Moscow, Russia, 10–16 August 1961. Oxford, UK: Pergamon Press.
- 14.de Duve C. 1967. Criteria of homogeneity and purity of mitochondria. In Methods in enzymology, vol. 10 (eds Estabrook RW, Pullman ME), pp. 7–18. New York, NY: Academic Press. [Google Scholar]
- 15.LaNoue KF, Bryla J, Williamson JR. 1972. Feedback interactions in the control of citric acid cycle activity in rat heart mitochondria. J. Biol. Chem. 247, 667–679. [PubMed] [Google Scholar]
- 16.Bishop PD. 1979. Factors affecting the regulation of mitochondrial respiration. Los Angeles, CA: The University of California. [Google Scholar]
- 17.Bishop PD, Atkinson DE. 1984. Adenine nucleotide control of the rate of oxygen uptake by rat heart mitochondria over a 15- to 20-fold range. Arch. Biochem. Biophys. 230, 335–344. ( 10.1016/0003-9861(84)90116-4) [DOI] [PubMed] [Google Scholar]
- 18.Jeneson JA, Wiseman RW, Westerhoff HV, Kushmerick MJ. 1996. The signal transduction function for oxidative phosphorylation is at least second order in ADP. J. Biol. Chem. 271, 27 995–27 998. ( 10.1074/jbc.271.45.27995) [DOI] [PubMed] [Google Scholar]
- 19.Denton RM, McCormack JG, Edgell NJ. 1980. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem. J. 190, 107–117. ( 10.1042/bj1900107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCormack JG, Denton RM. 1989. The role of Ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol. Cell Biochem. 89, 121–125. ( 10.1007/BF00220763) [DOI] [PubMed] [Google Scholar]
- 21.Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. 2001. Calcium activation of heart mitochondrial oxidative phosphorylation—Rapid kinetics of m(V) over dot (O2), NADH, and light scattering. J. Biol. Chem. 276, 2586–2599. ( 10.1074/jbc.M002923200) [DOI] [PubMed] [Google Scholar]
- 22.Territo PR, French SA, Balaban RS. 2001. Simulation of cardiac work transitions, in vitro: effects of simultaneous Ca2+ and ATPase additions on isolated porcine heart mitochondria. Cell Calcium 30, 19–27. ( 10.1054/ceca.2001.0211) [DOI] [PubMed] [Google Scholar]
- 23.Bose S, French S, Evans FJ, Joubert F, Balaban RS. 2003. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J. Biol. Chem. 278, 39 155–39 165. ( 10.1074/jbc.M306409200) [DOI] [PubMed] [Google Scholar]
- 24.Glancy B, Willis WT, Chess DJ, Balaban RS. 2013. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809. ( 10.1021/bi3015983) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beard DA. 2005. A biophysical model of the mitochondrial respiratory system and oxidative phosphorylation. PLoS Comput. Biol. 1, e36 ( 10.1371/journal.pcbi.0010036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu F, Yang F, Vinnakota KC, Beard DA. 2007. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J. Biol. Chem. 282, 24 525–24 537. ( 10.1074/jbc.M701024200) [DOI] [PubMed] [Google Scholar]
- 27.LaNoue KF, Jeffries FM, Radda GK. 1986. Kinetic control of mitochondrial ATP synthesis. Biochemistry 25, 7667–7675. ( 10.1021/bi00371a058) [DOI] [PubMed] [Google Scholar]
- 28.Wu F, Zhang EY, Zhang J, Bache RJ, Beard DA. 2008. Phosphate metabolite concentrations and ATP hydrolysis potential in normal and ischaemic hearts. J. Physiol. 586, 4193–4208. ( 10.1113/jphysiol.2008.154732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu F, Jeneson JA, Beard DA. 2007. Oxidative ATP synthesis in skeletal muscle is controlled by substrate feedback. Am. J. Physiol. Cell Physiol. 292, C115–C124. ( 10.1152/ajpcell.00237.2006) [DOI] [PubMed] [Google Scholar]
- 30.Wu F, Zhang J, Beard DA. 2009. Experimentally observed phenomena on cardiac energetics in heart failure emerge from simulations of cardiac metabolism. Proc. Natl Acad. Sci. USA 106, 7143–7148. ( 10.1073/pnas.0812768106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vinnakota KC, Dash RK, Beard DA. 2011. Stimulatory effects of calcium on respiration and NAD(P)H synthesis in intact rat heart mitochondria utilizing physiological substrates cannot explain respiratory control in vivo. J. Biol. Chem. 286, 30 816–30 822. ( 10.1074/jbc.M111.242529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinnakota KC, Singhal A, Van den Bergh F, Bagher-Oskouei M, Wiseman RW, Beard DA. In press. Open-loop stimulation by calcium contributes to control of oxidative phosphorylation in skeletal muscle mitochondria but not in cardiac mitochondria. Biophys. J. ( 10.1016/j.bpj.2015.12.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmström KM, et al. 2015. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 85, 178–182. ( 10.1016/j.yjmcc.2015.05.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan X, et al. 2013. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. ( 10.1038/ncb2868) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwong JQ, et al. 2015. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12, 15–22. ( 10.1016/j.celrep.2015.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinnakota KC, Bazil JN, Van den Bergh F, Wiseman RW, Beard DA. In press. Feedback regulation and time hierarchy of oxidative phosphorylation in cardiac mitochondria. Biophys. J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeneson JA, Westerhoff HV, Kushmerick MJ. 2000. A metabolic control analysis of kinetic controls in ATP free energy metabolism in contracting skeletal muscle. Am. J. Physiol. Cell Physiol. 279, C813–C832. [DOI] [PubMed] [Google Scholar]
- 38.Rorsman P, Braun M. 2013. Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 75, 155–179. ( 10.1146/annurev-physiol-030212-183754) [DOI] [PubMed] [Google Scholar]
- 39.Rorsman P, Renstrom E. 2003. Insulin granule dynamics in pancreatic beta cells. Diabetologia 46, 1029–1045. ( 10.1007/s00125-003-1153-1) [DOI] [PubMed] [Google Scholar]
- 40.Baldwin WM III, Su CA, Shroka TM, Fairchild RL. 2014. Experimental models of cardiac transplantation: design determines relevance. Curr. Opin. Organ Transplant 19, 525–530. ( 10.1097/MOT.0000000000000113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. 2011. Electrophysiology of pancreatic beta-cells in intact mouse islets of Langerhans. Prog. Biophys. Mol. Biol. 107, 224–235. ( 10.1016/j.pbiomolbio.2011.06.009) [DOI] [PubMed] [Google Scholar]
- 42.Ashcroft FM, Rorsman P. 2013. K(ATP) channels and islet hormone secretion: new insights and controversies. Nat. Rev. Endocrinol. 9, 660–669. ( 10.1038/nrendo.2013.166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frayn KN. 2010. Metabolic regulation: a human perspective, 3rd edn, 384 p. Oxford, UK: Wiley-Blackwell. [Google Scholar]
- 44.Ashcroft FM, Rorsman P. 2012. Diabetes mellitus and the beta cell: the last ten years. Cell 148, 1160–1171. ( 10.1016/j.cell.2012.02.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, Rorsman P. 2010. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 59, 1694–1701. ( 10.2337/db09-0797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. 2011. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diab. Obes. Metab. 13(Suppl 1), 95–105. ( 10.1111/j.1463-1326.2011.01450.x) [DOI] [PubMed] [Google Scholar]
- 47.Rorsman P, Braun M, Zhang Q. 2012. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 51, 300–308. ( 10.1016/j.ceca.2011.11.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. 2008. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628. ( 10.2337/db07-0991) [DOI] [PubMed] [Google Scholar]
- 49.Sherman A. 2011. Dynamical systems theory in physiology. J. Gen. Physiol. 138, 13–19. ( 10.1085/jgp.201110668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cha CY, Nakamura Y, Himeno Y, Wang J, Fujimoto S, Inagaki N, Earm YE, Noma A. 2011. Ionic mechanisms and Ca2+ dynamics underlying the glucose response of pancreatic beta cells: a simulation study. J. Gen. Physiol. 138, 21–37. ( 10.1085/jgp.201110611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gopel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renstrom E, Rorsman P. 1999. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β cells. J. Gen. Physiol. 114, 759–770. ( 10.1085/jgp.114.6.759) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riz M, Braun M, Pedersen MG. 2014. Mathematical modeling of heterogeneous elctrophysiological responses in human beta cells. PLoS Comput. Biol. 10, e1003389 ( 10.1371/journal.pcbi.1003389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. 2005. Pharmacological properties and functional role of Kslow current in mouse pancreatic beta-cells: SK channels contribute to Kslow tail current and modulate insulin secretion. J. Gen. Physiol. 126, 353–363. ( 10.1085/jgp.200509312) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanno T, Rorsman P, Göpel SO. 2002. Glucose-dependent regulation of rhythmic action potential firing in pancreatic beta-cells by KATP-channel modulation. J. Physiol. 545, 501–507. ( 10.1113/jphysiol.2002.031344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosengren AH, et al. 2012. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes 61, 1726–1733. ( 10.2337/db11-1516) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Unoki H, et al. 2008. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 40, 1098–1102. ( 10.1038/ng.208) [DOI] [PubMed] [Google Scholar]
- 57.McCarthy MI. 2008. Casting a wider net for diabetes susceptibility genes. Nat. Genet. 40, 1039–1040. ( 10.1038/ng0908-1039) [DOI] [PubMed] [Google Scholar]
- 58.Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET. 2013. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 23, 1554–1562. ( 10.1101/gr.150706.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruin JE, et al. 2014. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 12, 194–208. ( 10.1016/j.scr.2013.10.003) [DOI] [PubMed] [Google Scholar]
- 60.Almaca J, Molina J, Arrojo EDR, Abdulreda MH, Jeon WB, Berggren PO, Caicedo A, Nam HG. 2014. Young capillary vessels rejuvenate aged pancreatic islets. Proc. Natl Acad. Sci. USA 111, 17 612–17 617. ( 10.1073/pnas.1414053111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blandini F, Armentero MT. 2012. Animal models of Parkinson's disease. FEBS J. 279, 1156–1166. ( 10.1111/j.1742-4658.2012.08491.x) [DOI] [PubMed] [Google Scholar]
- 62.Langston JW, Ballard P. 1984. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): implications for treatment and the pathogenesis of Parkinson's disease. Can. J. Neurol. Sci. 11(Suppl 1), 160–165. [DOI] [PubMed] [Google Scholar]
- 63.Hartfield EM, Fernandes HJ, Vowles J, Cowley SA, Wade-Martins R. 2012. Cellular reprogramming: a new approach to modelling Parkinson's disease. Biochem. Soc. Trans. 40, 1152–1157. ( 10.1042/BST20120159) [DOI] [PubMed] [Google Scholar]
- 64.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. ( 10.1016/j.cell.2006.07.024) [DOI] [PubMed] [Google Scholar]
- 65.Hartfield EM, Yamasaki-Mann M, Ribeiro Fernandes HJ, Vowles J, James WS, Cowley SA, Wade-Martins R. 2014. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS ONE 9, e87388 ( 10.1371/journal.pone.0087388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Camnasio S, et al. 2012. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington's disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 46, 41–51. ( 10.1016/j.nbd.2011.12.042) [DOI] [PubMed] [Google Scholar]
- 67.Israel MA, et al. 2012. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 482, 216–220. ( 10.1038/nature10821) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karakikes I, et al. 2014. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 6, 6955 ( 10.1038/ncomms7955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karakikes I, Ameen M, Termglinchan V, Wu JC. 2015. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ. Res. 117, 80–88. ( 10.1161/CIRCRESAHA.117.305365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee DF, et al. 2015. Modeling familial cancer with induced pluripotent stem cells. Cell 161, 240–254. ( 10.1016/j.cell.2015.02.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ranga A, Gjorevski N, Lutolf MP. 2014. Drug discovery through stem cell-based organoid models. Adv. Drug Deliv. Rev. 69–70, 19–28. ( 10.1016/j.addr.2014.02.006) [DOI] [PubMed] [Google Scholar]
- 72.Chen V, Chen YH, Downing SE. 1987. An improved isolated working rabbit heart preparation using red cell enhanced perfusate. Yale J. Biol. Med. 60, 209–219. [PMC free article] [PubMed] [Google Scholar]
- 73.Podesser BK, Hallstrom S, Schima H, Huber L, Weisser J, Kroner A, Fürst W, Wolner E. 1999. The erythrocyte-perfused ‘working heart’ model: hemodynamic and metabolic performance in comparison to crystalloid perfused hearts. J. Pharmacol. Toxicol. Methods 41, 9–15. ( 10.1016/S1056-8719(99)00018-0) [DOI] [PubMed] [Google Scholar]
- 74.Schenkman KA, Beard DA, Ciesielski WA, Feigl EO. 2003. Comparison of buffer and red blood cell perfusion of guinea pig heart oxygenation. Am. J. Physiol. Heart Circ. Physiol. 285, H1819–H1825. ( 10.1152/ajpheart.00383.2003) [DOI] [PubMed] [Google Scholar]
- 75.Duvelleroy MA, Duruble M, Martin JL, Teisseire B, Droulez J, Cain M. 1976. Blood-perfused working isolated rat heart. J. Appl. Physiol. 41, 603–607. [DOI] [PubMed] [Google Scholar]
- 76.Bergmann SR, Clark RE, Sobel BE. 1979. An improved isolated heart preparation for external assessment of myocardial metabolism. Am. J. Physiol. 236, H644–H661. [DOI] [PubMed] [Google Scholar]
- 77.Chemnitius JM, Burger W, Bing RJ. 1985. Crystalloid and perfluorochemical perfusates in an isolated working rabbit heart preparation. Am. J. Physiol. 249, H285–H292. [DOI] [PubMed] [Google Scholar]
- 78.Segel LD, Rendig SV. 1982. Isolated working rat heart perfusion with perfluorochemical emulsion Fluosol-43. Am. J. Physiol. 242, H485–H489. [DOI] [PubMed] [Google Scholar]
- 79.Gillis AM, Kulisz E, Mathison HJ. 1996. Cardiac electrophysiological variables in blood-perfused and buffer-perfused, isolated, working rabbit heart. Am. J. Physiol. 271, H784–H789. [DOI] [PubMed] [Google Scholar]
- 80.MacDonald VW, Winslow RM. 1992. Oxygen delivery and myocardial function in rabbit hearts perfused with cell-free hemoglobin. J. Appl. Physiol. 72, 476–483. [DOI] [PubMed] [Google Scholar]
- 81.Schenkman KA. 2001. Cardiac performance as a function of intracellular oxygen tension in buffer-perfused hearts. Am. J. Physiol. Heart Circ. Physiol. 281, H2463–H2472. [DOI] [PubMed] [Google Scholar]
- 82.Wengrowski AM, Kuzmiak-Glancy S, Jaimes R III, Kay MW. 2014. NADH changes during hypoxia, ischemia, and increased work differ between isolated heart preparations. Am. J. Physiol. Heart Circ. Physiol. 306, H529–H537. ( 10.1152/ajpheart.00696.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tamura M, Oshino N, Chance B, Silver IA. 1978. Optical measurements of intracellular oxygen concentrations of rat heart in vitro. Arch. Biochem. Biophys. 191, 18–22. ( 10.1016/0003-9861(78)90062-0) [DOI] [PubMed] [Google Scholar]
- 84.Chess DJ, Billings E, Covian R, Glancy B, French S, Taylor J, de Bari H, Murphy E, Balaban RS. 2013. Optical spectroscopy in turbid media using an integrating sphere: mitochondrial chromophore analysis during metabolic transitions. Anal. Biochem. 439, 161–172. ( 10.1016/j.ab.2013.04.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wikstrom M, Morgan JE. 1992. The dioxygen cycle. Spectral, kinetic, and thermodynamic characteristics of ferryl and peroxy intermediates observed by reversal of the cytochrome oxidase reaction. J. Biol. Chem. 267, 10 266–10 273. [PubMed] [Google Scholar]
- 86.Heineman FW, Kupriyanov VV, Marshall R, Fralix TA, Balaban RS. 1992. Myocardial oxygenation in the isolated working rabbit heart as a function of work. Am. J. Physiol. 262, H255–H267. [DOI] [PubMed] [Google Scholar]
- 87.Arai AE, Kasserra CE, Territo PR, Gandjbakhche AH, Balaban RS. 1999. Myocardial oxygenation in vivo: optical spectroscopy of cytoplasmic myoglobin and mitochondrial cytochromes. Am. J. Physiol. 277, H683–H697. [DOI] [PubMed] [Google Scholar]
- 88.Cnota JF, Mays WA, Knecht SK, Kopser S, Michelfelder EC, Knilans TK, Claytor RP, Kimball TR. 2003. Cardiovascular physiology during supine cycle ergometry and dobutamine stress. Med. Sci. Sports Exerc. 35, 1503–1510. ( 10.1249/01.MSS.0000084436.15808.52) [DOI] [PubMed] [Google Scholar]
- 89.Mehrotra P, Labib SB, Schick EC. 2012. Differential effects of dobutamine versus treadmill exercise on left ventricular volume and wall stress. J. Am. Soc. Echocardiogr. 25, 911–918. ( 10.1016/j.echo.2012.05.002) [DOI] [PubMed] [Google Scholar]
- 90.La Gerche A, et al. 2012. Cardiac magnetic resonance imaging: a new gold standard for ventricular volume quantification during high-intensity exercise. Circ. Cardiovasc. Imaging 6, 329–338. ( 10.1161/CIRCIMAGING.112.980037) [DOI] [PubMed] [Google Scholar]
- 91.Claessen G, et al. In press. Accuracy of echocardiography to evaluate pulmonary vascular and right ventricular function during exercise. JACC Cardiovasc. Imaging. ( 10.1016/j.jcmg.2015.06.018) [DOI] [Google Scholar]
- 92.Chance B, Connelly CM. 1957. A method for the estimation of the increase in concentration of adenosine diphosphate in muscle sarcosomes following a contraction. Nature 179, 1235–1237. ( 10.1038/1791235a0) [DOI] [PubMed] [Google Scholar]
- 93.Hochachka PW, McClelland GB. 1997. Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J. Exp. Biol. 200, 381–386. [DOI] [PubMed] [Google Scholar]
- 94.Burt CT, Glonek T, Barany M. 1977. Analysis of living tissue by phosphorus-31 magnetic resonance. Science 195, 145–149. ( 10.1126/science.188132) [DOI] [PubMed] [Google Scholar]
- 95.Dawson J, Gadian DG, Wilkie DR. 1976. Proceedings: living muscle studied by 31P nuclear magnetic resonance. J. Physiol. 258, 82P–83P. [PubMed] [Google Scholar]
- 96.Ackerman JJ, Grove TH, Wong GG, Gadian DG, Radda GK. 1980. Mapping of metabolites in whole animals by 31P NMR using surface coils. Nature 283, 167–170. ( 10.1038/283167a0) [DOI] [PubMed] [Google Scholar]
- 97.Chance B, Eleff S, Leigh JS Jr. 1980. Noninvasive, nondestructive approaches to cell bioenergetics. Proc. Natl Acad. Sci. USA 77, 7430–7434. ( 10.1073/pnas.77.12.7430) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Radda GK. 1986. The use of NMR spectroscopy for the understanding of disease. Science 233, 640–645. ( 10.1126/science.3726553) [DOI] [PubMed] [Google Scholar]
- 99.Argov Z, Bank WJ, Maris J, Eleff S, Kennaway NG, Olson RE, Chance B. 1986. Treatment of mitochondrial myopathy due to complex III deficiency with vitamins K3 and C: A 31P-NMR follow-up study. Ann. Neurol. 19, 598–602. ( 10.1002/ana.410190615) [DOI] [PubMed] [Google Scholar]
- 100.Veech RL, Lawson JW, Cornell NW, Krebs HA. 1979. Cytosolic phosphorylation potential. J. Biol. Chem. 254, 6538–6547. [PubMed] [Google Scholar]
- 101.Taylor DJ, Styles P, Matthews PM, Arnold DA, Gadian DG, Bore P, Radda GK. 1986. Energetics of human muscle: exercise-induced ATP depletion. Magn. Reson. Med. 3, 44–54. ( 10.1002/mrm.1910030107) [DOI] [PubMed] [Google Scholar]
- 102.Sahlin K, Alvestrand A, Brandt R, Hultman E. 1978. Intracellular pH and bicarbonate concentration in human muscle during recovery from exercise. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 45, 474–480. [DOI] [PubMed] [Google Scholar]
- 103.Balaban RS. 1990. Regulation of oxidative phosphorylation in the mammalian cell. Am. J. Physiol. 258, C377–C389. [DOI] [PubMed] [Google Scholar]
- 104.Prompers JJ, Jeneson JA, Drost MR, Oomens CC, Strijkers GJ, Nicolay K. 2006. Dynamic MRS and MRI of skeletal muscle function and biomechanics. NMR Biomed. 19, 927–953. ( 10.1002/nbm.1095) [DOI] [PubMed] [Google Scholar]
- 105.Rodenburg JB, de Boer RW, Jeneson JA, van Echteld CJ, Bar PR. 1994. 31P-MRS and simultaneous quantification of dynamic human quadriceps exercise in a whole body MR scanner. J. Appl. Physiol. 77, 1021–1029. [DOI] [PubMed] [Google Scholar]
- 106.Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS, Wagner PD. 1995. Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J. Clin. Invest. 96, 1916–1926. ( 10.1172/JCI118237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jeneson JA, Bruggeman FJ. 2004. Robust homeostatic control of quadriceps pH during natural locomotor activity in man. FASEB J. 18, 1010–1012. ( 10.1096/fj.03-0762fje) [DOI] [PubMed] [Google Scholar]
- 108.Wasserman K, Whipp BJ, Casaburi R, Golden M, Beaver WL. 1979. Ventilatory control during exercise in man. Bull. Eur. Physiopathol. Respir. 15, 27–51. [PubMed] [Google Scholar]
- 109.Koyal SN, Whipp BJ, Huntsman D, Bray GA, Wasserman K. 1976. Ventilatory responses to the metabolic acidosis of treadmill and cycle ergometry. J. Appl. Physiol. 40, 864–867. [DOI] [PubMed] [Google Scholar]
- 110.Jeneson JA, Schmitz JP, Hilbers PA, Nicolay K. 2010. An MR-compatible bicycle ergometer for in-magnet whole-body human exercise testing. Magn. Reson. Med. 63, 257–261. [DOI] [PubMed] [Google Scholar]
- 111.Schmitz JP, van Dijk JP, Hilbers PA, Nicolay K, Jeneson JA, Stegeman DF. 2012. Unchanged muscle fiber conduction velocity relates to mild acidosis during exhaustive bicycling. Eur. J. Appl. Physiol. 112, 1593–1602. ( 10.1007/s00421-011-2119-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Brussel M, van Oorschot JWM, Schmitz JPJ, Nicolay K, Van Royen-Kerkhof A, Takken T, Jeneson JAL. 2015. Muscle metabolic responses during dynamic in-magnet exercise testing: a pilot study in children with an idiopathic inflammatory myopathy. Acad. Radiol. 22, 1443–1448. ( 10.1016/j.acra.2015.06.013) [DOI] [PMC free article] [PubMed] [Google Scholar]