

Abstract
Pore-forming toxins are ubiquitous cytotoxins that are exploited by both bacteria and the immune response of eukaryotes. These toxins kill cells by assembling large multimeric pores on the cell membrane. However, a quantitative understanding of the mechanism and kinetics of this self-assembly process is lacking. We propose an analytically solvable kinetic model for stepwise, reversible oligomerization. In biologically relevant limits, we obtain simple algebraic expressions for the rate of pore formation, as well as for the concentration of pores as a function of time. Quantitative agreement is obtained between our model and time-resolved kinetic experiments of Bacillus thuringiensis Cry1Ac (tetrameric pore), aerolysin, Staphylococcus aureus α-haemolysin (heptameric pores) and Escherichia coli cytolysin A (dodecameric pore). Furthermore, our model explains how rapid self-assembly can take place with low concentrations of oligomeric intermediates, as observed in recent single-molecule fluorescence experiments of α-haemolysin self-assembly. We propose that suppressing the concentration of oligomeric intermediates may be the key to reliable, error-free, self-assembly of pores.
Keywords: mathematical modelling, reaction kinetics, pore-forming toxins
1. Introduction
Pore-forming toxins (PFTs) are potent cytotoxic proteins that kill cells by creating pores on the cell membrane of targeted cells. These proteins are an integral part of eukaryotic immune response, whereby infected cells and bacteria can be attacked via pore generation [1–3]. Conversely, PFTs are commonly deployed by bacteria such as Streptococcus pneumoniae, group A and B streptococci, Staphylococcus aureus, Escherichia coli and Mycobacterium tuberculosis to attack host cells [4]. In fact, PFTs are a common attribute in antibiotic-resistant bacteria strains [5,6]. The absence of PFTs generally causes pathogenic bacteria to be less virulent or completely avirulent. Therefore, understanding PFTs is important for developing antimicrobial therapeutics. Interest in PFTs has also arisen from nanotechnological applications such as biosensing and single-molecule DNA sequencing [7].
Despite the profound biological and nanotechnological impacts of PFTs, the mechanism by which these multimeric membrane proteins self-assemble to form pores is not well understood. A wide variety of experimental techniques, including cell lysis [8–11], gel electrophoresis [12,13] and kinetic modelling [11], support a generic mechanism for β-barrel forming pores (figure 1). Monomers are first adsorbed from solution to the cell membrane and diffuse on the lipid bilayer. Subsequently, the monomers on the bilayer undergo oligomerization to form oligomeric intermediaries or pre-pores, followed by pore insertion into the membrane. The first step of monomer adsorption, and the last step of pore insertion with the associated structural rearrangements have received the most attention so far, with few quantitative studies focusing on the kinetics of oligomerization.
Figure 1.

Schematic diagrams illustrating monomer addition as a proposed pathway to pore formation. (Online version in colour.)
Our recent single-molecule fluorescence imaging experiments revealed that the assembly of staphylococcal PFT α-haemolysin is extremely rapid [14]—less than 5 ms is needed to go from monomers to a complete heptamer, with no lower order oligomeric intermediates detected. This rapid assembly is consistent with results from biochemical cross-linking experiments [15]. However, rapid self-assembly with no detectable intermediates poses a fundamental kinetic conundrum—rapid oligomerization without accumulation of intermediates seems to be at odds with mass-action kinetics. Dissecting the kinetics of this step may pave the way to designing agents that can disrupt or stall the action of PFTs.
In this paper, we will develop an analytically solvable kinetic model for stepwise, reversible, oligomerization. Applying mathematical techniques from perturbation theory that exploit features of the kinetic behaviour, we obtain simple expressions for the rate of pore formation, as well as the concentration of pores as a function of time. The results of this model will suggest a simple method that captures the key kinetic behaviour of a pore-forming system while bypassing the need for the complex numerical simulations that are used to model many processes (e.g. [11]). These algebraic expressions reveal that rapid self-assembly, as observed by single-molecule experiments, requires an intricate balance between the rate of polymerization and dissociation. We show that our theory is also able to reproduce time-resolved kinetic experiments for the self-assembly of Bacillus thuringiensis Cry1Ac (tetrameric pore), aerolysin, S. aureus α-haemolysin (heptameric pores) and E. coli cytolysin A (dodecameric pore). Furthermore, we suggest that suppressing the intermediate concentration could contribute to an important biological requirement of error minimization in the self-assembly of pores.
2. Kinetic model
We consider the kinetics of oligomerization where the formation of complexes takes place via stepwise association. The reaction mechanism is given by
| 2.1 |
where 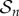 is an aggregate composed of n monomers,
is an aggregate composed of n monomers, 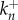 denotes the reaction rate of the nth oligomerization reaction and
denotes the reaction rate of the nth oligomerization reaction and 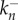 denotes the rate of the reverse reaction. We assume that a stable complex composed of N monomers is irreversibly formed in the final stage of the process at a different rate:
denotes the rate of the reverse reaction. We assume that a stable complex composed of N monomers is irreversibly formed in the final stage of the process at a different rate:
| 2.2 |
Note that the oligomerization reactions take place on the cell membrane, and we ignore the much faster step of monomer adsorption from the bulk. In some situations, for example, the formation of equinatoxin II, experiments suggest the route to stable complex formation occurs via the aggregation of monomers to stable dimers, which in turn aggregate to form tetramers and hexamers [16]. However, while such aggregation routes may also be followed, in many cases of interest the oligomers appear in much lower concentrations than the monomer and so catering for these reactions is expected to have little effect on the system behaviour (see [17] for more details).
The reaction system can be further simplified if we assume that all forward and backward rates are equal, so that 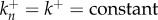 and
and 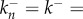 constant (though not necessarily the same as k+). This amounts to assuming that the binding energies of the monomer to successive oligomer intermediates are all equal to each other, and the dissociation energy of an oligomer as it releases a monomer are the same as each other. The various assumptions introduced here will be verified later by comparison to experiments. The resulting reaction system is
constant (though not necessarily the same as k+). This amounts to assuming that the binding energies of the monomer to successive oligomer intermediates are all equal to each other, and the dissociation energy of an oligomer as it releases a monomer are the same as each other. The various assumptions introduced here will be verified later by comparison to experiments. The resulting reaction system is
| 2.3 |
where the evolution of the concentrations of 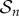 on the cell surface,
on the cell surface, 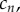 1 ≤ n ≤ N, is given by the law of mass action
1 ≤ n ≤ N, is given by the law of mass action
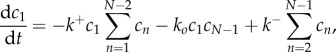 |
2.4 |
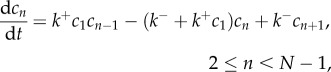 |
2.5 |
| 2.6 |
| 2.7 |
3. Mechanism of rapid pore formation
Single-molecule experiments reveal that oligomerization is rapid but the concentrations of intermediates are low [14]. Intuitively, the rate of oligomerization is controlled by the abundance of the intermediates as each successive step in mechanism (2.3) is bimolecular. To resolve this conundrum, we consider the quasi-steady-state behaviour of (2.4)–(2.7).
In the early-time regime, which is the focus of [14], there is a ready supply of monomers. As a result, the formation of complexes will only cause a slight change to the monomer concentration,  and we can consider the monomer concentration to be a constant. To elucidate the pertinent dynamics, we non-dimensionalize the system via
and we can consider the monomer concentration to be a constant. To elucidate the pertinent dynamics, we non-dimensionalize the system via
| 3.1 |
whence the governing equations (2.4)–(2.7), become
| 3.2 |
| 3.3 |
| 3.4 |
Here we have identified the key dimensionless parameters
| 3.5 |
which provide measures of the forward and final-complex-forming reaction rates, respectively.
In quasi-steady state, the concentration of intermediates, 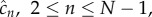 are constant, while stable complexes
are constant, while stable complexes 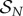 of concentration
of concentration  are formed at a rate
are formed at a rate 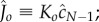 the time-dependent evolution of the system is considered in the next section. In this quasi-steady-state limit we can set the time derivatives in (3.2)–(3.3) to zero to obtain
the time-dependent evolution of the system is considered in the next section. In this quasi-steady-state limit we can set the time derivatives in (3.2)–(3.3) to zero to obtain
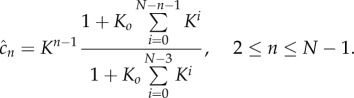 |
3.6 |
The stable complex 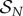 is therefore produced at a rate
is therefore produced at a rate
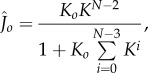 |
3.7 |
or, in dimensional units,
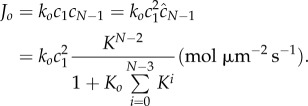 |
3.8 |
(We note that oligomerization occurs on the cell-membrane surface, hence the rate is in terms of surface density per unit time rather than bulk concentrations.) Thus, the average time needed for monomers to self-assemble into pores is given by
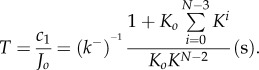 |
3.9 |
Equations (3.6) and (3.9) hold the key to resolving the aforementioned kinetic conundrum: equation (3.6) shows that the concentration of intermediates is minimal when 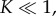 i.e. when the reaction equilibrium is biased towards dissociation of intermediates into monomers. In fact, in the limit
i.e. when the reaction equilibrium is biased towards dissociation of intermediates into monomers. In fact, in the limit 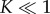 and
and 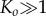 ,
,
| 3.10 |
and
| 3.11 |
and thus the concentration of intermediates decreases as a power law with K. However, in the same limit, the reaction time scales as
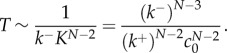 |
3.12 |
As one would expect, the reaction time is independent of the final rate of pore formation 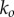 when the rate-limiting steps are the formation of intermediate oligomers. Importantly, equation (3.12) resolves the apparent paradox observed in single-molecule experiments [14]: ensuring
when the rate-limiting steps are the formation of intermediate oligomers. Importantly, equation (3.12) resolves the apparent paradox observed in single-molecule experiments [14]: ensuring 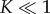 and thus keeping the concentration of intermediates low, the system can independently decrease reaction time by increasing
and thus keeping the concentration of intermediates low, the system can independently decrease reaction time by increasing 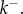 Crucially, the ability to form pores rapidly without accumulating intermediates is possible only if each step of the oligomerization is reversible, and perhaps counterintuitively, only if the reaction is biased towards dissociation rather than association.
Crucially, the ability to form pores rapidly without accumulating intermediates is possible only if each step of the oligomerization is reversible, and perhaps counterintuitively, only if the reaction is biased towards dissociation rather than association.
Although the biological rationale of rapid reaction is evident, why is low intermediate concentration favourable? We suggest that keeping the concentrations of intermediates low is a powerful error-avoiding mechanism. If the concentrations of intermediates are large, oligomers larger than the pore size will be formed due to collisions between smaller oligomer fragments. For the small pores that we are considering only oligomers of a defined size can cyclize and form a functional pore that is able to penetrate cell membranes. Formation of oligomers that are larger than the pore size is, at best, a parasitic kinetic process. At worst, the higher-order oligomers can form inactive or less potent cyclic structures or acyclic aggregates [16,18], and the process is thus actively harmful to the biological function of PFTs. By reducing the concentrations of intermediates, the system ensures that the only significant kinetic process is the sequential, stepwise, addition of monomers to oligomers. Therefore, provided 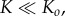 the reaction will terminate at the desired oligomer size.
the reaction will terminate at the desired oligomer size.
4. Formation of oligomer intermediates
The central conclusion of our model, namely that 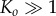 but
but 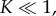 is better understood by considering the approach of the system to quasi-steady state. Motivated by equation (3.10), we can extract further analytical insights by rescaling the intermediates and time via
is better understood by considering the approach of the system to quasi-steady state. Motivated by equation (3.10), we can extract further analytical insights by rescaling the intermediates and time via
| 4.1 |
| 4.2 |
| 4.3 |
| 4.4 |
where 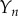 and
and  are quantities that are order-one in magnitude. The governing equations (2.4)–(2.7) then become
are quantities that are order-one in magnitude. The governing equations (2.4)–(2.7) then become
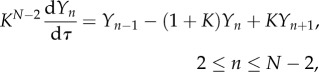 |
4.5 |
| 4.6 |
| 4.7 |
Thus we find that  is the appropriate timescale on which we observe accumulation of the final aggregate
is the appropriate timescale on which we observe accumulation of the final aggregate 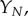 through the balance in equation (4.7). Upon taking the limit
through the balance in equation (4.7). Upon taking the limit 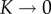 we verify that, on this timescale, the system is indeed in quasi-steady state, with equations (4.5) and (4.6) reducing to algebraic equations for the concentration of the intermediate aggregates. This leads to
we verify that, on this timescale, the system is indeed in quasi-steady state, with equations (4.5) and (4.6) reducing to algebraic equations for the concentration of the intermediate aggregates. This leads to 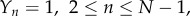 as predicted by the quasi-steady analysis in the limit
as predicted by the quasi-steady analysis in the limit 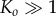 (equation (3.6)).
(equation (3.6)).
This asymptotic analysis allows us to investigate how the system approaches the quasi-steady state. Expressing system (4.5)–(4.7) back in terms of the original (fast) timescale, 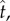 we obtain
we obtain
| 4.8a |
| 4.8b |
| 4.8c |
| 4.8d |
Taking the limit as 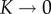 we are left with the system
we are left with the system
| 4.9a |
| 4.9b |
| 4.9c |
| 4.9d |
which may be solved subject to the initial conditions 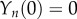 ,
, 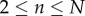 . This gives analytical expressions for the relaxation behaviour of the intermediate aggregates to the quasi-steady-state values (
. This gives analytical expressions for the relaxation behaviour of the intermediate aggregates to the quasi-steady-state values (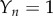 ),
),
 |
4.10 |
where
is the incomplete gamma function, and 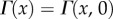 is the gamma function. Figure 2 shows that the asymptotic prediction (4.10) is in excellent agreement with full numerical solutions to (4.8).
is the gamma function. Figure 2 shows that the asymptotic prediction (4.10) is in excellent agreement with full numerical solutions to (4.8).
Figure 2.

Comparison between the numerical solution to the full system (4.8) for 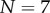 (solid curves) and the analytic asymptotic result (4.10) (dashed curves). We consider
(solid curves) and the analytic asymptotic result (4.10) (dashed curves). We consider 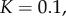 and plot
and plot 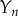 as a function of time, with
as a function of time, with 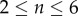 . Schematic diagrams illustrating monomer addition as a proposed pathway to pore formation.
. Schematic diagrams illustrating monomer addition as a proposed pathway to pore formation.
Equation (4.10) predicts that the kinetics of relaxation to quasi-steady state exhibit a surprising degree of universality: kinetic data for the build-up of oligomer intermediates of a given size should all follow the same relation (4.10) and thus could be collapsed onto one master curve by scaling time. Comparing any experimental measurements to equation (4.10) is a simple one-parameter fit. Therefore, our model identifies the kinetics of intermediate build-up as a possible ‘litmus test’ for the proposed kinetic scheme (2.3).
Although tracking the concentration of intermediates is not possible via single-molecule fluorescence imaging techniques for a heptameric α-haemolysin pore [14], we suggest that with greater time-resolution in the measurements, or experiments with larger pores, the dynamics of such intermediates can be tracked. Indeed, the inflection point in equation (4.10) is a measure of the characteristic time to approach the quasi-steady state (qualitatively the ‘lag time’ as shown in figure 2) and is given by 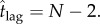 Thus, larger oligomeric intermediates take longer to reach the quasi-steady state, which is a prediction that can be used to experimentally investigate the dynamic formation of such intermediates.
Thus, larger oligomeric intermediates take longer to reach the quasi-steady state, which is a prediction that can be used to experimentally investigate the dynamic formation of such intermediates.
5. Late-stage dynamics
The quasi-steady state is maintained until the monomer is significantly depleted. To explore this nonlinear regime of (2.4)–(2.7), we develop an asymptotic method based on fixed-point iteration [19]. We observe that (2.4)–(2.7) can be written as a system of Volterra integral equations
 |
5.1 |
 |
5.2 |
 |
5.3 |
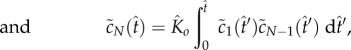 |
5.4 |
where we have taken the initial monomer concentration as the concentration scale, i.e. 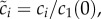 and defined
and defined
| 5.5 |
System (5.1)–(5.4) can be compactly denoted as
| 5.6 |
where 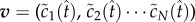 is the vector of concentrations and A is the corresponding nonlinear operator. Therefore, if we know a vector
is the vector of concentrations and A is the corresponding nonlinear operator. Therefore, if we know a vector  that is sufficiently close to
that is sufficiently close to 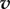 then by the contraction mapping theorem
then by the contraction mapping theorem
| 5.7 |
Although the above equation is exact, it also provides a strategy to systematically seek approximate solutions—if we can find a 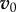 that is sufficiently close to the real solution, we can apply the operator A repeatedly to
that is sufficiently close to the real solution, we can apply the operator A repeatedly to 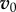 to obtain any desired degree of accuracy.
to obtain any desired degree of accuracy.
To find an approximate solution for the concentration of pores as a function of time using this method, we use equations (4.2) and (4.10) for an estimate for 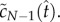 Substituting equation (4.10) into the right-hand side of equation (5.4), we obtain
Substituting equation (4.10) into the right-hand side of equation (5.4), we obtain
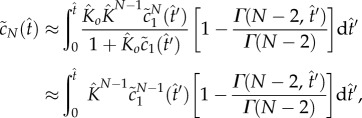 |
5.8 |
where in the second step we used the fact that 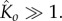 As
As  we can further estimate
we can further estimate 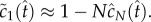 Substituting this estimate for
Substituting this estimate for 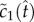 into equation (5.8), and converting the integral equation into a differential equation, we arrive at
into equation (5.8), and converting the integral equation into a differential equation, we arrive at
| 5.9 |
The solution of equation (5.9) with initial condition 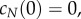 is
is
 |
5.10 |
The above equation is a crucial result as it provides an analytical expression for the concentration of product as a function of time that is valid for both the early-time, linear regime and the strongly nonlinear regime.
Figure 3 shows the predictions of equation (5.10). Pore formation is significant only after a ‘lag phase’ of building up the quasi-steady state. The lag time increases with increasing bias towards dissociation (i.e. decreasing 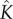 , figure 3a) and increasing number of monomer units needed to assemble the pore (figure 3b). After the quasi-steady state is attained, the pore concentration increases linearly in time, corresponding to a constant reaction flux. Pore formation is stalled eventually by monomer depletion. However, figure 3 shows that this last phase occurs over a much longer timescale. Although an appreciable amount of pores are produced by
, figure 3a) and increasing number of monomer units needed to assemble the pore (figure 3b). After the quasi-steady state is attained, the pore concentration increases linearly in time, corresponding to a constant reaction flux. Pore formation is stalled eventually by monomer depletion. However, figure 3 shows that this last phase occurs over a much longer timescale. Although an appreciable amount of pores are produced by 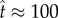 , the reaction is 90% complete only when
, the reaction is 90% complete only when 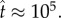 In dimensional units, assuming a typical value of
In dimensional units, assuming a typical value of 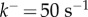 (figure 4),
(figure 4), 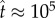 corresponds to 2000 s, or approximately half an hour.
corresponds to 2000 s, or approximately half an hour.
Figure 3.
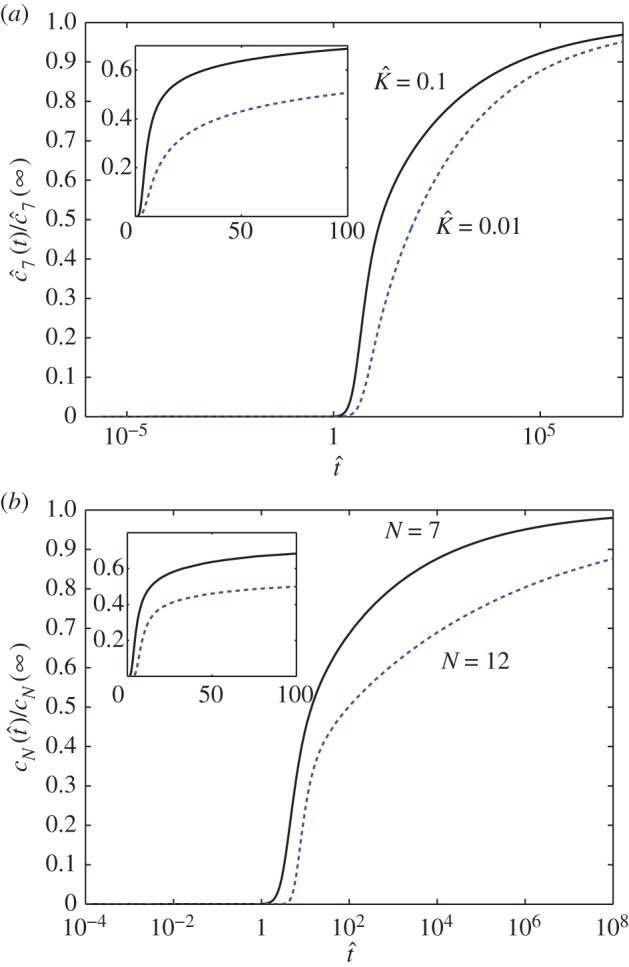
(a) The reaction yield as a function of time (on a logarithmic axis) for a heptameric pore (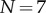 ) with
) with 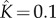 (solid curve) and
(solid curve) and 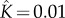 (dashed curve). (b) The reaction yield as a function of time (on a logarithmic axis) for heptameric (
(dashed curve). (b) The reaction yield as a function of time (on a logarithmic axis) for heptameric (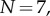 solid curve) and dodecameric (
solid curve) and dodecameric (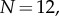 dashed curve) pore with
dashed curve) pore with 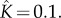 The insets show the early-time evolution of the system with respect to a linear time axis. (Online version in colour.)
The insets show the early-time evolution of the system with respect to a linear time axis. (Online version in colour.)
Figure 4.

Fitting equation (5.10) to experimental data. (a) Fitting of experimental data taken from [20] for Bacillus thuringiensis toxin Cry1Ac. Assuming that 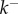 is concentration independent, the fitting yields
is concentration independent, the fitting yields 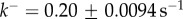 and
and 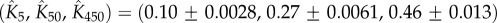 . (b) Fitting of experimental data taken from [21] for aerolysin (
. (b) Fitting of experimental data taken from [21] for aerolysin (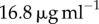 ). The fitting yields
). The fitting yields  and
and 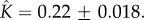 (c) Fitting of experimental data taken from [12] for α-haemolysin from Staphylococcus aureus. The two-parameter fit yields
(c) Fitting of experimental data taken from [12] for α-haemolysin from Staphylococcus aureus. The two-parameter fit yields 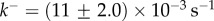 and
and 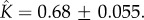 (d) Fitting of experimental data taken from [13] for cytolysin A from Escherichia coli. The two-parameter fit yields
(d) Fitting of experimental data taken from [13] for cytolysin A from Escherichia coli. The two-parameter fit yields 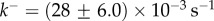 and
and 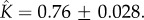 The fitting for all datasets was computed using the NonlinearModelFit package in Mathematica, with the uncertainties in the fitted values being quantified using the standard error (ParameterErrors in Mathematica). (Online version in colour.)
The fitting for all datasets was computed using the NonlinearModelFit package in Mathematica, with the uncertainties in the fitted values being quantified using the standard error (ParameterErrors in Mathematica). (Online version in colour.)
6. Comparison with experiments
To test the validity of our kinetic model, we compare experimental kinetic data of a range of pore-forming proteins with equation (5.10) using a two-parameter fit for 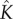 and the timescale
and the timescale 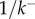 . The uncertainties in the fitted parameters will be quantified using the standard error. Note that the size of the final pore, N, is usually known a priori. While it is important to note that undoubtedly other, more complex aggregation mechanisms may also explain the experimental data, our reduced model demonstrates that this simple aggregation mechanism is able to reproduce the data accurately, validating our proposed pathway more generally for PFTs.
. The uncertainties in the fitted parameters will be quantified using the standard error. Note that the size of the final pore, N, is usually known a priori. While it is important to note that undoubtedly other, more complex aggregation mechanisms may also explain the experimental data, our reduced model demonstrates that this simple aggregation mechanism is able to reproduce the data accurately, validating our proposed pathway more generally for PFTs.
Figure 4a compares the experimental data taken from [20] for the tetramer (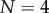 ) B. thuringiensis toxin Cry1Ac to equation (5.10) with fitted parameters
) B. thuringiensis toxin Cry1Ac to equation (5.10) with fitted parameters 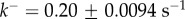 , and
, and 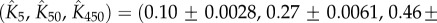
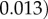 for initial monomer concentrations
for initial monomer concentrations 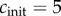 , 50 and 450 pmol mg–1, respectively. We have fitted the experimental curve for all three concentrations with the same value of
, 50 and 450 pmol mg–1, respectively. We have fitted the experimental curve for all three concentrations with the same value of 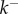 . Our fitting shows that, as expected,
. Our fitting shows that, as expected, 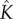 increases with the initial bulk concentration. However, the fact that
increases with the initial bulk concentration. However, the fact that 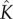 is not directly proportional to bulk monomer concentration suggests that there may be anti-cooperative binding of the monomer to the membrane, or monomer–monomer association in the bulk (recall that
is not directly proportional to bulk monomer concentration suggests that there may be anti-cooperative binding of the monomer to the membrane, or monomer–monomer association in the bulk (recall that 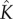 is directly proportional to
is directly proportional to 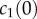 , the initial surface rather than bulk concentration of monomers). Our fitting thus suggests that the surface concentration of monomers is not directly proportional to the bulk concentration of monomers. In this experiment, the extent of oligomerization was monitored using an osmotic swelling assay [22]. To analyse the kinetic data, we normalize the percentage volume recovery relative to that at the end of the experiment.
, the initial surface rather than bulk concentration of monomers). Our fitting thus suggests that the surface concentration of monomers is not directly proportional to the bulk concentration of monomers. In this experiment, the extent of oligomerization was monitored using an osmotic swelling assay [22]. To analyse the kinetic data, we normalize the percentage volume recovery relative to that at the end of the experiment.
Figure 4b shows that kinetic data taken from [21] for the heptameric (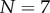 ) aerolysin can be fitted to equation (5.10) with parameters
) aerolysin can be fitted to equation (5.10) with parameters 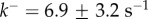 and
and 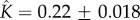 . The kinetic data were obtained via fluorescence spectroscopy, by monitoring the chloride efflux induced by the aerolysin pores from liposomes containing a dye that is quenched by the anion. To analyse the kinetic data, we normalize fluorescence intensity relative to the intensity at the end of the experiment.
. The kinetic data were obtained via fluorescence spectroscopy, by monitoring the chloride efflux induced by the aerolysin pores from liposomes containing a dye that is quenched by the anion. To analyse the kinetic data, we normalize fluorescence intensity relative to the intensity at the end of the experiment.
Figure 4c shows that experimental data taken from [12] for S. aureus
α-haemolysin can be fitted to equation (5.10) with parameters 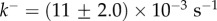 and
and 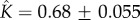 . In this experiment, the α-haemolysin monomer was incubated at 1 : 2000 molar protein : lipid ratio with mixed egg-yolk phosphatidylcholine/cholesterol liposomes, and the resulting pores were heptameric (
. In this experiment, the α-haemolysin monomer was incubated at 1 : 2000 molar protein : lipid ratio with mixed egg-yolk phosphatidylcholine/cholesterol liposomes, and the resulting pores were heptameric (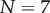 ). The reaction was monitored at 100 s intervals via an SDS-PAGE assay with silver staining, and the relative intensity between the monomer and heptamer bands was recorded. We assume the intensity of the silver stain is proportional to protein content, thus the relative intensity in our model is given by
). The reaction was monitored at 100 s intervals via an SDS-PAGE assay with silver staining, and the relative intensity between the monomer and heptamer bands was recorded. We assume the intensity of the silver stain is proportional to protein content, thus the relative intensity in our model is given by 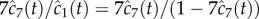 .
.
Figure 4d shows that the kinetics of self-assembly of E. coli cytolysin A, a larger dodecameric ( ) complex, can be fitted to our kinetic model with
) complex, can be fitted to our kinetic model with 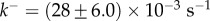 and
and 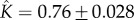 . The experimental data are taken from [13], where cytolysin A was incubated with n-dodecyl-β-d-maltopyranoside, and the reaction was tracked by SDS-PAGE assay with silver staining. The stain intensity, relative to the intensity at
. The experimental data are taken from [13], where cytolysin A was incubated with n-dodecyl-β-d-maltopyranoside, and the reaction was tracked by SDS-PAGE assay with silver staining. The stain intensity, relative to the intensity at 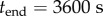 , was recorded (in our model this corresponds to tracking
, was recorded (in our model this corresponds to tracking 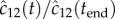 ). In both cases of experimental validation, the model is found to fit the data extremely well. The only discrepancy arises for small times, where the model appears to underpredict the concentration slightly, which we attribute to sensitivities in data acquisition at very low concentrations. We note that our asymptotic model is also able to replicate well the dynamics for larger pores, such as perfringolysin O, which are composed of up to 40 monomers. However, in these cases, the predicted value of K is no longer small and so such solutions fall outside the regime of validity of our asymptotic approximation. Nevertheless, in such cases, by using our theory for any dataset the predicted value of K required to fit the data can also suggest when the assumption of slow monomer accretion is no longer true, and thus when different aggregation kinetics are involved.
). In both cases of experimental validation, the model is found to fit the data extremely well. The only discrepancy arises for small times, where the model appears to underpredict the concentration slightly, which we attribute to sensitivities in data acquisition at very low concentrations. We note that our asymptotic model is also able to replicate well the dynamics for larger pores, such as perfringolysin O, which are composed of up to 40 monomers. However, in these cases, the predicted value of K is no longer small and so such solutions fall outside the regime of validity of our asymptotic approximation. Nevertheless, in such cases, by using our theory for any dataset the predicted value of K required to fit the data can also suggest when the assumption of slow monomer accretion is no longer true, and thus when different aggregation kinetics are involved.
7. Discussion and conclusion
Using a mean-field kinetic model for stepwise, reversible, self-assembly of multimeric PFTs, we have explained the counterintuitive observation by single-molecule fluorescence experiments that self-assembly of α-haemolysin is rapid, yet the concentrations of oligomeric intermediates are low. Our model singles out the ratio between the rates of monomer attachment and monomer detachment, 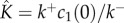 , as the parameter that determines the abundance of oligomer intermediates in the quasi-steady state. The rate of monomer detachment,
, as the parameter that determines the abundance of oligomer intermediates in the quasi-steady state. The rate of monomer detachment,  , is a separate parameter setting the overall timescale for the reaction. Asymptotic analysis of the model reveals that the approach to quasi-steady state follows a universal law that depends only on
, is a separate parameter setting the overall timescale for the reaction. Asymptotic analysis of the model reveals that the approach to quasi-steady state follows a universal law that depends only on 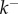 , thus our model identifies a powerful way to collapse experimental kinetic data onto a single master curve by scaling time.
, thus our model identifies a powerful way to collapse experimental kinetic data onto a single master curve by scaling time.
We suggest that the curious combination of low intermediate concentration and high reaction rate serves an important biological function of error minimization: by reducing the concentrations of intermediates, the system ensures that the only significant kinetic process is the stepwise addition of monomers to oligomers. This avoids the formation of oligomers that are larger than the pore size, which may be inactive or less potent. Indeed, larger pores, such as cholesterol-dependent cytolysins perfringolysin O and streptolysin O, usually display a degree of pore size heterogeneity [23–25], because the concentration of oligomer intermediates has to be large (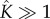 ) in order for pore formation to be complete within a biologically relevant timescale. From a reverse-engineering point of view, we speculate that exploiting the sensitivity of the self-assembly pathway to the concentration of intermediates may be a method to disrupt the self-assembly of pores and thus mechanism of virulence—a small change in
) in order for pore formation to be complete within a biologically relevant timescale. From a reverse-engineering point of view, we speculate that exploiting the sensitivity of the self-assembly pathway to the concentration of intermediates may be a method to disrupt the self-assembly of pores and thus mechanism of virulence—a small change in 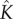 , the ratio between the rate of monomer attachment and monomer detachment, manifests itself in a large change in the concentration of intermediates in the quasi-steady state. Conversely, this ratio is an important factor to optimize in order to engineer artificial pores for nanotechnological purposes. We note that
, the ratio between the rate of monomer attachment and monomer detachment, manifests itself in a large change in the concentration of intermediates in the quasi-steady state. Conversely, this ratio is an important factor to optimize in order to engineer artificial pores for nanotechnological purposes. We note that 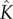 is effectively an equilibrium constant, and thus can be tuned via changing the temperature, or free energy gained from each monomer-attachment step.
is effectively an equilibrium constant, and thus can be tuned via changing the temperature, or free energy gained from each monomer-attachment step.
Beyond the quasi-steady state, we developed a systematic asymptotic technique in the strongly nonlinear regime that allows us to obtain an analytical formula for the concentration of product as a function of time. We showed that there are three kinetic stages: (i) a ‘lag phase’ corresponding to the approach to quasi-steady state; (ii) linear increase in product concentration once the quasi-steady state is established, with a constant flux of reactants converted into products; and (iii) breakdown of the quasi-steady state due to monomer depletion, and the reaction approaches completion only at a very long timescale. Our analytical result, equation (5.10), also compares well with experimental data for the self-assembly of pores of B. thuringiensis Cry1Ac (tetrameric pore), aerolysin and S. aureus α-haemolysin (heptameric pores) and E. coli cytolysin A (dodecameric pore). In these cases, in the absence of data for the concentration of intermediate aggregates, which could discriminate between assembly pathways, the positive comparisons drawn here support the possibility that such a pore-forming process may be more universal.
Acknowledgements
The authors would like to thank the Oxford Mathematics in Chemistry Study Group 2013 for useful discussions.
Authors' contributions
A.A.L. contributed to writing the article, and performed research for the model formulation and late-stage dynamics and model fitting. T.E.W. contributed to writing the article, and performed research for the model formulation and late-stage dynamics. M.J.S. and M.I.W. provided the biological motivation and background, and the resources for experimental validation. I.M.G. contributed to writing the article, and performed research of model formulation and the asymptotic analysis.
Competing interests
We declare we have no competing interests.
Funding
A.A.L. is supported by an EPSRC Research Fellowship and a Fulbright Scholarship. T.E.W. would like to thank St John's College Oxford for its financial support. M.I.W. is supported by the ERC (StG 309611 CoSMiC). M.J.S. is supported by the EPSRC. I.M.G. is supported by a Royal Society University Research Fellowship.
References
- 1.Voskoboinik I, Smyth MJ, Trapani JA. 2006. Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 6, 940–952. ( 10.1038/nri1983) [DOI] [PubMed] [Google Scholar]
- 2.Rosado CJ, et al. 2007. A common fold mediates vertebrate defense and bacterial attack. Science 317, 1548–1551. ( 10.1126/science.1144706) [DOI] [PubMed] [Google Scholar]
- 3.Iacovache I, Bischofberger M, van der Goot FG. 2010. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 20, 241–246. ( 10.1016/j.sbi.2010.01.013) [DOI] [PubMed] [Google Scholar]
- 4.Los FC, Randis TM, Aroian RV, Ratner AJ. 2013. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 77, 173–207. ( 10.1128/MMBR.00052-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325, 1089–1093. ( 10.1126/science.1176667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodford N, Livermore DM. 2009. Infections caused by Gram-positive bacteria: a review of the global challenge. J. Infect. 59, S4–S16. ( 10.1016/S0163-4453(09)60003-7) [DOI] [PubMed] [Google Scholar]
- 7.Branton D, et al. 2008. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 26, 1146–1153. ( 10.1038/nbt.1495) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris R, Sims P, Tweten R. 1991. Evidence that Clostridium perfringens theta-toxin induces colloid-osmotic lysis of erythrocytes. Infect. Immunity 59, 2499–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vandana S, Raje M, Krishnasastry MV. 1997. The role of the amino terminus in the kinetics and assembly of α-hemolysin of Staphylococcus aureus. J. Biol. Chem. 272, 24 858–24 863. ( 10.1074/jbc.272.40.24858) [DOI] [PubMed] [Google Scholar]
- 10.Skals M, Jorgensen NR, Leipziger J, Praetorius HA. 2009. α-Hemolysin from Escherichia coli uses endogenous amplification through P2X receptor activation to induce hemolysis. Proc. Natl Acad. Sci. USA 106, 4030–4035. ( 10.1073/pnas.0807044106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaidyanathan MS, Sathyanarayana P, Maiti PK, Visweswariah SS, Ayappa KG. 2014. Lysis dynamics and membrane oligomerization pathways for Cytolysin A (ClyA) pore-forming toxin. RSC Adv. 4, 4930 ( 10.1039/c3ra45159c) [DOI] [Google Scholar]
- 12.Bortoleto R, De Oliveira A, Ruller R, Arni R, Ward R. 1998. Tertiary structural changes of the α-hemolysin from Staphylococcus aureus on association with liposome membranes. Arch. Biochem. Biophys. 351, 47–52. ( 10.1006/abbi.1997.0550) [DOI] [PubMed] [Google Scholar]
- 13.Benke S, Roderer D, Wunderlich B, Nettels D, Glockshuber R, Schuler B. 2015. The assembly dynamics of the cytolytic pore toxin ClyA. Nat. Commun. 6, 6198 ( 10.1038/ncomms7198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson JR, Cronin B, Bayley H, Wallace MI. 2011. Rapid assembly of a multimeric membrane protein pore. Biophys. J. 101, 2679–2683. ( 10.1016/j.bpj.2011.09.054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawate T, Gouaux E. 2003. Arresting and releasing Staphylococcal α-hemolysin at intermediate stages of pore formation by engineered disulfide bonds. Protein Sci. 12, 997–1006. ( 10.1110/ps.0231203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subburaj Y, Ros U, Hermann E, Tong R, Garcia-Saez AJ. 2015. Toxicity of an α-pore-forming toxin depends on the assembly mechanism on the target membrane as revealed by single-molecule imaging. J. Biol. Chem. 290, 4856–4865. ( 10.1074/jbc.M114.600676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffiths IM, Breward CJW, Colegate DM, Dellar PJ, Howell PD, Bain CD. 2013. A new pathway for the re-equilibration of micellar surfactant solutions. Soft Matter 9, 853–863. ( 10.1039/C2SM27154K) [DOI] [Google Scholar]
- 18.Fahie M, Romano FB, Chisholm C, Heuck AP, Zbinden M, Chen M. 2013. A non-classical assembly pathway of Escherichia coli pore-forming toxin cytolysin A. J. Biol. Chem. 288, 31 042–31 051. ( 10.1074/jbc.M113.475350) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen SI, Vendruscolo M, Welland ME, Dobson CM, Terentjev EM, Knowles TP. 2011. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J. Chem. Phys. 135, 065105 ( 10.1063/1.3608916) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fortier M, Vachon V, Marceau L, Schwartz J-L, Laprade R. 2007. Kinetics of pore formation by the Bacillus thuringiensis toxin Cry1Ac. Biochim. Biophys. Acta (BBA) Biomembranes 1768, 1291–1298. ( 10.1016/j.bbamem.2007.02.013) [DOI] [PubMed] [Google Scholar]
- 21.Van der Goot FG, Pattus F, Wong KR, Buckley JT. 1993. Oligomerization of the channel-forming toxin aerolysin precedes insertion into lipid bilayers. Biochemistry 32, 2636–2642. ( 10.1021/bi00061a023) [DOI] [PubMed] [Google Scholar]
- 22.Carroll J, Ellar DJ. 1993. An analysis of Bacillus thuringiensis δ-endotoxin action on insect-midgut-membrane permeability using a light-scattering assay. Eur. J. Biochem. 214, 771–778. ( 10.1111/j.1432-1033.1993.tb17979.x) [DOI] [PubMed] [Google Scholar]
- 23.Palmer M, Valeva A, Kehoe M, Bhakdi S. 1995. Kinetics of streptolysin O self-assembly. Eur. J. Biochem. 231, 388–395. ( 10.1111/j.1432-1033.1995.tb20711.x) [DOI] [PubMed] [Google Scholar]
- 24.Czajkowsky DM, Hotze EM, Shao Z, Tweten RK. 2004. Vertical collapse of a cytolysin prepore moves its transmembrane β-hairpins to the membrane. EMBO J. 23, 3206–3215. ( 10.1038/sj.emboj.7600350) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert RJ, Mikelj M, Dalla Serra M, Froelich CJ, Anderluh G. 2013. Effects of MACPF/CDC proteins on lipid membranes. Cell. Mol. Life. Sci. 70, 2083–2098. ( 10.1007/s00018-012-1153-8) [DOI] [PMC free article] [PubMed] [Google Scholar]