

Abstract
Reconstructing and understanding the Human Physiome virtually is a complex mathematical problem, and a highly demanding computational challenge. Mathematical models spanning from the molecular level through to whole populations of individuals must be integrated, then personalized. This requires interoperability with multiple disparate and geographically separated data sources, and myriad computational software tools. Extracting and producing knowledge from such sources, even when the databases and software are readily available, is a challenging task. Despite the difficulties, researchers must frequently perform these tasks so that available knowledge can be continually integrated into the common framework required to realize the Human Physiome. Software and infrastructures that support the communities that generate these, together with their underlying standards to format, describe and interlink the corresponding data and computer models, are pivotal to the Human Physiome being realized. They provide the foundations for integrating, exchanging and re-using data and models efficiently, and correctly, while also supporting the dissemination of growing knowledge in these forms. In this paper, we explore the standards, software tooling, repositories and infrastructures that support this work, and detail what makes them vital to realizing the Human Physiome.
Keywords: Human Physiome, standards, repositories, service infrastructure, reproducible science, managing big data
1. Introduction
The Human Physiome consists of mathematical models that describe the focused intricacies of molecular transport, all the way up to the broader studies on health of populations of individuals over their entire lifetime. This requires model and data integration across nine orders of magnitude in space and 15 in time. Describing, data populating and integrating computational models over these vast spatio-temporal scales requires the interoperation of multiple disparate and often geographically separated data sources. The data and models are often embedded in proprietary software or databases. Extracting knowledge from such sources, even when the software or databases are readily available, is a difficult and demanding task—but one which scientists must frequently perform in order to reuse or integrate pre-existing knowledge into the common framework required to realize the Human Physiome.
One major challenge confronting the development of the Human Physiome is to reach a consensus agreement on measurements and valid performance criteria when dealing with process, data and differences in definitions/terminology. Standards are key for neutralizing many of these complexities by supporting each stage of an innovation process and by ensuring compatibility and interoperability of products and technologies through:
(i) formatting data and computer models consistently, so that people and software can identify the key information in the files and interrelate corresponding pieces of information;
(ii) providing metadata checklists (minimum information guideline standards), which ensure that all information that is vital for understanding, contextualising and securing validity and reproducibility of the data and models is available; and
(iii) providing controlled vocabularies and ontologies to describe components in data and models such that they can be unambiguously identified.
For the Human Physiome to be successful, all constituent parts that comprise it must be formatted, and described using accepted community standards. To enable the reuse, extension and composition of existing Human Physiome knowledge, this standardization needs to be applied to the mathematical models and their instantiation in computational simulations, individualized parametrization of the models and simulations, and all supporting or underlying data, including data that describes the context of the data and models (i.e. metadata). Furthermore, describing and cross-referencing the associations between these constituent parts is imperative as it provides the foundation for computationally automated reasoning facilitating the integration of these parts, a feat that is complex and error prone when completely human driven. This automation is further supported by repositories, which aid the discoverability and exploitation of accumulated knowledge. They also offer the ability to computationally query and identify relevant information for automated integration. We believe this greatly improves the quality and speed with which research into the Human Physiome, and even in broader biological sciences, can be driven.
In this paper, we will explore existing platforms and technologies that address these goals and examine how they are performing in regard to enabling the adoption of computational models as core laboratory instruments. We furthermore introduce new developments that allow scientists to not only reproduce previous work but also re-use it with confidence. These developments also start to allow the linkage between mathematical models and experimental and clinical data, which is an essential requirement for the realization of the Human Physiome. We then present a ‘worked example’ demonstrating how the Human Physiome can be realized through the composition and integration of these technologies.
2. Standards, resources and the Human Physiome
Standards in the context of the Human Physiome are agreed ways of structuring, and describing all of its constituent parts including data, models, methods and maps. They also assist with describing how these constituent parts interact together, or are linked. For the purposes of this paper, we focus on how key formatting standards, metadata checklists and ontologies support model and data handling. The implementation of these standards ensures that the data, models, methods and maps that are structured and described are FAIR: Findable, Accessible, Interoperable and Reusable [1].
Standards are often defined for communities on two levels: (i) through standardization communities at an official standards developing organization, such as the International Organization for Standardization (ISO), as well as national or regional (e.g. European) standardization bodies or (ii) bottom-up through grass roots activities emanating from the scientific communities within a field, such as the COMBINE Network (COmputational Modelling in BIology NEtwork).
The ISO technical committee for biotechnology standards (ISO/TC 276) has recently established a new working group for ‘Data processing and integration’ (WG5), which has the potential to impact the Human Physiome by defining an umbrella framework for modelling standards. Starting from existing community standards that are already used by specialists within the diverse sub-domains, this standardization panel is currently developing a meta-standard guideline for the consistent application of community standards. This new ISO standard will define a framework of minimal requirements and rules for the standardized and coherent formatting of biological data and resulting computer models, as well as for the standardized description of their corresponding metadata. This also will comprise standardized workflows for the structured processing, storing, integration and dissemination of the data, models and metadata.
Grass roots standards activities are often driven directly by the needs of the researchers within the research field themselves. Indeed, grass roots activities are often comprised of the researchers actively working in the field of research itself. One of the more prominent communities that affects the Human Physiome is the COMBINE (http://co.mbine.org/). COMBINE originates from a functional grouping of standards developing communities in biology [2], together they drive the establishment and adoption of a range of standards [3]. Part of the adoption process of standards is making sure that they are discoverable.
It is often difficult for researchers to be able to identify suitable standards for use within their field, as availability is not enough. Other researchers within the field also need to use the standards, and there has to be software to facilitate the generation and exchange of standardized data and models. Initiatives such as Biosharing.org assist with making standards more discoverable, by cataloguing and describing many of the standards available to bio-researchers. It has currently over 600 standards catalogued for life, environmental and biomedical sciences [4]. The experience of researchers within the field, however, is still often the most reliable source.
Here we introduce the standards most relevant to the Human Physiome, and its community. The use of standards in physiome-style modelling is well established for encoding some aspects of the mathematical models (e.g. [5–8]). These standards allow us to go beyond simple mathematical model descriptions, by enhancing the reproducibility of studies, which can be exchanged between tools and research groups.
2.1. The Human Physiome's key standards for modelling
The COMBINE initiative was established to coordinate community standards and formats for computational models for generation and adoption [2,3,9]. Some of the COMBINE core standards, such as CellML and SED-ML, are most relevant to this work, and the related FieldML standard is being developed to replace the range of ad hoc file formats currently used for sharing, archiving and exchanging field-based spatial models.
2.1.1. CellML
CellML ([10] http://cellml.org/) is an XML-based format for encoding mathematical models in a modular and composable manner. Primarily used for describing systems of differential algebraic equations, CellML provides the syntactic constructs to describe mathematical equations in a modular framework called components. Components can then be defined in one location and re-used in many other locations via an import mechanism [11]. CellML requires all variables and numerical quantities in a model to be associated with physical units (the dimensionless ‘unit’ is available for quantities that have no physical interpretation). This is an essential feature when composing a model from multiple child models, where the same quantity could be defined with compatible but different physical units [12].
2.1.2. The simulation experiment description markup language
The simulation experiment description markup language (SED-ML [13]; http://sed-ml.org/) is an XML-based format for encoding the description of a computational simulation. Typically used with an XML-based model description format (i.e. CellML or the systems biology markup language (SBML)), SED-ML allows for the description of applying a numerical algorithm to a mathematical model in order to perform a given task. Tasks may be nested to allow the composition of relatively simple tasks into increasingly complex simulations. Mechanisms exist in SED-ML to apply pre-processing steps to a model prior to executing a simulation task and also to apply post-processing to the raw simulation results. Rudimentary support is available in the SED-ML specification for describing the specific outputs desired from a simulation experiment (two- (2D) or three-dimensional (3D) plots and arbitrary reports). SED-ML is still in the early stages of development, and while it is able to encode common simulation experiments well, there is still much work to do in order to provide more comprehensive simulation experiment descriptions. Tool developers are encouraged to encode as much of their simulation experiment description as possible in SED-ML and make use of extensions to encode the remainder, and thus help the standard evolve to meet the needs of the community.
2.1.3. FieldML
FieldML [14,15] is a declarative language for building hierarchical models represented by generalized mathematical fields. FieldML can be used to represent the dynamic 3D geometry and solution fields from computational models of cells, tissues and organs. It enables model interchange for the bioengineering and general engineering analysis communities.
2.1.4. Pharmacometrics markup language
Pharmacometrics markup language (PharmML) [16] is an exchange format for encoding of models, associated tasks and their annotation as used in pharmacometrics, developed by the DDMoRe consortium (http://ddmore.eu/), an European Innovative Medicines Initiative (IMI) project (http://www.imi.europa.eu/). It provides the means to encode pharmacokinetic and pharmacodynamic (PK/PD) models, as well as clinical trial designs and modelling steps. Thus, ParmML, for example, allows the standardized mathematical description of the time course of effect intensity within a certain tissue or organ or even the whole body in response to administration of a drug dose.
2.1.5. Other standards
COMBINE has other core standards which are less directly relevant to the Human Physiome, but nevertheless important within the field. These include standards for biological pathway exchange (BioPAX, [17]), systems biology models (SBML, [18]), the graphical display of models (SBGN, [19]) and synthetic biology (SBOL, [20]). An overview about COMBINE formats and other community standards for modelling can be found in the NormSys registry (http://normsys.h-its.org), a freely available online resource that not only lists the standards, but also compares their major features, their possible fields of biological application and use cases (including model examples), as well as their relationships, commonalities and differences.
2.2. Describing the model
In the previous sections, we introduced the standards by which we are able to encode, archive, share and re-use mathematical models and numerical simulations (collectively called the computational model here) relevant to the Human Physiome. While data encoded in these formats provide the essential basis for realizing the quantitative Human Physiome, additional information is needed in order to better describe the computational models to allow others to correctly interpret or apply existing models in their own novel context. These descriptions are known as metadata, and there is a relevant checklist that provides guidelines on the minimum amount of information required in order to understand a model (MIRIAM [21])
A prime example of such additional information is the link between the computational model and the actual biology being modelled. An unambiguous description of the biology can be achieved through the annotation of entities in the computational model using widely used biological ontologies. For example, UniProt [22] or the Protein Ontology [23] can be used to uniquely identify proteins in a particular biological context which can then be linked to specific entities in the computational model. Similarly, the Gene Ontology [24,25] could be used to identify specific genes or cellular components and the Foundational Model of Anatomy (FMA, [26]) can be used to localize an entity in the computational model to specific spatial location or anatomical structure. The entities in the computational model may be specific variables, identified parts of a model, or even entire models. For some early examples, see [27–30].
Having a computational model annotated with its corresponding biological description then allows us to link entities in the computational model with experimental or clinical observations. By establishing such linkages, we are able to allow, for example, model parameters to have their value defined from an external (and potentially remote) database. Such external databases could be compilations of experimental recordings or real-world clinical data from electronic health record (EHR) systems or clinical registries of population health information.
The experimentally obtained kinetic data that describe the dynamics of biochemical reactions (e.g. metabolic or signalling reactions within a cellular network), for instance, can be obtained from databases such as SABIO-RK ([31]; http://sabiork.h-its.org). This manually curated database contains data which are either extracted by hand from the scientific literature [32] or directly submitted from laboratory experiments [33]. The database content includes kinetic parameters in relation to the reactions and biological sources, as well as experimental conditions under which they were obtained and is fully annotated to other resources, making the data easily accessible via web interface or web services for integration into the computational models. Through export of the data (together with its annotations and SABIO-RK identifiers for tracing back to the original dataset) in standardized data exchange formats like SBML this allows the direct data integration into models.
Four major resources of data can be identified: clinical data, laboratory data, sample data and trial data. Each of these groups implicates particular challenges with regard to data quality. The clinical data in health records are mainly recorded for billing and quality control, the ‘secondary use’ in clinical or biomedical research is not intended. Using these data in research calls for measures safeguarding completeness, documented provenance and appropriate accuracy [34]. Integration of EHR data requires considerable effort in mapping and alignment of the data [35]. The same applies for data from registries, as was exemplified for arthritis registries [36].
The quality of laboratory data is affected by metrological as well as methodological issues. Standard laboratory parameters are mostly validated by inter-laboratory tests which are meant to enable traceability and comparability of the reported results [37,38]. However, laboratory data are not necessarily comparable, particularly when multiple laboratories using different assays are involved [39]. Long-term observational studies additionally suffer from the lack of continuity in the laboratory methods applied [40,41]. Analysis of non-standard analytes or using not fully validated research methods aggravates the difficulties. Yet a major problem arises already before the samples have entered the laboratory. Sample collection, handling and processing can tremendously impact subsequent analysis and produce erroneous results. Extensive documentation and tight control of the preanalytical phase are imperative to enable later assessment of the quality of laboratory data [42]. When samples are obtained from sample repositories like biobanks, appropriate documentation is compulsory. For this purpose, data formats have been proposed already [43–45] but are rarely implemented. Occasionally, the integration of data from different studies is inevitable in order to achieve significant case numbers. Harmonization and integration of data from heterogeneous sources can pose substantial challenges demanding for powerful tools [46].
Considering the above enumerated problems, it is apparent that the quality issue in biomedical data can at present not be regarded as solved. Substantial effort is still required to develop standards, interfaces and specifications fostering data quality and encouraging their use. While these issues of data quality are important and highly relevant when using the data, we are able to begin linking entities in the computational models to the underpinning source or representative data.
In the biomedical informatics domain, health information are most commonly structured and codified by purpose specific standards (e.g. HL7, openEHR) and clinical terminologies (e.g. ICD, SNOMED CT, LOINC) [47,48]. While computational physiology and bioinformatics communities make good use of Semantic Web technologies for data representation, discovery, integration and inferencing, there is limited use in healthcare delivery. Therefore, the challenge is to make the connection with health information systems. For this to happen, use of shared ontologies for annotating both computational models and clinical information together with the use of clinical information modelling standards such as openEHR [49] will be important. An example would be linking of a medical observation, such as a pathology test result or a clinical diagnosis, that could be represented by a formal information model (an openEHR Archetype [50]) which can be encoded by both LOINC or SNOMED CT that defines clinical meaning and FMA that links to the COMBINE family of standards. This Archetype then can be used to find relevant clinical information given a biological meaning or a particular model metadata annotated by shared ontologies.
In achieving this, it is important that the underlying standards allow the computational model to be defined independent of a particular parametrization of the model. Thus, allowing mathematical models to be individualized to specific experimental or clinical data from a relevant data source. It would also be desirable for SED-ML to allow the selection of such parameter values to be defined as part of the simulation experiment—and in fact this is one of the key features being developed for future versions of SED-ML—but for now this falls into the realm of tool-specific functionality.
Similar to the parametrization of the computational model, the actual protocol being used in a given simulation experiment should be separate to the model. This allows the definition of common protocols which can then be applied to a range of models or even a particular model across a range of parametrizations (such as selections from a population distribution). One example of this in practice is the concept known as functional curation or virtual experiments [51–53].
We are now able to define computational models, describe the relevant biological information, and beginning to link the model to specific parametrizations and protocols. The final information that is required for all this to be really useful is the provenance—or origin—of the knowledge. In order to correctly apply the computational model and associated information in a given (potentially novel) scientific context it is often essential to know where, when, how and by whom a given piece of knowledge was obtained. Maintaining, presenting and curating such provenance information falls into the realm of repositories.
3. Discovering, sharing, archiving and reusing the Human Physiome
In the previous section, we introduced the wide range of standardization efforts relevant to the Human Physiome, including mathematical models, simulation experiments and their associated metadata. As the Human Physiome can only be realized by a large and collaborative community of scientists working in concert, a given piece of work or knowledge has to be efficiently distributed within this community. In the following, we present some recent developments that allow scientists to not only reproduce previous work, but also re-use it with confidence. These developments also start to allow the linkage between mathematical models and experimental and clinical data, which is an essential requirement for the realization of the Human Physiome.
Scientists archiving their work in accessible data repositories is the first step. Beyond archiving, scientists need to be able to discover, interrogate and use the wide range of data and knowledge being discovered throughout the Human Physiome community. Such discovery relies extensively on the semantic description of the computational data stored in the repositories, whereas the re-use relies on the data being encoded in a standard format. The semantic description allows not only the fact that some piece of knowledge exists, but potentially also the provenance and description of that data will provide scientists with further information as they judge whether the data are suitable for their needs. Thus, the infrastructural tools supporting the repositories need to provide access to a range of supporting information and metadata as well as the actual data.
Here we describe two large repositories commonly used in the Human Physiome community as well as introducing several related resources relevant to this context which address different aspects of the Human Physiome. A listing of further resources closely related to the Human Physiome community is available in table 1.
Table 1.
A listing of resources closely related to the Human Physiome community.
| resource | description | link |
|---|---|---|
| BioModels Database [54] | a repository of computational models of biological processes. Models described from the literature are manually curated and enriched with cross-references. The BioModels Database is a static archive of the models with periodic releases of the database made available in the Public Domain. | http://www.ebi.ac.uk/biomodels-main/ |
| JWS Online [55] | systems biology tool for the construction, modification and simulation of kinetic models and for the storage of curated models. | http://jjj.mib.ac.uk/ |
| The Drug Disease Model Resources (DDMoRe) Repository | provides a platform for sharing computational models used to describe the interactions between drugs and patients. This public, open-access repository allows users to encode their model in a single format that can be converted seamlessly and executed in commonly used software packages. The system can host both, publicly shared and private models and can store models in PharmML or MDL. | http://repository.ddmore.eu |
| The VPH-Share Portal | an online environment for the development, construction and storage of workflows for biomedical research and modelling. It is designed to support researchers, clinicians and software developers sharing their resources and provides an online infrastructure which allows users to share and access biomedical data, tools and workflows. VPH-Share was created through a 4-year, European FP7 funded project, which brought together 21 international partners from industry, academia and healthcare. | https://portal.vph-share.eu |
| The p-medicine portal | provides an integrative platform to clinicians, patients and researchers to collaborate, share data and expertise, and use tools and services to improve the personalized treatment of patients. The current version of the portal contains an initial functionality of the p-medicine security framework, data mining tools, p-medicine Workbench, Ontology Annotator as well as ObTiMA, an ontology-based clinical trial management system for designing and conducting clinical trials. | http://p-medicine.eu/tools/p-medicine-portal/ |
| The Quantiative Kidney DataBase [56,57]. | makes kidney-related physiological data easily available. The emphasis is on published experimental results relevant to quantitative renal physiology, with a particular focus on data relevant for evaluation of parameters in mathematical models of renal function. | http://physiome.ibisc.fr/qkdb/ |
| The Kidney Systems Biology Project | makes available curated transcriptomic and proteomic databases for the various renal tubule epithelia compiled from published papers. | http://helixweb.nih.gov/ESBL/Database/ |
3.1. Physiome model repository
The Physiome Model Repository (PMR, https://models.physiomeproject.org/) is a free and open repository for archiving, sharing and discovering data relevant to computational physiology and related fields [58,59]. All content in PMR is stored in version-controlled workspaces, and specific revisions of a workspace can be exposed with a persistent URL and customisable presentation via the web interface. Software clients are able to interact with PMR via a range of web services. An RDF-based knowledgebase is also available in PMR, which indexes identified annotations from all workspaces for the latest revision and across all exposed revisions. Queries can be submitted against this knowledgebase to discover and locate relevant data, models or other artefacts stored in the repository to which the user has access.
All data in PMR is located inside a workspace. PMR provides an access control framework that allows users to manage access options to their workspaces (private, shared with specific users or groups (with read and/or write access), publicly readable). The contents of a workspace are managed with a distributed version control system which enhances both the collaborative development of the artefacts stored therein and the management of the provenance of all data in the workspace [60]. The ability to embed workspaces within workspaces ensures that existing data can easily be re-used. Furthermore, such embedded workspaces are essentially recorded as a link to a specific revision of the source workspace and any change in the version linked to (such as updating to a newer revision) is recorded in the history of the workspace, thus ensuring that changes are accurately recorded in the workspace provenance.
The PMR also provides the ability to mark a specific revision of a workspace for publication as what is termed an exposure (in software engineering, this can be thought of as being similar to making a software release). When users create exposures they are able to flag specific artefacts in the workspace to be processed for display via the PMR website and made available via the webservice interface. Examples of such processing are: the generation of computational code from CellML models; converting various text formats to HTML for rendering in the web browser and rendering WebGL scenes in HTML pages to display interactive FieldML finite-element models. Users are able to manage access to their exposures in the same manner as for workspaces. If sufficient information are provided during the exposure creation process, COMBINE archives can also be generated and made available via the exposure.
The combination of community standards for encoding computational models together with semantic web and software development tools enables PMR to provide a unique resource for archiving, sharing, reusing and discovering a range of data relevant to the Human Physiome.
3.2. FAIRDOM
The FAIRDOM project is a European effort to establish integrated data and model management service facilities for Systems Biology and Synthetic Biology (http://fair-dom.org). The initiative works together with stakeholders towards FAIRer management; that is to make data, operating procedures and models better Findable, Accessible, Interoperable and thereby Reusable. It is, in part, an answer to the scientific community's need for reproducible research [61–63]. In a practical sense, it offers researchers software platforms and tools that assist with collecting and annotating data, integrating the data into models, and storing models in a central hub which functionally structures and links models, data and operating procedures using the Investigation > Study > Assay framework [64–66]. Laboratory data collection and post-processing is supported by openBis [67], while the processed data, model and operating procedure storage is handled by the FAIRDOMHub (http://www.fairdomhub.org), an instance of the software SEEK (http://www.seek4science.org/) [68]. The integration of gateways to other systems biology platforms allows, for example, seamless handling and simulation of models in SYCAMORE [69] or JWS Online [55], as well as visualization of networks in Cytoscape [70].
SEEK is applied as a central data management system for large-scale research initiatives like the German Virtual Liver Network (http://www.virtual-liver.de/) and European research networks like European Research Area Networks (e.g. ERA-Net for Systems Biology Applications), the former SysMO project (Systems Biology of Microorganisms) or NMTrypI (New Medicines for Trypanosomatidic Infections), as well as Synthetic Biology Centres at Manchester (SynBioChem) and Edinburgh (SynthSys). To this end the resource is ideal for supporting the Human Physiome, owing to its flexibility in collection, annotation, storage and distribution of data, models and operating procedures.
The FAIRDOM project also offers services for curation, training and assistance with data management plans for European research projects; runs workshops and summer schools to expand the expertise and knowledge about management strategies within the systems biology community; and contributes to public policy and standards setting for research asset management. FAIRDOM is a component of the initiative ISBE (Research Infrastructure for Systems Biology) as discussed later.
3.3. COMBINE archive and research object
The collections of data, models, operating procedures and associated descriptions that comprise published studies within projects such as the Human Physiome is growing in complexity. Exchanging these complex collections between researchers without compromising their meaning, and how they interrelate is difficult. The COMBINE archive is the concept of a single file in which the various documents required for a modelling and simulation study are bundled together, including all relevant information [71]. This provides a convenient method for transporting a reproducible study between software tools and scientists. Research Object (http://researchobject.org/) [72] is a general-purpose solution to contextualize the research findings of studies, compatible with the COMBINE archive [71]. Data, models, operating procedures, maps, metadata and more are packaged in a single archive. The archive includes descriptions of all the assets it contains, and how these are interrelated. This allows the export and transfer of information between researchers, without the risk of losing the original context, ensuring that researchers can be more confident in using the data/models/maps/operating procedures contained within it in a valid way.
3.4. All the support under one umbrella: infrastructure for systems biology Europe
The landscape that supports the success of projects like the Human Physiome is very complex. All of the standards, software, data and model management platforms, and expertise are very distributed, and often dependent on short-term funding. If a single cog of the support network fails, then vital research like the Human Physiome can experience delays and limitations in what it can achieve. In order to address this issue, Europe is looking towards establishing Research Infrastructures, which can support, maintain, assist with coordination and ensure sustainability of these key resources. ISBE (http://isbe.eu/) is one such emerging effort to establish a research infrastructure and works towards generating centralized services that support systems biology research across Europe. Once fully established, it will provide services to support a broad cross-section of systems biology research from storing, sharing and interoperating research assets; data generation for model integration; stewardship and standardization of research assets; modelling services; and, finally, education and training. This support will make it easier for researchers from a range of backgrounds to work together within the systems physiology community, as well as improving the quality of data and models constructed, published and archived in the long-term, in the physiology community, as well as systems biology in general. ISBE is currently in a ‘Light’ phase, building capacity and working towards full implementation. Over time infrastructures like ISBE will make realization of projects like the Human Physiome easier and more reliable.
4. Standardizing the Human Physiome: putting the resources into practice
4.1. Building multiscale models
CellML, as described above, allows a modular approach to model building via imports of separately defined component modules, facilitated by annotation of model variables and parameters so that imported components can be inserted automatically into the composite model [73,74]. In this section, we show how composite multiscale models can be constructed from CellML and FieldML models. We first deal with lumped parameter models based on ODEs and algebraic equations that can be encoded in CellML. The CellML models can be extended to include spatially distributed FieldML models where these are needed.
First, some definitions:
Compartments are well-mixed regions of cells, tissues or organs that represent either specific locations or more generic locations that can exist in multiple parts of the body. Variables are quantities that have units and fixed or variable numeric values and can be associated with compartments. Parameters are variables with fixed values. Models contain one or more equations in these variables and can stand alone or be associated with one or more compartments. Until filled with variables and equations, compartments are just empty containers.
Note that the term ‘well-mixed’ signifies that to an acceptable approximation in the model, the variables within a compartment are distributed homogeneously throughout that compartment. Of course no biological region is perfectly homogeneous and, if necessary, a well-mixed compartment can be replaced by more than one compartment or by a spatially distributed model where concentration gradients are governed by diffusion equations. However, it is often useful to start with a simple lumped parameter compartment model in order to understand basic mechanisms.
Modularity is an important aspect of multiscale modelling and we create composite models by importing other more basic models, as we will demonstrate. Model reduction can also be used to simplify a model for computational efficiency. But model expansion is equally important—there may be situations where more biophysical detail is called for under certain conditions, or spatial fields need to be considered as part of a model.
Reactions are the binding and unbinding of chemical species (almost always catalysed by an enzyme) and are always reversible under the appropriate conditions. When models include reactions, it is usually with equations that express mass conservation of chemical species and use either first order mass action kinetics or a steady state version in the form of an equilibrium equation. Note that it is often useful in diagrams to show the transfer of mass, energy or information across compartment boundaries and we use arrows to indicate the movement of variables across compartment boundaries, sometimes with the flux (time-varying flow of a variable) written beside the arrow. The direction of these arrows is just for explanatory reasons to show normal directions of movement—the fluxes could be in the reverse direction if conditions allowed it.
The formulation of a model and its encoding in CellML and/or FieldML is a completely separate exercise from the numerical solution of that model. Once a model is formulated, it can be solved by any solver that can handle the syntax of the model encoding languages and the range of mathematical operators expressed in a given model. The verification of the solver is an important but separate issue.
We now illustrate, via the example of acid–base physiology and the control of pH, how the use of standards and infrastructure described above can empower experimental biological research. Some of these models use molecular components, while others are treated in a more empirical ‘black-box’ fashion—possibly to be replaced at a later stage with a more mechanistic model when the physiological data become available to justify this, or the questions being asked of the model require this level of detail.
4.2. Acid–base physiology
Oxidative metabolism in tissue provides the bulk of the body's energy currency (ATP) by oxidizing carbohydrates and fats to carbon dioxide and water. To get rid of this metabolic waste product, CO2 is transferred from the tissues to the lungs via the blood, primarily in the form of bicarbonate (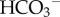 ) and carried primarily by red blood cells (RBCs). The formation of
) and carried primarily by red blood cells (RBCs). The formation of 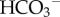 from CO2 via the CO2 hydration reaction is catalysed by carbonic anhydrase (CA):
from CO2 via the CO2 hydration reaction is catalysed by carbonic anhydrase (CA):
| 4.1 |
This generates large quantities of H+ in the blood. Buffering this H+—i.e. controlling pH within the narrow range compatible with life1—requires multiple weak acids and bases to be present in blood [75]. Understanding acid–base physiology, therefore, requires models of several organs: the muscles (as the major source of metabolism and hence metabolic waste products), the lungs (to exhale CO2) with the respiratory centre in the mid-brain to control respiration rate, the stomach where acid is excreted to maintain a low pH environment hostile to bacteria, the pancreas (where 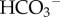 is excreted to neutralize stomach acid passed in the chime to the small intestines), the kidneys (to recover filtered
is excreted to neutralize stomach acid passed in the chime to the small intestines), the kidneys (to recover filtered 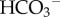 generate new
generate new 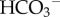 and excrete filtered buffers) and the arterial, capillary and venous components of the circulation system (each as homogeneous systems containing blood plasma and RBCs). These six organs or organ systems are illustrated, with their inputs and outputs, in figure 1.
and excrete filtered buffers) and the arterial, capillary and venous components of the circulation system (each as homogeneous systems containing blood plasma and RBCs). These six organs or organ systems are illustrated, with their inputs and outputs, in figure 1.
Figure 1.

A whole body model containing organ level models for ion or gas exchange in the kidneys, muscles, lungs (with respiratory control), stomach, pancreas and systemic circulation. (Online version in colour.)
There are many sources of H+ flux across a cell membrane. If JE is the rate of acid extrusion (generally at a metabolic cost) and JL is the rate of acid loading, the difference between them drives the time rate of change of pH in the cell:
| 4.2 |
where ρ (m−1) is the surface to volume ratio of the cell and β (mM pH−1 unit) is the buffering power.2 This is called the fundamental law of pH regulation [75]. Note that ρ/β has units of pH_unit m−1 mM−1, JE and JL have units of mol s−1 m−2 (or mM s−1 m) and dpHi/dt has units of pH_unit s−1.
For a particular cell the acid fluxes JL and JE are dependent on a range of protein exchangers, channels and pumps that reside in the cell membrane and are modelled individually. For the cells of interest here these are the acid extruding membrane transporters (NHE, NBC and NDCBE, all driven by the sodium gradient), the acid loading membrane transporters (NBCe1,2 and AE), and various leak channels and ATP-dependent pumps (such as HKA and V-type) shown individually in figure 2. Note that NHE, NBC and NDCBE depend on the sodium gradient which must be maintained by the ATP-dependent sodium–potassium exchange pump NCX, but as we are not tracking sodium or metabolism, that pump is not included.
Figure 2.

Membrane proteins (channels and transporters) divided into acid extruders (on the left) and acid loaders (on the right), shown here within a composite cell model. Note that the movement of ions across the cell membrane is driven by a sodium gradient (NHE, NBC, NDCBE), a chloride gradient (AE) or a metabolically dependent process (ATP label). Each channel and transporter is modelled with a separate CellML model on the PMR website (https://models.physiomeproject.org/workspace/27e). (Online version in colour.)
The equations governing these exchangers and pumps are provided in the PMR (https://models.physiomeproject.org/workspace/27e).
To illustrate how these CellML protein models are linked with tissue level processes such as those represented by equations (4.1) and (4.2), we consider three processes that are each modelled in CellML with separate components.
(1) One is the cellular mechanism of
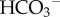 reabsorption in the kidney. The filtered
reabsorption in the kidney. The filtered 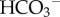 is combined with H+ excreted from the epithelial cell (via the ATP-dependent H-pump and HKA pump, and the Na-dependent exchanger NHE) to form CO2 and water (via the CA reaction at the tubule membrane) that diffuses back into the epithelial cell to then regenerate
is combined with H+ excreted from the epithelial cell (via the ATP-dependent H-pump and HKA pump, and the Na-dependent exchanger NHE) to form CO2 and water (via the CA reaction at the tubule membrane) that diffuses back into the epithelial cell to then regenerate 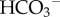 in the blood (via the CA reaction in the epithelial cell). The kidney reclaims virtually all filtered HCO3−3
in the blood (via the CA reaction in the epithelial cell). The kidney reclaims virtually all filtered HCO3−3(2) The second is ammonium excretion. The kidney synthesizes
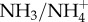 as a urinary buffer to assist with the excretion of H+. Filtered H+ in the tubule titrates NH3 excreted from the epithelial cell following glutamine metabolism (which produces both
as a urinary buffer to assist with the excretion of H+. Filtered H+ in the tubule titrates NH3 excreted from the epithelial cell following glutamine metabolism (which produces both 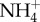 and OH−, thereby also generating new
and OH−, thereby also generating new 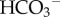 into the blood). Conversion of the amino acid glutamine to urea and ammonia, is catalysed by glutaminase in both the liver and the kidney. Under conditions of blood acidosis,
into the blood). Conversion of the amino acid glutamine to urea and ammonia, is catalysed by glutaminase in both the liver and the kidney. Under conditions of blood acidosis, 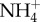 excretion in the kidney is enhanced, by upregulating glutaminase production to increase NH3 production, and glutaminase production is correspondingly downregulated in the liver.
excretion in the kidney is enhanced, by upregulating glutaminase production to increase NH3 production, and glutaminase production is correspondingly downregulated in the liver.(3) The third is titratable acid formation. When the excreted H+ combines with filtered buffers other than
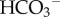 or ammonia, the outcome is the urinary loss of uncharged weak acid HA and the generation of new
or ammonia, the outcome is the urinary loss of uncharged weak acid HA and the generation of new  into the blood.4
into the blood.4
These three processes are shown in figure 3 together with the protein level ion exchange models. The corresponding models are available (https://models.physiomeproject.org/workspace/27e).
Figure 3.

Cellular mechanisms in the kidney: 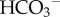 reabsorption, ammonium excretion and titratable acid formation. (Online version in colour.)
reabsorption, ammonium excretion and titratable acid formation. (Online version in colour.)
Similar tissue models can be defined for all the other modules shown in figure 1. The assembly of these into the overall composite model for acid–base physiology is shown in figure 4.
Figure 4.

Block diagram showing how models relate to each other. (Online version in colour.)
5. Conclusion
Here we have introduced the standardization efforts most closely aligned with the Human Physiome along with many aspects of the supporting infrastructure. These standards ensure that the models can be curated (i.e. that they work, have consistent units and obey the laws of physics where appropriate) and are modular and annotated with supporting knowledge (to ensure reusability). Along with the standards comes the need for open source software and web-accessible repositories built around these standards. We have furthermore provided an example demonstrating the application of these standards and supporting infrastructure to a well-known physiological phenomenon.
Biophysically based models of physiological processes capture physiological phenotype in a quantitative and predictive form. They should underpin research in physiology in the same way that nucleotide sequence underpins research in genetics, as there is no other way to deal with the complexity of physiological systems in a quantitative fashion. Most importantly, biophysical models bring with them the constraints on physiological behaviour imposed by the conservation laws of physics. Physiological function depends on these laws of nature as much as it depends on molecular biology.
Harmonization and interfacing on the level of data formats and structures, descriptors and metadata represent just one side of the coin. The quality of the data provided is an issue of fundamental importance which, as we have described above, has not yet been resolved satisfactorily. The diversity of data sources precludes any straightforward and coherent strategy for maintaining and documenting the quality of the data. Data quality implies not only fit for use of the data but also metrological traceability, repeatability, reproducibility, consistency and comparability. At best some but not all of the requirements are met by the prevailing data standards. More work is required to ensure that the experimental and clinical data underlying the computational models is not only appropriate for the context in which it is being used, but is also of sufficient quality for that purpose.
The modelling and simulation standards and their associated infrastructure, on the other hand, are now well understood and the largely technical barriers impeding implementations are rapidly crumbling. The challenge now is threefold: (i) incentives are needed to persuade modellers to use the standards in order to ensure that their models are reusable by others, (ii) a framework is needed to ensure that the multiscale models developed by independent research groups can be connected into integrative models of whole body physiology for application to healthcare, and (iii) a concerted effort is now needed to link the integrative models to electronic health records so that they can be used in the interpretation of patient data and clinical decision workflows—not least because the epidemic of chronic diseases that confronts the developed world can only be addressed by methods that deal with highly complex physiological processes involving multiple organs and tissue types linked to molecular mechanisms.
To address these three related challenges, three new developments are being explored. One is to establish a new journal, to be called the Physiome Journal, the second is to create a new web portal, based on models from the journal, that provides access to integrative Human Physiome models, and the third is to join forces with the openEHR organization5 to develop the means to link VPH/Physiome models with patient data via electronic health records.
With these new developments we hope to see the adoption of computational modelling as a standard laboratory instrument throughout the Human Physiome community, and indeed the wider biological research field. The new web portal will ensure that it is easy for scientists, and the public alike, to navigate and exploit the content of the repositories described here in novel ways that empower experimental biological research. The Physiome Journal will provide the scientific ‘credit’ that often holds back scientists from adopting or sharing their work in standard formats or public repositories. Finally, linking closely with the openEHR organization will enable tighter and more automated integration between computational modelling and clinical data and practices.
Funding for the standardization and service infrastructures described here has traditionally relied on a small number of individuals or relatively short-term projects. In achieving the goals described above it is hoped that more diverse and long-term funding sources can be established. The Physiome Journal and associated web portal, in particular, could provide a central core able to attract funding independent of specific individuals or organizations. Furthermore, the increasing awareness of reproducibility issues throughout the scientific community, especially within the funding bodies and governmental organizations, has seen the creation of new funding streams that may contribute to the maintenance and further development of this crucial aspect of the VPH endeavour.
Endnotes
Binding proteins are heavily influenced by the dissociation of acidic or basic amino acids, especially histidine, and are therefore dependent on ambient pH. Intracellular metabolism is also influenced by pH—for example, glycolysis and lactic acid formation are inhibited by acidosis (and, conversely, stimulated by alkalosis). K+ channels in a wide variety of cells are inactivated by acidosis. H+ affects smooth muscle in blood vessels (vasoconstriction in pulmonary arterioles and vasodilation in the systemic circulation) and reduces Ca2+ binding by plasma proteins.
A buffer is any substance that tends to minimize changes in pH by reversely producing or consuming H+. The buffering power of a solution is measured by the amount of added acid or base (per unit volume of solution) needed to decrease or increase the solution pH by one pH unit. i.e. a buffer with a high value of β requires a lot of additional acid or base to produce a unit change in pH. Without 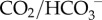 , the buffering power of whole blood is about 25 mM pH−1 unit (called the non-
, the buffering power of whole blood is about 25 mM pH−1 unit (called the non-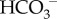 buffering power). Eighty per cent of this non-
buffering power). Eighty per cent of this non-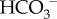 buffering power is associated with the cellular components of blood. [75].
buffering power is associated with the cellular components of blood. [75].
At 180 l of filtered blood per day with [HCO3−] = 24 mM, this is 4.32 moles of HCO3− per day [75].
Note that about 70 mmol/day of new 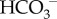 is needed to neutralise the approximately 70 mmol/day of non-volatile acid—comprised of approximately 40 mmol/day of non-volatile acid produced (in addition to CO2) by metabolism, approximately 20 mmol/day of dietary strong acid, and the intestinal loss of approximately 10 mmol/day of
is needed to neutralise the approximately 70 mmol/day of non-volatile acid—comprised of approximately 40 mmol/day of non-volatile acid produced (in addition to CO2) by metabolism, approximately 20 mmol/day of dietary strong acid, and the intestinal loss of approximately 10 mmol/day of 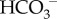 . There must also be a corresponding excretion of approximately 70 mmol/day of H+ into the urine [76].
. There must also be a corresponding excretion of approximately 70 mmol/day of H+ into the urine [76].
Author contributions
D.N. and P.H. conceived and coordinated the work. P.H. and B.d.B. developed the acid-base demonstration and D.N. implemented the demonstration. All authors contributed to the work and to the drafting of the manuscript. All authors approved the final version of the manuscript.
Competing interests
We declare we have no competing interests.
Funding
We acknowledge the support of the openEHR Foundation. D.N. is supported by The Virtual Physiological Rat Project (NIH P50-GM094503) and the Maurice Wilkins Centre for Molecular Biodiscovery. N.S. and C.G. would like to thank BBSRC BB/M013189/1 (DMMCore). M.G., B.R. and S.H. received funding for this work from the German Federal Ministry for Economic Affairs and Energy (BMWi) via the NormSys project (grant FKZ 01FS14019-FKZ01FS14021). M.G. and W.M. also received additional funding from the German Federal Ministry of Education and Research (BMBF) through the Virtual Liver Network (FKZ 0315749), as well as from the Klaus Tschira Foundation (KTS).
References
- 1.Force11. 2015. Guiding principles for findable, accessible, interoperable and re-usable data publishing version B1.0. See https://www.force11.org/node/6062 (accessed 30 Nov 2015).
- 2.Hucka M, et al. 2015. Promoting coordinated development of community-based information standards for modeling in biology: the COMBINE initiative. Front. Bioeng. Biotechnol. 3, 19 ( 10.3389/fbioe.2015.00019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber F, et al. 2015. Specifications of standards in systems and synthetic biology. J. Integr. Bioinform. 12, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maguire E, Gonzalez-Beltran A, Rocca-Serra P, Sansone S-A, Thurston M.2015. BioSharing—a web-based curated and searchable registry of content standards, database and policies in the life sciences. See http://figshare.com/articles/BioSharing_a_web_based_curated_and_searchable_registry_of_content_standards_database_and_policies_in_the_life_sciences/1285388. (accessed 17 Nov 2015).
- 5.Nickerson D, Nash M, Nielsen P, Smith N, Hunter P. 2006. Computational multiscale modeling in the IUPS Physiome Project: modeling cardiac electromechanics. IBM J. Res. Dev. 50, 617–630. ( 10.1147/rd.506.0617) [DOI] [Google Scholar]
- 6.Nickerson DP, Terkildsen JR, Hamilton KL, Hunter PJ. 2011. A tool for multi-scale modelling of the renal nephron. Interface Focus 1, 417–425. ( 10.1098/rsfs.2010.0032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nickerson DP, Ladd D, Hussan JR, Safaei S, Suresh V, Hunter PJ, Bradley CP. 2015. Using CellML with OpenCMISS to simulate multi-scale physiology. Front. Bioeng. Biotechnol. 2, 79 ( 10.3389/fbioe.2014.00079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith NP, Nickerson DP, Crampin EJ, Hunter PJ. 2004. Multiscale computational modelling of the heart. Acta Numer. 13, 371–431. ( 10.1017/S0962492904000200) [DOI] [Google Scholar]
- 9.Waltemath D, et al. 2014. Meeting report from the fourth meeting of the Computational Modeling in Biology Network (COMBINE). Stand. Genomic Sci. 9, 1285–1301. ( 10.4056/sigs.5279417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuellar AA, Lloyd CM, Nielsen PF, Bullivant DP, Nickerson DP, Hunter PJ. 2003. An Overview of CellML 1.1, a biological model description language. SIMULATION 79, 740–747. ( 10.1177/0037549703040939) [DOI] [Google Scholar]
- 11.Nickerson D, Buist M. 2008. Practical application of CellML 1.1: the integration of new mechanisms into a human ventricular myocyte model. Prog. Biophys. Mol. Biol. 98, 38–51. ( 10.1016/j.pbiomolbio.2008.05.006) [DOI] [PubMed] [Google Scholar]
- 12.Terkildsen JR, Niederer S, Crampin EJ, Hunter P, Smith NP. 2008. Using Physiome standards to couple cellular functions for rat cardiac excitation–contraction. Exp. Physiol. 93, 919–929. ( 10.1113/expphysiol.2007.041871) [DOI] [PubMed] [Google Scholar]
- 13.Waltemath D, et al. 2011. Reproducible computational biology experiments with SED-ML—the simulation experiment description markup language. BMC Syst. Biol. 5, 198 ( 10.1186/1752-0509-5-198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Britten RD, Christie GR, Little C, Miller AK, Bradley C, Wu A, Yu T, Hunter P, Nielsen P. 2013. FieldML, a proposed open standard for the Physiome project for mathematical model representation. Med. Biol. Eng. Comput. 51, 1191–1207. ( 10.1007/s11517-013-1097-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christie GR, Nielsen PMF, Blackett SA, Bradley CP, Hunter PJ. 2009. FieldML: concepts and implementation. Phil. Trans. R. Soc. A 367, 1869–1884. ( 10.1098/rsta.2009.0025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swat M, et al. 2015. Pharmacometrics markup language (PharmML): opening new perspectives for model exchange in drug development. CPT Pharmacomet. Syst. Pharmacol. 4, 316–319. ( 10.1002/psp4.57) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demir E, et al. 2010. BioPAX—a community standard for pathway data sharing. Nat. Biotechnol. 28, 935–942. ( 10.1038/nbt.1666) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hucka M, et al. 2003. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531. ( 10.1093/bioinformatics/btg015) [DOI] [PubMed] [Google Scholar]
- 19.Le Novère N, et al. 2009. The systems biology graphical notation. Nat. Biotechnol. 27, 735–741. ( 10.1038/nbt.1558) [DOI] [PubMed] [Google Scholar]
- 20.Galdzicki M, et al. 2014. The synthetic biology open language (SBOL) provides a community standard for communicating designs in synthetic biology. Nat. Biotechnol. 32, 545–550. ( 10.1038/nbt.2891) [DOI] [PubMed] [Google Scholar]
- 21.Novère NL, et al. 2005. Minimum information requested in the annotation of biochemical models (MIRIAM). Nat. Biotechnol. 23, 1509–1515. ( 10.1038/nbt1156) [DOI] [PubMed] [Google Scholar]
- 22.Consortium TU. 2015. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. ( 10.1093/nar/gku989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Natale DA, et al. 2014. Protein ontology: a controlled structured network of protein entities. Nucleic Acids Res. 42, D415–D421. ( 10.1093/nar/gkt1173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner M, et al. 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. ( 10.1038/75556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium TGO. 2015. Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. ( 10.1093/nar/gku1179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosse C, Mejino JLV Jr. 2003. A reference ontology for biomedical informatics: the foundational model of anatomy. J. Biomed. Inform. 36, 478–500. ( 10.1016/j.jbi.2003.11.007) [DOI] [PubMed] [Google Scholar]
- 27.Terkildsen J, Nickerson DP.2015. Renal Na+/H+ antiporter (NHE3) model. See https://models.physiomeproject.org/e/236. (accessed 23 Dec 2015).
- 28.Lloyd CM, Nickerson DP.2015. Thiazide-sensitive Na-Cl cotransporter. See https://models.physiomeproject.org/e/25b. (accessed 23 Dec 2015).
- 29.Lloyd CM, Nielsen H, Nickerson DP.2015. Renal SGLT2 model. See https://models.physiomeproject.org/e/233. (accessed 23 Dec 2015).
- 30.Terkildsen J, Nickerson DP.2015. Renal SGLT1 model. See https://models.physiomeproject.org/e/232. (accessed 23 Dec 2015)
- 31.Wittig U, et al. 2012. SABIO-RK—database for biochemical reaction kinetics. Nucleic Acids Res. 40, D790–D796. ( 10.1093/nar/gkr1046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wittig U, et al. 2014. Challenges for an enzymatic reaction kinetics database. FEBS J. 281, 572–582. ( 10.1111/febs.12562) [DOI] [PubMed] [Google Scholar]
- 33.Swainston N, et al. 2010. Enzyme kinetics informatics: from instrument to browser. FEBS J. 277, 3769–3779. ( 10.1111/j.1742-4658.2010.07778.x) [DOI] [PubMed] [Google Scholar]
- 34.Richesson RL, Horvath MM, Rusincovitch SA. 2014. Clinical research informatics and electronic health record data. Yearb. Med. Inform. 9, 215–223. ( 10.15265/IY-2014-0009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zunner C, Ganslandt T, Prokosch H-U, Bürkle T. 2014. A reference architecture for semantic interoperability and its practical application. Stud. Health Technol. Inform. 198, 40–46. [PubMed] [Google Scholar]
- 36.Verstappen SMM, et al. 2015. Methodological challenges when comparing demographic and clinical characteristics of international observational registries. Arthritis Care Res. 67, 1637–1645. ( 10.1002/acr.22661) [DOI] [PubMed] [Google Scholar]
- 37.Panteghini M. 2009. Traceability as a unique tool to improve standardization in laboratory medicine. Clin. Biochem. 42, 236–240. ( 10.1016/j.clinbiochem.2008.09.098) [DOI] [PubMed] [Google Scholar]
- 38.White GH. 2011. Metrological traceability in clinical biochemistry. Ann. Clin. Biochem. 48, 393–409. ( 10.1258/acb.2011.011079) [DOI] [PubMed] [Google Scholar]
- 39.Manley SE, Stratton IM, Clark PM, Luzio SD. 2007. Comparison of 11 human insulin assays: Implications for clinical investigation and research. Clin. Chem. 53, 922–932. ( 10.1373/clinchem.2006.077784) [DOI] [PubMed] [Google Scholar]
- 40.Copeland BE, Pesce AJ. 1992. The medical heritage concept: a model for assuring comparable laboratory results in long-term longitudinal studies. Ann. Clin. Lab. Sci. 22, 110–124. [PubMed] [Google Scholar]
- 41.Cull CA, Manley SE, Stratton IM, Neil HA, Ross IS, Holman RR, Turner RC, Matthews DR. 1997. Approach to maintaining comparability of biochemical data during long-term clinical trials. Clin. Chem. 43, 1913–1918. [PubMed] [Google Scholar]
- 42.Ransohoff DF. 2007. How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J. Clin. Epidemiol. 60, 1205–1219. ( 10.1016/j.jclinepi.2007.04.020) [DOI] [PubMed] [Google Scholar]
- 43.Caboux E, Plymoth A, Hainaut P. 2007. Common minimum technical standards and protocols for biological resource centres dedicated to cancer research. 150 cours Albert Thomas, 69372 Lyon Cedex 08, France: International Agency for Research on Cancer.
- 44.Moore HM, et al. 2011. Biospecimen reporting for improved study quality (BRISQ). J. Proteome Res. 10, 3429–3438. ( 10.1021/pr200021n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinlan PR, Mistry G, Bullbeck H, Carter A. 2014. A data standard for sourcing fit-for-purpose biological samples in an integrated virtual network of biobanks. Biopreserv. Biobank. 12, 184–191. ( 10.1089/bio.2013.0089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fortier I, et al. 2010. Quality, quantity and harmony: the DataSHaPER approach to integrating data across bioclinical studies. Int. J. Epidemiol. 39, 1383–1393. ( 10.1093/ije/dyq139) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martínez-Costa C, Kalra D, Schulz S. 2014. Improving EHR semantic interoperability: future vision and challenges. Stud. Health Technol. Inform. 205, 589–593. [PubMed] [Google Scholar]
- 48.Atalag K, Paton C, Kingsford D, Warren J. 2010. Putting health record interoperability standards to work. Electron. J. Health Inform. 5, e1. [Google Scholar]
- 49.Kalra D, Beale T, Heard S. 2005. The openEHR Foundation. Stud. Health Technol. Inform. 115, 153–173. [PubMed] [Google Scholar]
- 50.Beale T. 2002. Archetypes: constraint-based domain models for future-proof information systems. In Eleventh OOPSLA workshop on behavioral semantics: serving the customer, pp. 16–32. Seattle, WA: Northeastern University. [Google Scholar]
- 51.Cooper J, Vik JO, Waltemath D. 2015. A call for virtual experiments: accelerating the scientific process. Prog. Biophys. Mol. Biol. 117, 99–106. ( 10.1016/j.pbiomolbio.2014.10.001) [DOI] [PubMed] [Google Scholar]
- 52.Cooper J, Spiteri RJ, Mirams GR. 2015. Cellular cardiac electrophysiology modeling with Chaste and CellML. Comput. Physiol. Med. 5, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cooper J, Mirams GR, Niederer SA. 2011. High-throughput functional curation of cellular electrophysiology models. Prog. Biophys. Mol. Biol. 107, 11–20. ( 10.1016/j.pbiomolbio.2011.06.003) [DOI] [PubMed] [Google Scholar]
- 54.Juty N, et al. 2015. BioModels: content, features, functionality, and use. CPT Pharmacomet. Syst. Pharmacol. 4, 55–68. ( 10.1002/psp4.3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olivier BG, Snoep JL. 2004. Web-based kinetic modelling using JWS Online. Bioinformatics 20, 2143–2144. ( 10.1093/bioinformatics/bth200) [DOI] [PubMed] [Google Scholar]
- 56.Dzodic V, Hervy S, Fritsch D, Khalfallah H, Thereau M, Thomas SR. 2004. Web-based tools for quantitative renal physiology. Cell Mol. Biol. 50, 795–800. [PubMed] [Google Scholar]
- 57.Ribba B, Tracqui P, Boix J-L, Boissel J-P, Thomas SR. 2006. QxDB: a generic database to support mathematical modelling in biology. Phil. Trans. R. Soc. A 364, 1517–1532. ( 10.1098/rsta.2006.1784) [DOI] [PubMed] [Google Scholar]
- 58.Yu T, et al. 2011. The physiome model repository 2. Bioinformatics 27, 743–744. ( 10.1093/bioinformatics/btq723) [DOI] [PubMed] [Google Scholar]
- 59.Lloyd CM, Lawson JR, Hunter PJ, Nielsen PF. 2008. The CellML model repository. Bioinformatics 24, 2122–2123. ( 10.1093/bioinformatics/btn390) [DOI] [PubMed] [Google Scholar]
- 60.Miller AK, et al. 2011. Revision history aware repositories of computational models of biological systems. BMC Bioinform. 12, 22 ( 10.1186/1471-2105-12-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buck S. 2015. Solving reproducibility. Science 348, 1403 ( 10.1126/science.aac8041) [DOI] [PubMed] [Google Scholar]
- 62.Collaboration OS. 2015. Estimating the reproducibility of psychological science. Science 349, aac4716. ( 10.1126/science.aac4716) [DOI] [PubMed] [Google Scholar]
- 63.McNutt M. 2014. Journals unite for reproducibility. Science 346, 679 ( 10.1126/science.aaa1724) [DOI] [PubMed] [Google Scholar]
- 64.Rocca-Serra P, et al. 2010. ISA software suite: supporting standards-compliant experimental annotation and enabling curation at the community level. Bioinformatics 26, 2354–2356. ( 10.1093/bioinformatics/btq415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.González-Beltrán A, Maguire E, Sansone S-A, Rocca-Serra P. 2014. linkedISA: semantic representation of ISA-Tab experimental metadata. BMC Bioinform. 15(Suppl. 14), S4 ( 10.1186/1471-2105-15-S14-S4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rocca-Serra P, Walls R, Parnell J, Gallery R, Zheng J, Sansone S-A, Gonzalez-Beltran A. 2015. Modeling a microbial community and biodiversity assay with OBO Foundry ontologies: the interoperability gains of a modular approach. Database 2015, bau132. ( 10.1093/database/bau132) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bauch A, et al. 2011. openBIS: a flexible framework for managing and analyzing complex data in biology research. BMC Bioinform. 12, 468 ( 10.1186/1471-2105-12-468) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolstencroft K, et al. 2015. SEEK: a systems biology data and model management platform. BMC Syst. Biol. 9, 33 ( 10.1186/s12918-015-0174-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weidemann A, et al. 2008. SYCAMORE—a systems biology computational analysis and modeling research environment. Bioinformatics 24, 1463–1464. ( 10.1093/bioinformatics/btn207) [DOI] [PubMed] [Google Scholar]
- 70.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwukowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. ( 10.1101/gr.1239303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergmann FT, et al. 2014. COMBINE archive and OMEX format: one file to share all information to reproduce a modeling project. BMC Bioinform. 15, 369 ( 10.1186/s12859-014-0369-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bechhofer S, et al. 2013. Why linked data is not enough for scientists. Future Gen. Comput. Syst. 29, 599–611. ( 10.1016/j.future.2011.08.004) [DOI] [Google Scholar]
- 73.Neal ML, Gennari JH, Arts T, Cook DL. 2009. Advances in semantic representation for multiscale biosimulation: a case study in merging models. Pac. Symp. Biocomput. 2009, 304–315. [PMC free article] [PubMed] [Google Scholar]
- 74.Neal ML, Cooling MT, Smith LP, Thompson CT, Sauro HM, Carlson BE, Cook DL, Gennari JH. 2014. A reappraisal of how to build modular, reusable models of biological systems. PLoS Comput. Biol. 10, e1003849 ( 10.1371/journal.pcbi.1003849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boron WF, Boulpaep ELJB. 2012. Medical physiology a cellular and molecular approach. Philadelphia, PA: Elsevier Saunders. [Google Scholar]
- 76.Occhipinti R, Boron WF. 2015. Mathematical modeling of acid-base physiology. Prog. Biophys. Mol. Biol. 117, 43–58. ( 10.1016/j.pbiomolbio.2015.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]