

Abstract
Research development is burgeoning for genetic and cellular therapy for retinal dystrophies. These dystrophies are the focus of many research efforts due to the unique biology and accessibility of the eye, the transformative advances in ocular imaging technology that allows for in vivo monitoring, and the potential benefit people would gain from success in the field – the gift of renewed sight. Progress in the field has revealed the immense complexity of retinal dystrophies and the challenges faced by researchers in the development of this technology. This study reviews the current trials and advancements in genetic and cellular therapy in the treatment of retinal dystrophies and also discusses the current and potential future challenges.
Keywords: Cell Therapy, Genetic Therapy, Retinal Dystrophies
INTRODUCTION
The last few years have shown rapid growth in genetic and cellular therapy advancement for the treatment of retinal dystrophies. Development of effective therapies may revolutionize ophthalmology since there are currently no available treatments for these diseases, and many patients are destined to lose substantial vision during their lifetime. However, extensive research has further revealed the genetic and phenotypic complexity of retinal dystrophies. Causative mutations have been discovered for many different retinal dystrophies affecting multiple new genes, complex biologic pathways, as well as retinal pigment epithelium (RPE), and photoreceptor cell function. These intricate connections and functions offer researchers many challenges in therapeutic development.
The eye represents a unique target organ for gene therapy due to the immune privilege afforded by the blood-ocular barrier, the ability to directly visualize, access and locally treat the target tissue and with regard to clinical trials, there is the benefit of a simultaneous control provided by the other eye. In this review, we discuss the current status of both gene therapy and cell therapy for retinal dystrophies as well as the challenges that researchers face in the development of these therapies for human use.
OVERVIEW OF RETINAL DYSTROPHIES
Retinal dystrophies are a diverse group of disorders, both genetically and phenotypically affecting the function of the retina. Inheritance patterns include X-linked, autosomal dominant and recessive, and mitochondrial forms. Phenotypic categories include retinitis pigmentosa (RP) and related disorders such as Bardet–Biedl and Usher syndromes, Leber's congenital amaurosis (LCA), rod-cone dystrophy, cone or cone-rod dystrophy, Stargardt's macular dystrophy, congenital stationary night blindness, and choroideremia. Currently, 278 genetic loci and 238 genes have been identified affecting a variety of pathways and mechanisms in retinal disease (RetNet, https://www.sph.uth.edu/retnet/sum-dis.htm). The complexity of the neuronal pathways, the structure of each cell, and the diversity of functions of each retinal layer create many challenges in therapeutic development for these diseases. For example, mechanisms affecting diseases of RPE include visual cycle reactions, phagocytic activity, membrane trafficking, and ion transport. Functional rod and cone photoreceptors cells require phototransduction, connecting cilium structure and transport, inner segment protein and vesicle trafficking, chaperone function, lipid metabolism, transcription and RNA splicing, and synaptic transmission. All of these roles must be restored to regain appropriate retinal function.
GENE THERAPY
Until recently, there has been little hope for curing or slowing the progression of these blinding retinal degenerations. Gene therapy offers a therapeutic option for these disorders [Table 1]. In many cases, mouse models have paved the way for our understanding of the major pathologic pathways involved in retinal degenerations. The more recent successful treatment of large animal models marked a turning point in gene therapy and the advent of treatment options in humans.1,2
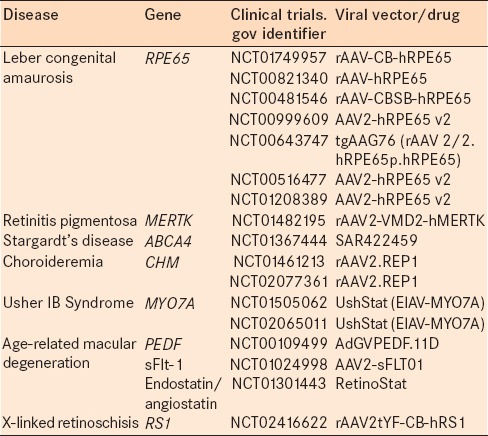
Table 1.
Current clinical trials of gene therapy for retinal degenerations
Photoreceptors and RPE are the primary targets for gene therapy as these cells play a key role in the visual cycle and are prone to a large number of potential single gene defects. RPE is a monolayer of epithelial cells between the choroid and the neurosensory retina. It is involved in many aspects of photoreceptor maintenance including the replenishment of 11-cis-retinal and the phagocytosis of rod and cone outer segment discs. The inherent phagocytic ability of the RPE facilitates gene delivery. A significant portion of retinal disease arises from genetic defects affecting the photoreceptors as these are the primary light sensitive cells of retina. Delivering gene therapy to the photoreceptors is more challenging as their lipid-rich discs act as physical barriers between the subretinal space, the site of delivery, and the cell nucleus in the outer nuclear layer.
Various vector technologies have, however, enabled adequate delivery to the photoreceptors. The principle of gene therapy is based on the transfer of a therapeutic gene to the eye using viral or nonviral vectors. Adeno-associated virus (AAV)-based vectors are currently the gold standard for retinal gene therapy and enable cell type specific transgene expression in RPE cells and photoreceptor cells via subretinal injection.3
PREVIOUS AND CURRENT CLINICAL TRIALS
RPE65 for leber's congenital amaurosis type-2
LCA is a severe, early onset form of RP and is defined by severe visual impairment in the first 3 years of life.4 Currently, three RPE expressed genes are known to be implicated in LCA and account for up to 17% of LCA cases.5
RPE65 is an RPE-specific gene encoding the isomerohydrolase that recycles bleached all-trans-retinal into photosensitive 11-cis-retinal. Mutations in the RPE65 gene account for 5–10% of all LCA cases and cause a severe phenotype called LCA type 2. In this disease, severe visual loss is present at birth or within the 1st months of life.6,7
Gene therapy for LCA began in an RPE65 “knockout” mouse model, and the concept was then carried to a canine model of RPE65 deficiency.2,8,9 The first three human clinical trials investigating gene therapy for LCA due to RPE65 gene mutations were initiated in 2007.10,11,12 All three trials used a subretinal injection of recombinant rAAV2 vector to deliver human RPE65 cDNA to target RPE cells. None of the trials showed adverse local or systemic effects, and there were improvements in the perception of light as noted by increased pupillary response to light and self-reported increased visual sensitivity.10,11 Longer follow-up studies showed that improvement was noted to peak at 6–12 months and then slightly decline, suggesting that perhaps these initial trials did not meet the full demand for RPE65 in affected individuals.10,11 These groundbreaking studies were the first to demonstrate proof of concept for gene therapy in human retinal disease while improving the lives of many families with children suffering from this devastating disease. The remarkable success of the RPE65 studies has paved the way for other clinical trials, particularly using AAV-mediated gene delivery; however, there are challenges to delivering an effective and enduring dose to halt or inhibit further cell death or degeneration.
MER proto-oncogene tyrosine kinase (RP38) for retinitis pigmentosa
RP is one of the most common types of retinal degenerations and is responsible for vision loss in 1 in 4000 people worldwide.13 RP is classically characterized by ophthalmoscopic findings of abnormal dark bone-spicule pigmentation, attenuated blood vessels, and optic atrophy. The most prominent initial symptoms are nyctalopia and peripheral field loss due to photoreceptor dysfunction with the rods being affected first. Eventually, central visual loss occurs due to cone involvement. So far, more than 60 gene defects have been identified.14,15,16
In the Royal College of Surgeons (RCS) rat, a classic model of RP, the MER proto-oncogene tyrosine kinase (MERTK) gene was identified as the causative gene. MERTK is activated by ligands on outer segment disc membranes and triggers outer segment phagocytosis by RPE cells.5,17 Mutations of MERTK cause outer segment debris to accumulate in the subretinal space leading to photoreceptor degeneration. MERTK knockout mice showed impaired ingestion of photoreceptor outer segments by RPE.18 In one study, an AAV vector was used to transfer MERTK to the subretinal space of RCS rats, leading to restoration of phagocytic function with decrease in outer segment debris.19 Electroretinogram (ERG) results were transiently improved, and photoreceptor survival was prolonged, for about 12 weeks. A gene therapy trial has recently been started at the King Khaled Eye Hospital in Saudi Arabia to treat patients with RP caused by MERTK mutations (NCT01482195).
ABCA4 for Stargardt's disease
Stargardt's disease, also known as Stargardt's macular degeneration or fundus flavimaculatus, is the most common form of juvenile macular degeneration.20 It is usually inherited as an autosomal recessive disease caused by mutations in the ABCA4 gene.21 The ABCA4 protein localizes to photoreceptor outer segments and aides in the translocation of N-retinylidene-phosphatidylethanolamine (PE), an intermediate in the visual cycle, from the lumen of the disc to the photoreceptor cytoplasm.22,23
During ABCA4 dysfunction, N-retinylidene-PE accumulates in the outer segment discs and reacts with all-trans-retinal leading to the formation of apolipoprotein 2E (A2E), a major toxic component of lipofuscin. High levels of lipofuscin cause the photoreceptor dysfunction seen in Stargardt's disease.24 A mouse model of the disease was created by Weng et al., which shared several features seen in humans such as delayed rod dark adaptation, delayed clearance of all-trans-retinal, and increased N-retinylidene-PE. There was additionally increased lipofuscin and A2E within cells.25
The size of the ABCA4 gene is too large to be packaged in the more commonly used AAV vector. Currently, a commercial trial led by Oxford Biomedica is using equine infectious anemia virus (EIAV) to deliver ABCA4 (NCT01367444). Beyond the size of the gene and the difficulty of delivering ABCA4 transgene to the photoreceptors, the phenotypic heterogeneity makes Stargardt's disease a challenging condition to treat with gene therapy.
CHM for choroideremia (Rab-escort protein 1 vector)
Choroidermia is a condition leading to the degeneration of the choroid, RPE, and neural retina affecting 1 in 50,000 individuals.26 The disease is X-linked recessive and arises from mutations affecting the CHM gene. CHM encodes Rab-escort protein 1 (REP 1), resulting in a prenylation deficiency and leading to the retinal degeneration seen in choroideremia.27
A current multicenter clinical trial is ongoing (NCT01461213) in which AAV REP1 is administered as a subfoveal injection. The preliminary results of 6 enrolled patients showed that 2 patients gained between 2 and 4 lines of vision while 4 patients recovered 1–3 letters, for a mean visual acuity gain of 3.8 letters at 6 months. Maximal sensitivity measured with dark-adapted microperimetry also increased but not significantly.28
MYO7A for Usher 1B
Usher syndrome is characterized by the dual sensory impairment of the visual and audiovestibular systems.29 It is a heterogeneous disease with 9 associated genes, affecting 1 in 25,000 people.30,31 The syndrome is divided into three subtypes based on the severity. Usher I patients are usually born with deafness and vestibular problems.
Mutations in MYO7A causes Usher IB syndrome. The gene encodes myosin VIIA, a protein essential for transport along cilia such as those between photoreceptor inner and outer segments. Usher 1B disease is a good candidate for gene therapy as the syndromic features of deafness and some photoreceptor dysfunction are present at birth, but evidence of retinal degeneration does not manifest until adolescence, thereby giving a therapeutic window to intervene.
There is currently a clinical trial in phase I by Oxford Biomedica (NCT01505062) evaluating MYO7A therapy. One challenge in the treatment of this disease with gene therapy is the large size of the transgene required to express myosin VIIA. An equine lentiviral vector, which can accommodate the larger sized gene, is used in this case rather than the adenovirus vector.
Gene therapy in neovascular age-related macular degeneration
Age-related macular degeneration (AMD) is a complex disease with many potential therapeutic targets. Gene therapy may be used to augment the expression of endogenous antiangiogenic factors to suppress neovascularization (NV) in wet AMD. Pigment epithelium-derived factor (PEDF), vascular endothelial growth factor (VEGF) binding proteins, endostatin, and angiostatin are all potential antiangiogenic therapeutics in this disease and are currently investigated using gene therapy.
PEDF is a serine protease inhibitor, which is both neurotrophic and antiangiogenic.32,33,34 Intravitreous or subretinal injection of an adenoviral vector expressing human PEDF suppressed the development of retinal or subretinal NV and caused regression of established NV.35,36,37,38 These results led to a phase I clinical trial in patients with advanced NVAMD. Twenty-eight patients were given various doses of PEDF. About 71% of patients in the high-dose group and 55% in low dose group had improvement or stability in size of NV lesion. This study provided proof of concept for the strategy of gene transfer for antiangiogenesis.39
The secreted extracellular domain of VEGF receptor-1, sFlt-1 is a naturally occurring protein antagonist of VEGF.40 Intraocular injections of adenovirus vector associated sFlt-1 in mice and monkeys suppressed subretinal NV and intravenous injections suppressed retinal NV in primates.41,42,43 Intravitreal AAV2-sFlt01 is under development by Genzyme, and subretinal AAV2-sFlt01 is under development by Avalanche Biotechnologies. Avalanche has completed a phase 1 and phase 2a study.
Endostatin is a cleavage product of collagen XVIII, and angiostatin is a cleavage product of fibrinogen, each one inhibiting tumor angiogenesis. Endostatin plays a role in regulating retinal vascular development and the physiologic regression of the hyaloid vasculature.44 In mice models, gene transfer of endostatin blocked vascular leakage, suppressed NV, and inhibited ischemic-induced retinal NV. Inducible expression of VEGF in adult mice causes severe proliferative retinopathy and retinal detachment.45 Intraocular expression of endostatin reduces VEGF-induced retinal vascular permeability, NV, and retinal detachment.38,46 Oxford Biomedica has currently undertaken a phase I trial for gene transfer of both endostatin and angiostatin in equine infectious anemia viral vectors in patients with advanced NVAMD (NCT01301443).
X-linked retinoschisis (RS1) – adeno-associated virus-RS1
X-linked retinoschisis is a leading cause of hereditary visual loss in young males. This degeneration is caused by mutations in the RS1 gene encoding retinoschisin, a protein implicated in cell adhesion and maintenance of retinal integrity.47 It displays an X-linked mode of inheritance and is characterized by foveo-macular schisis and intraretinal cystic degeneration resulting in the progressive loss of macular photoreceptors. The peripheral changes consist of the separation of retinal layers.
Mouse models of retinoschisis showed that subretinal or intravitreal delivery of AAV-mediated gene therapy led to increased expression of RS1 protein, improved retinal integrity, and increased ERG b-wave.48,49,50 Currently, there is an ongoing phase I trial to examine the safety and efficacy of rAAV-hRS1 in patients with X-linked retinoschisis (NCT02416622).
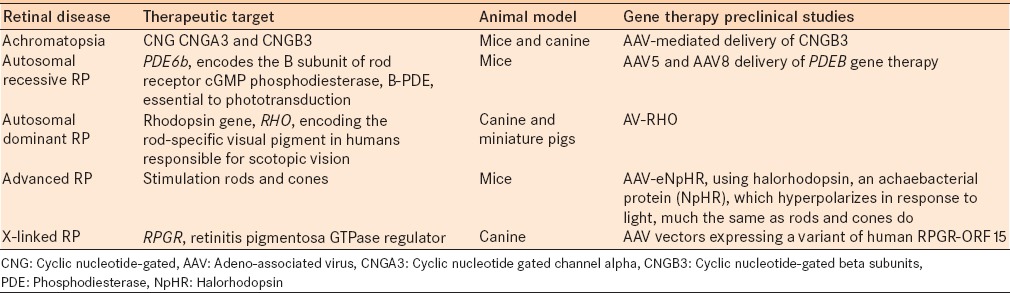
Other than the above mentioned clinical studies, various other studies are currently underway [Table 2]. AAV-derived vectors have led to significant advances in gene transfer therapeutics. However, there are several challenges in the delivery and effectiveness of these treatments.
Table 2.
Promising preclinical gene therapy studies
CHALLENGES IN GENE THERAPY
Vector development and delivery present challenges.51,52 AAV2- and EIAV-vectors currently predominate in retinal gene therapy research due in part to their low immunogenicity and long-term expression.53 Benefits of AAV2-based vectors include their efficiency in transducing postmitotic cells and these vectors are nonintegrating. However, the cargo capacity of AAV2-based vectors is 4.7 kb and its use is restricted to the delivery of small genes. As discussed above, there are many genes that are likely too big for delivery by AAV, including ABCA4 in Stargardt's disease and MYO7A in Usher syndrome. Other delivery options are being pursued to accommodate larger cargo, including dual AAV vectors, EIA-based vectors, and nonviral methods for gene delivery. Dual AAV vectors deliver partial cDNAs that then assemble full-length coding sequences in vivo via trans-splicing. EIA vectors can accommodate cargo up to 8 kb and are currently studied as a potential option in ABCA4-related Stargardt's disease. Nonviral vectors include liposomes, lipocomplexes, polyplexes, nanoparticles, microparticles, or a combination of these and have an unlimited carrying capacity.51,54 Although nonviral vectors may be safer than viral vectors, they transfect less effectively and have shorter lives than the viral vectors.55
Delivery of the vectors remains the next challenge. The main locations of delivery are intravitreal, subretinal, and suprachoroidal. Intravitreal delivery is less invasive, can be done in an office setting, and can potentially deliver viral particles across the entire retina.53,56 Dilution of AAV once in the vitreous, the necessity for the construct to cross the internal limiting membrane, as well as the relative abundance of AAV receptors that capture vectors after intravitreal administration can affect gene transduction.57,58,59 Subretinal injections are technically challenging and complications have been reported in some of the LCA2 clinical trials. These included foveal thinning, macular hole, choroidal effusion, and ocular hypo- and hyper-tension.10,60 In addition, subretinal injection in extrafoveal regions can generate new “pseudofoveas” from vector transduction.61
Optimal vectors and method of delivery do not guarantee success. Gene therapy relies on the presence of viable target cells. Some patients with LCA or achromatopsia are suitable patients for gene therapy as they have preserved retinal structure for many years after the diagnosis. In addition, in these diseases, the functional component of the disease can be tested in a reasonable time frame in the context of clinical trials. Instead, many forms of RP have mild phenotypes because of the slow progression of the disease which makes it less suitable from a clinical trial perspective point.
CELL THERAPY
Cell therapy is also a potential landmark regenerative therapy for retinal dystrophies. Especially, in advanced-stage disease, cell or tissue transplantation may provide a potential option for restoration of vision. AMD is the leading cause of blindness in people older than 50 years of age in the Western World. Severe vision loss occurs in patients with the advanced dry form (geographic atrophy) and patients with the wet form. Anti-VEGF injections offer significant visual benefits for patients with the wet form. However, there is no effective treatment for the advanced dry form or for the patients who develop atrophy after repeated anti-VEGF injections. These patients may benefit from cell therapy.
The eye is thought to be an ideal organ for cell transplantation as well. The optical media are clear, allowing for the intraoperative and postoperative view to the area of transplantation, and its immune privilege status make it especially attractive for such treatments. The retinal detail that can be displayed with the state of the art imaging such as optical coherence tomography has improved the evaluation of eyes following cell transplantation.62 Research subjects can be more easily monitored for improvements in retinal structure, maintenance of retinal layers, integration of cells in the correct location, and cell and tissue survival in vivo.
Depending on the retinal disease, current cell transplant therapy is directed at replacing the retinal pigment epithelial cells or the photoreceptor cells, as well as implanting cells for the trophic factors that they produce. In diseases primarily affecting the RPE, such as AMD, RPE compromise eventually leads to photoreceptor loss. RPE replacement in such diseases could also prevent secondary photoreceptor degeneration and vision decline.
RPE TRANSPLANTATION
Human RPE cells were first isolated and characterized over 30 years ago.63,64 Since then, the understanding of the role of the RPE cell layer in the pathophysiology of retinal disease has shown significant progress. As an attraction in cell transplant research, the RPE cells do not require synaptic connections to perform their role, unlike other cell types in the retina.65 However, the ability to perform its essential functions is dependent on the RPE layer being a confluent monolayer with tight junctions and the RPE cells maintaining polarity for ion transport.65
Seminal experiments by Li and Turner demonstrated proof of principle of RPE transplantation in an animal model of retinal dystrophy with abnormal RPE phagocytosis function. They demonstrated that subretinal injection of healthy RPE cells allows preservation of the outer nuclear, outer plexiform, and photoreceptor inner/outer segment layers.66,67
Improvements in surgical strategies for RPE transplantation that have progressed since the Submacular Surgery Trial in 1998 have allowed for innovative methods of RPE transplantation.68 Macular translocation in which the neurosensory retina is surgically detached and repositioned to an area of healthy RPE previously showed some improvements in visual acuity and quality of life.69,70,71 The procedure was also associated with many complications including torsional diplopia, retinal detachment, proliferative vitreoretinopathy (PVR), and macular hole. Therefore, this procedure has largely been abandoned.70 Transplantation of autologous RPE patch grafts on a bed of Bruch's membrane and partial thickness choroid to the submacular space has also been demonstrated by several groups.72,73,74 Some studies have shown some slight improvement of vision, but this may just signify the ability to detect a fixation target on the fovea.73 This procedure has also shown significant complications including subretinal bleeding, PVR, and mal-positioned graft. More recently, van Zeeburg et al. published the long-term outcomes of 133 patients who underwent CNV excision followed by autologous RPE-choroid grafting. The postoperative PVR rate was 10% and visual acuity outcomes were modest. However, 5% of the patients did show the best corrected visual acuity better than 20/40.75
An alternative method for transplantation is a submacular injection of an RPE cell suspension in the location of an RPE defect. Studies have shown the importance of a healthy Bruchs membrane in the ability of these cells to successfully attach to Bruchs and restore RPE continuity.76 Several different cell sources have been studied as potential options in RPE transplant therapy, including autologous RPE cells aspirated from a different location in the retina,76 autologous iris pigment epithelium,77,78 in vitro cultured allogenic cells, human embryonic stem cell-derived (hESC) RPE,79,80,81,82,83,84 and induced pluripotent stem cell-derived (iPSC) RPE.85,86,87,88,89 The medium and long-term safety and efficacy of the use of hESC-derived RPE in patients with AMD and Stargardt's macular dystrophy in two open-label phase I/II studies were recently reported (NCT01345006 – Stargardt's macular dystrophy and NCT01344993 – AMD).90,91 None of the eighteen patients showed evidence of adverse cell proliferation, rejection or serious ocular, or systemic safety issues. Complications reported were related to the vitreoretinal surgery and immunosuppression including progression of cataract, localized damage to RPE, and intraocular inflammation, and one patient had staphylococcus endophthalmitis.91 Best-corrected visual acuity was improved in ten eyes, maintained in seven eyes, and declined in one eye.91 However, it is emphasized that these studies did not include a control group, and patients and investigators were not masked to treatment.
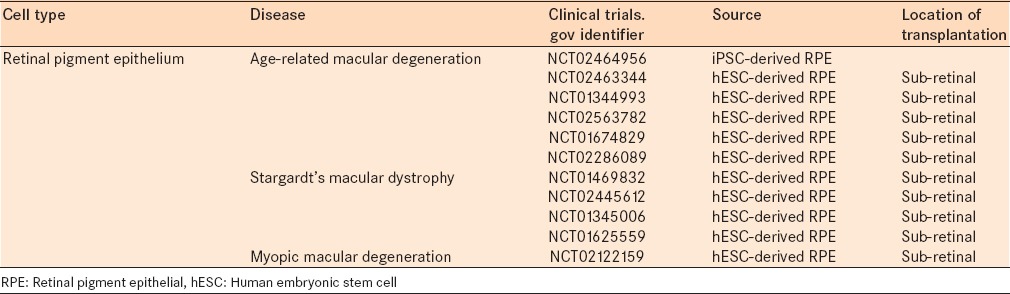
There are currently eleven clinical trials posted on clinical trials.gov evaluating the use of RPE transplantation in retinal disease [Table 3]. These trials are evaluating the use of RPE cells from several different sources, including transplantation of autologous RPE, translocation of autologous RPE, iPSC-derived RPE, and hESC-derived RPE, and also in the treatment of several different retinal dystrophies – AMD, Stargardt's macular dystrophy, and myopic macular degeneration. The majority of these trials are planning to primarily evaluate the safety and tolerability of RPE transplantation in human subjects while simultaneously evaluating visual outcomes. Future studies will confirm the safety of RPE transplantation from stem cell sources and will further evaluate the efficacy and outcomes while continuing to advance the technology.
Table 3.
Current clinical trials in cell therapy for retinal degenerations
PHOTORECEPTOR CELL TRANSPLANTATION
Progressive loss of photoreceptor cells resulting in decreased vision occurs in RP and other retinal degenerations. Transplantation of these photoreceptor cells and restoration of the function of the outer nuclear layer would provide a treatment for these currently untreatable blinding diseases. Previously blind patients have shown the ability to read with stimulation by subretinal electrodes in advanced macular degeneration suggesting that the downstream circuitry of the human inner retina retains the capacity to be reactivated to restore visual function long after the absence of photoreceptors.92,93 Different from RPE transplantation, in photoreceptor replacement, newly introduced precursors would have to resume their developmental program after transplantation to form a polarized outer nuclear layer with formation of light-sensitive outer segments containing enzymes critical for light sensitivity and then would have to reconnect synaptically with downstream retinal neurons in order for light-evoked signals to send information down the visual pathway.
Protocols for differentiating photoreceptors from ESCs have been extensively optimized by Osakada et al.80,94 and Lamba et al.84,95 to allow ESC-derived photoreceptors to be a potential cell source for transplantation.84,96 Fetal retinal cells or fetal retinal tissue are other potential cell sources.97,98,99,100 After transplantation, the cells have shown survival and improvement in visual function.98,101 However, the major disadvantages of ESCs and fetal cells are ethical restrictions and availability. Therefore, other groups evaluate the use of adult photoreceptor or stem cell sources for transplantation.102,103 Many of the earlier studies evaluated photoreceptor survival in nondiseased or early diseased retina, but more recent studies have also shown restoration of visual function and survival of photoreceptor cells in retina with more advanced disease as well (which would be more characteristic of its need in cell therapy for human retinal diseases).104
Some patients suffer from loss of both RPE and photoreceptors. Patients with advanced macular degeneration have RPE disease that later leads to photoreceptor cell death. In the future, these patients may benefit from a combined RPE and photoreceptor transplant. Whether this would be a step-wise approach with RPE replacement and then photoreceptors transplantation or whether the two could be transplanted together is to be discovered. Zhu et al. showed that the survival of photoreceptor progenitor cells was increased when co-cultured with hESC-derived RPE,105 suggesting that this dual replacement is likely to be a promising strategy. Other recent studies have shown the formation of entire optic cups from both mouse ESC and hESC in minimal media conditions.96,106 Therefore, stratified neural retina and RPE in a single complex may also be a potential route to develop a dual RPE/PR graft that can be used in individuals with end-stage disease with severe retinal RPE and PR loss.
Prosthetic devices are another approach investigated and optimized for the restoration of sight in those blind from retinal degenerations. The Argus II Retinal Prosthesis System (Second Sight Medical Products, Inc., Sylmar, CA) has gained Food and Drug Administration approval and a CE mark in Europe after positive early results from the Argus II clinical trial for patients with RP.107 This is the first and only retinal implant to have obtained both of these approvals. The 3-year results of the clinical trial evaluating the safety, reliability, and benefit of this retinal implant supported the long-term safety profile and benefit of this system in low vision patients with RP.107 Twenty-nine of the 30 patients had functioning implants after 3 years and the subjects as a group performed significantly better on vision testing with the system on compared to when it was off. Eleven patients did experience device or surgical complications but these were addressed with standard ophthalmic techniques.107 These promising results open new and hopeful opportunities for visual function in patients with RP and also other retinal degenerations.
CHALLENGES WITH CELL THERAPY
One of the challenges in cell therapy for treatment of retinal dystrophies is choosing the source of the cells. As discussed above, many different cell sources have been studied for the use in retinal disease, including fetal and adult cells, ESCs, autologous RPE or iris pigment epithelial cells, iPSCs, or other stem cells. ESCs and retinal fetal cells have been used in many studies, but the major disadvantages of these cell sources are ethical restrictions and availability. Autologous cell transplants or more recently developed iPSCs from the individual requiring the transplant are ideal in terms of issues with allogeneic immune rejection and availability; however, there are concerns about differentiation and maturation that could pose a risk of immature teratoma formation.108
Once the cell source is determined, the mechanism for delivering the cells is the next challenge. Subretinal transplantation is the more commonly used technique. However, it is a more complex surgical procedure requiring a skilled retinal surgeon with experience in subretinal surgery and has a higher risk of surgical complications, including hemorrhage, PVR, graft dislocation, and NV.74,76,109 With this technique, the cells are delivered to the intended location and, therefore, better integration and differentiation is observed. Intravitreal injections have also been tried since they are a common procedure in a retina clinic for macular degeneration and are associated with fewer complications.110 The concern is that the host retina may act as a barrier and prevent the transplanted cells from migrating and integrating into the retinal tissue in the correct location. Especially, if using a cell sheet or an RPE-photoreceptor-scaffold complex, a subretinal approach would be necessary since transplants of this size could not traverse the inner retina.
Next, ensuring that the cells integrate into the correct location in the retina and differentiate to establish their natural connections and functions is important. Normally, the photoreceptor inner segments are linked to Müller cells through tight junctions and form synapses with bipolar cells. Fully differentiated photoreceptor cells may not form these necessary connections whereas photoreceptor precursors may differentiate after transplantation and create these important connections. If more than one type of cell is needed to restore the natural retinal cell layers, the question will then be whether the layers should be transplanted sequentially or if a retinal complex including the necessary layers would be optimal for restoration of visual function.
CONCLUSION
Research evaluating potential treatments for retinal dystrophies has progressed significantly over the last few years. For blinding diseases that currently have no available treatments, success in the development of cutting-edge therapies for these diseases would change the lives of many individuals. Mutation-specific genetic therapies for retinal dystrophies are currently being developed. Stem cell transplantation for retinal degenerations has now transitioned from over a decade of preclinical studies to phase I/II clinical trials. The potential benefits are a tremendous motivation to ensure that retinal dystrophy translational research reaches its full potential.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Stieger K, Schroeder J, Provost N, Mendes-Madeira A, Belbellaa B, Le Meur G, et al. Detection of intact rAAV particles up to 6 years after successful gene transfer in the retina of dogs and primates. Mol Ther. 2009;17:516–23. doi: 10.1038/mt.2008.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–5. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 3.Surace EM, Auricchio A. Versatility of AAV vectors for retinal gene transfer. Vision Res. 2008;48:353–9. doi: 10.1016/j.visres.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 4.Leber T. Ueber retinitis pigmentosa und angeborene amaurose. Graefes Arch Clin Exp Ophthalmol. 1869;15:1–25. [Google Scholar]
- 5.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell. 2005;122:449–59. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005;102:12413–8. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narfström K, Bragadóttir R, Redmond TM, Rakoczy PE, van Veen T, Bruun A. Functional and structural evaluation after AAV. RPE65 gene transfer in the canine model of Leber's congenital amaurosis. Adv Exp Med Biol. 2003;533:423–30. doi: 10.1007/978-1-4615-0067-4_54. [DOI] [PubMed] [Google Scholar]
- 9.Le Meur G, Stieger K, Smith AJ, Weber M, Deschamps JY, Nivard D, et al. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther. 2007;14:292–303. doi: 10.1038/sj.gt.3302861. [DOI] [PubMed] [Google Scholar]
- 10.Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–8. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–90. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231–9. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 13.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 14.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–75. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Sahel J, Bonnel S, Mrejen S, Paques M. Retinitis pigmentosa and other dystrophies. Dev Ophthalmol. 2010;47:160–7. doi: 10.1159/000320079. [DOI] [PubMed] [Google Scholar]
- 16.Anasagasti A, Irigoyen C, Barandika O, López de Munain A, Ruiz-Ederra J. Current mutation discovery approaches in retinitis pigmentosa. Vision Res. 2012;75:117–29. doi: 10.1016/j.visres.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Hall MO, Obin MS, Heeb MJ, Burgess BL, Abrams TA. Both protein S and Gas6 stimulate outer segment phagocytosis by cultured rat retinal pigment epithelial cells. Exp Eye Res. 2005;81:581–91. doi: 10.1016/j.exer.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 18.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–11. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 19.Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR. AAV-Mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8:188–95. doi: 10.1016/s1525-0016(03)00144-8. [DOI] [PubMed] [Google Scholar]
- 20.Stargardt KB. Über familiäre, progressive degeneration in der makulagegend des auges. Albrecht Von Graefes Arch Ophthalmol. 1909;71:534–50. [Google Scholar]
- 21.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 22.Azarian SM, Travis GH. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR) FEBS Lett. 1997;409:247–52. doi: 10.1016/s0014-5793(97)00517-6. [DOI] [PubMed] [Google Scholar]
- 23.Illing M, Molday LL, Molday RS. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem. 1997;272:10303–10. doi: 10.1074/jbc.272.15.10303. [DOI] [PubMed] [Google Scholar]
- 24.Burke TR, Rhee DW, Smith RT, Tsang SH, Allikmets R, Chang S, et al. Quantification of peripapillary sparing and macular involvement in Stargardt disease (STGD1) Invest Ophthalmol Vis Sci. 2011;52:8006–15. doi: 10.1167/iovs.11-7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 26.Sankila EM, Tolvanen R, van den Hurk JA, Cremers FP, de la Chapelle A. Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nat Genet. 1992;1:109–13. doi: 10.1038/ng0592-109. [DOI] [PubMed] [Google Scholar]
- 27.Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: Deficiency of rab geranylgeranyl transferase. Science. 1993;259:377–81. doi: 10.1126/science.8380507. [DOI] [PubMed] [Google Scholar]
- 28.MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet. 2014;383:1129–37. doi: 10.1016/S0140-6736(13)62117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Usher C. On the inheritance of retinitis pigmentosa with notes of cases. Roy Lond Ophthalmol Hosp Rep. 1914;19:130–236. [Google Scholar]
- 30.Möller CG, Kimberling WJ, Davenport SL, Priluck I, White V, Biscone-Halterman K, et al. Usher syndrome: An otoneurologic study. Laryngoscope. 1989;99:73–9. doi: 10.1288/00005537-198901000-00014. [DOI] [PubMed] [Google Scholar]
- 31.Spandau UH, Rohrschneider K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch Clin Exp Ophthalmol. 2002;240:495–8. doi: 10.1007/s00417-002-0485-8. [DOI] [PubMed] [Google Scholar]
- 32.Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, et al. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science. 1999;285:245–8. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 33.Tombran-Tink J. PEDF in angiogenic eye diseases. Curr Mol Med. 2010;10:267–78. doi: 10.2174/156652410791065336. [DOI] [PubMed] [Google Scholar]
- 34.Cao W, Tombran-Tink J, Elias R, Sezate S, Mrazek D, McGinnis JF. In vivo protection of photoreceptors from light damage by pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2001;42:1646–52. [PubMed] [Google Scholar]
- 35.Mori K, Duh E, Gehlbach P, Ando A, Takahashi K, Pearlman J, et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001;188:253–63. doi: 10.1002/jcp.1114. [DOI] [PubMed] [Google Scholar]
- 36.Mori K, Gehlbach P, Ando A, McVey D, Wei L, Campochiaro PA. Regression of ocular neovascularization in response to increased expression of pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2002;43:2428–34. [PubMed] [Google Scholar]
- 37.Mori K, Gehlbach P, Yamamoto S, Duh E, Zack DJ, Li Q, et al. AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1994–2000. [PubMed] [Google Scholar]
- 38.Auricchio A, Behling KC, Maguire AM, O’Connor EM, Bennett J, Wilson JM, et al. Inhibition of retinal neovascularization by intraocular viral-mediated delivery of anti-angiogenic agents. Mol Ther. 2002;6:490–4. doi: 10.1006/mthe.2002.0702. [DOI] [PubMed] [Google Scholar]
- 39.Campochiaro PA, Nguyen QD, Shah SM, Klein ML, Holz E, Frank RN, et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: Results of a phase I clinical trial. Hum Gene Ther. 2006;17:167–76. doi: 10.1089/hum.2006.17.167. [DOI] [PubMed] [Google Scholar]
- 40.Kendall RL, Wang G, Thomas KA. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun. 1996;226:324–8. doi: 10.1006/bbrc.1996.1355. [DOI] [PubMed] [Google Scholar]
- 41.Lai YK, Shen WY, Brankov M, Lai CM, Constable IJ, Rakoczy PE. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002;9:804–13. doi: 10.1038/sj.gt.3301695. [DOI] [PubMed] [Google Scholar]
- 42.Lukason M, DuFresne E, Rubin H, Pechan P, Li Q, Kim I, et al. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther. 2011;19:260–5. doi: 10.1038/mt.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai CM, Shen WY, Brankov M, Lai YK, Barnett NL, Lee SY, et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12:659–68. doi: 10.1016/j.ymthe.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 44.Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, et al. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 2002;21:1535–44. doi: 10.1093/emboj/21.7.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohno-Matsui K, Hirose A, Yamamoto S, Saikia J, Okamoto N, Gehlbach P, et al. Inducible expression of vascular endothelial growth factor in adult mice causes severe proliferative retinopathy and retinal detachment. Am J Pathol. 2002;160:711–9. doi: 10.1016/S0002-9440(10)64891-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi K, Saishin Y, Saishin Y, Silva RL, Oshima Y, Oshima S, et al. Intraocular expression of endostatin reduces VEGF-induced retinal vascular permeability, neovascularization, and retinal detachment. FASEB J. 2003;17:896–8. doi: 10.1096/fj.02-0824fje. [DOI] [PubMed] [Google Scholar]
- 47.Vijayasarathy C, Takada Y, Zeng Y, Bush RA, Sieving PA. Organization and molecular interactions of retinoschisin in photoreceptors. Adv Exp Med Biol. 2008;613:291–7. doi: 10.1007/978-0-387-74904-4_34. [DOI] [PubMed] [Google Scholar]
- 48.Min SH, Molday LL, Seeliger MW, Dinculescu A, Timmers AM, Janssen A, et al. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of x-linked juvenile retinoschisis. Mol Ther. 2005;12:644–51. doi: 10.1016/j.ymthe.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Zeng Y, Takada Y, Kjellstrom S, Hiriyanna K, Tanikawa A, Wawrousek E, et al. RS-1 gene delivery to an adult rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2004;45:3279–85. doi: 10.1167/iovs.04-0576. [DOI] [PubMed] [Google Scholar]
- 50.Park TK, Wu Z, Kjellstrom S, Zeng Y, Bush RA, Sieving PA, et al. Intravitreal delivery of AAV8 retinoschisin results in cell type-specific gene expression and retinal rescue in the Rs1-KO mouse. Gene Ther. 2009;16:916–26. doi: 10.1038/gt.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright AF. Gene therapy for the eye. Br J Ophthalmol. 1997;81:620–3. doi: 10.1136/bjo.81.8.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson DA, Ali RR, Banin E, Branham KE, Flannery JG, Gamm DM, et al. Advancing therapeutic strategies for inherited retinal degeneration: Recommendations from the Monaciano Symposium. Invest Ophthalmol Vis Sci. 2015;56:918–31. doi: 10.1167/iovs.14-16049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vandenberghe LH, Auricchio A. Novel adeno-associated viral vectors for retinal gene therapy. Gene Ther. 2012;19:162–8. doi: 10.1038/gt.2011.151. [DOI] [PubMed] [Google Scholar]
- 54.Han Z, Conley SM, Makkia RS, Cooper MJ, Naash MI. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J Clin Invest. 2012;122:3221–6. doi: 10.1172/JCI64833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hangai M, Kaneda Y, Tanihara H, Honda Y. In vivo gene transfer into the retina mediated by a novel liposome system. Invest Ophthalmol Vis Sci. 1996;37:2678–85. [PubMed] [Google Scholar]
- 56.Dalkara D, Sahel JA. Gene therapy for inherited retinal degenerations. C R Biol. 2014;337:185–92. doi: 10.1016/j.crvi.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Bennett J, Wilson J, Sun D, Forbes B, Maguire A. Adenovirus vector-mediated in vivo gene transfer into adult murine retina. Invest Ophthalmol Vis Sci. 1994;35:2535–42. [PubMed] [Google Scholar]
- 58.Li T, Adamian M, Roof DJ, Berson EL, Dryja TP, Roessler BJ, et al. In vivo transfer of a reporter gene to the retina mediated by an adenoviral vector. Invest Ophthalmol Vis Sci. 1994;35:2543–9. [PubMed] [Google Scholar]
- 59.Dalkara D, Kolstad KD, Caporale N, Visel M, Klimczak RR, Schaffer DV, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17:2096–102. doi: 10.1038/mt.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simonelli F, Ziviello C, Testa F, Rossi S, Fazzi E, Bianchi PE, et al. Clinical and molecular genetics of Leber's congenital amaurosis: A multicenter study of Italian patients. Invest Ophthalmol Vis Sci. 2007;48:4284–90. doi: 10.1167/iovs.07-0068. [DOI] [PubMed] [Google Scholar]
- 61.Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, et al. Vision 1 year after gene therapy for Leber's congenital amaurosis. N Engl J Med. 2009;361:725–7. doi: 10.1056/NEJMc0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joeres S, Llacer H, Heussen FM, Weiss C, Kirchhof B, Joussen AM. Optical coherence tomography on autologous translocation of choroid and retinal pigment epithelium in age-related macular degeneration. Eye (Lond) 2008;22:782–9. doi: 10.1038/sj.eye.6702761. [DOI] [PubMed] [Google Scholar]
- 63.Flood MT, Gouras P, Kjeldbye H. Growth characteristics and ultrastructure of human retinal pigment epithelium in vitro . Invest Ophthalmol Vis Sci. 1980;19:1309–20. [PubMed] [Google Scholar]
- 64.Boulton ME, Marshall J, Mellerio J. Human retinal pigment epithelial cells in tissue culture: A means of studying inherited retinal diseases. Birth Defects Orig Artic Ser. 1982;18:101–18. [PubMed] [Google Scholar]
- 65.Alexander P, Thomson HA, Luff AJ, Lotery AJ. Retinal pigment epithelium transplantation: Concepts, challenges, and future prospects. Eye (Lond) 2015;29:992–1002. doi: 10.1038/eye.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li LX, Turner JE. Transplantation of retinal pigment epithelial cells to immature and adult rat hosts: Short- and long-term survival characteristics. Exp Eye Res. 1988;47:771–85. doi: 10.1016/0014-4835(88)90044-9. [DOI] [PubMed] [Google Scholar]
- 67.Li LX, Turner JE. Inherited retinal dystrophy in the RCS rat: Prevention of photoreceptor degeneration by pigment epithelial cell transplantation. Exp Eye Res. 1988;47:911–7. doi: 10.1016/0014-4835(88)90073-5. [DOI] [PubMed] [Google Scholar]
- 68.Bressler NM, Bressler SB, Hawkins BS, Marsh MJ, Sternberg P, Jr, Thomas MA Submacular Surgery Trials Pilot Study Investigators. Submacular surgery trials randomized pilot trial of laser photocoagulation versus surgery for recurrent choroidal neovascularization secondary to age-related macular degeneration: I. Ophthalmic outcomes submacular surgery trials pilot study report number 1. Am J Ophthalmol. 2000;130:387–407. doi: 10.1016/s0002-9394(00)00729-7. [DOI] [PubMed] [Google Scholar]
- 69.da Cruz L, Chen FK, Ahmado A, Greenwood J, Coffey P. RPE transplantation and its role in retinal disease. Prog Retin Eye Res. 2007;26:598–635. doi: 10.1016/j.preteyeres.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 70.Chen FK, Patel PJ, Uppal GS, Tufail A, Coffey PJ, Da Cruz L. Long-term outcomes following full macular translocation surgery in neovascular age-related macular degeneration. Br J Ophthalmol. 2010;94:1337–43. doi: 10.1136/bjo.2009.172593. [DOI] [PubMed] [Google Scholar]
- 71.Cahill MT, Stinnett SS, Banks AD, Freedman SF, Toth CA. Quality of life after macular translocation with 360 degrees peripheral retinectomy for age-related macular degeneration. Ophthalmology. 2005;112:144–51. doi: 10.1016/j.ophtha.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 72.Peyman GA, Blinder KJ, Paris CL, Alturki W, Nelson NC, Jr, Desai U. A technique for retinal pigment epithelium transplantation for age-related macular degeneration secondary to extensive subfoveal scarring. Ophthalmic Surg. 1991;22:102–8. [PubMed] [Google Scholar]
- 73.Stanga PE, Kychenthal A, Fitzke FW, Halfyard AS, Chan R, Bird AC, et al. Retinal pigment epithelium translocation and central visual function in age related macular degeneration: Preliminary results. Int Ophthalmol. 2001;23:297–307. doi: 10.1023/a:1014482025960. [DOI] [PubMed] [Google Scholar]
- 74.Van Meurs JC, Van Den Biesen PR. Autologous retinal pigment epithelium and choroid translocation in patients with exudative age-related macular degeneration: Short-term follow-up. Am J Ophthalmol. 2003;136:688–95. doi: 10.1016/s0002-9394(03)00384-2. [DOI] [PubMed] [Google Scholar]
- 75.van Zeeburg EJ, Maaijwee KJ, Missotten TO, Heimann H, van Meurs JC. A free retinal pigment epithelium-choroid graft in patients with exudative age-related macular degeneration: Results up to 7 years. Am J Ophthalmol. 2012;153:120–7.e2. doi: 10.1016/j.ajo.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 76.Binder S, Stolba U, Krebs I, Kellner L, Jahn C, Feichtinger H, et al. Transplantation of autologous retinal pigment epithelium in eyes with foveal neovascularization resulting from age-related macular degeneration: A pilot study. Am J Ophthalmol. 2002;133:215–25. doi: 10.1016/s0002-9394(01)01373-3. [DOI] [PubMed] [Google Scholar]
- 77.Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–17. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 78.Aisenbrey S, Lafaut BA, Szurman P, Hilgers RD, Esser P, Walter P, et al. Iris pigment epithelial translocation in the treatment of exudative macular degeneration: A 3-year follow-up. Arch Ophthalmol. 2006;124:183–8. doi: 10.1001/archopht.124.2.183. [DOI] [PubMed] [Google Scholar]
- 79.Lund RD, Wang S, Klimanskaya I, Holmes T, Ramos-Kelsey R, Lu B, et al. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells. 2006;8:189–99. doi: 10.1089/clo.2006.8.189. [DOI] [PubMed] [Google Scholar]
- 80.Osakada F, Ikeda H, Mandai M, Wataya T, Watanabe K, Yoshimura N, et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 2008;26:215–24. doi: 10.1038/nbt1384. [DOI] [PubMed] [Google Scholar]
- 81.Idelson M, Alper R, Obolensky A, Ben-Shushan E, Hemo I, Yachimovich-Cohen N, et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell. 2009;5:396–408. doi: 10.1016/j.stem.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 82.Lu B, Malcuit C, Wang S, Girman S, Francis P, Lemieux L, et al. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells. 2009;27:2126–35. doi: 10.1002/stem.149. [DOI] [PubMed] [Google Scholar]
- 83.Wang NK, Tosi J, Kasanuki JM, Chou CL, Kong J, Parmalee N, et al. Transplantation of reprogrammed embryonic stem cells improves visual function in a mouse model for retinitis pigmentosa. Transplantation. 2010;89:911–9. doi: 10.1097/TP.0b013e3181d45a61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;4:73–9. doi: 10.1016/j.stem.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 86.Comyn O, Lee E, MacLaren RE. Induced pluripotent stem cell therapies for retinal disease. Curr Opin Neurol. 2010;23:4–9. doi: 10.1097/WCO.0b013e3283352f96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jin ZB, Okamoto S, Mandai M, Takahashi M. Induced pluripotent stem cells for retinal degenerative diseases: A new perspective on the challenges. J Genet. 2009;88:417–24. doi: 10.1007/s12041-009-0063-5. [DOI] [PubMed] [Google Scholar]
- 88.Parameswaran S, Balasubramanian S, Babai N, Qiu F, Eudy JD, Thoreson WB, et al. Induced pluripotent stem cells generate both retinal ganglion cells and photoreceptors: Therapeutic implications in degenerative changes in glaucoma and age-related macular degeneration. Stem Cells. 2010;28:695–703. doi: 10.1002/stem.320. [DOI] [PubMed] [Google Scholar]
- 89.Buchholz DE, Hikita ST, Rowland TJ, Friedrich AM, Hinman CR, Johnson LV, et al. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells. 2009;27:2427–34. doi: 10.1002/stem.189. [DOI] [PubMed] [Google Scholar]
- 90.Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, et al. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet. 2012;379:713–20. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- 91.Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt's macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet. 2015;385:509–16. doi: 10.1016/S0140-6736(14)61376-3. [DOI] [PubMed] [Google Scholar]
- 92.Zrenner E, Bartz-Schmidt KU, Benav H, Besch D, Bruckmann A, Gabel VP, et al. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proc Biol Sci. 2011;278:1489–97. doi: 10.1098/rspb.2010.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Medeiros NE, Curcio CA. Preservation of ganglion cell layer neurons in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2001;42:795–803. [PubMed] [Google Scholar]
- 94.Osakada F, Ikeda H, Sasai Y, Takahashi M. Stepwise differentiation of pluripotent stem cells into retinal cells. Nat Protoc. 2009;4:811–24. doi: 10.1038/nprot.2009.51. [DOI] [PubMed] [Google Scholar]
- 95.Lamba DA, Karl MO, Ware CB, Reh TA. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103:12769–74. doi: 10.1073/pnas.0601990103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472:51–6. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 97.Aramant RB, Seiler MJ, Ball SL. Successful cotransplantation of intact sheets of fetal retina with retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1999;40:1557–64. [PubMed] [Google Scholar]
- 98.Radtke ND, Aramant RB, Seiler MJ, Petry HM, Pidwell D. Vision change after sheet transplant of fetal retina with retinal pigment epithelium to a patient with retinitis pigmentosa. Arch Ophthalmol. 2004;122:1159–65. doi: 10.1001/archopht.122.8.1159. [DOI] [PubMed] [Google Scholar]
- 99.MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, et al. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444:203–7. doi: 10.1038/nature05161. [DOI] [PubMed] [Google Scholar]
- 100.Pearson RA, Barber AC, Rizzi M, Hippert C, Xue T, West EL, et al. Restoration of vision after transplantation of photoreceptors. Nature. 2012;485:99–103. doi: 10.1038/nature10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Radtke ND, Aramant RB, Petry HM, Green PT, Pidwell DJ, Seiler MJ. Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am J Ophthalmol. 2008;146:172–82. doi: 10.1016/j.ajo.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 102.Gust J, Reh TA. Adult donor rod photoreceptors integrate into the mature mouse retina. Invest Ophthalmol Vis Sci. 2011;52:5266–72. doi: 10.1167/iovs.10-6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wohl SG, Schmeer CW, Isenmann S. Neurogenic potential of stem/progenitor-like cells in the adult mammalian eye. Prog Retin Eye Res. 2012;31:213–42. doi: 10.1016/j.preteyeres.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Singh MS, Charbel Issa P, Butler R, Martin C, Lipinski DM, Sekaran S, et al. Reversal of end-stage retinal degeneration and restoration of visual function by photoreceptor transplantation. Proc Natl Acad Sci U S A. 2013;110:1101–6. doi: 10.1073/pnas.1119416110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu D, Deng X, Spee C, Sonoda S, Hsieh CL, Barron E, et al. Polarized secretion of PEDF from human embryonic stem cell-derived RPE promotes retinal progenitor cell survival. Invest Ophthalmol Vis Sci. 2011;52:1573–85. doi: 10.1167/iovs.10-6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10:771–85. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 107.Ho AC, Humayun MS, Dorn JD, da Cruz L, Dagnelie G, Handa J, et al. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology. 2015;122:1547–54. doi: 10.1016/j.ophtha.2015.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 109.Ma Z, Han L, Wang C, Dou H, Hu Y, Feng X, et al. Autologous transplantation of retinal pigment epithelium-Bruch's membrane complex for hemorrhagic age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2975–81. doi: 10.1167/iovs.08-2573. [DOI] [PubMed] [Google Scholar]
- 110.Castanheira P, Torquetti L, Nehemy MB, Goes AM. Retinal incorporation and differentiation of mesenchymal stem cells intravitreally injected in the injured retina of rats. Arq Bras Oftalmol. 2008;71:644–50. doi: 10.1590/s0004-27492008000500007. [DOI] [PubMed] [Google Scholar]